
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRevealing the exchange kinetics of thiol-capped Au25 nanoclusters with alkynyl ligands
Yuping
Chen†
a,
Guoqing
Bian†
b,
Zhikun
Wu
 *b and
Qing
Tang
*a
*b and
Qing
Tang
*a
aSchool of Chemistry and Chemical Engineering, Chongqing Key Laboratory of Chemical Theory and Mechanism, Chongqing University, Chongqing 401331, China. E-mail: qingtang@cqu.edu.cn
bKey Laboratory of Materials Physics, Anhui Key Laboratory of Nanomaterials and Nanotechnology, CAS Center for Excellence in Nanoscience, Institute of Solid State Physics, Chinese Academy of Sciences, Hefei, Anhui 230031, China. E-mail: zkwu@issp.ac.cn
First published on 8th September 2025
Abstract
Ligand exchange is an important strategy to functionalize metal nanoclusters (NCs) for enhanced properties. Thiolates and alkynyls have been widely used for NC protection; however, the possibility for alkynyl-for-thiolate exchange as well as the kinetics for the ligand exchange process have remained largely unexplored. Herein, we have reported for the first time a kinetic investigation into the alkynyl-for-thiolate exchange through the reaction of thiolated Au25(SR)18 with the incoming alkynyl ligands. Interestingly, our simulations revealed the electronic and steric effects of alkynyl ligands and the precursor cluster's charge state collectively govern the exchange efficiency and regioselectivity. Notably, the alkynyl-for-thiolate exchange is highly facile when reacting with the nucleophilic lithium or gold(I)–alkynyl complex (Au(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CR) or Li(CCR)), but fails when using HCCR as the exchange ligand. The Au(CCPh) complex exhibits charge-state-dependent exchange at the S1 or S2 position, whereas the sterically bulky Au(CCtBu) complex decelerates the exchange kinetics and universally targets the S1 position as the exchange product. By contrast, the lithium–alkynyl complex (Li(CCPh) or Li(CCtBu)) preferentially leads to the
CR) or Li(CCR)), but fails when using HCCR as the exchange ligand. The Au(CCPh) complex exhibits charge-state-dependent exchange at the S1 or S2 position, whereas the sterically bulky Au(CCtBu) complex decelerates the exchange kinetics and universally targets the S1 position as the exchange product. By contrast, the lithium–alkynyl complex (Li(CCPh) or Li(CCtBu)) preferentially leads to the  exchange isomer, driven by the ionic Li–C bonding that enhances the CC π*-electron density and alkynyl nucleophilicity. Our predictions are further validated by the ligand exchange experiments between phenyl ethanethiol (PET) protected [Au25(PET)18]− and Au(CCtBu). The electrospray ionization mass spectra (ESI-MS) unambiguously confirm the successful substitution of 4, 5 and 6 PET ligands by the –CCtBu ligand, and the absorption spectrum drastically changes upon alkynyl exchange. This work establishes an important atomic-level understanding of the alkynyl-for-thiolate exchange mechanism, offering a convenient strategy for realizing alkynyl and thiolate co-protected gold clusters under mild conditions.
exchange isomer, driven by the ionic Li–C bonding that enhances the CC π*-electron density and alkynyl nucleophilicity. Our predictions are further validated by the ligand exchange experiments between phenyl ethanethiol (PET) protected [Au25(PET)18]− and Au(CCtBu). The electrospray ionization mass spectra (ESI-MS) unambiguously confirm the successful substitution of 4, 5 and 6 PET ligands by the –CCtBu ligand, and the absorption spectrum drastically changes upon alkynyl exchange. This work establishes an important atomic-level understanding of the alkynyl-for-thiolate exchange mechanism, offering a convenient strategy for realizing alkynyl and thiolate co-protected gold clusters under mild conditions.
Introduction
Atomically precise gold nanoclusters (Au NCs) protected by organic ligands have emerged as a central focus in nanomaterials research due to their well-defined atomic structures, unique physicochemical properties and tunable reactivity.1–4 These properties are highly sensitive to factors such as the cluster size, shape, the metal–ligand interface, and the ligand environment, making Au NCs ideal candidates for various tailored applications.5,6 By rationally modifying the core and surface or interface structures, the electronic and catalytic properties of Au NCs can be fine-tuned to the optimal level. In this context, the protecting ligands play a dual role, which on one hand, stabilize the cluster framework from agglomeration, while on the other hand, modulate the interface properties and coordination environment of the metal active sites.4 In particular, as a powerful post-synthesis strategy, ligand exchange provides a versatile tool for the modification and functionalization of metal NCs.7–10 This technique has been widely applied to enhance the cluster properties such as photoemission,11,12 chirality,13–15 and solubility,16–19 thereby expanding the potential applications of Au NCs in fields ranging from biomedicine20,21 to nanocatalysis22 to cluster self-assembly.23 In terms of ligand exchange, recent efforts have mainly concentrated on the exchange reaction of thiolate-protected gold NCs with free thiol or thiolated metal complexes,24–27 wherein some of the surface ligands are replaced with new thiolate ligands to control the surface chemical functionalization of Au NCs.9,28–31 It has been demonstrated that the thiol exchange reaction is enabled by the flexibility and reactivity of the Au–S interface, in which the ligand-exchange position and kinetics are largely controlled by the electronic and steric effects of the incoming thiol ligands.In addition to the widely studied thiolate (–SR) ligands, the alkynyl ligands (–CCR) have recently emerged as new alternatives for surface functionalization, and have gained special attention for their ability to interact with gold through electronic conjugation as well as versatile interface interactions to form stable organogold nanoclusters.32 These interactions offer new opportunities for tuning the electronic and catalytic properties of Au NCs. In 2011, Tsukuda et al. reported the first experimental synthesis of a series of homoleptic alkynyl-protected Au NCs by direct ligand exchange of preformed polyvinylpyrrolidone (PVP)-stabilized Au NCs with phenylacetylene (PhCCH)33 under thermal conditions in a biphasic solvent system. Later in 2015, Konishi et al. introduced two alkynyl ligands on the surface of an icosahedral Au13 skeleton ([Au13(dppe)5(CCPh)2]3+) by the ligand exchange reaction of a dichloro-substituted Au13 cluster ([Au13(dppe)5Cl2]3+) with a free PhCCH ligand under basic conditions.34 Besides the post-synthetic ligand exchange method, the alkynyl gold NCs are more often achieved by the bottom-up reduction of gold(I) alkynyl precursors in solution,35 and thus far, more than two dozen alkynyl gold NCs with well-defined compositions and structures (e.g., Au19, Au23, Au24, Au25, Au36, Au38, Au44, and Au144) have been prepared via the direct reduction route.36 Among them, some alkynyl protected Au NCs even share the same structures and ligand-to-metal ratio as their thiolate protected counterparts. A representative example is Au36(CCPh)24 and Au44(CCPh)28, which are, respectively, isostructural to their thiolated Au36(SR)24 and Au44(SR)28 counterparts.37 Another intriguing example is [Au25(CCR)18]−,38 which has the same icosahedral Au13 core as in [Au25(SR)18]−, but the peripheral six V-shaped dimeric staple motifs are arranged differently. Moreover, recent determination of the ligand effect in alkynyl vs. thiolate has demonstrated that the alkynyl-protected metal NCs exhibit better stability against oxidation and much enhanced catalytic performances in comparison with the thiolated counterparts.39,40 This indicates that there might exist a similar but quite different parallel universe between the alkynyl-protected Au NCs and the thiolated ones.
In particular, the structural similarity between thiolate- and alkynyl-protected metal NCs suggests that the alkynyl ligands might possibly replace the thiolates in the protecting motifs via the ligand-exchange process. Noteworthily, the alkynyl and thiolate ligands have similar but different coordination preferences, both being negatively charged and forming strong interactions with the metals, but the thiolate usually adopts the σ-only coordination modes, whereas the alkynyl with its CC bond would coordinate with the metals via σ- or/and π-bonding modes. Their distinct metal–ligand interactions provide new avenues for controlling the cluster reactivity and stability, it thus would be highly desirable to realize the exchange reactions between alkynyl and thiolate ligands to produce potentially new properties in the atomically precise Au NCs.32,36 However, the exchange of alkynyl for thiolate ligands is challenging, and thus far only a few studies have conducted the alkynyl-for-thiolate (or thiolate-for-alkynyl) exchange on Au NCs. In this context, the first attempt towards the alkynyl-for-thiolate exchange was reported in 2020 by the Ackerson group.41 Their results revealed that the thiolate-protected Au NCs such as Au25(SR)18 can readily undergo the alkynyl-for-thiolate ligand exchange under mild conditions when a lithium–phenylacetylide or gold(I)–phenylacetylide complex was used as the incoming ligand.41 They further showed that the reverse thiolate-for-alkynyl exchange is also facile when using the free thiol as the incoming ligand to the alkynyl-protected Au25(CCAr)18 NCs. However, due to the possible large number of competing exchange products, the distribution of the reaction products is very complicated, and no specific composition with well-defined formulae or crystal structure was isolated successfully at that time. In a more recent report, Nakamura et al. has demonstrated that partial ligand exchange with alkynyl groups on Au25(SR)18 is crucial for enabling the dual catalytic activity,42 which greatly promotes the photocatalytic cross-dehydrogenative coupling of terminal alkynes and tertiary amines. Despite the impressive progress, examples of alkynyl-for-thiolate exchange reactions or vice versa are, currently, still very rare, and the microscopic details of the exchange mechanism remain obscure. A molecular-level understanding of the transformation mechanism and the preferred reactive sites in the ligand exchange process will enable more precise engineering of surface microenvironments. Therefore, it is essential to clarify the alkynyl-for-thiolate exchange mechanism and develop strategies for controlling regioselectivity during this exchange process to design mixed-ligand metal NCs with new functionalities.
In this work, using the widely studied Au25(SR)18 as the parent cluster, we performed systematic first-principles simulations to probe the reactivity of phenyl ethanethiol (PET)-protected Au25(PET)18 with the alkynyl ligands (Scheme 1), including free phenylacetylene (PhCCH) and the alkynyl metal compounds (gold(I)–acetylide or lithium(I)–acetylide) as the incoming ligands. As shown in Scheme 1a, the protecting motif of Au25(PET)18 NC features an elegant assembly of six dimeric [RS–Au(I)–SR–Au(I)–SR] units around the icosahedral Au13 core.43 In this dimeric motif, one thiolate ligand is positioned at the apex (denoted as the “S2” site), while two other thiolates are anchored to the surface of Au13 core, which are differentiated by the different orientations of SR groups. The thiolate that orients in a cis-configuration with the apex SR is denoted as “S1”, while that orienting in a trans-configuration with the apex SR is denoted as “ ”. This unique arrangement gives rise to three possible distinct regioisomers (S1, S2, and
”. This unique arrangement gives rise to three possible distinct regioisomers (S1, S2, and  ) in the ligand-exchanged products (Scheme 1b).
) in the ligand-exchanged products (Scheme 1b).
