
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWatch out electrons!: positron binding redefines chemical bonding in Be2
Rafael Porras-Roldan a,
Jorge Charryb,
Felix Moncadac,
Roberto Flores-Morenod,
Márcio T. do N. Varellae and
Andrés Reyes*a
a,
Jorge Charryb,
Felix Moncadac,
Roberto Flores-Morenod,
Márcio T. do N. Varellae and
Andrés Reyes*a
aDepartment of Chemistry, Universidad Nacional de Colombia, Av. Cra 30 45-03, Bogotá, Colombia
bLuxembourg Researchers Hub asbl, 223 Rue de Luxembourg, L-4222, Esch-sur-Alzette, Luxembourg
cDepartment of Physics, AlbaNova University Center, Stockholm University, 106 91, Stockholm, Sweden
dDepartamento de Química, Universidad de Guadalajara, Blvd. Marcelino García Barragán 1421, Col Olímpica, Guadalajara Jal., C.P. 44430, Mexico
eInstituto de Física, Universidade de São Paulo, Rua do Matão 1731, 05508-090 São Paulo, Brazil. E-mail: areyesv@unal.edu.co
First published on 27th October 2025
Abstract
Positrons, the antiparticles of electrons, serve as unique probes for fundamental interactions and are crucial in diverse applications. We present a new mechanism in chemical bonding: the formation of a positron-driven bond that fundamentally alters electronic bonding interactions. Investigating the Be2 dimer with Quantum Monte Carlo (QMC) simulations, we construct the potential energy curve of e+:Be2. Our analysis reveals a significant energetic stabilization of the Be–Be bond upon positron attachment, a result that challenges conventional understanding. We show this stabilization arises from a novel, two-stage mechanism: at longer distances, a positron bond forms via internuclear positron accumulation, similar to that in positron–anion systems. However, as the atoms approach equilibrium, the positron density undergoes a unique redistribution, moving out of the internuclear region to accumulate in the outer molecular vicinity. This distinct positron localization, combined with an otherwise repulsive electronic component, leads to overall system stabilization as the electron density dynamically follows the positron. This work expands our understanding of chemical bonding by strongly suggesting how an antiparticle can profoundly influence molecular stability.
1 Introduction
The positron, the electron's antiparticle, annihilates upon contact with an electron, yielding lifetimes ranging from 10−1 to 102 nanoseconds.1,2 This pair annihilation serves as a key signature of positron presence in the matter world and is exploited across physics,2,3 materials science,4–6 chemistry,1,7 and various technological and medical applications.3,8–10 In recent years, positronium imaging11,12 has emerged as a new medical technique, complementary to positron emission tomography (PET), based on measuring differences in positronium lifetimes to gather information about the surrounding environment of positronium in living tissues. Therefore, it is crucial to simulate positron–matter interactions to refine the interpretation of emerging positron-based applications. Before annihilation, positrons can form bound states with matter systems, either free electrons, atoms, or molecules. Indirect detection via the vibrational Feshbach resonance (VFR) technique has identified positron binding energies, hereafter called positron affinities (PAs), up to 275 meV in approximately 100 molecules, including nonpolar species.13–15 The technique exploits the fact that higher electron–positron annihilation rates are observed when a vibrationally excited positron–molecule complex is formed, i.e., when the kinetic energy of the positron resonates with the vibrational frequencies of the complex, based either on dipolar16 or non-dipolar17 positron-molecule coupling. Consequently, PAs can be inferred from redshifts of the annihilation resonances with respect to the infrared vibrational spectrum of the isolated molecule.Although the VFR technique cannot be used to measure atomic PAs, due to the absence of vibrational degrees of freedom, theoretical predictions suggest a broader range of positron-binding species. In general, accurate predictions of positron affinities remain challenging, requiring high-level computational methods to reliably determine the magnitude of the PA.18–24 In some cases, such as nonpolar molecules, even predicting the existence of binding can itself be difficult.22 Theoretical studies have also extended the findings to polar molecules not considered in experiments, revealing significantly higher positron affinities, up to 1 eV, in highly dipolar systems such as alkali hydrides.25 In all those cases, the complexes are formed by positron binding to stable atomic or molecular targets accompanied by a mild relaxation of the underlying electronic structure.
Recent investigations have expanded from positron binding to positron bonding. In the latter case, otherwise unstable purely electronic systems are stabilized by the formation of positronic bonds. Those studies have pointed out that positronic bonding interactions are similar to the electronic counterparts, enabling the binding of repelling anions, occasionally forming thermodynamically stable molecules. The formation of one-positron (1e+) bonding is predicted between pairs of anions such as F−, Cl−, Br−,26,27 CN−, and CNO−.27 Similarly, predictions exist for 1e+ bonding between two H− anions;23,27–30 however, exact DMC calculations by Bressanini31 have demonstrated that the e+[H22−] system is energetically favorable compared to its dissociation into H− + PsH, with a binding energy of 23.5 (1) mhartree. Below 3.2 bohrs, the system becomes unstable and decays into H2 + Ps−. Additionally, two-positron (2e+) bonds have been reported in dihydride,32,33 trihydride,34 and dihalide complexes.35
The discovery of positron-bonded systems has attracted considerable interest among physicists and chemists, who are actively investigating the nature of these bonds,32,33 developing theoretical approaches to predict new systems, and designing experimental strategies for their production and detection. A major breakthrough occurred in 2023 with the potential experimental formation of the F−e+F− complex as part of the mechanism for molecular ion desorption from LiF crystals irradiated with positrons,36 in consistency with the prediction made by some of us in 2020.26
However, the experimental realization and detection of e+-bonded systems in the gas phase remain challenging. The manipulation of anions and positrons to form bonded complexes presents significant technical difficulties. Additionally, detecting these complexes via the VFR technique is not possible because it requires energy transfer to vibrational modes of the purely electronic molecule, which are absent in the repulsive dianionic systems.
Taking currently available experimental techniques into account, such as VFR, the realization of positron-bonded systems would be more feasible for positively charged complexes, i.e., neutral electronic fragments stabilized by a positron bond. While these bonds have so far been primarily predicted for negatively charged electronic fragments, neutral molecules emerge as viable systems for detection of positron bonding, at least in principle. A key advantage lies in considering non-reactive, neutral systems that typically do not tend to form conventional chemical bonds but are rather stabilized by intermolecular interactions. Such systems would favor the observation of positron bonds, which generally display lower energies than regular electronic bonds, between 10 to 20 kcal mol−1.26,28 Furthermore, studying positron bonding in neutral systems could circumvent difficulties encountered with their anionic counterparts. Anionic positron-bonded species require bonding species with positive positronium binding energies (PsBE > 0) for thermodynamic stability, as this condition prevents dissociation into free positronium. This stringent requirement significantly narrows the range of viable candidates for anionic positron-bonded systems.
Although neutral apolar molecules emerge as the natural starting point in the quest to identify neutral candidates for positron bonding, the positronic complexes formed with covalently bonded molecules usually show considerably delocalized positronic densities around the molecular core, for example, with aliphatic or aromatic complexes.15
This poses a significant challenge because positron bonding relies on positron localization in the internuclear regions.26,28 Insufficient localization may lead to low bond energies, comparable in magnitude with dispersion-dominated interactions, which are typically in the range of 2–10 kcal mol−1,37,38 thus making it difficult to distinguish positron bonding from electronic intermolecular forces.
In contrast, neutral polar molecules, characterized by positive–negative (p–n) poles, exhibit higher PAs and positronic densities located around the negative pole, for example, in alkali hydrides or alkaline earth oxides.39 In that case, the interaction of a positron with two polar molecules could result in the formation of an energetically stable p–n⋯e+⋯n–p bond; the stabilization from the positron interaction would need to counterbalance the sum of the dipole–dipole interactions (n–p⋯n–p) and the PA of the dipole-bonded complex, e+:n–p⋯n–p. As a consequence, positronic bond formation in such a strongly dipole–dipole bonded compound would require positron-induced structural rearrangements, similar to those found in the electron-induced binding in MX–MX systems.40 The rearrangements could involve high-energy barriers, as well as time scales comparable to the annihilation lifetimes, thus hindering positron bond formation and detection.
Alkaline-earth (AE) atoms are recognized for their ability to form stable dimers.41–44 Typically, AE dimers exhibit van der Waals (vdW) interactions at large internuclear separations (more complex bonding mechanisms emerge at shorter separations), with a binding energy of less than 5 kcal mol−1. AE atoms also have positive PAs,45 thus making AE dimers attractive candidates for positron bonding in neutral systems. However, these dimers pose challenges to theory and computation. Accurate calculations of their atomic PAs are inherently difficult due to the weak electron–positron correlation effects at long-range separation from the nuclei,23,45–48 and even obtaining potential energy curves (PECs) for the diatomic complexes demands advanced electron–electron correlated methods to account for both static and dynamical correlation.42,49–51
Recent work by Upadhyay et al.45 highlights the challenges associated with calculating positron affinities (PA) for some neutral apolar systems, including small AE complexes, where a Hartree–Fock (HF) reference is well-known to provide unbound states. They primarily examined the suitability of various configuration-interaction (CI) trial wavefunctions for Diffusion Monte Carlo (DMC), finding that DMC with a single determinant (SD) and a Jastrow factor can properly predict the PAs in Ben, Mgn systems compared to multi-determinant (MD) ansatzes. It is important to note, however, that their study did not extend to an investigation of the physicochemical properties of these systems themselves nor the positron binding mechanisms in those systems.
To delve into a possible positron bond in the Be2 dimer we first derive an expression to estimate the change in the bond energy (BE) of the Be2 dimer (dissociating into Be + Be) after binding a positron to form the positronic complex e+:Be2 (dissociating into e+:Be + Be). We define the PA for the beryllium atom as
| PA[Be] = E[Be] − E[e+:Be] | (1) |
| BE[Be2(R)] = 2E[Be] − E[Be2(R)] | (2) |
| BE[e+:Be2(R)] = E[Be] + E[e+:Be] − E[e+:Be2(R)] | (3) |
| BE[e+:Be2(R)] − BE[Be2(R)] = PA[Be2(R)] − PA[Be] | (4) |
Such an unexpected result raises fundamental inquiries about its origin as well as the effects of positron binding on other properties of the diatomic system, including equilibrium distance, force constant, vibrational states, electronic structure, electron density, and positron density distribution. In this work, we investigate some of these unexplored aspects to describe the bonding mechanism in the positronic Be dimer, e+:Be2, using QMC methods.
We initially determine potential energy curves (PECs) for Be2 and e+:Be2 to assess the impact of positron binding on equilibrium distances, binding energies, force constants, vibrational states, and changes in PA along the PEC. Subsequently, we perform a simple energy decomposition of the QMC total energies of Be2 and e+:Be2 to elucidate the nature of positron affinity along the PEC. This analysis helps us to distinguish electronic bonding interactions from positron binding ones. Finally, we evaluate the impact of positron binding on the electron density along the PEC and investigate the positron density itself along the PEC. Together with energy component analysis, this helps us discern whether stabilization is due to the formation of positron bonds or other binding mechanisms.
2 Methods
This study utilizes diverse theoretical approaches previously reported by some of the authors in related positronic studies.23 Here, we briefly review the key elements of such methods.2.1 Hamiltonian
The non-relativistic Hamiltonian of a system comprised of N−e electrons, one positron, and Nc classical nuclei can be written under the Born-Oppenheimer approximation as:
 | (5) |
2.2 Quantum Monte Carlo
The time-independent Schrödinger equation for the above molecular electron-positron Hamiltonian can be numerically solved via stochastic techniques52–54 through the integration of a given trial wavefunction. Among the most common techniques, variational Monte Carlo (VMC) and diffusion Monte Carlo (DMC) methods for electron-positron molecular systems were previously extended and implemented in the QMeCha code.23,55In VMC, the stochastic integration of the energy functional is carried out with the Metropolis-Hastings algorithm56,57 over a trial variational wavefunction.
For this work, we employed the molecular orbital ansatz as the trial wavefunction,23 which consists of a Slater determinant for the electronic part, a Jastrow factor to describe explicit correlation between all particles, and a positronic orbital as
 | (6) |
 is the Jastrow factor. A more detailed description of the Jastrow factor is found in ref. 23. Here we summarize five terms describing, respectively, the electron-nucleus
is the Jastrow factor. A more detailed description of the Jastrow factor is found in ref. 23. Here we summarize five terms describing, respectively, the electron-nucleus  , positron-nucleus
, positron-nucleus 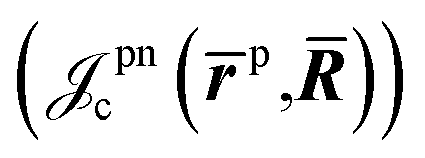 , electron–electron
, electron–electron  , and electron-positron
, and electron-positron  cusps, and a term that describes the dynamical correlation between the fermionic particles in the field of the nuclei
cusps, and a term that describes the dynamical correlation between the fermionic particles in the field of the nuclei  , which is an extension of the one defined in ref. 58. For particle pairs with the same charge, the Jastrow is built with slowly decaying functions, while for particles with opposite charges, a faster decaying cusp function is employed. Finally, the dynamical Jastrow factor23,58 is written as a linear combination of products of non-normalized atomic orbitals.
, which is an extension of the one defined in ref. 58. For particle pairs with the same charge, the Jastrow is built with slowly decaying functions, while for particles with opposite charges, a faster decaying cusp function is employed. Finally, the dynamical Jastrow factor23,58 is written as a linear combination of products of non-normalized atomic orbitals.
 | (7) |
All the parameters of the wavefunctions are variationally optimized with the stochastic reconfiguration method.59,60 The optimized VMC wavefunction is then used as the DMC52 trial wavefunction to better describe the dynamical correlation among the particles. In the long-time limit, DMC can converge towards the exact ground state through a time evolution of the wavefunction in imaginary time.52 Nevertheless, it suffers from the sign problem, which appears for fermionic systems, making it necessary to constrain the nodal surface of the wavefunction, commonly known as the fixed-node approximation.61 Yet, it can be shown that FN-DMC, at least in the formalism employed here, is variational with respect to the quality of nodal surfaces, which here are defined by the optimized trial wavefunctions.