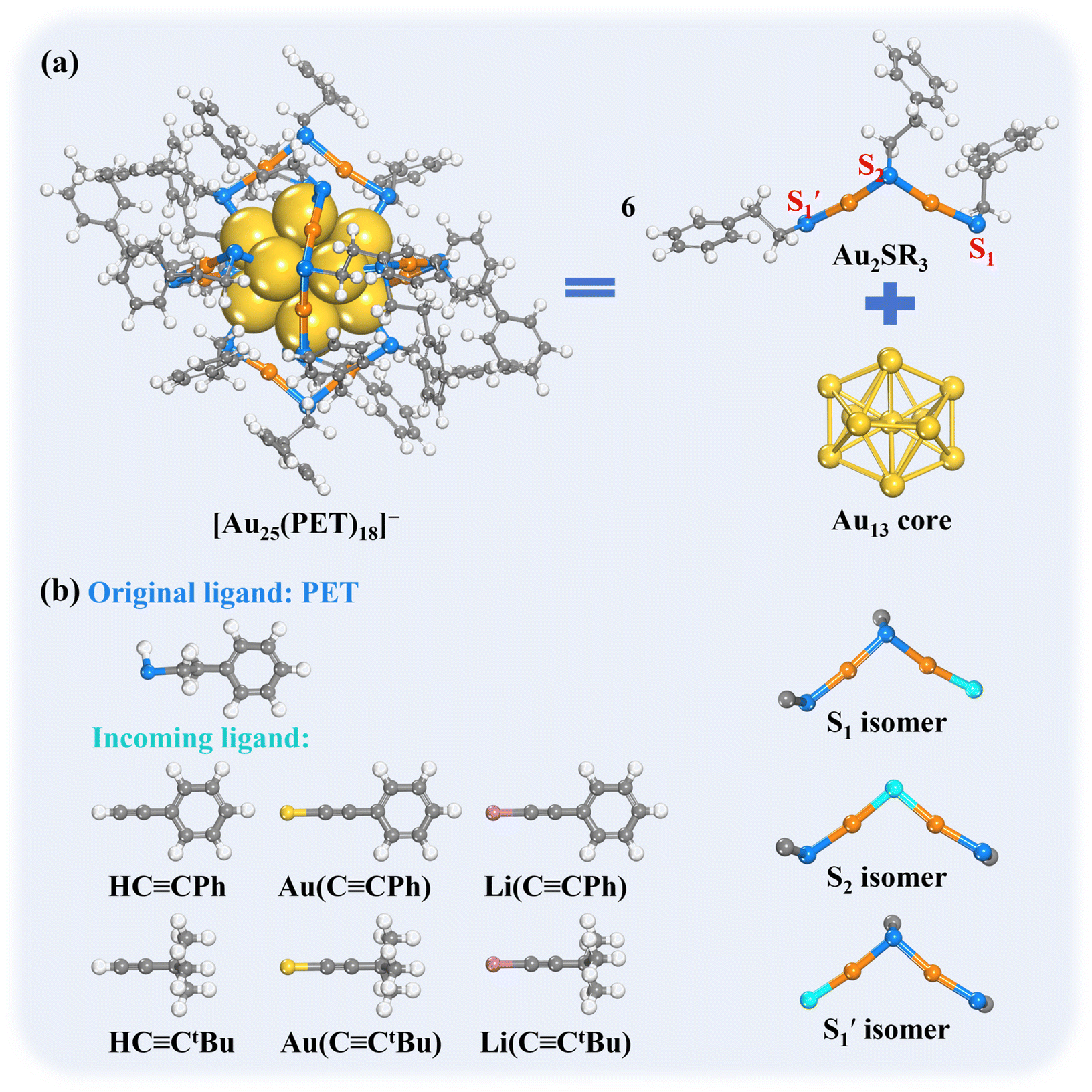 | ||
| Scheme 1 (a) Structure of [Au25(PET)18]− and three possible exchange sites in the ligand-exchange reaction. (b) The original PET ligand and the incoming alkynyl ligand, including HCCPh, Au(CCPh), Li(CCPh), HCCtBu, Au(CCtBu) and Li(CCtBu), as well as the three possible regioisomers formed after the ligand-exchange reaction (the site of the exchanged alkynyl ligand is colored in green). Color code: core Au – yellow (Au25 cluster); staple Au – orange (Au25 cluster); S – blue; C – gray; incoming ligand – sapphire; Li – purple; H – white. | ||
Based on the above Au25(PET)18 model cluster, we then pursued an atomic-scale mechanism governing the alkynyl-for-thiolate exchange efficiency and regioselectivity, focusing on the influence of ligand electronic characteristics, steric interactions and the charge state of Au25q (q = −1, 0, and +1) NCs. The free energy calculations with enhanced sampling revealed the fundamental energy constraints (>3 eV) for the direct HCCR-for-thiolate exchange, a consequence of the highly localized and rigid CC π-bond causing mismatch with the cluster frontier orbitals. Notably, the alkynyl–metal complexes (e.g., Au/Li(CCPh)) overcome these constraints and initiate the exchange reactions through the π-system polarization. The Au(CCPh) species characterized by the d–π* back-bonding between the occupied Au d orbitals and the π* orbitals of the CC bond exhibits charge-state-dependent regioselectivity, in which the negatively charged [Au25(SR)18]− favors the S1 isomer formation, while the neutral Au25(SR)18 promotes the selective substitution at the apex S2 site. Differently, the bulkier Au(CCtBu) universally targets the S1 position across the different charge states, wherein the bulkier tert-butyl ligand slows down the exchange kinetics relative to the phenyl analogues (–CCPh) while thermodynamically stabilizing the exchange products. By comparison, the Li-coordinated alkynyl complexes (Li(CCR)) universally generate the  isomers, driven by the ionic Li–C bonding that enhances the CC π*-electron density. Importantly, the Au center in Au(CCR) actively participates in the exchange, whereas the Li center in Li(CCR) functions solely as a transient mediator. This non-incorporative mediation promotes ligand exchange without inducing significant Au25 structural reorganization. Our predictions are further validated by the ligand exchange experiments. The electrospray ionization mass spectrum (ESI-MS) quantification confirms the replacement of 4, 5 and 6 PET ligands by –CCtBu on [Au25(PET)18]−. These insights establish clear mechanistic guidelines for the atom-precise alkynyl-for-thiolate engineering of Au25(PET)18 clusters, signifying the importance of electronic and steric effects as well as the precursor charge states in collectively dictating the substitution pathways.
isomers, driven by the ionic Li–C bonding that enhances the CC π*-electron density. Importantly, the Au center in Au(CCR) actively participates in the exchange, whereas the Li center in Li(CCR) functions solely as a transient mediator. This non-incorporative mediation promotes ligand exchange without inducing significant Au25 structural reorganization. Our predictions are further validated by the ligand exchange experiments. The electrospray ionization mass spectrum (ESI-MS) quantification confirms the replacement of 4, 5 and 6 PET ligands by –CCtBu on [Au25(PET)18]−. These insights establish clear mechanistic guidelines for the atom-precise alkynyl-for-thiolate engineering of Au25(PET)18 clusters, signifying the importance of electronic and steric effects as well as the precursor charge states in collectively dictating the substitution pathways.
Methods
Computational modeling
The [Au25(PET)18]q (q = −1, 0, +1) NCs were initially placed within a cubic simulation box measuring 30 Å × 30 Å × 30 Å, followed by optimization to obtain the most stable configuration. To simulate the ligand exchange process with varying alkynyl ligand (e.g., HCCPh) concentrations at 10 or 40 equivalents, 10 or 40 alkynyl ligand molecules were incorporated around the [Au25(PET)18]q cluster during the unconstrained molecular dynamics (MD) simulations. This setup allowed for the exploration of ligand exchange dynamics at different ligand concentrations. For the constrained MD simulations and free energy calculations, only a single alkynyl ligand was incorporated around the cluster to model the exchange of the specific PET ligand at the designated position.
DFT calculations
The ab initio molecular dynamics (AIMD) simulations and constrained MD (c-MD) simulations were performed using the CP2K package (version 2023),44 based on the PBE functional and a hybrid Gaussian/Plane-Wave scheme (GPW).45 The GTH pseudopotentials were chosen to describe the core electrons. The wave functions were expanded in optimized double-ζ Gaussian basis sets, and the plane waves were expanded with a cutoff energy of 400 Rydberg.46 Dispersion correction was applied in all calculations with the DFT-D3(BJ) method.47 The simulations were sampled by the canonical (NVT) ensemble employing Nose–Hoover thermostats with a time step of 1.0 fs at a finite temperature of 298.15 K for more than 10 ps.48 Constrained molecular dynamics (c-MD) simulations were calculated based on the thermodynamic integration (TI) method,49,50 where the reaction free energy and kinetic barrier were obtained by applying a holonomic constraint on the reaction coordinate (ζ) during MD simulations and integrating over the average unbiased force associated with the reaction coordinate, as shown in the following equation:where ΔA(ζa, ζb) is the free energy difference between two reaction coordinates (ζa and ζb) and F(ζ) is the averaged constrained force. ζ is the velocity of transformation, and the free energy gradient
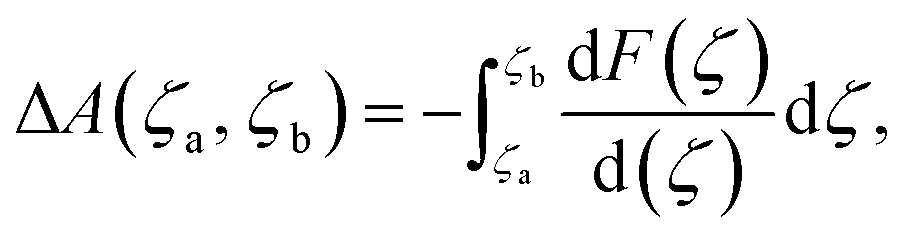
 can be computed along c-MD using the SHAKE algorithm.50 The growth speed (dζ) was set as 0.0008, and the tolerance for the Shake/Rattle constraint algorithm was set as 0.0001. The reaction barriers were obtained by integrating the free-energy gradients to compute the free energy profiles based on thermodynamic integrations.
can be computed along c-MD using the SHAKE algorithm.50 The growth speed (dζ) was set as 0.0008, and the tolerance for the Shake/Rattle constraint algorithm was set as 0.0001. The reaction barriers were obtained by integrating the free-energy gradients to compute the free energy profiles based on thermodynamic integrations.
Molecular fragment calculations
The molecular fragments were modeled using the Gaussian 16 software package.51 Geometry optimizations were performed with the B3LYP functional, employing the 6-31G(d) basis set for Li, C, N, and H atoms52 and the Stuttgart/Dresden (SDD) effective core potential for the Au atom.53 To account for the dispersion interactions, the D3 correction with Becke–Johnson damping was applied.54 Molecular orbital analysis, Hirshfeld/CM5 population analysis, and Mayer bond order analysis55–57 were carried out using the Multiwfn program,58 based on the converged wavefunctions obtained from the DFT calculations.Experimental section
Chemicals
All chemicals are commercially available and were used as received. Tetrachloroauric(III) acid (HAuCl4·4H2O, >99.8%), triethylamine (Net3, ≥99.5%) and sodium borohydride (NaBH4, 99.8%) were purchased from Shanghai Chemical Reagent Co., Ltd. Tetraoctylammonium bromide (TOAB, ≥98.0%), dimethyl sulfide (DMS, ≥99%) and 3,3-dimethyl-1-butyne ((CH3)3CCCH, ≥98.0%) were purchased from Aladdin. Solvents dichloromethane (DCM), dimethyl sulfoxide (DMSO), petroleum ether, tetrahydrofuran, toluene, acetone and methanol were purchased from Shanghai Chemical Reagent Co., Ltd. The water used in all experiments was ultrapure (resistivity: 18.2 MΩ cm), produced using a Milli-Q NANO pure water system. Me2SAuCl was prepared according to the literature methods.59
ESI-MS data collection
Electrospray ionization (ESI) mass spectra were recorded on a Waters Q-TOF mass spectrometer using a Z-spray source. The sample was first dissolved in toluene (∼0.5 g L−1) and then diluted (6/1 v/v) with a dimethyl sulfoxide (DMSO) solution containing 50 mmol of CsOAc. The sample was directly infused into the chamber at 5 μL min−1. The source temperature was maintained at 70 °C, the spray voltage was 2.20 kV and the cone voltage was adjusted to 60 V.Synthesis of TOA[Au25(PET)18]
A previously published method was adapted for the synthesis of TOA[Au25(PET)18].60 Specifically, in a 100 mL three-necked flask, 674.8 mg of TOAB and 30 mL of tetrahydrofuran solution were added and rapidly stirred for 10 minutes. Then, 500 mg of HAuCl4·4H2O was added to the solution. After stirring at medium speed for 30 minutes, the solution changed from yellow to burgundy. Afterwards, 1020 μL of PET was added to the solution, and the stirring was continued at a low speed for 4 hours. The solution gradually changed to colorless. The flask was placed in an ice bath and stirred rapidly for 17 hours after adding 528.2 mg of aqueous NaBH4. The solution was washed three times with methanol and centrifuged. The resulting precipitate was dissolved in 8 mL of toluene and mixed with 32 mL of methanol in a 50 mL beaker. After one day, a large number of black crystals of TOA[Au25(PET)18] could be observed at the bottom of the beaker.Synthesis of Au(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e002.gif) CtBu)
CtBu)
50 mg of chloro(dimethylsulfanyl)gold(I) was added to 5 mL of acetone, 60 μL of 3,3-dimethyl-1-butyne, and 30.5 μL of triethylamine in a 20 mL round-bottomed flask and stirred in the dark for two hours. The mixed solution can be used directly for ligand exchange without purification.
Exchange of –CCtBu with TOA[Au25(PET)18]
Ligand exchange was performed by adding 1 mL of mixed solution containing Au(CCtBu) and 12 mg of TOA[Au25(PET)18] into 3 mL of dichloromethane in a 20 mL round-bottomed flask. The solution was stirred for three hours at room temperature in darkness. Then, the product was washed with methanol two times and extracted with DCM. The crude products dissolved in 2 mL of DCM were smeared on ten pieces of preparative thin-layer chromatography (PTLC) plates (10 cm × 20 cm) and separated using an eluent (DCM/petroleum ether = 10/15 v/v). The brown band in the PTLC plate was cut off by using a knife and extracted by DCM. The DCM solution was dried by rotary evaporation for ESI-MS analysis.