3 Computational details
VMC and DMC calculations were performed using the QMeCha quantum Monte Carlo package,23,55 which is available on GitHub under an academic license.The electronic and positronic wavefunctions for all systems were constructed using a (15s5p2d) primitive Gaussian-type function (GTFs) basis, contracted to [3s3p2d]. The initial electronic basis set parameters were adopted from the aug-cc-pVTZ basis set.62 Then, wavefunction optimization was carried out for all variational parameters on each system independently, using the stochastic reconfiguration method.58 This process involved variationally optimizing 414 non-zero parameters for Be2 and 562 non-zero parameters for e+:Be2. For VMC stochastic integration, 128 walkers were employed over 100 blocks, with each block consisting of 1000 steps.
Fixed-node DMC calculations were performed with an imaginary time step of 0.001 a.u., using 6400 walkers with 5000 blocks of length 100. DMC density plots were generated by counting the number of particles within each weighted configuration on a three-dimensional grid. For DMC, we performed an energy decomposition analysis, which required storing the weighted averages for each local energy component, according to the estimated MC expectation values of the operators of the molecular Hamiltonian (eqn (5)). We note that the distribution of the DMC energetic components exhibits large fluctuations, as indicated by their estimated error bars, due to the divergences on the kinetic and Coulomb potential operators, which compensate each other when computing the total energies. Therefore, only general trends should be interpreted along the PECs.
4 Results and discussion
4.1 Be and e+:Be energy data
We start by analyzing positron binding to a beryllium atom from the energies of Be and e+:Be obtained with the VMC and DMC methods.The calculated energies and PAs, along with the data from previous computational studies, are presented in Table 1. Our calculated VMC and DMC affinities for atomic Be are 56 ± 6 meV, 104 ± 4 meV, respectively. Although our DMC value is in agreement with the reported DMC value of 96 ± 5 meV, obtained from single-determinant (SD) reference, it is slightly larger than the best multi-determinant (MD) DMC value of 91 ± 4 meV also reported by – Deible et al.,50 indicating a small lack of static correlation. Our SD-DMC PA result is also statistically indistinguishable from the SD-DMC result of Mella et al. (100 ± 5)63 and also from the DMC SD/HF//SD/NO rSDCIb result (104 ± 4) reported by Upadhyay et al.45 Despite the accurate SD/DMC results, there is an overestimation of around 31(3) meV (22% relative error) on the Be–Be binding energy due to the lack of multireference character description in Be2 with respect to Be. Thus, this energy difference propagates to the PA of Be2, which is overestimated by 33 (10) meV (7% relative error) compared to the more accurate MD/DMC results of Upadhyay et al.45
| Method | E[Be] | E[e+:Be] | PA |
|---|---|---|---|
| SVM64 | −14.667106 | −14.669042 | 46 |
| FSVM65 | — | — | 86 |
| RXCHF66 | — | −14.5746 | 82 |
| ECG67 | −14.667338 | −14.670519 | 87 |
| CI68 | — | — | 84 |
| MBT69 | — | — | 290 |
| SD/DMC45,50 | −14.65730 (4) | −14.66100 (3) | 96 (5) |
| MD/DMC45,50 | −14.66725 (1) | −14.67059 (4) | 91 (4) |
| CCSD(T)48 | — | — | 214 |
| Present SD/VMC | −14.64974 (12) | −14.65183 (13) | 57 (6) |
| Present SD/DMC | −14.65727 (6) | −14.66108 (8) | 104 (3) |
4.2 Potential energy curves for Be2 and e+:Be2
The beryllium dimer (Be2), despite having only 8 electrons, represents a significant challenge for accurate electronic structure calculations. The complexity primarily arises from two aspects: the near-degeneracy of the 2s and 2p subshells in the Be monomers, which makes the system multi-reference in nature, and the substantial dynamical correlation effects that demand large basis sets in perturbative or excitation expansions. The binding interaction in Be2 has been extensively analyzed. The potential energy curve (PEC) deviates significantly from conventional potential models such as Morse and Lennard-Jones, typical of covalent or van der Waals interactions.70,71 This intricate potential shape arises from the evolution of configurational mixing as the atoms approach, resulting in complex orbital hybridization as the weak chemical bond is formed.51 Theoretical analyses indicate a delicate balance between distinct energetic contributions to the stabilization at the equilibrium distance: those typically associated with covalent-like electronic rearrangements (chemical interactions) and those deriving from dispersion forces and electrostatic polarization effects (physical interactions).72–74 The quality of computed PECs is generally assessed in terms of the reported bond energies (BE), equilibrium distances (Re), harmonic force constants (ke), and vibrational state energies.Fig. 1 displays the computed PECs for Be2 and e+:Be2. These DMC energies were fitted to a sum of fifth-order polynomial and inverse power terms.75 Our results and other theoretical results for e+:Be2 are collected in Table 2, which also includes the experimental data for Be2. VMC results are reported for comparison purposes and to qualitatively validate the trial wavefunction ansatz. The VMC properties, as well as the VMC PECs in SI, show that VMC correctly predicts electronic and positronic bound states for all the studied systems.
 | ||
| Fig. 1 Potential energy curve for Be2 (green) and e+:Be2 (blue) computed at the DMC level. Error bars represent one standard error on the mean. | ||
| Property | Method | Be2 | e+:Be2 |
|---|---|---|---|
| a Equilibrium distances (Re) in Å, total energies in a.u., harmonic force constants (ke) in a.u. ×103, binding energies (BE and D0) and positron affinities (PA) in meV, harmonic frequencies (ωe), fundamental vibrational transition (ν0→1) and anharmonic constant (ωeχe) in cm−1. | |||
| R e | Present SD/VMC | 2.531 (9) | 2.415 (7) |
| Present SD/DMC | 2.447 (7) | 2.404 (6) | |
| SD/LRDMC49 | 2.46 (3) | — | |
| Exp70 | 2.453603 | — | |
| E(Re) | Present SD/VMC | −29.30134(2) | −29.3172(1) |
| Present SD/DMC | −29.3197(2) | −29.3376(1) | |
| PA | Present SD/VMC | 431 (9) | — |
| Present SD/DMC | 478 (4) | — | |
| SD(HF)/DMC45 | 456 (11) | — | |
| MD/DMC45 | 445 (9) | — | |
| k e | SD/VMC | 10.9 (9) | 17 (2) |
| SD/DMC | 12 (1) | 18 (1) | |
| Exp70 | 11.20 | — | |
| ω e | Present SD/VMC | 221 (10) | 317 (14) |
| Present SD/DMC | 260 (9) | 329 (9) | |
| Exp70 | 256.8 | — | |
| BE | Present SD/VMC | 52 (5) | 426 (8) |
| Present SD/DMC | 140 (3) | 524 (3) | |
| SD/LRDMC49 | 143 (6) | — | |
| SD(HF)/DMC45,50 | 87 (3) | 456 (11) | |
| SD(LDA)/DMC50 | 140 (9) | — | |
| MD/DMC45,50 | 109 (1) | 467 (10) | |
| Exp70 | 115.3 | — | |
| ν 0→1 | Present SD/VMC | 181(11) | 289(14) |
| Present SD/DMC | 222(9) | 321(9) | |
| Exp70 | 222.6 | — | |
| D 0 | Present SD/VMC | 39(5) | 407(8) |
| Present SD/DMC | 125(4) | 503(3) | |
| Exp70 | 100.1 | ||
| ω e χ e | Present SD/VMC | 20(2) | 14(1) |
| Present SD/DMC | 19(1) | 4.0(2) | |
Our analysis of the DMC fitted potential energy curve for neutral Be2 reveals a bond length of 2.448 Å, which closely agrees with the experimental value of 2.454 Å. However, the calculated binding energy is 140(3) meV, deviating from the experimental value of 115.3 meV by 25(3) meV due to the use of a single-determinant reference. On the other hand, deviations in the BE in other DMC-SD methods50 are around 2.7 meV.
In turn, analysis of the DMC fitted potential energy curve for e+:Be2 shows an equilibrium distance of 2.404 Å closely matching that of the neutral Be2 (2.448 Å). The computed bonding energy for the Be + e+:Be dissociation channel is 520(4) meV, consistent with Upadhyay's et al. MD/CISD//MD/rSDTCI DMC prediction of 466.6 meV using PA data from ref. 45 and eqn (4).
We note that the chemical bonding in the e+:Be2 system differs fundamentally from other single-positron-bond systems, such as e+H22− (ref. 23,30,31) and e+Li22−.76 In these examples, the lowest dissociation channel is the formation of Positronium (Ps), a process driven by the inherent instability of the corresponding dianion dimers. In striking contrast, the e+:Be2 does not favor Ps formation because the energy of the Be2 dimer is lower than the Ps + Be2+ dissociation channel at all internuclear distances, as confirmed by the most accurate electronic PECs found in the literature77,78 (See SI).
It is evident that positron binding to Be2 increases its BE by a factor of three, compared to the neutral dimer.
Analysis of the PECs in Fig. 1 indicates that e+:Be2 maintains significant BEs even at larger separations, as evidenced by the deeper and wider minimum well, which decays slower than that of Be2. For instance, e+:Be2 reaches the BE of Be2 around 6.0 Å, and a BE of 50 meV around 10.0 Å, whereas neutral Be2 reaches a comparable value at 3.0 Å.
To further analyze the physical properties of Be2 and e+:Be2 we estimated the vibrational properties, namely the force constants (ke) and harmonic frequencies (ωe) employing the fitted PECs in Fig. 1.
We also solved numerically the 1D Schrödinger equation for the vibrational levels and fundamental frequency (ν![[0 with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/char_0030_20d1.gif) 1) by solving the eigenvalue problem of a Toeplitz tridiagonal matrix.79 All results were calculated employing a generalized Morse potential from ref. 70 or the fitted form from DMC results.
1) by solving the eigenvalue problem of a Toeplitz tridiagonal matrix.79 All results were calculated employing a generalized Morse potential from ref. 70 or the fitted form from DMC results.
There is a reduction from the harmonic frequency and the first vibrational transition of 34.2 and 38.5 cm−1 for reference values70 and our fitted potential. Deviations of just 0.34 × 10−3 a.u. and 3.6 cm−1 for ke and ωe, respectively, were estimated. Furthermore, the difference in ν1 of 0.9 cm−1 and 24.6 cm−1 for D0 provides agreement with respect to experimental data and supports the use of our DMC-SD methodology for physical properties of Be2. Additionally, there is a marked deviation between harmonic frequency and the fundamental vibrational transition due to the anharmonic character of the dimer interaction.
The effect of positron binding to Be2 is reflected in a 6.89 × 10−3 a.u. increase in harmonic force constant, also seen qualitatively in the PEC widths of each system (Fig. 1). On the other hand, both ωe and ν0→1 are 68.2 and 99.4 cm−1 larger in the e+:Be2 complex. The difference between ωe and ν0→1 is also smaller in the positronic complex (7.7 cm−1) than in the purely electronic dimer (38.5 cm−1).
The obtained anharmonic constant, ωeχe, is lower in e+Be2 than Be2, which generally suggests the vibrational well is deeper and the bond is stronger and less likely to break upon reaching higher vibrational states due to the slower approach to the dissociation limit.
4.3 Vertical positron affinities
We define the geometry-dependent vertical positron affinity (VPA(R)) of Be2 as:| VPA(R) = E[Be2(R)] − E[e+:Be2(R)] | (8) |
 | ||
| Fig. 2 Be2 vertical positron affinity at the DMC level (in green line). The dashed line indicate the Be atom positron affinity. Error bars represent one standard error on the mean. | ||
Rearranging eqn (4) (from the previous section) results in:
| BE[e+:Be2(R)] = BE[Be2(R)] + (VPA(R) − PA[Be]) | (9) |
4.4 Energy decomposition analysis
To investigate the source of the marked rise in the BE[Be2] due to positron binding, we performed an energy decomposition analysis (EDA) on the DMC total energies of neutral dimer Be2 (E[Be2], also referred to as Ee−[Be2]) and positron-bound complex e+:Be2 (E[e+:Be2]). We computed the virial theorem to support the EDA (In SI), observing it is roughly satisfied along the PEC, in particular near the equilibrium distance and dissociation limit, considering it's a weakly bound system compared to regular electronic covalent bonds, on top of the estimated DMC error bars.The total energy of the positronic complex, E[e+:Be2], is decomposed into two components:
| E[e+:Be2(R)] = Ee−[e+:Be2(R)] + Ee+[e+:Be2(R)] | (10) |
The first energy component, Ee−[e+:Be2], represents the electronic energy of e+:Be2. This term encompasses the nuclear repulsion (Vnn), nuclear-electron attraction (Vne−), electron–electron repulsion (Ve−e−), and electron kinetic energy (Ke−).
The second energy component, Ee+[e+:Be2], is the positronic energy of e+:Be2. This term includes the positron kinetic energy (Ke+), positron–electron attraction (Ve−e+), and positron-nuclear repulsion (Vne+).