Results and discussion
Ligand exchange between Au25 and Au/Li(CCPh)
To explore the dynamic exchange mechanism in thiol-capped Au25 NCs with alkynyl ligands, we used the well-characterized [Au25(PET)18]q (q = −1, 0, and 1) as a template (Fig. 1a) and introduced a series of alkynyl ligands (Scheme 1b) to study the ligand exchange process via AIMD simulations. We start with the negatively charged [Au25(PET)18]− as the parent cluster. Our initial simulations with the free phenylacetylene (HCCPh) as the incoming ligand did not induce any exchange on [Au25(PET)18]−, even at an elevated temperature of 373 K (Fig. S1), where the cluster structure remained intact. The constrained AIMD (c-MD) simulations further revealed an extremely high energy barrier of 3.03 eV for the HCCPh-for-PET ligand displacement (Fig. S2b), affirming the robustness of Au–S coordination towards the incoming HCCPh ligand. Although the weak π-interactions were observed between the CC bond and the staple Au atoms, these interactions were insufficient to rival the robust Au–S bonds. In contrast, introducing alkynyl as the gold(I)–phenylacetylide complex (Au(CCPh)) enabled efficient ligand exchange on [Au25(PET)18]−. Compared with free HCCPh, the Au–C σ-bond in Au(CCPh) has increased electron density, resulting in a reduction in LUMO energy primarily due to 79.79% contribution from the Au atom (Fig. 1b). This greatly narrows the HOMO–LUMO gap to 2.68 eV in Au(CCPh), as compared to 5.51 eV in HCCPh (Fig. S2a), enhancing the reactivity. Fig. 1c presents the AIMD snapshots at 298 K detailing the key stages in this exchange process, which displays the dynamic changes in bond lengths and Mayer bond order (MBO). Initially, a strong attraction forms between the active Au363 atom in Au(CCPh) and the apex S20 atom on the staple motif (stage (i)). This attraction leads to reinforcement of the Au363⋯S20 interaction, evidenced by a reduced bond length (<3 Å) and an MBO of 0.74 within the first 1 ps (Fig. 1c). Concurrently, the staple Au11–S20 bond weakens, as reflected by the dropping of its MBO to 0.51. At around 3 ps, a covalent bond forms between Au363 and S20 (MBO = 0.74), while the staple Au11–S20 bond and the surface Au12–S21 bond elongate to ∼3 Å, with their MBOs falling to zero, marking a significant structural reconfiguration (stage (ii)). At around 9 ps, the C350 atom from the alkynyl CC group coordinates with the surface Au12 atom, forming a stable δ-bond interaction that completes the ligand exchange process (stage (iii); MBO = 0.70). This exchange process demonstrates a characteristic associative SN2-like mechanism,30,61 in which both the incoming and outgoing ligands transiently coordinate with Au atoms, facilitating the smooth ligand substitution. The CM5 charge analysis (Fig. S3) captures the transient charge redistribution during the exchange reaction, showing temporary electron fluctuations as ligands exchange within the Au25 framework. This exchange culminates in a stable “S1” regioisomer, characterized by [Au–SR] units being directly replaced with Au(CCPh) fragments (Fig. 1c). These findings underscore the critical role of Au+ ions in driving the SN2-like ligand substitution and reveal the intricate dynamics of ligand exchange in metal clusters, highlighting the unique reactivity of Au(CCPh) complexes.
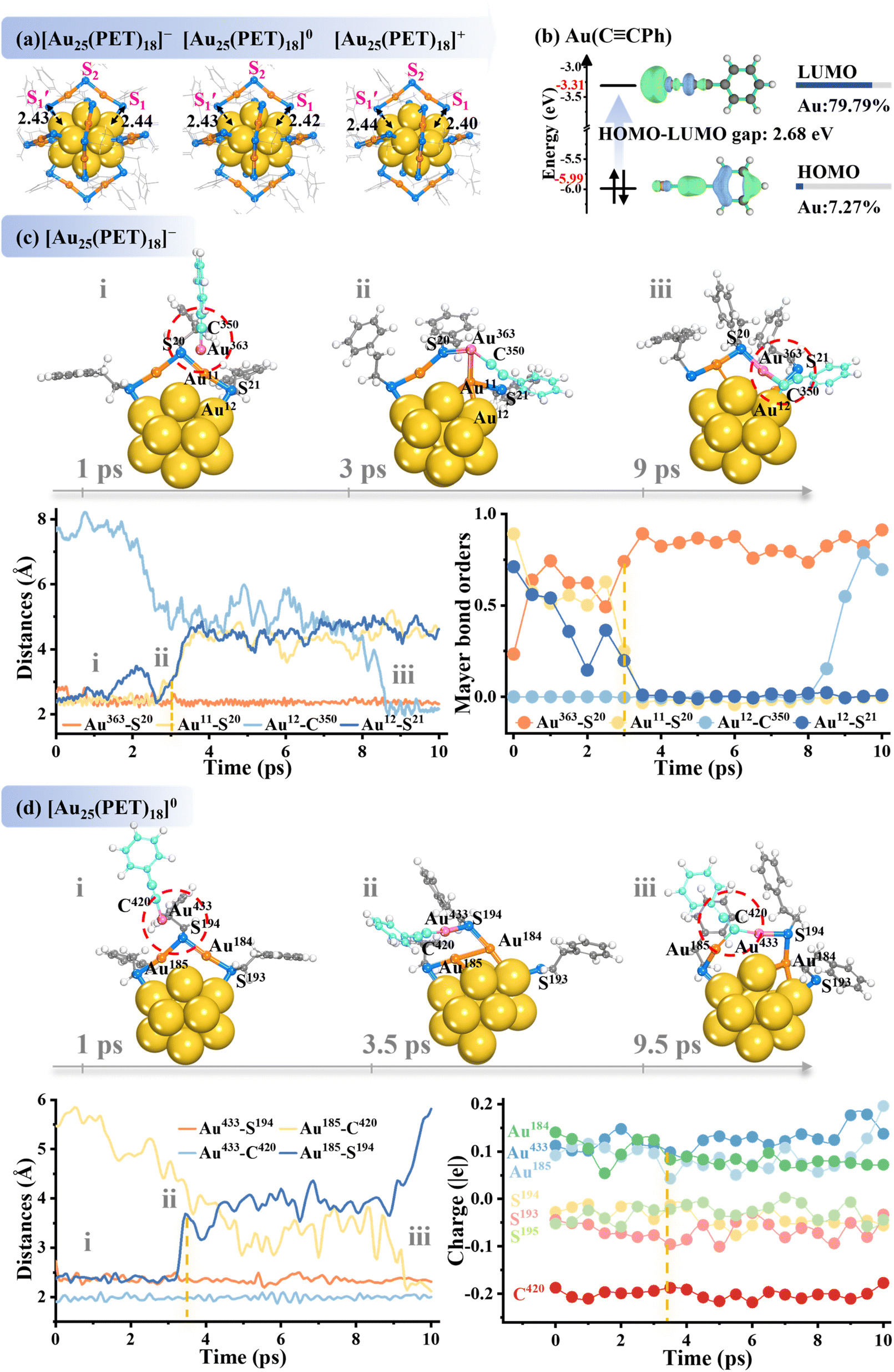 | ||
| Fig. 1 (a) Structures of [Au25(PET)18]q clusters (q = −1, 0, 1; PET = 2-phenylethanethiol), where the surface Au–S bond lengths are inset. (b) Frontier orbitals of the Au(CCPh) fragment. (c) AIMD snapshots at 298 K depicting the key stages of Au(CCPh) ligand exchange on [Au25(PET)18]−, displaying the bond length and Mayer bond order variations. (d) Snapshot structures, bond length dynamics, and CM5 charges for Au(CCPh) exchange on neutral [Au25(PET)18]0. For clarity, only the Au13 core and one dimeric staple motif that reacts with the incoming Au(CCPh) ligand are shown in [Au25(PET)18]− (c) and [Au25(PET)18]0 (d), while other staple motifs are omitted. Color code: C – sapphire (incoming ligand); Au – pink (incoming ligand); C – gray (Au25 cluster); S – blue; core Au – yellow (Au25 cluster); staple Au – orange (Au25 cluster); H – white. | ||
Moreover, it is well known that the charge state of metal NC could significantly modulate its electronic structure and, consequently, its reactivity.62,63 Interestingly, Au25 NCs can readily interconvert their charged states between Au25− and Au025 under mild conditions in the presence of oxygen or thiols.62,64 However, the influence of charge state on the ligand-exchange mechanism in Au25 has not been clarified. To probe this, we performed the alkynyl-for-thiol exchange simulations on the neutral [Au25(PET)18]0 NC. Intriguingly, different from the preferred “S1” regioisomer in [Au25(PET)18]−1, the “S2” isomer emerged as the primary exchange product in [Au25(PET)18]0 (Fig. 1d), suggesting a charge-mediated effect on the regioselectivity. In the neutral [Au25(PET)18]0 NC, the Au433 atom in the Au(CCPh) complex moves closer to the apex S194 (3.50 Å, stage (i)), destabilizing the staple Au185–S194 bond (2.81 Å) by drawing the electron density away from the S, which subsequently dissociated at around 3.5 ps (stage (ii)). In particular, this bond disruption allows the gradual formation of a covalent Au185–C420 bond, progressively shortening from 4.37 Å to 2.18 Å as the electron density redistributes from the Au center to the CC group (stage (iii)). The formation of the Au185–C420 bond not only stabilizes the incoming alkynyl ligand but also imposes significant steric strain on the adjacent [S194–Au184–S193] unit. This dynamic interaction results in the displacement of the S2R fragment and ultimately expels the sterically crowded [Au184–S193] unit, favoring the formation of the S2 regioisomer as the major exchange product. Note that the symmetrical geometry of [Au25(PET)18]0 always promotes selective substitution at the apex S2 site, independent of the initial location of the incoming Au(CCPh) ligand (Fig. S4). Herein, the observed regioselectivity is attributed to the unique electronic properties of the Au–CC bond, characterized by the d–π* back-bonding between the occupied Au d orbitals and the π* orbitals of the CC bond. This interaction enhances the CC bond's reactivity, thereby promoting the ligand substitution. In addition, we also investigated the ligand exchange of Au(CCPh) with positively charged [Au25(SR)18]+. Our simulations revealed that the Au(CCPh) ligand would favorably attack and insert into the staple Au–S(apex) bond, which forms an elongated and distorted trimeric [RS–Au–PhCC–Au–SR–Au–SR] motif, limiting effective alkynyl ligand replacement (Fig. S5). This interesting charge-dependent transformation demonstrates how the charge state modulates the exchange dynamics and regioselectivity in the alkynyl-for-thiolate exchange reaction. These insights underscore the great promise for using charge-state control to drive regioselective substitution in gold NCs with finely tuned reactivity and stability.