Fig. 3 compares the electronic energy (Ee−[e+:Be2]) and positronic energy (Ee+[e+:Be2]) across the 2–16 Å interval. As shown, the PEC for Ee−[e+:Be2] is entirely repulsive and lacks an energy minimum, unlike E[Be2] in Fig. 1. This indicates an absence of electronic bonding in the complex, suggesting that positron binding to Be2 eliminates any trace of the electronic bonding interactions in the e+:Be2 complex.
 | ||
| Fig. 3 Electronic energy (Ee−[e+:Be2]), and positronic energy (Ee+[e+:Be2]) contributions. Error bars represent one standard error on the mean. | ||
Consistent with the EDA eqn (10), the observed bonding nature in e+:Be2, as evidenced by the energy minimum in its PEC E[e+:Be2] in Fig. 1, arises solely from the contribution of the positronic energy Ee−[e+:Be2].
The analysis of eqn (4) suggested that the bonding interaction present in Be2 was further stabilized when the VPA(R) exceeded that of atomic Be, which was the case across the distance range analyzed in Fig. 2. In other words, it postulated that positron binding to Be2 further reinforced its electronic bond.
However, our EDA analysis contradicts this assumption. Instead, it confirms that positron binding stabilizes the Be2 bond by first eliminating the electronic bonding interaction. This process arises from a significant deformation of the electronic structure of Be2 leading to a substantial increase in Ee−[e+:Be2] compared to Ee−[Be2], thereby rendering the electronic interaction repulsive. Consequently, to minimize the total energy of the positron complex, E[e+:Be2], and to produce a bonding PEC, this energetic rise in the electronic component must be compensated by a highly stabilizing positronic energy component, Ee+[e+:Be2]. The electronic energy destabilization in e+:Be2 is highly unusual for an apolar molecule binding a positron. Beyond Ps formation, it is commonly accepted that positron binding typically induces only minor perturbations to the electronic structure of such systems.18–24
The magnitude of the stabilizing effect provided by the positronic energy component, Ee+[e+:Be2], can be quantified by rearranging the terms of the VPA and the energy decomposition definition in eqn (10). Recalling that VPA is defined as VPA(R) = E[Be2(R)] − E[e+:Be2(R)], and that the total energy of the complex is decomposed into its electronic and positronic components as E[e+:Be2(R)] = Ee−[e+:Be2(R)] + Ee+[e+:Be2(R)], we can write:
| VPA(R) = E[Be2(R)] − Ee−[e+:Be2(R)] − Ee+[e+:Be2(R)] | (11) |
| Edf(R) = Ee−[e+:Be2(R)] − E[Be2(R)] | (12) |
| VPA(R) = −Edf(R) − Ee+[e+:Be2(R)] | (13) |
Solving for the positronic energy component, we obtain
| −Ee+[e+:Be2(R)] = VPA(R) + Edf(R) | (14) |
This equation implies that the magnitude of the stabilizing positronic energy component |Ee+[e+:Be2]| (where Ee+[e+:Be2] is a negative value for a bound state) must be greater than VPA because it must overcome the initial electronic deformation energy Edf to provide a net stabilization.
We now try to gain a more in-depth understanding of how this electron deformation causes the destabilization of the electronic energy and how this destabilization is compensated by the positron energy. Let us focus first on the origin of the destabilization of the electronic energy. Fig. 4 reports the deformation energy differences, Edf, along with their kinetic and potential energy contributions. This figure evidences that the electronic energy destabilization of Be2 upon positron binding comes mostly from the destabilization of the potential energy component. Positron binding to Be2 results in reduced Ke− and Ve−e−, and a less negative Ve−n.Across all points of the curve, changes in Ve−e− term are negative, indicating a stabilization of the positron system, while positive Ve−n differences destabilize the system. The sum of the latter terms results in a positive value, indicating an overall destabilization of electronic energy upon positron binding. These energy variations suggest that positron binding promotes electron density delocalization of Be2.
 | ||
| Fig. 4 Deformation energy contributions, differences between Ee−[e+:Be2] − Ee−[Be2] electronic energy components. Error bars represent one standard error on the mean. | ||
Next, we analyze the origin of the energy compensation caused by the positron energy. Fig. 5 illustrates the positron energy with its energy components, positron kinetic energy Ke+[e+:Be2], and the summed positron nuclear Vne+[e+:Be2] and positron electron Ve−e+[e+:Be2] interactions.
 | ||
| Fig. 5 Decomposition of positron energy into kinetic and potential components. Error bars represent one standard error on the mean. | ||
As expected, the high negative values of positron energy arise from the electron-positron attraction, which is intensified by electronic deformation, eqn (14). Here, it is remarkable how the potential energy terms exhibit a larger and faster stabilizing decay than the flatter kinetic term with respect to the dissociation region. Moreover, the positronic kinetic term does not display any lowering or minimum near the bonding region, contrary to what is observed in regular electronic bonds.80,81
To further investigate the energy stabilization mechanisms of the Be2 bond in e+:Be2, we examine in detail the PECs of Ee−[e+:Be2] and Ee+[e+:Be2]. An ‘anomalous’ variation emerges in both curves between 3.0 and 4.5 Å, and on their energetic components suggesting a shift in the nature of the interaction. To gain deeper insight into the fundamental nature of the interaction, we also evaluate the corresponding changes in electron and positron densities.
4.5 Positron and electron density analysis
To shed light on the origin of the bonding interactions in e+:Be2 we report in Fig. 6 1D and 2D positron density, ρe+, plots at different internuclear distances.Analyzing these density plots in parallel with the PEC of e+:Be2 in Fig. 1, we observe distinct bonding regimes. From large distances and up to approximately 3.0–4.5 Å, the reduction in total energy as the distance shortens can be attributed to the gradual accumulation of positron density in the internuclear region. This phenomenon is similar to what has been observed in the bonding of anions with positrons,28,32,33 where it is understood as the formation of a positron bond, characterized by internuclear positron density accumulation. This similarity strongly suggests that the stabilization in this internuclear distance regime arises from the formation of a positron bond. | ||
| Fig. 6 Comparison of the positronic density in e+Be2 computed with DMC at representative internuclear separation. The bottom panel shows one-dimensional cuts of the density along the internuclear axis (X) with Z = Y = 0. Black dots indicate the position of the Be nuclei. | ||
Crucially, when combining this analysis with the PEC of Ee−[e+:Be2] shown in Fig. 3, which exhibits repulsive behavior across this distance range, we can conclude that the binding of a positron effectively vanishes the intrinsic electronic bonding interaction of Be2, completely replacing it with a positron bonding interaction. To our knowledge, this represents a unique manifestation where positron binding fundamentally alters the nature of the molecular interaction, replacing conventional electronic bonding with a positron-driven bonding mechanism.
As the internuclear distance continues to shorten towards the equilibrium distance, we observe a significant shift in the positron density distribution. The positron density is gradually pushed out of the internuclear region and instead accumulates in the outer regions of the complex, primarily along the internuclear axis. This novel mode of positron localization appears to be responsible for the further stabilization of the bonding interaction at these shorter distances. Here again, the repulsive behavior observed in Ee−[e+:Be2] PEC indicates that the electronic bonding is also absent in this region. To our knowledge, this is the first manifestation of a bonding interaction in a neutral system where the electronic bonding is supplanted by a unique stabilization mechanism arising from positron accumulation in the outer, rather than internuclear, region of the molecule.