Fig. 2 summarizes the overall structural variation of [Au25(PET)18]−, [Au25(PET)18]0 and [Au25(PET)18]+ NCs from stage (i) to stage (iii) during the ligand exchange process within the 10 ps AIMD simulations at 298 K. Note that the simulations were conducted by reacting one Au25 NC with 10 equivalents of Au(CCPh) molecules, and we observed that two Au(CCPh) ligands can exchange onto [Au25(PET)18]0 in a pair of symmetrical staple motifs, while only one Au(CCPh) ligand is added onto [Au25(PET)18]− and [Au25(PET)18]+. Noteworthily, the SR ligand replacement in Au25 NCs consistently occurs at the exposed Au3 faces of the uncapped icosahedron, independent of the charge state (note that the icosahedra has 20 triangular faces in total, the formation of six staple motifs leaves eight Au3 faces that are not capped, and herein only two of the adjacent Au3 triangles are shown shaded in green and red, wherein the three surface Au–Au bond lengths are inset in each shaded triangle). As shown in Fig. 2, the change in the charge state of Au25q NCs has a minimal effect on the Austaple–S bond lengths but significantly alters the Aucore–S bonding at the “S1” position (stage (i)), which shortens as the cluster charge increases (from 2.44 Å in Au25− to 2.40 Å in Au25+). Furthermore, increasing the charge state elongates the Au–Au bonds within the Au3 faces, particularly in the exposed “pockets” marked by the red Au3 triangle close to the “S1” position. The induced local strain as well as the longer Aucore–S bond thus offer enhanced flexibility and facilitate the reaction at the “S1” position. Moreover, during the ligand exchange reaction, the icosahedral Au13 core undergoes considerable reconstruction (Fig. S6). Initially having an approximate D2h symmetry, the stability of the icosahedron core is intensively sensitive to the ligand bonding modes.43 Rigid alkynyl ligands like –CCPh would induce atomic rearrangement of surface atoms around the exchange site, which transiently destabilizes the core upon coexistence with alkynyl ligands until the stability is restored upon complete alkynyl integration (stage (iii)). In the anionic Au25− NC, the shorter Au–Au bonds on the surface of Au13 core impart greater stability, with minimal structural deviation in the mono-alkynyl-coordinated cluster. In contrast, the neutral Au025 NC shows slight distortion in the post-exchange process, while the cationic Au25+ NC experiences further amplified distortion in the Au13 core. Obviously, as the cluster charge progresses from −1 to +1, the charge-driven rearrangements increasingly influence the ligand exchange pathway, underscoring its critical role in controlling the structural stability and specificity in the substitution site.
 | ||
| Fig. 2 The overall structural variation of [Au25(PET)18]−, [Au25(PET)18]0 and [Au25(PET)18]+ NCs from stage (i) to stage (ii) and to stage (iii) during the 10 ps AIMD simulations at 298 K in the ligand exchange process. Color code: C – sapphire (incoming ligand); Au – pink (incoming ligand); C – gray (Au25 cluster); S – blue; core Au – yellow (Au25 cluster); staple Au – orange (Au25 cluster); H – white. | ||
Building on our above investigation of the exchange reaction between Au25 and Au(CCPh), we further examined the exchange of lithium phenylacetylide (Li(CCPh)) on Au25(PET)18 to understand how the change in the metal center affects the exchange reaction dynamics. Lithium, with a smaller ionic radius (1.52 Å) and higher electropositivity (0.98), forms a significantly more ionic Li–C bond compared to the covalent Au–C bond in Au(CCPh). This ionic character alters the electronic environment of the alkynyl group, reducing the electron delocalization and thereby increasing the HOMO and LUMO energies of the CC bond (−5.05 and −1.38 eV, respectively, Fig. 3a), driven by a high contribution (99.05%) from Li in the LUMO, as compared to Au(CCPh). The resulting electron-rich CC bond displays greater nucleophilicity, enhancing its coordination ability with the Au site in the cluster's staple motif, facilitating a more efficient ligand exchange. In Fig. 3b, the –CCPh moiety initially coordinates with the staple Au180 of [Au25(PET)18]−, prompting the structural rearrangement that enhances the nucleophilic interaction. This coordination strengthens the Au180–C448 bond, stabilizing at 2.5 Å around 2.5 ps, while concurrently the Li–C bond weakens to 2.0 Å, allowing the Li atom to approach S190, which in turn induces steric and electronic strain that weakens the surface Au179–S190 bond, extending it to 2.85 Å. At 6.5 ps (Fig. 3b, stage (ii)), the increased interaction between Au180 and C448 (2.07 Å; MBO = 0.86; Fig. S7) facilitates the dissociation of the staple Au180–S190 bond, as indicated by the MBO reduction to 0.15. This bond reconfiguration induces the formation of a polar covalent Li–S bond with significant ionic character (2.51 Å; MBO = 0.32), anchoring the formed RS–Li complex to the cluster surface. Simultaneously, the Li–C bond weakens, as evidenced by its elongation (2.21 Å). This progression ultimately releases the –CCPh moiety, allowing it to replace the original SR ligand and form the “ ” isomer (stage (iii), Fig. 3b). This exchange mechanism demonstrates that the ionic nature of the Li–C bond, in combination with the nucleophilic feature of the –CCPh moiety, promote an efficient ligand exchange in [Au25(PET)18]−.
” isomer (stage (iii), Fig. 3b). This exchange mechanism demonstrates that the ionic nature of the Li–C bond, in combination with the nucleophilic feature of the –CCPh moiety, promote an efficient ligand exchange in [Au25(PET)18]−.
 | ||
| Fig. 3 (a) Frontier orbitals of the Li(CCPh) fragment. (b) AIMD snapshots depicting key stages of Li(CCPh) ligand exchange on [Au25(PET)18]−, as well as the bond length variations during the 10 ps simulations at 298 K. The variation of Mayer bond order in ligand exchange of Li(CCPh) on [Au25(PET)18]0 (c) and [Au25(PET)18]+ (d) during the 10 ps AIMD simulations at 298 K, where the final snapshot structures are shown in the inset. The Li atom in the incoming ligand is colored in purple. Color code: C – sapphire (incoming ligand); C – gray (Au25 cluster); S – blue; core Au – yellow (Au25 cluster); staple Au – orange (Au25 cluster); Li – purple; H – white. | ||
In addition, similar to the case in [Au25(PET)18]−, both [Au25(PET)18]0 (Fig. 3c) and [Au25(PET)18]+ (Fig. 3d) also preferably produce the exchange isomer at the “ ” site when interacting with the incoming Li(CCPh) (Fig. S8 and S9). Notably, despite the greater MBO fluctuations in Au25+ indicating the higher structural sensitivity, the cluster's structural integrity remains stable across different charge states during the exchange process. Interestingly, the Li atom in Li(CCPh) primarily serves as a mediator in the alkynyl-for-thiolate exchange, facilitating the exchange but without integrating into the cluster's structure. This may explain why the overall structural reorganization of Au25 is less pronounced than that in the exchange with Au(CCPh). Overall, the ionic Li–C bond facilitates the dynamic adjustments, promoting the regioselective substitution with minimal steric hindrance. By comparison, the covalent Au–C bond in Au(CCPh) stabilizes the interactions with Au25 but restricts exchange flexibility, highlighting a key distinction in their reactivity. These findings illustrate that the metals in acetylide with distinct bonding characteristics (e.g., ionic versus covalent) can significantly affect the ligand exchange dynamics. Leveraging these differences opens up new avenues for designing functionalized gold NCs with tailored properties, enabling precise control over cluster stability and reactivity.
” site when interacting with the incoming Li(CCPh) (Fig. S8 and S9). Notably, despite the greater MBO fluctuations in Au25+ indicating the higher structural sensitivity, the cluster's structural integrity remains stable across different charge states during the exchange process. Interestingly, the Li atom in Li(CCPh) primarily serves as a mediator in the alkynyl-for-thiolate exchange, facilitating the exchange but without integrating into the cluster's structure. This may explain why the overall structural reorganization of Au25 is less pronounced than that in the exchange with Au(CCPh). Overall, the ionic Li–C bond facilitates the dynamic adjustments, promoting the regioselective substitution with minimal steric hindrance. By comparison, the covalent Au–C bond in Au(CCPh) stabilizes the interactions with Au25 but restricts exchange flexibility, highlighting a key distinction in their reactivity. These findings illustrate that the metals in acetylide with distinct bonding characteristics (e.g., ionic versus covalent) can significantly affect the ligand exchange dynamics. Leveraging these differences opens up new avenues for designing functionalized gold NCs with tailored properties, enabling precise control over cluster stability and reactivity.
Ligand exchange between Au25 and Au/Li(CCtBu)
Furthermore, based on the insights from the –CCPh exchange, we additionally considered the bulky tert-butyl group –CCtBu to examine how the increased steric hindrance impacts the ligand exchange in Au25 NCs. The tert-butyl group introduces considerable steric effects and unique electronic characteristics, providing an opportunity to understand how the presence of larger substituents modifies the exchange behavior. As shown in Fig. S10a, HCCtBu has a significantly wider HOMO–LUMO gap of 8.73 eV compared to 5.51 eV in HCCPh, primarily due to the electron localization. Incorporating Au or Li into the –CCtBu group notably reduces the LUMO energy (Fig. 4a), resulting in a narrower HOMO–LUMO gap of 3.40 eV and 4.06 eV, respectively. Compared to their Au/Li(CCPh) analogues (2.68 and 3.66 eV), the bulkier Au/Li(CCtBu) complexes demonstrate an increased HOMO–LUMO gap, underscoring the combined effects of metal incorporation and steric bulkiness in fine-tuning the electronic properties.
 | ||
Fig. 4 (a) Frontier orbitals of Au(CCtBu) and Li(CCtBu) fragments. (b) Free energy barriers for the ligand exchange reaction between Au(CCtBu) and [Au25(PET)18]q (q = −1, 0, and 1) at S1, S2,  , S2 (near , S2 (near  ), and ), and  sites during the c-MD simulations. (c) Reaction mechanism and free energy diagram for the Au(CCtBu) ligand exchange on [Au25(PET)18]− at the S1 site. Color code: C – sapphire (incoming ligand); Au – pink (incoming ligand); C – -gray (Au25 cluster); S – blue; core Au – yellow (Au25 cluster); staple Au – orange (Au25 cluster); H – white. sites during the c-MD simulations. (c) Reaction mechanism and free energy diagram for the Au(CCtBu) ligand exchange on [Au25(PET)18]− at the S1 site. Color code: C – sapphire (incoming ligand); Au – pink (incoming ligand); C – -gray (Au25 cluster); S – blue; core Au – yellow (Au25 cluster); staple Au – orange (Au25 cluster); H – white. | ||
We first assessed the exchange feasibility of the HCCtBu ligand. From Fig. S10b, our c-MD simulations at 298 K revealed a high free energy barrier of 3.48 eV for –CCtBu binding with the staple Au in [Au25(PET)18]− to realize the “ ” exchange, indicating the significant challenge of the HCCtBu exchange. In the case of Au(CCtBu), the unconstrained AIMD simulations around 10 ps at 298 K in various Au25 charge states (−1, 0, and +1) showed that the incoming Au(CCtBu) ligand would be adsorbed and bonded to the S atom of the original thiolate ligand, but no noticeable ligand exchange was observed (Fig. S11). Note that this finding is drastically distinct from that of the prior Au(CCPh) ligand where the Au(CCPh)-for-thiolate exchange occurs spontaneously in the unconstrained 10 ps AIMD simulations. This suggests that the incorporation of the bulkier (–CCtBu) group would greatly decelerate the exchange reaction kinetics. To capture the activation barrier for the ligand exchange reaction, we then employed the c-MD free energy calculations to analyze the formation of an exchange isomer in Au25 when it reacts with the bulkier Au(CCtBu) complex. Herein, we investigated three potential exchange sites: S1, S2 and
” exchange, indicating the significant challenge of the HCCtBu exchange. In the case of Au(CCtBu), the unconstrained AIMD simulations around 10 ps at 298 K in various Au25 charge states (−1, 0, and +1) showed that the incoming Au(CCtBu) ligand would be adsorbed and bonded to the S atom of the original thiolate ligand, but no noticeable ligand exchange was observed (Fig. S11). Note that this finding is drastically distinct from that of the prior Au(CCPh) ligand where the Au(CCPh)-for-thiolate exchange occurs spontaneously in the unconstrained 10 ps AIMD simulations. This suggests that the incorporation of the bulkier (–CCtBu) group would greatly decelerate the exchange reaction kinetics. To capture the activation barrier for the ligand exchange reaction, we then employed the c-MD free energy calculations to analyze the formation of an exchange isomer in Au25 when it reacts with the bulkier Au(CCtBu) complex. Herein, we investigated three potential exchange sites: S1, S2 and  , where S1 is located within the pocket region, as described previously. Note that our above studies on the Au(CCPh) exchange have highlighted the critical role of S1 site in the ligand exchange, particularly within the pocket region. Based on these findings, we also considered additional sites: the
, where S1 is located within the pocket region, as described previously. Note that our above studies on the Au(CCPh) exchange have highlighted the critical role of S1 site in the ligand exchange, particularly within the pocket region. Based on these findings, we also considered additional sites: the  position within the pocket and the S2 position adjacent to
position within the pocket and the S2 position adjacent to  , hereafter referred to as ‘S2 (near
, hereafter referred to as ‘S2 (near  )’ (Fig. 4b, inset). This broader analysis provides a more comprehensive view of ligand exchange dynamics in and around the pocket site, as revealed by the c-MD simulations. The calculated energy barriers for the ligand exchange at the five designated positions—S1, S2,
)’ (Fig. 4b, inset). This broader analysis provides a more comprehensive view of ligand exchange dynamics in and around the pocket site, as revealed by the c-MD simulations. The calculated energy barriers for the ligand exchange at the five designated positions—S1, S2,  , S2 (near
, S2 (near  ), and
), and  —are presented in Fig. 4b. Note that the substitution at the S2 and S2 (near
—are presented in Fig. 4b. Note that the substitution at the S2 and S2 (near  ) positions is hindered by the steric effects (Fig. S12–S14), resulting in incomplete exchange despite the relatively lower barriers. In contrast, the S1 position consistently exhibits the lowest energy barriers across all the charge states (0.84 eV for Au25−, 1.43 eV for Au025, and 1.2 eV for Au25+), highlighting the energetic preference of ligand exchange at the S1 site. The higher barriers and significant steric effects at other positions (e.g., S2 and
) positions is hindered by the steric effects (Fig. S12–S14), resulting in incomplete exchange despite the relatively lower barriers. In contrast, the S1 position consistently exhibits the lowest energy barriers across all the charge states (0.84 eV for Au25−, 1.43 eV for Au025, and 1.2 eV for Au25+), highlighting the energetic preference of ligand exchange at the S1 site. The higher barriers and significant steric effects at other positions (e.g., S2 and  ) limit their accessibility, making S1 isomer as the most favorable exchange product across all the charge states (Fig. S12–S14). Consequently, the bulkier Au(CCtBu) incoming ligand exhibits a pronounced regioselectivity toward the S1 position near the pocket, while Au(CCPh) shows charge-dependent regioselectivity.