Complementary to this, an analysis of the change in the electron density (ρe−) of Be2 upon positron binding reveals a crucial counteracting effect. As observed in Fig. 7, there is a gradual but minor accumulation of electron density in the internuclear region as the distance shortens, similarly to how the electronic density accumulates when two Be atoms interact to form Be2. This extra accumulation, however, leads to an increase in the electronic energy, causing the system to become electronically repulsive. This electronic repulsion is a direct consequence of the positron's influence, deforming the electron cloud in a way that is energetically unfavorable for electronic bonding. Moreover, around the equilibrium distance at 2.4 Å, for e+:Be2 there is an additional redistribution of the electronic density accumulation in the outer regions of the complex. Thus, by combining the analysis in Fig. 6 and 7, it is evident that the electronic density roughly follows the positronic one, leading to an increase in the electron-positron attractive potential, and thereby contributing to the stabilization of the entire system.
 | ||
| Fig. 7 Comparison of the DMC electronic density changes. Top: electronic density changes upon the addition of positron to Be2, the red and green surfaces indicate regions of electronic density accumulation and depletion in e+Be2 compared to Be2, respectively. Bottom: electronic density changes upon bound state formation of Be2 from 2 Be atoms, the red and green surfaces indicate regions of electronic density accumulation and depletion in Be2 compared to 2Be, respectively. Black dots indicate the position of the Be atoms. | ||
5 Conclusions
Our study uncovers a fascinating and unprecedented interplay between positron binding and molecular interactions in the Be2 dimer, revealing two distinct positron-driven bonding mechanisms that entirely supplant conventional electronic interactions.Our energy decomposition analysis (EDA) reveals that the electronic energy component (Ee−[e+:Be2]) becomes entirely repulsive across the entire internuclear range. This unequivocally indicates that positron binding effectively eliminates the intrinsic electronic bonding present in neutral Be2. Consequently, the observed bonding in the e+:Be2 complex arises solely from the highly stabilizing contribution of the positronic energy component (Ee+[e+:Be2]). This electronic energy destabilization is highly atypical for apolar molecules interacting with positrons, as minor electronic perturbations are usually expected.18–24 We propose that the positron induces a significant deformation and delocalization of the electron cloud, rendering the electronic interaction unfavorable for bonding. Crucially, this electronic deformation simultaneously enhances the electron-positron attractive potential, thereby compensating for the electronic repulsion and providing the net stabilization.
In the context of electronic bond elimination, we identify two distinct positron-driven stabilization regimes. At internuclear distances beyond approximately 3.5–4.0 Å, the binding of the positron vanishes the weak electronic interaction and significantly increases the vertical positron affinity (VPA) of Be2. This is accompanied by an accumulation of positron density in the internuclear region, consistent with established positron bonding phenomena observed in systems like positron-anion complexes.28 This internuclear positron accumulation is thus identified as the primary stabilizing factor for the Be2 dimer in this long-range regime.
For distances shorter than 3.5 Å, our positron density analysis rules out conventional internuclear positron bond formation. Instead, we observe a significant and novel shift in the positron density distribution: the positron density is largely pushed out of the internuclear region and accumulates in the outer regions of the complex, primarily along the internuclear axis. Despite this, the VPA continues to increase, leading to a rise in binding energy (BE) until reaching a minimum at the equilibrium distance of 2.4 Å. This highlights a unique and previously unobserved short-range stabilization mechanism for neutral systems.
To the best of our knowledge, our findings provide the first confirmation of the stabilization of weakly bonded complexes through these two unique positron bonding mechanisms. This study represents a significant advance, challenging the conventional understanding of how positrons influence molecular structure and stability by demonstrating the complete replacement of electronic bonding with positron-driven interactions. The neutral nature of Be2 suggests high feasibility for experimental confirmation. Given that Be2 can form dimers at ultracold temperatures and the calculated binding energy of the e+-bonded Be2 system aligns with typical vibrational excitation energies, it stands as a promising candidate for experimental detection. Such a detection would mark the pioneering observation of a positron bond in neutral atomic systems, a phenomenon previously overlooked, and would represent a significant scientific advance in the long history of vdW-bonded system studies.
The complete replacement of electronic bonding with positron-driven stabilization, particularly the novel “outer-region” positron accumulation, opens unprecedented avenues for exploring exotic chemical bonds. For instance, methods as multicomponent quantum theory of atoms in molecules (MC-QTAIM)29,82 or interference energy analysis83 might elucidate deeper characteristics of these new exotic bonds. Future research will explore positron binding in other dimers. Investigating the dynamical aspects of positron-induced electron cloud deformation could also provide crucial insights into the transient processes involved. Ultimately, these findings contribute significantly to the field of positron chemistry, offering a fresh perspective on how positrons can mediate and even dictate molecular interactions.
Author contributions
Conceptualization: J. C., F. M., A. R. Formal analysis: J. C., F. M., A. R. Investigation: R. P. R., J. C., A. R. Funding acquisition: M. T. N. V., A. R. Methodology: R. P. R., J. C., A. R. Project administration: A. R. Validation: R. F. M., M. T. N. V., A. R. Resources: J. C., A. R. Software: R. P. R., J. C., F. M., R. F. M., A. R. Supervision: R. F. M, M. T. N. V., A. R. Visualization: R. P. R., J. C. Writing – original draft: R. P. R., J. C., F. M., R. F. M., M. T. N. V., A. R. Writing – review & editing: R. P. R., J. C., F. M., R. F. M., M. T. N. V., A. R.Conflicts of interest
The authors declare no conflict of interest.Data availability
Data for this paper, including basis set, QMeCha optimised wave function files, density cube files, tabulated raw energetic data, outputs, and postprocessing scripts, are available in the Zenodo archive at https://doi.org/10.5281/zenodo.16582423.Supplementary information: potential energy curve for Be2 and e+:Be2 at the VMC level. See DOI: https://doi.org/10.1039/d5sc05711f.
Acknowledgements
The QMC calculations presented in this paper were carried out using the HPC facilities of the University of Luxembourg84 (see https://hpc.uni.lu). The authors acknowledge Dr Matteo Barborini (E-mail: matteo.barborini@uni.lu) for the support on QMeCha code, the implementation of the energy decomposition, discussions, and for providing the resources to perform the QMC calculations. MTNV acknowledges support from CNPq (Grant no.306285/2022-3). AR acknowledges support from Universidad Nacional de Colombia (HERMES 63579).Notes and references
- Y. C. Jean, P. E. Mallon and D. M. Schrader, Principles and Applications of Positron and Positronium Chemistry, World Scientific, 2003 Search PubMed.
- Physics with Many Positrons, ed. A. Dupasquier, A. P. Mills, R. S. Brusa and R. S. Brusa, IOS Press, 2010 Search PubMed.
- S. D. Bass, S. Mariazzi, P. Moskal and E. Stepien, Rev. Mod. Phys., 2023, 95, 021002 CrossRef CAS.