) limit their accessibility, making S1 isomer as the most favorable exchange product across all the charge states (Fig. S12–S14). Consequently, the bulkier Au(CCtBu) incoming ligand exhibits a pronounced regioselectivity toward the S1 position near the pocket, while Au(CCPh) shows charge-dependent regioselectivity.
Moreover, analogous to the Au(CCPh)-for-thiolate exchange observed in the unconstrained MD simulations, the Au(CCtBu)-for-thiolate exchange follows a two-step mechanism. In the first step, the bulkier Au(CCtBu) ligand adsorbs at the S site. The second step is determined by the ligand's binding mode: coordination to the Au13 core leads to the formation of S1 or  isomers, while binding to the Austaple results in S2 or S2 (near
isomers, while binding to the Austaple results in S2 or S2 (near  ) isomers (Fig. S12–S14). The energy barriers shown in Fig. 4b correspond to three distinct pathways: (i) formation of the S1 isomer, (ii) formation of the S2 and S2 (near
) isomers (Fig. S12–S14). The energy barriers shown in Fig. 4b correspond to three distinct pathways: (i) formation of the S1 isomer, (ii) formation of the S2 and S2 (near  ) isomers, and (iii) formation of the
) isomers, and (iii) formation of the  and
and  isomers, as illustrated in Fig. 4c and S15. Taking the formation of the S1 isomer on the Au25− cluster as an example, the S–Au bond first cleaves with a low energy barrier (≤0.57 eV), facilitating stable adsorption of the –CCtBu ligand onto the staple Au moiety (Fig. 4c). In the subsequent step (the rate-determining step), the –CCtBu moiety forms a C–Au α-bond with the Au13 core, requiring a higher energy barrier (≥0.84 eV) to complete the exchange. However, the moderate overall barriers (≤1.5 eV) indicate that both steps are experimentally accessible. Notably, the rate-limiting formation of the C–Au α-bond occurs most readily in the [Au25(PET)18]− cluster (0.84 eV), followed by [Au25(PET)18]+ (1.20 eV) and [Au25(PET)18]0 (1.43 eV) (Fig. 4b). Additionally, the exchange mechanisms for the S2 (near
isomers, as illustrated in Fig. 4c and S15. Taking the formation of the S1 isomer on the Au25− cluster as an example, the S–Au bond first cleaves with a low energy barrier (≤0.57 eV), facilitating stable adsorption of the –CCtBu ligand onto the staple Au moiety (Fig. 4c). In the subsequent step (the rate-determining step), the –CCtBu moiety forms a C–Au α-bond with the Au13 core, requiring a higher energy barrier (≥0.84 eV) to complete the exchange. However, the moderate overall barriers (≤1.5 eV) indicate that both steps are experimentally accessible. Notably, the rate-limiting formation of the C–Au α-bond occurs most readily in the [Au25(PET)18]− cluster (0.84 eV), followed by [Au25(PET)18]+ (1.20 eV) and [Au25(PET)18]0 (1.43 eV) (Fig. 4b). Additionally, the exchange mechanisms for the S2 (near  ) and
) and  isomer formations (ii and iii) on [Au25(PET)18]− are depicted in Fig. S15, along with the corresponding key geometric structures.
isomer formations (ii and iii) on [Au25(PET)18]− are depicted in Fig. S15, along with the corresponding key geometric structures.
Fig. S16 illustrates the structural evolution of [Au25(PET)18]−, [Au25(PET)18]0 and [Au25(PET)18]+ NCs from the initial state (IS) through an intermediate state to the final state (FS) during the ligand exchange via c-MD simulations. The results confirm that the SR replacement is restricted to the exposed “pockets” marked by the red Au3 triangle, regardless of the charge state. Variations in the charge state primarily affect the local bonding environment at the Aucore–S1 and  positions (IS). Specifically, as the charge of Au25q NCs increases, the Aucore–S1 bond elongates from 2.36 Å in Au25− to 2.47 Å in Au25+, while the
positions (IS). Specifically, as the charge of Au25q NCs increases, the Aucore–S1 bond elongates from 2.36 Å in Au25− to 2.47 Å in Au25+, while the  bond extends from 2.39 Å to 2.43 Å. These changes are accompanied by the slight elongation of Au–Au bonds within the Au3 face within the pocket region (red triangle, Fig. S16). The Aucore–S bond at “S1” is consistently longer than that at “
bond extends from 2.39 Å to 2.43 Å. These changes are accompanied by the slight elongation of Au–Au bonds within the Au3 face within the pocket region (red triangle, Fig. S16). The Aucore–S bond at “S1” is consistently longer than that at “ ”, inducing tensile strain localized at the S1 site. This strain enhances the dynamic flexibility of the S1 site, weakening the Aucore–S covalent interaction and collectively facilitating ligand exchange with the sterically demanding Au(CCtBu) ligands. Notably, a slight reconstruction of the Au13 core occurs during the exchange process, which is caused by the surface atom rearrangements driven by the bulky alkynyl ligands temporarily destabilizing the core structure. This instability is mitigated once the –CCtBu moiety fully integrates into the cluster framework in the final state (FS). Note that the higher charge states amplify these structural distortions, reducing the bond length uniformity across the cluster. These findings also highlight the significant impact of charge states on the ligand exchange at the single-ligand level.
”, inducing tensile strain localized at the S1 site. This strain enhances the dynamic flexibility of the S1 site, weakening the Aucore–S covalent interaction and collectively facilitating ligand exchange with the sterically demanding Au(CCtBu) ligands. Notably, a slight reconstruction of the Au13 core occurs during the exchange process, which is caused by the surface atom rearrangements driven by the bulky alkynyl ligands temporarily destabilizing the core structure. This instability is mitigated once the –CCtBu moiety fully integrates into the cluster framework in the final state (FS). Note that the higher charge states amplify these structural distortions, reducing the bond length uniformity across the cluster. These findings also highlight the significant impact of charge states on the ligand exchange at the single-ligand level.
Furthermore, in contrast to Au(CCtBu), the interaction of Li(CCtBu) with [Au25(PET)18]0, shown in Fig. 5a, reveals a facile spontaneous ligand exchange during the unconstrained AIMD simulations at 298 K. In fact, the strong ionic C–Li interaction increases the nucleophilicity of the alkynyl group, promoting its adsorption onto the staple Au6 motif and facilitating the formation of the Au–C α-bond between the –CCtBu group and the Au6 site (2.19 Å, (ii)) at around 2.2 ps. Concurrently, the Li ion gradually coordinates with the  atom (∼3 Å), and as the Au6–C366 and Li–S16 bonds strengthen, the Au6–S16 bond progressively weakens and eventually breaks (3.5 Å, (iii)) at around 10 ps, completing the exchange process. To further confirm that the –CCtBu group can adsorb onto the Au13 core, we present a free energy calculation for the subsequent
atom (∼3 Å), and as the Au6–C366 and Li–S16 bonds strengthen, the Au6–S16 bond progressively weakens and eventually breaks (3.5 Å, (iii)) at around 10 ps, completing the exchange process. To further confirm that the –CCtBu group can adsorb onto the Au13 core, we present a free energy calculation for the subsequent  isomer formation on [Au25(PET)18]0, following the exchange with 10 equivalents of Li(CCtBu) molecules via c-MD simulations (Fig. 5b). The low barrier (0.64 eV) indicates that the exchanged alkynyl group can readily coordinate with the unsaturated Au atom on the icosahedron surface, stabilizing the Au13 core and forming newly mixed-ligand capped Au025 NCs. In the case of the negatively charged [Au25(PET)18]− cluster, the c-MD simulations (Fig. 5c) reveal the initially spontaneous adsorption of the –CCtBu moiety onto the staple Au site with a minimal energy barrier (0.05 eV, Fig. S17). This is followed by the ligand exchange at the
isomer formation on [Au25(PET)18]0, following the exchange with 10 equivalents of Li(CCtBu) molecules via c-MD simulations (Fig. 5b). The low barrier (0.64 eV) indicates that the exchanged alkynyl group can readily coordinate with the unsaturated Au atom on the icosahedron surface, stabilizing the Au13 core and forming newly mixed-ligand capped Au025 NCs. In the case of the negatively charged [Au25(PET)18]− cluster, the c-MD simulations (Fig. 5c) reveal the initially spontaneous adsorption of the –CCtBu moiety onto the staple Au site with a minimal energy barrier (0.05 eV, Fig. S17). This is followed by the ligand exchange at the  position, which occurs with a lower energy barrier (1.08 eV) compared to the S1 site (1.57 eV, Fig. S18), confirming the preferred regioselectivity of the S1 isomer. For the positively charged [Au25(PET)18]+ cluster, the exchange barrier at the
position, which occurs with a lower energy barrier (1.08 eV) compared to the S1 site (1.57 eV, Fig. S18), confirming the preferred regioselectivity of the S1 isomer. For the positively charged [Au25(PET)18]+ cluster, the exchange barrier at the  position is 0.94 eV (Fig. 5d); nevertheless, the final state (FS) exhibits greater geometric distortion and instability in the icosahedral Au13 core. These findings highlight that the incorporation of Li into the –CCtBu moiety ensures 100% selective formation of the
position is 0.94 eV (Fig. 5d); nevertheless, the final state (FS) exhibits greater geometric distortion and instability in the icosahedral Au13 core. These findings highlight that the incorporation of Li into the –CCtBu moiety ensures 100% selective formation of the  isomer. The exchange with the Li(CCtBu) group to form the
isomer. The exchange with the Li(CCtBu) group to form the  isomer is energetically more facile on [Au25(SR)18]0, followed by [Au25(PET)18]− and [Au25(PET)18]+ NCs. These results underscore the potential to engineer metal clusters with tailored steric and electronic properties, offering precise control over the ligand exchange efficiency and reactivity.
isomer is energetically more facile on [Au25(SR)18]0, followed by [Au25(PET)18]− and [Au25(PET)18]+ NCs. These results underscore the potential to engineer metal clusters with tailored steric and electronic properties, offering precise control over the ligand exchange efficiency and reactivity.
 | ||
Fig. 5 (a) AIMD snapshots depicting key stages of Li(CCtBu) ligand exchange on [Au25(PET)18]0, along with the bond length variations during the 10 ps unconstrained AIMD simulations at 298 K. (b) Mechanistic pathway and free energy diagram of subsequent  isomer formation on the [Au25(PET)18]0 cluster upon exchange with 10 equivalents of Li(CCtBu) molecules. Mechanistic step and free energy diagram of isomer formation on the [Au25(PET)18]0 cluster upon exchange with 10 equivalents of Li(CCtBu) molecules. Mechanistic step and free energy diagram of  isomer formation during Li(CCtBu) exchange with (c) [Au25(PET)18]− and (d) [Au25(PET)18]+ clusters. Color code: C – sapphire (incoming ligand); C – gray (Au25 cluster); S – blue; core Au – yellow (Au25 cluster); staple Au – orange (Au25 cluster); Li – purple; H – white. isomer formation during Li(CCtBu) exchange with (c) [Au25(PET)18]− and (d) [Au25(PET)18]+ clusters. Color code: C – sapphire (incoming ligand); C – gray (Au25 cluster); S – blue; core Au – yellow (Au25 cluster); staple Au – orange (Au25 cluster); Li – purple; H – white. | ||
Furthermore, to confirm the applicability of AIMD in simpler systems (such as mononuclear complexes) and different reaction systems, we simulated the PET-to-TBBT (TBBT = 4-tert-butylbenzenethiolate) conversion in Au25. As shown in Fig. S19, AIMD simulations of this process successfully identified a high energy barrier of 1.91 eV. The reaction proceeds through a concerted attack by H and S of TBBT on the S and Austaple of Au25, respectively (IS), leading to TBBT adsorption onto Austaple (intermediate state), ultimately leading to PET dissociation (FS). Crucially, the PET-to-TBBT conversion offers a structurally less complex and core-stable way compared to the original SR-to-alkynyl exchange. These simulations confirm that AIMD can effectively capture the key dynamic features (such as energy barriers) in ligand displacement processes.
Experimental validation
In order to verify the alkynyl-for-thiolate exchange on the Au25 clusters, [Au25(PET)18]− was used as the starting material, and the ligand exchange products were detected by electrospray ionization mass spectroscopy (ESI-MS) measurements. TOA[Au25(PET)18] (TOA[Au25], TOA = tetraoctylammonium) was synthesized in accordance with the previous report.60 The ligand-exchange reaction of [Au25]− with –CCtBu was performed by adding excess Au(CCtBu) to a dichloromethane (DCM) solution of TOA[Au25] at room temperature in darkness for 3 hours. After the ligand exchange process, the 695 nm peak in the UV/vis/NIR absorption spectra of [Au25]− red-shifted to 715 nm (Fig. S20). In positive mode ESI-MS, three discrete peaks in the range m/z 3650–3750 can be discerned at m/z3662.67, 3690.67 and 3718.65, respectively (Fig. 6a). These complexes can be assigned to [Au25(PET)18−x(CCtBu)x + 2Cs]2+ with the x values of 6, 5, and 4, respectively. Within the range m/z7160 to 7340 , another three peaks are observed at m/z7190.14, 7247.10 and 7303.05, respectively (Fig. 6b). The three products can be designated as [Au25(PET)18−x(CCtBu)x + Cs]+ with x values of 6, 5, and 4, respectively. The ESI-MS data of the two different valence products indicate that 4, 5 and 6 PET ligands have been exchanged for –CCtBu on [Au25]− during the process of ligand exchange. The isotopic pattern also matched well with [Au25(PET)13(CCtBu)5 + 2Cs]2+ and [Au25(PET)13(CCtBu)5 + Cs]+ (Fig. 6c and d). The remaining isotopic patterns matched well with the simulations (Fig. S21 and S22), and Au25(PET)18−x(CCtBu)x can be further auto-oxidized to [Au25(PET)18−x(CCtBu)x]+. As shown in Fig. S23, three discrete peaks in the range m/z 7040 and 7181 can be discerned at m/z7058.14, 7114.22 and 7170.26, respectively. These complexes can be assigned to [Au25(PET)18−x(CCtBu)x]+ with x values of 6, 5, and 4, respectively. Altogether, these pieces of experimental evidence clearly show that it is possible to exchange the PET ligand with the new –CCtBu ligand on the [Au25(PET)18]− nanocluster under ambient conditions.
 | ||
| Fig. 6 Positive-ion mode ESI-MS spectra of the product clusters (a) [Au25(PET)18−x(CCtBu)x + 2Cs]2+ and (b) [Au25(PET)18−x(CCtBu)x + Cs]+ after the ligand exchange of PET with –CCtBu, and x represents the number of exchanged ligands. (c and d) Experimental (dark lines) and simulated (red lines) isotopic pattern comparisons of [Au25(PET)13(CCtBu)5 + 2Cs]2+ and [Au25(PET)13(CCtBu)5 + Cs]+, respectively. | ||
For the synthesis of Au(CCtBu), it is essential to retain the acetone solvent and avoid its loss. Removal of acetone would prevent the complete re-dissolution of Au(CCtBu) and subsequent ligand exchange. Furthermore, during the ligand exchange reaction between Au(CCtBu) and TOA[Au25(PET)18], significant insoluble precipitation was observed following PTLC separation after solvent removal. These observations collectively demonstrate that the solvent is essential for maintaining the stability of both Au(CCtBu) and the ligand-exchanged product Au25(PET)18−x(CCtBu)x. Besides Au(CCtBu), Au(CCPh) has also been shown to undergo ligand exchange with TOA[Au25(PET)18].41 Unfortunately, the product has not been successfully identified so far. Due to the difficult access to Li(CCR) in our lab, the related ligand exchange process was only theoretically investigated.
It is noted that our experimental optimization established that 20–40 ligand equivalents are required for Au(CCtBu)-for-PET exchange in the Au25− cluster, indicating tolerance to ligand concentration within this range. Our AIMD simulations employed ligand concentrations of 10–40 equivalents, encompassing the core experimental range (20–40 equivalents) while extending to 10 equivalents to investigate concentration-dependent trends. Simulations revealed consistent ligand binding efficiency, reaction pathways, and exchange outcomes across 10–40 equivalents. This demonstrates system insensitivity to ligand concentration within this window and aligns with experiments showing that concentration primarily governs exchange kinetics—not the fundamental mechanism. Thus, our simulation design directly corresponds to experimental conditions, with the extended range validating result reliability under experimental concentrations. Moreover, computational constraints limited our simulations to single Au(CCtBu)-for-PET exchange in Au25−, yet experimental findings reveal up to six ligand substitutions. To resolve this discrepancy, we propose a possible stepwise substitution mechanism supported by bond length and Bader charge analyses (Fig. S24). The Au25(PET)18 framework features six dimeric [RS–Au(I)–SR–Au(I)–SR] units surrounding the icosahedral Au13 core, yielding three possible distinct regioisomers (S1, S2, and  ) upon ligand exchange. Structural characterization and Bader charge analysis reveal persistently maintained dual S–Au bonds and a more positive charge localized at the central S2 site in mono-substituted clusters, conferring exchange resistance at this position. While following a single Au(CCtBu)-for-PET exchange in Au25−, five S sites (red) exhibit elongated S–Au bonds (Au–S >2.45 Å) and acquire a negative charge. These activated sites originating from distinct Au2(SR)3 units demonstrate greater attraction toward Au atoms in the incoming Au(CCtBu) ligands. Collectively, the progressive structural and electronic modifications induced by initial substitution facilitate subsequent exchanges, thereby accounting for the observed multi-ligand substitution pathway.
) upon ligand exchange. Structural characterization and Bader charge analysis reveal persistently maintained dual S–Au bonds and a more positive charge localized at the central S2 site in mono-substituted clusters, conferring exchange resistance at this position. While following a single Au(CCtBu)-for-PET exchange in Au25−, five S sites (red) exhibit elongated S–Au bonds (Au–S >2.45 Å) and acquire a negative charge. These activated sites originating from distinct Au2(SR)3 units demonstrate greater attraction toward Au atoms in the incoming Au(CCtBu) ligands. Collectively, the progressive structural and electronic modifications induced by initial substitution facilitate subsequent exchanges, thereby accounting for the observed multi-ligand substitution pathway.
Discussion
Electronic effects
Our above results on the kinetics of –CCR-for-thiolate exchange in [Au25(PET)18]q NCs (q = −1, 0, and +1) unveil a highly innovative and unique mechanism governing the reactivity and selectivity. Our findings highlight the decisive role of metal–alkynyl complexes (e.g., Au(CCPh) and Li(CCPh)) in driving the alkynyl-for-thiolate exchange, a metathesis reaction involving thiolate and alkynyl groups. The metal fundamentally alters the electronic properties of the –CCR group, significantly enhancing their reactivity compared to the rigid HCCR molecule. The incorporation of Au (rAu = 1.36 Å) or Li (rLi = 1.28 Å) enhances the spatial flexibility of the Au–C (1.926 Å) and Li–C (1.896 Å) bonds, while the increased electron-donating capability of CC moiety facilitates efficient electronic reorganization, thereby facilitating the ligand exchange. The Electron Localization Function (ELF) analysis confirms this mechanism, where the electron localization around the CC moiety of HCCPh and HCCtBu is concentrated and rigid, limiting the interactions with Au25(SR)18 (Fig. S25a and b). By contrast, Au incorporation into the –CCR group induces significant electron redistribution (Fig. S25c and d), reducing electron localization around the Au–C bond and creating an asymmetric electron distribution along the CC bond. The Localization Molecular Orbital (LMO) analysis (Fig. S26) further reveals that this redistribution originates from the d–π* interactions, involving back-donation of the occupied Au d-orbital electrons into the π* orbitals of the CC moiety. This back-donation not only strengthens the covalent character of the Au–C bond but also redistributes and polarizes the π*-electron density of the alkynyl group, thereby significantly enhancing its reactivity. Frontier molecular orbital analysis (Fig. 1b and 4a) further supports this mechanism, with the LUMO dominated by the Au contributions (79.79% and 79.42%), highlighting the Au-driven electronic polarization and structural activation of the alkynyl ligand. Moreover, for the Li–alkynyl complex (Fig. S25e and f), the ELF analysis confirms the predominantly ionic nature of the Li–C bond with limited charge transfer. Nevertheless, this ionic interaction intensifies the π*-electron density around the CC bond, boosting the alkynyl nucleophilicity, as reflected in the LMO analysis (Fig. S26). Intriguingly, while Li does not directly participate in the exchange reaction, its capacity to polarize the –CCR electronic structure plays a critical role for the ligand exchange. Together, these findings establish that the distinct yet complementary roles of Au and Li in alkynyl-to-thiolate exchange arise from their distinct contribution to the electronic structure, with Au driving the covalent activation and Li inducing the ionic polarization.
Steric hindrance effects
Steric hindrance critically governs the dynamics and regioselectivity of the ligand exchange. The bulkier HCCtBu with a kinetic diameter of 6.99 Å, imposes greater spatial constraints than HCCPh (4.03 Å, Fig. S27). The Interaction Region Indicator (IRI) analysis reveals pronounced steric clashes between –CCtBu and PET ligands on Au25, restricting the binding-site accessibility and slowing the exchange kinetics (Fig. S28). The compact –CCPh experiences minimal steric interference, forming weak cluster–surface interactions. Taking Au(CCR)-for-thiolate exchange as an example, this accessibility permits selective exchange at the S1 or S2 positions depending on the charge state, while the bulky –CCtBu limits the exchange process only to the less hindered S1 site across all the charge states. These comparative analyses demonstrate how the ligand bulkiness dictates the exchange efficiency and positional selectivity through steric modulation of binding-site accessibility and surface interaction dynamics.
Charge state of Au25 clusters
The charge-state variations in Au25 NCs modulate the structural stability, which in turn regulates the ligand binding dynamics. These effects stem from the quantum size properties of Au25 NCs, which govern their electronic and absorptive behavior.43 The anionic [Au25(PET)18]− demonstrates the maximal stability, minimizing the reorganization during interaction with Au/Li(CCR) (Fig. 2 and S16). Variations in charge states induce the structural adjustments in Aucore–S and Au–Au bond lengths within the pocket region of Au25(PET)18, which directly influence the regioselectivity. For Au(CCPh) with lower steric hindrance, the stable Au25− NC selectively substitutes the [Au–S1R] unit to form the S1 isomer. The neutral Au025 undergoes moderate structural rearrangement, compressing the [Au–S1R] unit to yield the S2 isomer. Whereas the instability of the Au13 core in Au25+ induces significant structural distortion and impedes the exchange. For the bulkier –CCtBu ligand, the Aucore–S bond lengths provide a steric advantage to the less hindered S1 position. Notably, the Li(CCR) complexes exhibit the charge-state-independent reactivity, achieving nearly 100%  selectivity due to the ionic Li–C bond's insensitivity to the electrostatic modulation. These results reflect the exchange regioselectivity as a synergistic interplay of steric effects and charge-driven structural dynamics.
selectivity due to the ionic Li–C bond's insensitivity to the electrostatic modulation. These results reflect the exchange regioselectivity as a synergistic interplay of steric effects and charge-driven structural dynamics.