- D. W. Gidley, H.-G. Peng and R. S. Vallery, Annu. Rev. Mater. Res., 2006, 36, 49–79 CrossRef CAS.
- A. N. Singh, Appl. Spectrosc. Rev., 2016, 51, 359–378 CrossRef.
- G. Consolati, D. Nichetti and F. Quasso, Polymers, 2023, 15, 3128 CrossRef CAS PubMed.
- M. J. Brunger, D. B. Cassidy, S. Dujko, D. Marić, J. Marler, J. P. Sullivan and J. Fedor, Eur. Phys. J. D, 2020, 74, 158 CrossRef CAS.
- J. J. Vaquero and P. Kinahan, Annu. Rev. Biomed. Eng., 2015, 17, 385–414 CrossRef CAS PubMed.
- T. Jones and D. Townsend, J. Med. Imaging, 2017, 4, 011013 CrossRef.
- M. M. Shokoya, B.-M. Benkő, K. Süvegh, R. Zelkó and I. Sebe, Pharmaceuticals, 2023, 16, 252 CrossRef CAS PubMed.
- P. Moskal, B. Jasińska, E. Ł. Stępień and S. D. Bass, Nat. Rev. Phys., 2019, 1, 527–529 CrossRef.
- P. Moskal, K. Dulski, N. Chug, C. Curceanu, E. Czerwiński, M. Dadgar, J. Gajewski, A. Gajos, G. Grudzień, B. C. Hiesmayr, K. Kacprzak, Ł. Kapłon, H. Karimi, K. Klimaszewski, G. Korcyl, P. Kowalski, T. Kozik, N. Krawczyk, W. Krzemień, E. Kubicz, P. Małczak, S. Niedźwiecki, M. Pawlik-Niedźwiecka, M. Pędziwiatr, L. Raczyński, J. Raj, A. Ruciński, S. Sharma, S. R. Y. Shopa, M. Silarski, M. Skurzok, E. Ł. Stępień, M. Szczepanek, F. Tayefi and W. Wiślicki, Sci. Adv., 2021, 7, eabh4394 CrossRef CAS PubMed.
- J. R. Danielson, A. C. L. Jones, M. R. Natisin and C. M. Surko, Phys. Rev. Lett., 2012, 109, 113201 CrossRef CAS PubMed.
- J. Danielson, S. Ghosh and C. Surko, Phys. Rev. A, 2022, 106, 032811 CrossRef CAS.
- J. R. Danielson, E. Arthur-Baidoo and C. M. Surko, Phys. Rev. A, 2025, 111, 042809 CrossRef CAS.
- G. F. Gribakin and C. M. R. Lee, Phys. Rev. Lett., 2006, 97, 193201 CrossRef CAS PubMed.
- M. R. Natisin, J. R. Danielson, G. F. Gribakin, A. R. Swann and C. M. Surko, Phys. Rev. Lett., 2017, 119, 113402 CrossRef CAS PubMed.
- D. M. Schrader and J. Moxom, in Antimatter Compounds, ed. C. M. Surko and F. A. Gianturco, Springer Netherlands, Dordrecht, 2001, pp. 263–290 Search PubMed.
- G. F. Gribakin, J. A. Young and C. M. Surko, Rev. Mod. Phys., 2010, 82, 2557–2607 CrossRef CAS.
- J. P. Coe and M. J. Paterson, Chem. Phys. Lett., 2016, 645, 106–111 CrossRef CAS.
- A. R. Swann and G. F. Gribakin, J. Chem. Phys., 2018, 149, 244305 CrossRef CAS PubMed.
- J. Hofierka, B. Cunningham, C. M. Rawlins, C. H. Patterson and D. G. Green, Nature, 2022, 606, 688–693 CrossRef CAS PubMed.
- J. A. Charry Martinez, M. Barborini and A. Tkatchenko, J. Chem. Theory Comput., 2022, 18, 2267–2280 CrossRef CAS PubMed.
- G. Cassella, W. M. C. Foulkes, D. Pfau and J. S. Spencer, Nat. Commun., 2024, 15, 5214 CrossRef CAS PubMed.
- F. A. Gianturco, J. Franz, R. J. Buenker, H.-P. Liebermann, L. Pichl, J.-M. Rost, M. Tachikawa and M. Kimura, Phys. Rev. A: At., Mol., Opt. Phys., 2006, 73, 022705 CrossRef.
- F. Moncada, L. Pedraza-González, J. Charry, M. T. do N. Varella and A. Reyes, Chem. Sci., 2020, 11, 44–52 RSC.
- J. P. Cassidy, J. Hofierka, B. Cunningham and D. G. Green, J. Chem. Phys., 2024, 160, 084304 CrossRef CAS PubMed.
- J. Charry, M. T. d. N. Varella and A. Reyes, Angew. Chem., Int. Ed., 2018, 57, 8859–8864 CrossRef CAS PubMed.
- M. Goli and S. Shahbazian, ChemPhysChem, 2019, 20, 831–837 CrossRef CAS PubMed.
- S. Ito, D. Yoshida, Y. Kita and M. Tachikawa, J. Chem. Phys., 2020, 153, 224305 CrossRef CAS PubMed.
- D. Bressanini, J. Chem. Phys., 2021, 154, 224306 CrossRef CAS PubMed.
- D. Bressanini, J. Chem. Phys., 2021, 155, 054306 CrossRef CAS PubMed.
- M. Goli, D. Bressanini and S. Shahbazian, Phys. Chem. Chem. Phys., 2023, 25, 29531–29547 RSC.
- J. Charry, F. Moncada, M. Barborini, L. Pedraza-González, M. T. d. N. Varella, A. Tkatchenko and A. Reyes, Chem. Sci., 2022, 13, 13795–13802 RSC.
- D. Archila-Peña, F. Moncada, J. Charry, M. T. do N. Varella, R. Flores-Moreno, F. J. Torres and A. Reyes, Chem. - Eur. J., 2024, 30, e202402618 CrossRef PubMed.
- T. Tachibana, D. Hoshi and Y. Nagashima, Phys. Rev. Lett., 2023, 131, 143201 CrossRef CAS.
- B. B. Gerbelli, S. V. Vassiliades, J. E. U. Rojas, J. N. B. D. Pelin, R. S. N. Mancini, W. S. G. Pereira, A. M. Aguilar, M. Venanzi, F. Cavalieri, F. Giuntini and W. A. Alves, Macromol. Chem. Phys., 2019, 220, 1900085 CrossRef.
- Non-Covalent Interactions in Quantum Chemistry and Physics: Theory and Applications, ed. A. Otero de la Roza and G. A. DiLabio, Elsevier, Amsterdam, Netherlands, 2017 Search PubMed.
- J. Romero, J. a Charry, R. Flores-Moreno, M. T. D. N. Varella and A. Reyes, J. Chem. Phys., 2014, 141, 114103 CrossRef PubMed.
- J. Simons and K. D. Jordan, Chem. Rev., 1987, 87, 535–555 CrossRef CAS.
- K. Patkowski, R. Podeszwa and K. Szalewicz, J. Phys. Chem. A, 2007, 111, 12822–12838 CrossRef CAS PubMed.
- J. J. Bao, L. Gagliardi and D. G. Truhlar, J. Phys. Chem. Lett., 2019, 10, 799–805 CrossRef CAS PubMed.
- A. Das and E. Arunan, Phys. Chem. Chem. Phys., 2022, 24, 28913–28922 RSC.
- J. T. Boronski, Dalton Trans., 2024, 53, 33–39 RSC.
- S. Upadhyay, A. Benali and K. D. Jordan, J. Chem. Theory Comput., 2024, 20, 9879–9893 CrossRef CAS PubMed.
- V. A. Dzuba, V. V. Flambaum, W. A. King, B. N. Miller and O. P. Sushkov, Phys. Scr., 1993, 1993, 248 CrossRef.
- J. Mitroy, M. W. J. Bromley and G. G. Ryzhikh, J. Phys. B: At., Mol. Opt. Phys., 2002, 35, R81 CrossRef CAS.
- C. Harabati, V. Dzuba and V. Flambaum, Phys. Rev. A, 2014, 89, 022517 CrossRef.
- M. Marchi, S. Azadi, M. Casula and S. Sorella, J. Chem. Phys., 2009, 131, 154116 CrossRef PubMed.
- M. J. Deible, M. Kessler, K. E. Gasperich and K. D. Jordan, J. Chem. Phys., 2015, 143, 084116 CrossRef PubMed.
- M. El Khatib, G. L. Bendazzoli, S. Evangelisti, W. Helal, T. Leininger, L. Tenti and C. Angeli, J. Phys. Chem. A, 2014, 118, 6664–6673 CrossRef CAS PubMed.
- W. M. C. Foulkes, L. Mitas, R. J. Needs and G. Rajagopal, Rev. Mod. Phys., 2001, 73, 33–83 CrossRef CAS.
- M. H. Kalos and P. A. Whitlock, in Quantum Monte Carlo, John Wiley & Sons, Ltd, 2000, ch. 8 Search PubMed.
- F. Becca and S. Sorella, Quantum Monte Carlo approaches for correlated systems, Cambridge University Press, 2017, pp. 1–274 Search PubMed.
- M. Barborini, Quantum Mecha (QMeCha) package β (private repository Jan-12th, 2025, https://github.com/mbarborini/QMeCha_beta.
- N. Metropolis, A. W. Rosenbluth, M. N. Rosenbluth, A. H. Teller and E. Teller, J. Chem. Phys., 1953, 21, 1087–1092 CrossRef CAS.
- W. K. Hastings, Biometrika, 1970, 57, 97–109 CrossRef.
- M. Casula and S. Sorella, J. Chem. Phys., 2003, 119, 6500–6511 CrossRef CAS.
- S. Sorella, Phys. Rev. B:Condens. Matter Mater. Phys., 2001, 64, 024512 CrossRef.
- S. Sorella, Phys. Rev. B:Condens. Matter Mater. Phys., 2005, 71, 241103 CrossRef.
- P. J. Reynolds, D. M. Ceperley, B. J. Alder and W. A. Lester, J. Chem. Phys., 1982, 77, 5593–5603 CrossRef CAS.
- R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- M. Mella, M. Casalegno and G. Morosi, J. Chem. Phys., 2002, 117, 1450–1456 CrossRef CAS.
- G. G. Ryzhikh, J. Mitroy and K. Varga, J. Phys. B: At., Mol. Opt. Phys., 1998, 31, 3965–3996 CrossRef CAS.
- J. Mitroy and G. G. Ryzhikh, J. Phys. B: At., Mol. Opt. Phys., 2001, 34, 2001–2007 CrossRef CAS.
- K. R. Brorsen, M. V. Pak and S. Hammes-Schiffer, J. Phys. Chem. A, 2017, 121, 515–522 CrossRef CAS PubMed.
- S. Bubin and O. V. Prezhdo, Phys. Rev. Lett., 2013, 111, 193401 CrossRef PubMed.
- M. W. J. Bromley and J. Mitroy, Phys. Rev. A, 2001, 65, 012505 CrossRef.
- J. Hofierka, B. Cunningham, C. M. Rawlins, C. H. Patterson and D. G. Green, Gaussian-basis many-body theory calculations of positron binding to negative ions and atoms, 2023, https://arxiv.org/abs/2311.13066.
- J. M. Merritt, V. E. Bondybey and M. C. Heaven, Science, 2009, 324, 1548–1551 CrossRef CAS PubMed.
- X. Sheng, X. Kuang, P. Li and K. Tang, Phys. Rev. A: At., Mol., Opt. Phys., 2013, 88, 022517 CrossRef.
- I. Röeggen, K. Morokumi and K. Yamashita, Chem. Phys. Lett., 1987, 140, 349–354 CrossRef.
- S. Evangelisti, G. L. Bendazzoli and L. Gagliardi, Chem. Phys., 1994, 185, 47–56 CrossRef CAS.
- A. Krapp, F. M. Bickelhaupt and G. Frenking, Chem. - Eur. J., 2006, 12, 9196–9216 CrossRef CAS PubMed.
- The final form of the fit was
 . Coefficients were estimated by least squares minimization.
. Coefficients were estimated by least squares minimization. - S. Ito, D. Yoshida, Y. Kita, T. Shimazaki and M. Tachikawa, J. Chem. Phys., 2023, 158, 204303 CrossRef CAS PubMed.
- H. Li, H. Feng, W. Sun, Y. Zhang, Q. Fan, K. A. Peterson, Y. Xie and H. F. Schaefer, Mol. Phys., 2013, 111, 2292–2298 CrossRef CAS.
- A. Kalemos, J. Chem. Phys., 2016, 145, 214302 CrossRef PubMed.
- S. Noschese, L. Pasquini and L. Reichel, Numer. Lin. Algebra Appl., 2013, 20, 302–326 CrossRef.
- D. S. Levine and M. Head-Gordon, Nat. Commun., 2020, 11, 4893 CrossRef CAS PubMed.
- T. Clark, P. Politzer and J. S. Murray, Phys. Chem. Chem. Phys., 2022, 24, 12116–12120 RSC.
- M. Goli and S. Shahbazian, Theor. Chem. Acc., 2013, 132, 1365 Search PubMed.
- T. M. Cardozo, D. W. O. D. Sousa, F. Fantuzzi and M. A. C. Nascimento, Comprehensive Computational Chemistry, Elsevier, Amsterdam, Netherlands, 2024, vol. 1, pp. 552–588 Search PubMed.
- S. Varrette, P. Bouvry, H. Cartiaux and F. Georgatos, Proc. of the 2014 Intl. Conf. on High Performance Computing & Simulation (HPCS 2014), Bologna, Italy, 2014, pp. 959–967 Search PubMed.

| This journal is © The Royal Society of Chemistry 2025 |