While this work focuses on PET-protected Au25(SR)18− clusters, the revealed alkynyl-for-thiolate exchange mechanism may exhibit broader physicochemical implications. Given the shared structural principles governing noble metal nanoclusters, we anticipate its potential applicability to other systems featuring comparable metallic cores and bonding motifs. For homologous gold clusters such as Au38(PET)2465 which contains six dimeric Au2(SR)3 staple motifs, structurally analogous to Au25, mechanistic commonalities in the ligand exchange process are highly probable. Similarly, Ag25 (e.g., [Ag25(SPhMe2)18]−)66 and their alloyed analogues (e.g., AuxAg25−x)67–69 possess analogous Au25-like core–shell structures. Notably, alloying-induced core rigidity may reduce exchange kinetics while enhancing structural stability during ligand exchange. Furthermore, variations in the ligand environment, such as thiolate chain length or functional groups, could critically modulate exchange efficiency through steric or electronic effects.9 Longer thiolate chains are expected to impose greater steric hindrance, while different functional groups (e.g., –NHCs70,71 and –COOH72,73) may significantly alter electronic donation/withdrawal effects, thereby influencing the affinity for alkynyl ligands. We emphasize that validating these hypotheses requires integrated approaches combining computational modeling, atomically precise structural characterization, and system-specific in situ spectroscopy techniques. This delineates clear and necessary avenues for future research.
Conclusion
In summary, we have performed extensive AIMD simulations to reveal the alkynyl-for-thiolate exchange mechanism in the atomically precise Au25(SR)18 prototype cluster. By systematically probing the electronic and steric effects of the exchange ligand as well as the charge state of Au25q (q = −1, 0, and +1) NCs, we identify that the electronic characteristics of the alkynyl ligands in the metal–alkynyl complex (e.g., Au(CCPh) or Li(CCPh)) play the decisive role in determining the nucleophilicity and initiating the exchange reactions. In the dynamic exchange process with the Au(CCR) complex, the anionic [Au25(SR)18]− favors to form the S1 isomer, whereas the S2 isomer is the dominant product when the neutral Au25(SR)18 is used as the precursor. Differently, the sterically bulky Au(CCtBu) complex preferentially targets the S1 position, attributed to the steric hindrance with PET ligands that restricts its access to the binding sites. Interestingly, the alkynyl-for-thiolate exchange ensures 100% selective formation of the  isomer when using the lithium–alkynyl complex (Li(CCR)) as the incoming ligand. Our experimental validation further confirms the successful alkynyl-for-thiolate exchange on the model cluster [Au25(PET)18]−, where up to 6 PET ligands can be substituted by the alkynyl –CCtBu ligands. Overall, these findings greatly deepen our understanding of the exchange reaction between thiolates and alkynyl ligands, leading to the insightful design guidelines for the creation of novel gold nanoclusters with new chemical functions and interface properties.
isomer when using the lithium–alkynyl complex (Li(CCR)) as the incoming ligand. Our experimental validation further confirms the successful alkynyl-for-thiolate exchange on the model cluster [Au25(PET)18]−, where up to 6 PET ligands can be substituted by the alkynyl –CCtBu ligands. Overall, these findings greatly deepen our understanding of the exchange reaction between thiolates and alkynyl ligands, leading to the insightful design guidelines for the creation of novel gold nanoclusters with new chemical functions and interface properties.
Author contributions
Yuping Chen: conceptualization, data curation, formal analysis, investigation, methodology, writing – original draft; Guoqing Bian: investigation, methodology; Zhikun Wu: supervision, resources, writing – review & editing; Qing Tang: conceptualization, supervision, resources, writing – review & editing.Conflicts of interest
The authors declare no conflict of interests.Data availability
The data supporting this article have been included as part of the SI. Supplementary information: Additional simulation models, reaction coordinates for constrained AIMD simulations, free energy profiles, HOMO and LUMO, CM5 atomic charges, Electron Localization Function (ELF) maps, Localized Molecular Orbitals (LMOs), Interaction Region Indicator (IRI) plots, and UV/vis/NIR absorption spectra. See DOI: https://doi.org/10.1039/d5sc04701c.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22473017) and the Chongqing Science and Technology Commission (CSTB2024NSCQ-MSX0250). Z. K. Wu acknowledges the support from National Natural Science Foundation of China (No. 22471275, 21925303, 21829501, and 21771186) and the Collaborative Innovation Program of Hefei Science Center, CAS (No. 2020HSC-CIP005 and 2022HSC-CIP018).References
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Atomically Precise Colloidal Metal Nanoclusters and Nanoparticles: Fundamentals and Opportunities, Chem. Rev., 2016, 116(18), 10346–10413 CrossRef CAS PubMed
.
- Y. Zhu, H. Qian, B. A. Drake and R. Jin, Atomically Precise Au25(SR)18 Nanoparticles as Catalysts for the Selective Hydrogenation of α, β-Unsaturated Ketones and Aldehydes, Angew. Chem., Int. Ed., 2010, 7(49), 1295–1298 CrossRef PubMed
- S. Li, D. Alfonso, A. V. Nagarajan, S. D. House, J. C. Yang, D. R. Kauffman, G. Mpourmpakis and R. Jin, Monopalladium Substitution in Gold Nanoclusters Enhances CO2 Electroreduction Activity and Selectivity, ACS Catal., 2020, 10(20), 12011–12016 CrossRef CAS
- W. Jing, H. Shen, R. Qin, Q. Wu, K. Liu and N. Zheng, Surface and Interface Coordination Chemistry Learned from Model Heterogeneous Metal Nanocatalysts: From Atomically Dispersed Catalysts to Atomically Precise Clusters, Chem. Rev., 2022, 123(9), 5948–6002 CrossRef PubMed
- B. Zhang, J. Chen, Y. Cao, O. J. H. Chai and J. Xie, Ligand Design in Ligand-Protected Gold Nanoclusters, Small, 2021, 17(27), 2004381 CrossRef CAS PubMed
- M. Cui, Y. Shi, X. Ma, Q. Li, L. Chen, L. Zhang, J. Wu, H. Yu and M. Zhu, The Pivotal Radical Intermediate [Au21(SR)15]+ in the Ligand-Exchange-Induced Size-Reduction of [Au23(SR)16]− to Au16(SR)12, ACS Nano, 2024, 18(8), 6591–6599 CrossRef CAS PubMed
- V. Truttmann, C. Herzig, I. Illes, A. Limbeck, E. Pittenauer, M. Stöger-Pollach, G. Allmaier, T. Bürgi, N. Barrabés and G. Rupprechter, Ligand Engineering of Immobilized Nanoclusters on Surfaces: Ligand Exchange Reactions with Supported Au11(PPh3)7Br3, Nanoscale, 2020, 12(24), 12809–12816 RSC
- Y. Cao, V. Fung, Q. Yao, T. Chen, S. Zang, D.-e. Jiang and J. Xie, Control of Single-Ligand Chemistry on Thiolated Au25 Nanoclusters, Nat. Commun., 2020, 11(1), 5498 CrossRef CAS PubMed
- W. Suzuki, R. Takahata, Y. Chiga, S. Kikkawa, S. Yamazoe, Y. Mizuhata, N. Tokitoh and T. Teranishi, Control over Ligand-Exchange Positions of Thiolate-Protected Gold Nanoclusters using Steric Repulsion of Protecting Ligands, J. Am. Chem. Soc., 2022, 144(27), 12310–12320 CrossRef CAS PubMed
- G. Salassa, A. Sels, F. Mancin and T. Burgi, Dynamic Nature of Thiolate Monolayer in Au25(SR)18 Nanoclusters, ACS Nano, 2017, 11(12), 12609–12614 CrossRef CAS PubMed
- Y. Zeng, S. Havenridge, M. Gharib, A. Baksi, K. L. D. M. Weerawardene, A. R. Ziefuß, C. Strelow, C. Rehbock, A. Mews and S. Barcikowski,
et al., Impact of Ligands on Structural and Optical Properties of Ag29 Nanoclusters, J. Am. Chem. Soc., 2021, 143(25), 9405–9414 CrossRef CAS PubMed
- J. Dong, Z. Gan, W. Gu, Q. You, Y. Zhao, J. Zha, J. Li, H. Deng, N. Yan and Z. Wu, Synthesizing Photoluminescent Au28(SCH2Ph-tBu)22 Nanoclusters with Structural Features by Using a Combined Method, Angew. Chem., Int. Ed., 2021, 133(33), 18076–18080 CrossRef
- H. Yao, Chiral Ligand-Protected Gold Nanoclusters: Considering the Optical Activity from a Viewpoint of Ligand Dissymmetric Field, Prog. Nat. Sci., 2016, 26(5), 428–439 CrossRef CAS
- C. A. Hosier and C. J. Ackerson, Regiochemistry of Thiolate for Selenolate Ligand Exchange on Gold Clusters, J. Am. Chem.
Soc., 2019, 141(1), 309–314 CrossRef CAS PubMed
- Y. Li, T. Higaki, X. Du and R. Jin, Chirality and Surface Bonding Correlation in Atomically Precise Metal Nanoclusters, Adv. Mater., 2020, 32(41), 1905488 CrossRef CAS PubMed
- L. Cseh and G. H. Mehl, The Design and Investigation of Room Temperature Thermotropic Nematic Gold Nanoparticles, J. Am. Chem. Soc., 2006, 128(41), 13376–13377 CrossRef CAS PubMed
- Q. Yao, X. Yuan, Y. Yu, Y. Yu, J. Xie and J. Y. Lee, Introducing Amphiphilicity to Noble Metal Nanoclusters via Phase-Transfer Driven Ion-Pairing Reaction, J. Am. Chem. Soc., 2015, 137(5), 2128–2136 CrossRef CAS PubMed
- W. Ndugire and M. Yan, Synthesis and Solution Isomerization of Water-Soluble Au9 Nanoclusters Prepared by Nuclearity Conversion of [Au11(PPh3)8Cl2]Cl, Nanoscale, 2021, 13(39), 16809–16817 RSC
- H. Shen, Z. Xu, M. S. A. Hazer, Q. Wu, J. Peng, R. Qin, S. Malola, B. K. Teo, H. Häkkinen and N. Zheng, Surface Coordination of Multiple Ligands Endows N-Heterocyclic Carbene-Stabilized Gold Nanoclusters with High Robustness and Surface Reactivity, Angew. Chem., Int. Ed., 2021, 60(7), 3752–3758 Search PubMed
- Z. Luo, K. Zheng and J. Xie, Engineering Ultrasmall Water-Soluble Gold and Silver Nanoclusters for Biomedical Applications, Chem. Commun., 2014, 50(40), 5143–5155 Search PubMed
- A. Retnakumari, S. Setua, D. Menon, P. Ravindran, H. Muhammed, T. Pradeep, S. Nair and M. Koyakutty, Molecular-Receptor-Specific, Non-Toxic, Near-Infrared-Emitting Au Cluster-Protein Nanoconjugates for Targeted Cancer Imaging, Nat. Nanotechnol., 2009, 21(5), 055103 CrossRef PubMed
- M. Sera, S. Hossain, S. Yoshikawa, K. Takemae, A. Ikeda, T. Tanaka, T. Kosaka, Y. Niihori, T. Kawawaki and Y. Negishi, Atomically Precise Au24Pt (thiolate)12(dithiolate)3 Nanoclusters with Excellent Electrocatalytic Hydrogen Evolution Reactivity, J. Am. Chem. Soc., 2024, 146(43), 29684–29693 CrossRef CAS PubMed
- T. Lahtinen, E. Hulkko, K. Sokołowska, T.-R. Tero, V. Saarnio, J. Lindgren, M. Pettersson, H. Häkkinen and L. Lehtovaara, Covalently Linked Multimers of Gold Nanoclusters Au102(p-MBA)44 and Au∼250(p-MBA)n, Nanoscale, 2016, 8(44), 18665–18674 RSC
- Q. Zhu, X. Huang, Y. Zeng, K. Sun, L. Zhou, Y. Liu, L. Luo, S. Tian and X. Sun, Controllable Synthesis and Electrocatalytic Applications of Atomically Precise Gold Nanoclusters, Nanoscale Adv., 2021, 3(22), 6330–6341 RSC
- J. Zhao, A. Ziarati, A. Rosspeintner, Y. Wang and T. Bürgi, Engineering Ligand Chemistry on Au25 Nanoclusters: from Unique Ligand Addition to Precisely Controllable Ligand Exchange, Chem. Sci., 2023, 14(28), 7665–7674 RSC
- Y. Chen, C. Liu, Q. Tang, C. Zeng, T. Higaki, A. Das, D.-e. Jiang, N. L. Rosi and R. Jin, Isomerism in Au28(SR)20 Nanocluster and Stable Structures, J. Am. Chem. Soc., 2016, 138(5), 1482–1485 CrossRef CAS PubMed
- Y. Negishi, H. Horihata, A. Ebina, S. Miyajima, M. Nakamoto, A. Ikeda, T. Kawawaki and S. Hossain, Selective Formation of [Au23(SPhtBu)17]0, [Au26Pd(SPhtBu)20]0 and [Au24Pt(SC2H4Ph)7(SPhtBu)11]0 by Controlling Ligand-exchange Reaction, Chem. Sci., 2022, 13(19), 5546–5556 RSC
- P. D. Jadzinsky, G. Calero, C. J. Ackerson, D. A. Bushnell and R. D. Kornberg, Structure of a Thiol Monolayer-Protected Gold Nanoparticle at 1.1 Å Resolution, Science, 2007, 318(5849), 430–433 CrossRef CAS PubMed
- M. W. Heaven, A. Dass, P. S. White, K. M. Holt and R. W. Murray, Crystal Structure of the Gold Nanoparticle [N(C8H17)4][Au25(SCH2CH2Ph)18], J. Am. Chem. Soc., 2008, 130(12), 3754–3755 CrossRef CAS PubMed
- M. J. Hostetler, A. C. Templeton and R. W. Murray, Dynamics of Place-Exchange Reactions on Monolayer-Protected Gold Cluster Molecules, Langmuir, 1999, 15(11), 3782–3789 CrossRef CAS
- H. Xiang, H. Yan, J. Liu, R. Cheng, C.-Q. Xu, J. Li and C. Yao, Identifying the Real Chemistry of the Synthesis and Reversible Transformation of AuCd Bimetallic Clusters, J. Am. Chem. Soc., 2022, 144(31), 14248–14257 CrossRef CAS PubMed
- X. Ma, Y. Tang, G. Ma, L. Qin and Z. Tang, Controllable Synthesis and Formation Mechanism Study of Homoleptic Alkynyl-Protected Au Nanoclusters: Recent Advances, Grand Challenges, and Great Opportunities, Nanoscale, 2021, 13(2), 602–614 RSC
- P. Maity, H. Tsunoyama, M. Yamauchi, S. Xie and T. Tsukuda, Organogold Clusters Protected by Phenylacetylene, J. Am. Chem. Soc., 2011, 133(50), 20123–20125 CrossRef CAS PubMed
- M. Sugiuchi, Y. Shichibu, T. Nakanishi, Y. Hasegawa and K. Konishi, Cluster–π Electronic Interaction in a Superatomic Au13 Cluster Bearing σ-Bonded Acetylide Ligands, Chem. Commun., 2015, 51(70), 13519–13522 Search PubMed
- X.-K. Wan, Q. Tang, S.-F. Yuan, D.-e. Jiang and Q.-M. Wang, Au19 Nanocluster Featuring a V-Shaped Alkynyl–Gold Motif, J. Am. Chem. Soc., 2015, 137(2), 652–655 Search PubMed
- Z. Lei, X.-K. Wan, S.-F. Yuan, Z.-J. Guan and Q.-M. Wang, Alkynyl Approach toward the Protection of Metal Nanoclusters, Acc. Chem. Res., 2018, 51(10), 2465–2474 CrossRef CAS PubMed
- X. K. Wan, Z. J. Guan and Q. M. Wang, Homoleptic Alkynyl-Protected Gold Nanoclusters: Au44(PhC≡C)28 and Au36(PhC≡C)24, Angew. Chem., Int. Ed., 2017, 56(38), 11494–11497 CrossRef CAS PubMed
- J.-J. Li, Z.-J. Guan, Z. Lei, F. Hu and Q.-M. Wang, Same Magic Number but Different Arrangement: Alkynyl-Protected Au25 with D3 Symmetry, Angew. Chem., Int. Ed., 2019, 58(4), 1083–1087 CrossRef CAS PubMed
- X.-K. Wan, J.-Q. Wang, Z.-A. Nan and Q.-M. Wang, Ligand Effects in Catalysis by Atomically Precise Gold Nanoclusters, Sci. Adv., 2017, 3(10), e1701823 CrossRef PubMed
- X. Li, S. Takano and T. Tsukuda, Ligand Effects on the Hydrogen Evolution Reaction Catalyzed by Au13 and Pt@Au12: Alkynyl vs. Thiolate, J. Phys. Chem. C, 2021, 125(42), 23226–23230 CrossRef CAS
- C. A. Hosier, I. D. Anderson and C. J. Ackerson, Acetylide-for-Thiolate and Thiolate-for-Acetylide Exchange on Gold Nanoclusters, Nanoscale, 2020, 12(11), 6239–6242 RSC
- K. Isozaki, K. Iseri, R. Saito, K. Ueda and M. Nakamura, Dual Catalysis of Gold Nanoclusters: Photocatalytic Cross-Dehydrogenative Coupling by Cooperation of Superatomic Core and Molecularly Modified Staples, Angew. Chem., Int. Ed., 2024, 136(2), e202312135 CrossRef
- M. Zhu, C. M. Aikens, F. J. Hollander, G. C. Schatz and R. Jin, Correlating the Crystal Structure of a Thiol-Protected Au25 Cluster and Optical Properties, J. Am. Chem. Soc., 2008, 130(18), 5883–5885 CrossRef CAS PubMed
- J. VandeVondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, Quickstep: Fast and Accurate Density Functional Calculations using a Mixed Gaussian and Plane Waves Approach, Comput. Phys. Commun., 2005, 167(2), 103–128 CrossRef CAS
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77(18), 3865 CrossRef CAS PubMed
- C. Hartwigsen, S. Gœdecker and J. Hutter, Relativistic Separable Dual-Space Gaussian Pseudopotentials from H to Rn, Phys. Rev. B:Condens. Matter Mater. Phys., 1998, 58(7), 3641 CrossRef CAS
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the Damping Function in Dispersion Corrected Density Functional Theory, J. Comput. Chem., 2011, 32(7), 1456–1465 CrossRef CAS PubMed
- G. J. Martyna, M. L. Klein and M. Tuckerman, Nosé–Hoover Chains: The Canonical Ensemble via Continuous Dynamics, J. Chem. Phys., 1992, 97(4), 2635–2643 CrossRef
- E. A. Carter, G. Ciccotti, J. T. Hynes and R. Kapral, Constrained Reaction Coordinate Dynamics for the Simulation of Rare Events, Chem. Phys. Lett., 1989, 156(5), 472–477 CrossRef CAS
- M. Sprik and G. Ciccotti, Free Energy from Constrained Molecular Dynamics, J. Chem. Phys., 1998, 109(18), 7737–7744 CrossRef CAS
-
M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, G. Petersson and H. Nakatsuji, Gaussian 16, Revision A. 03, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed
-
S. Huzinaga, J. Andzelm, E. Radzio-Andzelm, Y. Sakai, H. Tatewaki and M. Klobukowski, Gaussian Basis Sets for Molecular Calculations, Elsevier, 2012 Search PubMed
- D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Energy-Adjustedab Initio Pseudopotentials for the Second and Third Row Transition Elements, Theor. Chim. Acta, 1990, 77(2), 123–141 CrossRef CAS
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A Consistent and Accurate ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu, J. Chem. Phys., 2010, 132(15), 154104 CrossRef PubMed
- J. Stevenson, B. Sorenson, V. H. Subramaniam, J. Raiford, P. P. Khlyabich, Y.-L. Loo and P. Clancy, Mayer Bond Order as a Metric of Complexation Effectiveness in Lead Halide Perovskite Solutions, Chem. Mater., 2017, 29(6), 2435–2444 CrossRef CAS
- I. Mayer, Charge, Bond Order and Valence in the ab Initio SCF Theory, Chem. Phys. Lett., 1983, 97(3), 270–274 CrossRef CAS
- I. Mayer, Bond Order and Valence: Relations to Mulliken's Population Analysis, Int. J. Quantum Chem., 1984, 26(1), 151–154 CrossRef CAS
- T. Lu and F. Chen, Multiwfn: A Multifunctional Wavefunction Analyzer, J. Comput. Chem., 2012, 33(5), 580–592 CrossRef CAS PubMed
- Z. Lei, J. J. Li, X. K. Wan, W. H. Zhang and Q. M. Wang, Isolation and Total Structure Determination of an All-Alkynyl-Protected Gold Nanocluster Au144, Angew. Chem., Int. Ed., 2018, 57(28), 8639–8643 CrossRef CAS PubMed
- L. Liao, S. Zhou, Y. Dai, L. Liu, C. Yao, C. Fu, J. Yang and Z. Wu, Mono-Mercury Doping of Au25 and the HOMO/LUMO Energies Evaluation Employing Differential Pulse Voltammetry, J. Am. Chem. Soc., 2015, 137(30), 9511–9514 CrossRef CAS PubMed
- C. L. Heinecke, T. W. Ni, S. Malola, V. Makinen, O. A. Wong, H. Hakkinen and C. J. Ackerson, Structural and Theoretical Basis for Ligand Exchange on Thiolate Monolayer Protected Gold Nanoclusters, J. Am. Chem. Soc., 2012, 134(32), 13316–13322 CrossRef CAS PubMed
- M. Zhu, C. M. Aikens, M. P. Hendrich, R. Gupta, H. Qian, G. C. Schatz and R. Jin, Reversible Switching of Magnetism in Thiolate-Protected Au25 Superatoms, J. Am. Chem. Soc., 2009, 131(7), 2490–2492 CrossRef CAS PubMed
- S. Antonello, N. V. Perera, M. Ruzzi, J. A. Gascón and F. Maran, Interplay of Charge State, Lability, and Magnetism in the Molecule-like Au25(SR)18 Cluster, J. Am. Chem. Soc., 2013, 135(41), 15585–15594 CrossRef CAS PubMed
- M. Zhu, W. T. Eckenhoff, T. Pintauer and R. Jin, Conversion of Anionic [Au25(SCH2CH2Ph)18]− Cluster to Charge Neutral Cluster via Air Oxidation, J. Phys. Chem. C, 2008, 112(37), 14221–14224 CrossRef CAS
- H. Qian, W. T. Eckenhoff, Y. Zhu, T. Pintauer and R. Jin, Total Structure Determination of Thiolate-Protected Au38 Nanoparticles, J. Am. Chem. Soc., 2010, 132(24), 8280–8281 CrossRef CAS PubMed
- C. P. Joshi, M. S. Bootharaju, M. J. Alhilaly and O. M. Bakr, [Ag25(SR)18]−: The “Golden” Silver Nanoparticle, J. Am. Chem. Soc., 2015, 137(36), 11578–11581 CrossRef CAS PubMed
- L. He, Z. Gan, N. Xia, L. Liao and Z. Wu, Alternating Array Stacking of Ag26Au and Ag24Au Nanoclusters, Angew. Chem., Int. Ed., 2019, 131(29), 10002–10006 CrossRef
- D. Liu, W. Du, S. Chen, X. Kang, A. Chen, Y. Zhen, S. Jin, D. Hu, S. Wang and M. Zhu, Interdependence between nanoclusters AuAg24 and Au2Ag41, Nat. Commun., 2021, 12(1), 778 CrossRef CAS PubMed
- X. Liu, J. Yuan, C. Yao, J. Chen, L. Li, X. Bao, J. Yang and Z. Wu, Crystal and Solution Photoluminescence of MAg24(SR)18 (M = Ag/Pd/Pt/Au) Nanoclusters and Some Implications for the Photoluminescence Mechanisms, J. Phys. Chem. C, 2017, 121(25), 13848–13853 CrossRef CAS
- H. Shen, Z. Xu, M. S. A. Hazer, Q. Wu, J. Peng, R. Qin, S. Malola, B. K. Teo, H. Häkkinen and N. Zheng, Surface Coordination of Multiple Ligands Endows N-Heterocyclic Carbene-Stabilized Gold Nanoclusters with High Robustness and Surface Reactivity, Angew. Chem., Int. Ed., 2021, 133(7), 3796–3802 CrossRef
- M. R. Narouz, K. M. Osten, P. J. Unsworth, R. W. Man, K. Salorinne, S. Takano, R. Tomihara, S. Kaappa, S. Malola and C.-T. Dinh, N-Heterocyclic Carbene-Functionalized Magic-Number Gold Nanoclusters, Nat. Chem., 2019, 11(5), 419–425 CrossRef CAS PubMed
- W.-D. Tian, W.-D. Si, S. Havenridge, C. Zhang, Z. Wang, C. M. Aikens, C.-H. Tung and D. Sun, Biomimetic Crystallization for Long-Pursued –COOH-Functionalized Gold Nanocluster with Near-Infrared Phosphorescence, Sci. Bull., 2024, 69(1), 40–48 CrossRef CAS PubMed
- S. M. Han, M. Park, J. Kim and D. Lee, Boosting the Electroreduction of CO2 to CO by Ligand Engineering of Gold Nanoclusters, Angew. Chem., Int. Ed., 2024, 63(31), e202404387 CrossRef CAS PubMed
Footnote |
| † These two authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |