
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The organophosphorus synthesis triangle: introducing methods for the missing quaternization and de-quaternization routes
Anna C.
Vetter
 ,
Yannick
Ortin
,
Kirill
Nikitin
* and
Declan G.
Gilheany
*
,
Yannick
Ortin
,
Kirill
Nikitin
* and
Declan G.
Gilheany
*
School of Chemistry, University College Dublin, Belfield, Dublin 4, Ireland. E-mail: kirill.nikitin@ucd.ie; declan.gilheany@ucd.ie
First published on 24th October 2025
Abstract
In organophosphorus chemistry, several established reactions, such as the conversion of phosphorus trichloride into tertiary phosphines, followed by oxidation and quaternization to form phosphine oxides and phosphonium salts, are widely recognized and routinely applied. In contrast, other potentially valuable transformations, including reverse or complementary versions of these standard synthetic routes, remain largely unexplored or technically challenging. This work introduces two new reaction pathways that broaden the scope of organophosphorus synthesis. The first involves a P–C bond-forming process that enables interconversion of symmetrical phosphine oxides, such as triphenylphosphine oxide (Ph3PO), into P-stereogenic phosphine oxides and quaternary phosphonium salts. The second transformation is based on the distinctive reactivity of methoxymethyl (MOM)-substituted quaternary phosphonium salts. These compounds undergo a P–C bond cleavage reaction that results in de-quaternization, allowing the synthesis of mixed-substituent tertiary phosphines from triphenylphosphine as a common precursor. Together, these two processes provide multiple efficient synthetic routes to phosphines, phosphine oxides, and quaternary phosphonium salts. The overall synthetic approach is flexible, so that the target compounds can be obtained through several pathways using different substituent combinations as starting materials.
Introduction
In the context of the importance of phosphorus compounds in Nature, industrial materials and synthesis,1 the synthetic chemistry of organophosphorus compounds assumed a standing from the earliest days of organic synthesis2 and today is explored extensively.3 However, despite all of that activity, there are still a few basic interconversions that are difficult or impossible to achieve and some phosphorus substitution patterns that are inaccessible. To illustrate the issues, we introduce here the Organophosphorus Synthesis Triangle (or P-triangle, Scheme 1). This reflects that, historically, organophosphorus synthesis developed around a trio of key molecular motifs: P(III) compounds 1 and their P(V) quaternized, 2, and oxidized derivatives, 3 (Scheme 1).2b,3d,f Structure 1 is represented largely by tertiary phosphines R3P but also includes halophosphines and certain P–O structures: for example phosphonous acid derivatives (R = organic group, Y = leaving group, e.g. alkoxy, halide). Similarly, structure 3 describes both phosphine oxides (PO) and phosphon(in)ic acid derivatives. Structure 2 is mostly associated with quaternary phosphonium salts, (QPS, R = organic group) but also encompasses their oxo- and halo-analogues. The interconnectivity, both between 1–3 (transformations A–C) and within these three classes (replacing Y with an organic R, transformations D–F), has served many lines of synthetic chemistry in accessing, for example: important building blocks in organic synthesis,4 indispensable ligands5 for transition metal catalysis and, notably, chiral non-racemic variants of such ligands along with their organo- and phase transfer counterparts.6 Thus, adding new, and developing existing, efficient routes within the P-triangle is an important and topical task.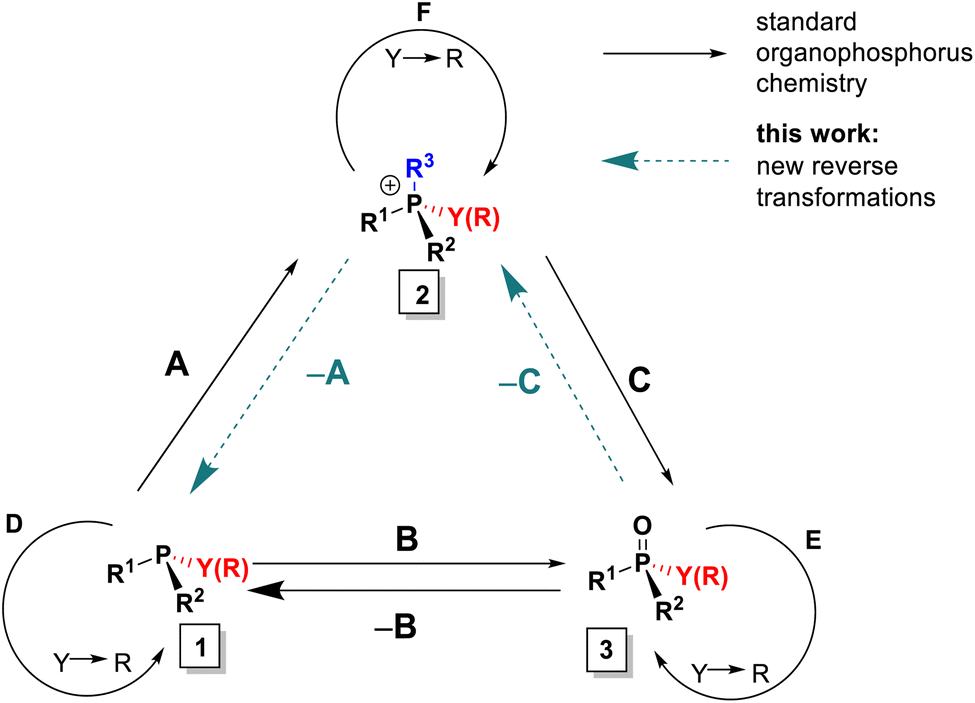 | ||
| Scheme 1 The “P-triangle” interconnects P(III) and P(V) derivatives: 1 (e.g. phosphines, halophosphines), 2 (phosphonium salts) and 3 (e.g. phosphine oxides, phosphinates). Y – a leaving group; R – a C-linked organic group. Transformations between classes: A – quaternization; B – oxidation; C – olefination or hydrolysis. | ||
When we examine the P-triangle in more detail, it illustrates the close synthetic relationships of the key organophosphorus classes 1–3. Triorganophosphines 1, being often the entry point, are commonly, albeit not exclusively,7 accessed from chlorophosphines or, more recently, arylphosphines, by sequential introduction of organic groups R according to path D.8 From this point, established forward quaternization A9a–c and oxidation B9d processes allow relatively easy access to tetrahedral 2 and 3, respectively. In turn, facile cleavage of P–C bonds in 2, by, for example, hydrolysis or Wittig reaction10 path C, can also be used to access 3. The set of available P(V) derivatives 3 including P-stereogenic ones can be further expanded through replacement of suitable leaving groups Y (halide, alkoxide and even Ar) via path E.11 Analogous interconversions of cationic species 2, path F, play a pivotal role in highly selective substitution reactions at the phosphorus centre.12
In practice, there are several critical caveats associated with implementing certain reactions of the P-triangle. The first limitation is poor availability of mixed-group phosphines PR1R2R3 from phosphorus trihalides, especially in the case of compact, sterically innocent substituents R.8 As a redress for this, versatile alternative methods involving inverse polarity P–C coupling of phosphides have been introduced to access such mixed-group 1.13 However, these alternatives often suffer from unwanted quaternization of the desired P(III) target compound, whereas analogous inverse polarity approaches to mixed P(V) structures 3 are generally less problematic.14 The second limitation is that the direct quaternization of phosphines 1, path A, is often quite slow and, for less reactive electrophiles, requires extreme conditions or the use of metal catalysts.15
The significance of the P-triangle to the present work is that it illustrates the relationships between direct and reverse interconversions of compound types 1–3. In almost all cases, the reverse reactions –A, –B and –C, are much more demanding than their forward counterparts, which makes their study both more challenging and potentially rewarding. Of these, by far the best known and studied is the deoxygenation –B which is thermochemically disfavored (due to the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond enthalpy: 130–150 kcal mol−1
O bond enthalpy: 130–150 kcal mol−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 16). This has been, and remains, a significant challenge in organophosphorus chemistry to which many workers,17 including ourselves18 have contributed solutions that have led to a variety of symmetrical and P-stereogenic 1 and 2.19–21
16). This has been, and remains, a significant challenge in organophosphorus chemistry to which many workers,17 including ourselves18 have contributed solutions that have led to a variety of symmetrical and P-stereogenic 1 and 2.19–21
This study focuses on two additional reverse transformations. Although there are many efficient synthetic routes to compounds 1 and 2,8–12,22 the applications of reverse transformation “–A” remains underdeveloped. Classical electrochemical reductive cleavage of phosphonium salts to phosphines is well known,9a,23,24 but it is rarely applied preparatively for unsymmetrical cases due to uncertainty regarding which group is cleaved.9a More recently, reports of debenzylation and dearylative reactions25 at phosphorus include examples of –A transformations that provide mixed-group compounds 1 with improved selectivity. The second transformation, –C, involves the conversion of phosphine oxides 3 to quaternary structures 2 (particularly where Y = R). This reaction would enable direct utilization of phosphine oxides, which are typically inert by-products of stoichiometric10,12a–e,26 and catalytic19–21,27 processes rather than reducing PO to P(III) structures 1. Prior to this work, such a transformation had not been demonstrated.
Here, we present and develop solutions to the missing synthetic transformations –A and –C which arise through two new reactions discovered in our laboratory (Scheme 2(I and II)). The first new reaction, described in a preliminary communication,28 involves Umpolung quaternization of phosphine oxides 3. This transformation belongs to class –C shown in Scheme 1 and is a general reverse route that enables efficient conversion of phosphine oxides 3 into quaternary salts 2via a P-chlorophosphonium cation intermediate followed by treatment with a Grignard reagent. When combined with standard P–C bond cleavage such as hydrolysis or Wittig reaction, this approach allows controlled, stepwise interconversion between different types of achiral and P-stereogenic quaternary salts 2 and phosphine oxides 3.
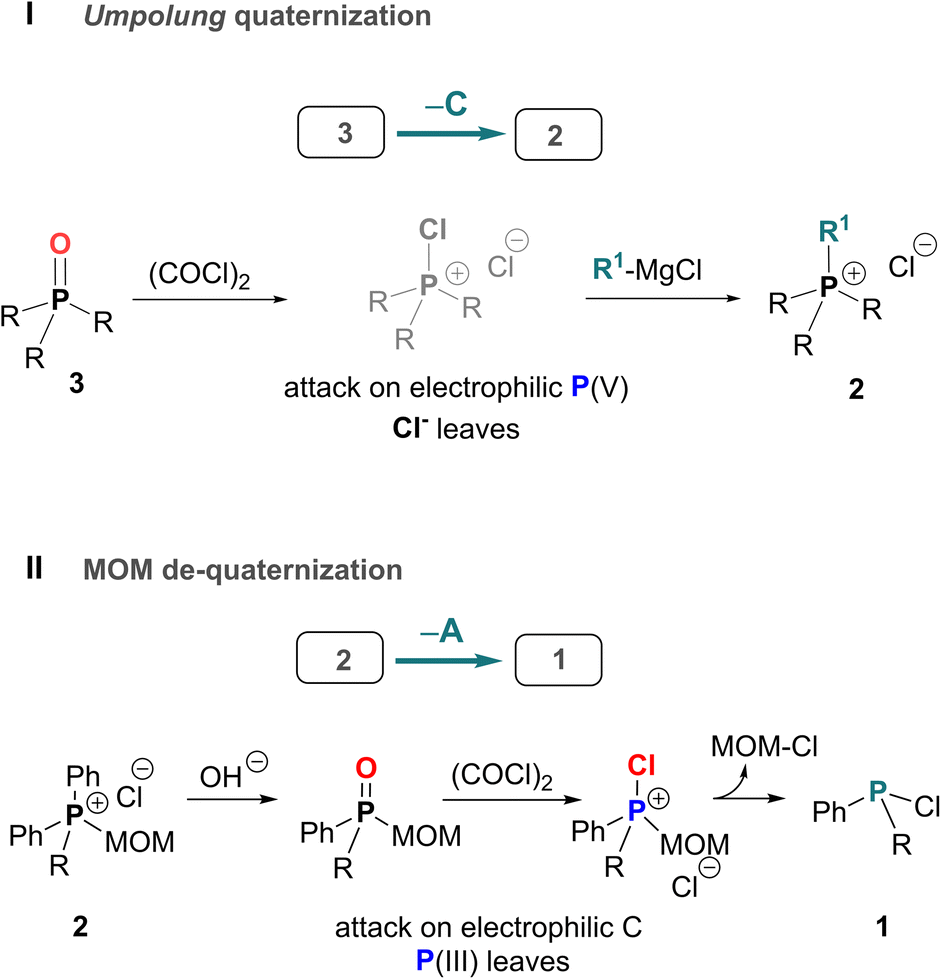 | ||
| Scheme 2 Novel reverse transformations presented in this report: (I) – Umpolung quaternization “–C” of phosphine oxides 3; (II) – spontaneous reverse quaternization “–A” (de-quaternization) of MOM-phosphonium salts 2. | ||
The second transformation (Scheme 2(II)), involves a direct spontaneous de-quaternization (reversal of quaternization corresponding to path –A) of MOM-derived phosphonium cations 2. Hydrolytic treatment of the quaternary salt (MOM-protected phosphine), followed by chlorination and cleavage of the MOM–P bond (MOM deprotection), produces mixed-group P-chlorophosphines 1. This new high-yielding one-pot transformation enables stepwise replacement of the organic groups of triarylphosphines Ar3P with other organic groups and is at the core of our novel approach to formal organic group-substitution at phosphorus25 connecting the initial Ar3P-based triangle to other parallel P-triangles, including P-stereogenic variants. From an environmental standpoint, an important feature of these reverse transformations is that they use readily available starting materials such as the widely available Ph3P 1.0-a and its commonly discarded oxide Ph3PO 3.0-a.
Results and discussion
Design of reaction sequence to access mixed-group structures
The Umpolung quaternization provides an effective route for constructing P–C bonds and, as demonstrated here, for synthesizing mixed aryl–alkyl P(V) compounds, including quaternary salts 2 and oxides 3. This approach can be combined with classical transformation “C”, involving selective cleavage of existing P–C bonds through hydrolysis10a or Wittig reaction.10b Controlled P–C bond cleavage has a long-established role10c,d in organophosphorus synthesis and has been used to good effect recently.10e–g Clearly, P–C cleavage gets the synthesis off the ground when combined with the Umpolung quaternization allowing the formation of mixed-group and P-stereogenic compounds in a controlled stepwise fashion. This process is exemplified by the transformation of Ph2n-BuPO 3.1-a into the two related phosphonium structures 2.1-a and 2.2-a, and back, as shown in Scheme 3. | ||
| Scheme 3 Complementary approaches to phosphine oxide 3.1-a using hydrolysis (H, red) or Wittig olefination (W, black) are mirrored by its Umpolung quaternization (U, blue) with different nucleophiles. | ||
The alkaline hydrolysis of phosphonium cation Ph3n-BuP+2.1-a (compound type PAB3) proceeds through hydrolytic cleavage (H, red arrow) of the group most stable as carbanion10a – a Ph-substituent. In contrast, the Wittig olefination (W, black arrow) of the quaternary cation 2.2-a (type PA2B2) can only result in de-alkylation ultimately yielding the same parent oxide 3.1-a. Through Umpolung quaternization of 3.1-a, it becomes possible to regenerate either of the starting cations, 2.1-a or 2.2-a, from this common intermediate by employing two different Grignard reagents (U, blue arrows) in high yields.
The combination of H, W, and U steps provides a versatile synthetic approach for the controlled formation and cleavage of P–C bonds. This sequence enables access to a wide variety of mixed-group phosphonium salts and phosphine oxides from a single common precursor. To demonstrate the scope of this method, several examples of the sequence illustrated in Scheme 4 were carried out. The stepwise replacement of different groups can be viewed in the 2D aryl–alkyl space as shown in Scheme 4 (which we ironically termed the “Umpolung Walk” in our laboratory). The readily available Ph3PO 3.0-a was selected as the common starting material, a compound often regarded as a “phosphorus sink” in synthetic chemistry.29 The sequence of H, W, and U steps allows straightforward preparation of diverse phosphonium salts and phosphine oxides. Notably, the reactions proceed in high yield, exhibit good selectivity, and avoid the need to handle or isolate air-sensitive intermediates.
 | ||
| Scheme 4 2D-sequence reaction featuring Umpolung quaternization in the aryl–alkyl coordinates provides access to a wide variety of quaternary phosphonium salts 2 and phosphine oxides 3 (digit after the dot indicates alkyl group count). P-stereogenic structures are marked with an asterisk. | ||
The first step of the sequence, U1, converts 3.0-a into a generic alkyltriphenylphosphonium salt Ph3R1P+Cl−2.1. Alkaline hydrolysis H1 of this salt yields Ph2R1PO (3.1), which serves as the substrate for a second quaternization U2, producing a mixed-group salt 2.2 (type PABC2). The subsequent hydrolysis step H2 affords a dialkylphenyl phosphine oxide PhR1R2PO (3.2), which becomes P-stereogenic when R1 ≠ R2. The following U3 step generates a P-stereogenic salt PhR1R2R3P+ (2.3), and the subsequent H3 and U4 steps yield the P-stereogenic oxide 3.3 and the tetraalkylphosphonium salt 2.4, respectively. This sequence of high-yielding quaternization and hydrolysis reactions enables the preparation of P-stereogenic compounds from the commonly discarded Ph3PO.
An alternative route, starting from the opposite end of the sequence, Alk4P+ (2.4), produces, via dealkylative olefination W1, the corresponding symmetrical phosphine oxide Alk3P(O) 3.3. This oxide can then be used to prepare phosphonium salts Ar1Alk3P+ (2.3, where Ar1 = Ph in Scheme 4) via the U5 reaction with an aromatic Grignard reagent Ar1MgCl. Extending this sequence through steps U6–U7 affords mixed aryl–alkyl salts Ar1Ar2Ar3AlkP+Cl− (2.1), which upon olefination yield P-stereogenic triarylphosphine oxides 3.0 containing, in principle, three distinct aromatic groups Ar1–Ar3.
The structural diversity accessible through this approach is extensive and extends beyond the scope of the present study. The examples below provide a representative overview of the Umpolung transformation sequence, emphasizing substitution patterns that were previously difficult to achieve.
Exploring the sequence from all-aryl structures 3.0
In our earlier report28 the quaternization U1 was examined and shown to be an effective method for forming P–C bonds, proceeding efficiently at low or ambient temperature to yield monoalkylphosphonium salts 2.1 of the general formula Ar3RP+Cl−. The present study extends this approach through Umpolung steps U2–U4, enabling the preparation of di-, tri-, and tetraalkylphosphonium salts 2.2–2.4, including asymmetric (P-stereogenic) derivatives.Diarylalkylphosphine oxides 3.1a–3.1c were prepared quantitatively (R1 = n-Bu, Et, Me; see SI for details). These well-known 3.1 species were formally synthesized via step H1 and subsequently used in the quaternization U2 reaction to generate a series of mixed Ph2Alk2P+2.2 through the corresponding intermediates 4.1a–c (Scheme 5). As shown in Scheme 5, most U2 reactions produced the desired salts 2.2 in high yields. The slightly reduced yield observed for 2.2-f is likely due to partial deprotonation at the benzylic position. The U1–H1–U2 sequence demonstrates good scalability; for instance, compound 2.2-i was obtained in 75% yield on a 14 mmol scale by sequential introduction of n-Bu and n-Pr groups.
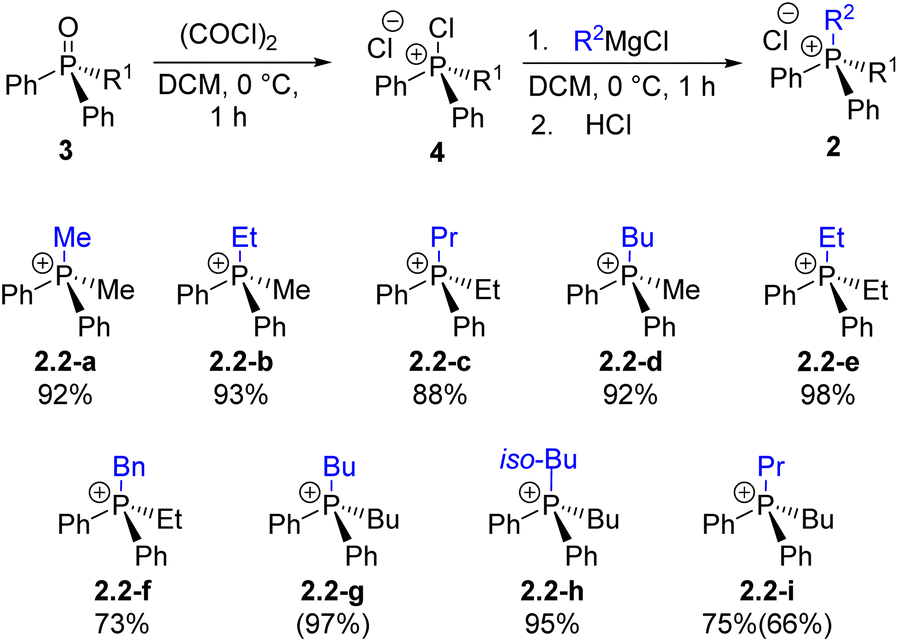 | ||
| Scheme 5 Exploration of step U2: preparation of quaternary phosphonium chlorides 2via the stage of chlorophosphonium species 4 followed by the introduction of an alkyl substituent R2; yield determined by 31P NMR spectroscopy (isolated yield). | ||
A representative example of the alternative routes available through the new transformation sequence is shown in Scheme 6 for compound 2.2-b. Notably, this target can be obtained in high yield through two distinct convergent pathways, both originating from the inexpensive and readily available Ph3PO. This outcome underscores the synthetic flexibility and efficiency of the Umpolung-based methodology.
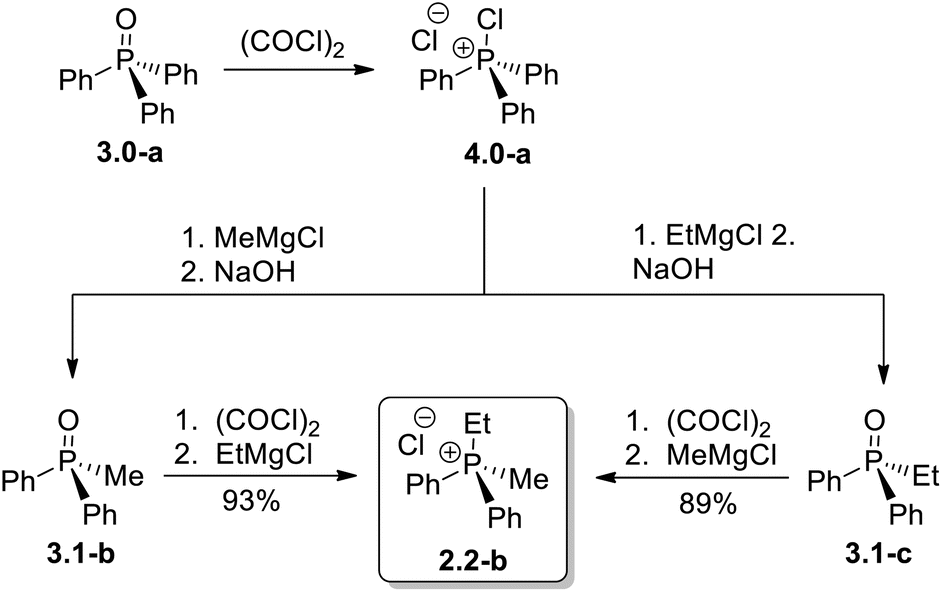 | ||
| Scheme 6 Two alternative Umpolung approaches to mixed dialkyldiaryl phosphonium chloride 2.2-b from the same parent phosphine oxide 3.0-a. | ||
Formerly inaccessible synthetic targets
With the establishment of Umpolung-hydrolysis sequence (Scheme 4) described so far, a range of potential compounds can now be accessed using the U and H transformations. To illustrate this capability, several targets that were previously considered particularly challenging were selected for synthesis.Previously, asymmetric (P-stereogenic) organophosphorus compounds have often featured PhMeP- or tert-BuMe-type motifs. While exceptions exist, these groups were favoured because they could be introduced relatively easily from readily available PhPCl2 or t-BuPCl2via the Mislow route10c involving Arbuzov rearrangement of the corresponding phosphonous methyl (or ethyl) esters followed by nucleophilic displacement of the remaining ester group. This approach allowed the introduction of a single Me group (or Et with more difficulty) and overcame the substantial challenge of selectively substituting one chlorine atom in RPCl2 with a sterically unencumbered nucleophile. Incorporation of substituents other than Ph, Me, or tert-Bu required additional steps such as quaternization, hydrolysis, or Wittig reactions of the intermediate salts to phosphine oxides, followed by reduction to phosphines. These multistep processes often involved handling air-sensitive intermediates. The Umpolung sequence now removes many of these limitations.
Alkaline hydrolysis (H2) was applied to selectively cleave one aryl group from the mixed alkyl–aryl phosphonium salts 2.2-g and 2.2-i, generating phosphine oxides Phn-Bu2PO (3.2-a) and Phn-Bun-PrPO (3.2-b) in high yield. The latter is an example of a species previously not easily accessible.30
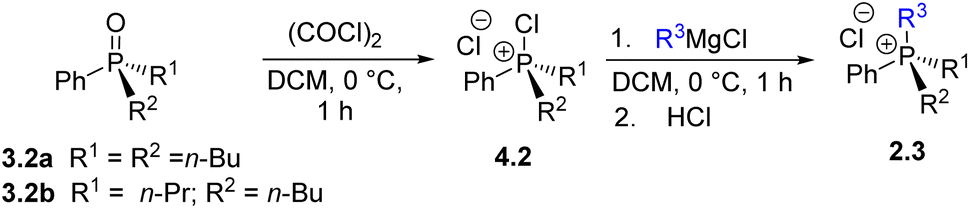
The oxides 3.2-a and 3.2-b were then subjected to step U3via the corresponding chlorophosphonium intermediates 4.2-a and 4.2-b, using four different Grignard reagents to produce phosphonium salts 2.3-a through 2.3-d. As illustrated in Table 1, step U3 successfully furnished mixed-substituent phosphonium salts ArAlk3PCl. For example, reactions with n-BuMgCl and iso-BuMgCl yielded salts 2.3-a and 2.3-b (entries 1 and 2) in high yields, while the use of bulkier iso-PrMgCl (entry 3) resulted in a lower yield (60%) of 2.3-c. In contrast, the synthesis of the asymmetric phosphonium salt 2.3-d, containing a phenyl and three distinct n-alkyl groups, proceeded in high yield (entry 4). The overall yield of 2.3-d (68%) from Ph3PO over five steps, corresponding to an average of 93% per transformation, demonstrates the practicality and efficiency of the Umpolung sequence. The quaternary salt 2.3-a can also be obtained by quaternization of commercially available but air-sensitive n-Bu3P, whereas 2.3-b and 2.3-c would require prior synthesis of Phn-Bu2P from PhPCl2. For compound 2.3-d, however, the quaternization U3 route we propose appears to be the only practical synthetic pathway.
|
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | Producta | Yieldb, % |
| a Using 2 equivalents of Grignard reagent, see SI for details. b Yields determined by 31P NMR analysis, isolated yields in brackets. c 31P NMR analysis indicated the presence of n-Bu2PhP 1.2-a (20%). | |||||
| 1 | n-Bu | n-Bu | n-Bu | 2.3-a | 89 (80) |
| 2 | n-Bu | n-Bu | iso-Bu | 2.3-b | 91 (84) |
| 3 | n-Bu | n-Bu | iso-Pr | 2.3-c | 60c |
| 4 | n-Bu | n-Pr | Et | 2.3-d | 90 |
Steps H3 and U4 (Scheme 4) formally complete the Umpolung transformation sequence, leading to all-alkyl P-stereogenic structures 2.4 (Scheme 4, R1–R4 = Alk). Alkaline hydrolysis (H3) of 2.3-a (n-Bu3PhPCl) proceeded in high yield to give n-Bu3PO, which was then subjected to quaternization U4via the corresponding chlorophosphonium intermediate 4.3-a using a vinyl Grignard reagent, affording the trialkylvinyl cation 2.3-e in high yield.
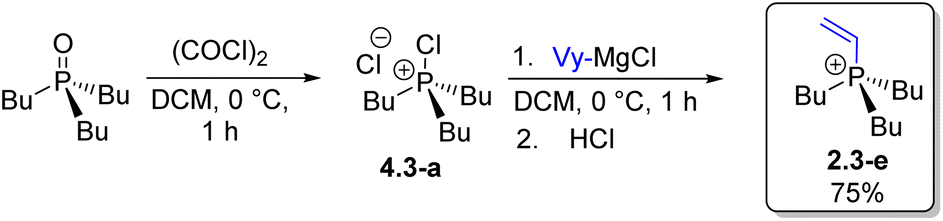
The example of the vinyl group is significant because vinylphosphonium salts are valuable intermediates in organic synthesis but are typically difficult to prepare.31 Standard quaternization of phosphines with vinyl halides is generally not feasible, except in certain cases under Pd catalysis. The main method of choice is Peterson reaction of alpha-silyl phosphonium ylides,31 which often produces Wittig-type side products and cannot be applied to the unsubstituted vinyl case achieved here.32 The Umpolung quaternization method now provides a more straightforward and general route to a variety of vinylphosphonium salts.
Alkaline hydrolysis, H3, of the asymmetric (P-stereogenic) 2.3-d required a 7.5-fold excess of NaOH at 75 °C resulting in selective cleavage of the Ph–P bond and formation of the phosphine oxide n-BuEtn-PrPO (3.3-a) in 93% yield (Scheme 7A). The transformation sequence can be accessed at multiple points (Scheme 4); the synthesis of 3.3-a described here proceeds in six steps from Ph3PO, while a shorter five-step route can be envisioned from commercially available Ph2PCl33 entering the sequence at compound 2.2 through classical organometallic treatment followed by quaternization.
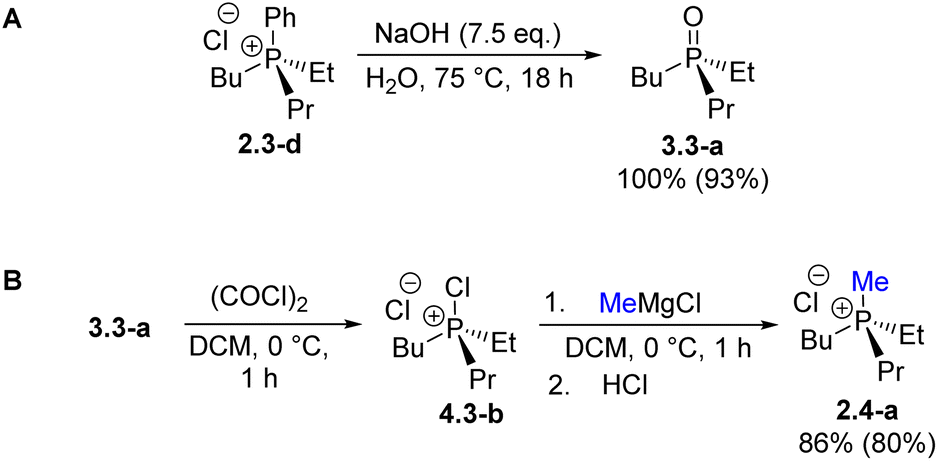 | ||
| Scheme 7 First synthesis of phosphonium salt 2.4-a. (A) hydrolysis of 2.3-d leads to the trialkyl structure 3.3-a. (B) Umpolung quaternization of 3.3-a furnished 2.4-a in high yield. | ||
The phosphine oxide 3.3-a enables access to uncommon P-stereogenic phosphonium structures of the all-alkyl type (PABCD). Umpolung quaternization (U4) of 3.3-a with methyl Grignard reagent shown in Scheme 7B afforded n-butyl(ethyl)(methyl)(n-propyl)phosphonium chloride 2.4-b in 86% yield (54% overall from Ph3PO). This represents the first authentic preparation of the smallest P-stereogenic tetra-n-alkylphosphonium cation.34 While the motivation for phosphonium structure 2.4-b may appear trivial, its synthesis via known quaternization procedures is challenging because the required P-stereogenic trialkylphosphines R1R2R3P are difficult to access even as racemic materials.
Exploring the sequence from the all-alkyl structure 3.3
For illustrative purposes, we explored this solely with phenyl Grignard reagent. As shown in Scheme 8, the sequence begins with symmetrical tetraalkylphosphonium structures such as n-Bu4PCl (2.4-b). If desired, a simple olefination with acetaldehyde (W1) quantitatively affords n-Bu3PO (3.3-b). From this intermediate, reaction U5 with PhMgBr produces the phosphonium chloride 2.3-a in 95% yield,28 and its subsequent olefination W2 (see SI) proceeds in similarly high yield to give the corresponding oxide 3.2-a. | ||
| Scheme 8 Exploration of the sequence featuring Umpolung quaternization from the all-alkyl side: alternative (see Scheme 4) preparation of phosphonium salts 2 and oxides 3 from symmetrical 3.3-b. | ||
Further along the sequence, the unoptimized Umpolung steps show somewhat lower yields. For example, the diaryldialkylphosphonium salt 2.2-g was obtained in 66% yield (U6, Scheme 8), and its olefination produced the oxide 3.1-a in 88% yield. The final quaternization of 3.1-a (U7) afforded the triaryl structure 2.1-a in 33% yield. Although the later steps exhibit modest, unoptimized performance these results clearly demonstrate that the sequence proceeding from the all-alkyl structure 3.3 is feasible and it mirrors the transformations observed when starting from Ph3PO.
The Umpolung transformation sequence offers several advantages that challenge conventional expectations in synthetic organophosphorus chemistry. First, the reactions proceed as selective single-step processes, typically providing high yields of the desired products. Second, the methodology is highly flexible, allowing optimization of a specific synthetic route by varying the order of Umpolung quaternization U, olefination W and hydrolysis H steps. This adaptability enables alternative pathways that complement traditional synthetic strategies. A third and particularly important advantage is the self-limiting nature of each step: once the formation or cleavage of a specific P–C bond is achieved, the resulting products remain stable and do not undergo further transformation under the chosen reaction conditions. Another practical benefit is that both the target compounds and intermediates can be purified by standard recrystallization or chromatographic methods without the need for air-free techniques. Dry conditions are required only when conventional Grignard reagents are employed. Finally, the sequence relies on readily available and easy-to-handle starting materials such as Ph3PO or n-Bu3PO, further enhancing its practicality and accessibility for synthetic applications.
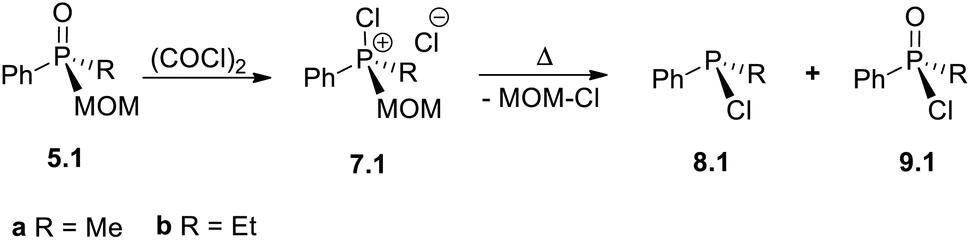
Spontaneous reverse quaternization of certain MOM-phosphonium salts (transformation –A)
During the investigation of the Umpolung quaternization sequence, an important question arose: can methoxymethyl (MOM)-derived phosphine oxides 5 and quaternary phosphonium salts 6 serve as viable intermediates within the sequence? Until now, the use of MOM-derived phosphonium salts, such as Ph3PMOM+Cl−6.0-a, in synthesis has been largely confined to aldehyde homologation and chain-extension reactions.35 In this study, it is demonstrated for the first time that the MOM group can be readily removed as MOM–Cl, effectively acting as a temporary substituent at the phosphorus centre. This behaviour parallels the conventional protecting-group function of MOM in organic synthesis, establishing its role as a removable placeholder within the phosphorus framework.The MOM-derived 5.0-a (Scheme 9) was obtained in 95% yield via hydrolysis of the easily accessible366.0-a. When 5.0-a was treated with oxalyl chloride at −20 °C (see SI) it furnished the corresponding kinetic product – chlorophosphonium species 7.0-a. At ambient temperature, this MOM-derived phosphonium cation underwent a spontaneous transformation yielding the well-known P(III) derivative, chlorodiphenylphosphine 8.0-a as thermodynamic product, and releasing the free MOM–Cl. This reaction represents an unprecedented, facile collapse of a P(V) compound to a P(III) chlorophosphine as the sole product, with no direct analogues.37 Unlike recently reported de-arylation processes,25 such clean unassisted de-alkylation at the phosphorus centre is the exact reversal of standard quaternization process A and thus corresponds to the “–A” pathway in Scheme 1. Due to its mechanistic resemblance to the classical Arbuzov collapse of alkoxyphosphonium species,38 the new –A process could also be termed a pseudo-Arbuzov collapse, as it involves nucleophilic cleavage of a C–P rather than a C–O bond. Treatment of 8.0-a with R1MgCl produces mixed-group phosphines 1.1 (the second “1” denoting one alkyl group R1). Conversely, at temperatures below 0 °C, where 7.0-a remains kinetically stable, reaction with R1MgX under Umpolung quaternization conditions affords new phosphonium structures 6.1.
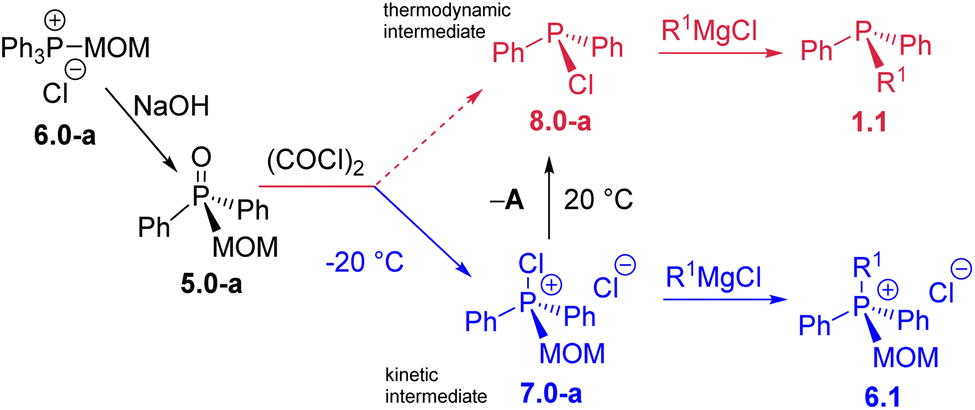 | ||
| Scheme 9 Unexpected P(V)–P(III) dichotomy: at low temperature the kinetic P(V) intermediate 7.0-a leads to salts 6.1; at ambient temperature, the thermodynamic intermediate 8.0-a leads to monoalkylphosphines 1.1. | ||
This temperature-dependent behavior, illustrated in Scheme 9, can be described as an intriguing example of phosphorus oxidation-state thermal dichotomy.39 There is a notable parallel between the two alternative transformations of 7.0-a. At room temperature, the reaction pathway results in the net conversion of Ph3P (1.0-a) into monoalkyldiphenylphosphines 1.1, whereas at low temperature, the process proceeds from the MOM-protected triphenylphosphonium species 6.0-a to new quaternary structures 6.1 in which one phenyl group is replaced by an alkyl substituent. This dual reactivity underscores the fine balance between kinetic and thermodynamic control in phosphorus-centered transformations and highlights the versatility of the Umpolung framework in accessing distinct oxidation states and substitution patterns from a single precursor.
The novelty of the pseudo-Arbuzov collapse and its potential synthetic significance warranted a more detailed study. When the collapse of 7.0-a was monitored by 31P NMR spectroscopy it demonstrated first-order kinetic as illustrated in Fig. 1. The reaction is dramatically accelerated at higher temperature such that at 30 °C (blue line) the rate constant k(30) = 1.87 × 10−4 s−1i.e. some 180 times greater than at 0 °C.
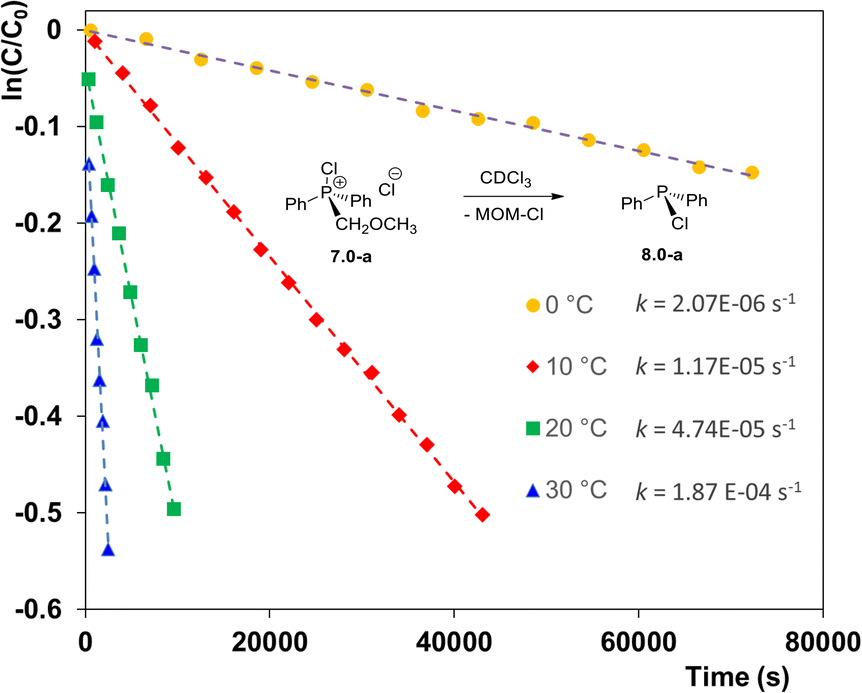 | ||
| Fig. 1 Pseudo-Arbuzov collapse of MOM-derived phosphonium chloride 7.0-a at different temperatures (solvent CDCl3). | ||
Eyring analysis of the kinetic data provided an activation enthalpy ΔH‡ = 24.0 kcal mol−1, which appears to be the dominant contributor to the activation barrier within the temperature range studied. This is consistent with a small positive activation entropy ΔS‡ = 3.7 cal K−1 mol−1, indicating that the transition state 7.0-TS is slightly more disordered than the relatively compact ion pair 7.0-a in the ground state.
This assumption is supported by the results of a DFT computational study of the transition state 7.0-TS (Fig. 2) which is characterized by a bent geometry with the bond angle P–C–Cl ca. 152° and elongated P–C and C–Cl bonds as shown. The more uniform charge distribution in the transition state is further evidenced by solvent effects: in more polar solvents, such as CH3CN, the reaction proceeds significantly more slowly than in CDCl3, yielding only a small amount of chlorodiphenylphosphine 8.0-a after 18 hours. Overall, these findings confirm that the pseudo-Arbuzov collapse of the MOM-derived chlorophosphonium species 7.0-a proceeds via an intramolecular nucleophilic mechanism, representing a well-defined transformation within phosphorus chemistry.
 | ||
| Fig. 2 DFT (B3LYP-631G*) calculated structure of transition state 7.0-TS: (a) space-fill view; (b) the nearly planar MOM fragment is marked by significantly elongated P–C and C–Cl bonds. | ||
Exploring the new reverse transformation –A: diverse reactivity of MOM derivatives
The spontaneous de-quaternization of 7.0-a (Scheme 8) complements the P-triangle (Scheme 1) with the missing reverse transformation –A. Here we show that this novel reaction is potentially general and, apart from a purely academic interest, is highly applicable as it leads from quaternary phosphonium P(V) structures back to P(III) derivatives. Importantly, this means that collapse of mixed-group chlorophosphonium species 7.1 (“1” – one alkyl group) would afford non-symmetrical monochlorophosphines R1PhPCl 8.1 and can provide a selective route to P(III) derivatives incorporating different small organic groups. Formerly, such species comprising sterically innocent groups were virtually inaccessible preparatively in good yields.8aTo examine this possibility, the required alkyl–aryl MOM-derived phosphine oxides 5.1 were prepared by MOM-protection of alkyldiphenylphosphines 1.1-a (R1 = Me) and 1.1-b (R1 = Et) followed by hydrolytic cleavage of the Ph. Treatment of 5.1-a/b with oxalyl chloride (Table 2) quantitatively gives methyl-, 7.1-a, and ethyl-substituted 7.1-b. The thermal collapse of the ethyl species 7.1-b was too slow at ambient temperature (Table 2, entry 1) and at 40 °C. The unwanted P(V) compound 9.1-b was mainly observed using dry chloroform as solvent (entry 2). Switching to the less polar benzene and toluene (entries 3 and 4) led to improved conversion to chlorophosphine EtPhPCl 8.1-b. However, using thoroughly degassed chloroform as solvent the collapse gave 8.1-b in excellent yield (sealed tube, 90 °C, 6 h, entry 5). Under such conditions, the collapse of Me-substituted 8.1-a gives chloride PhMePCl 9.1-a in 95% yield too (95 °C, 18 h, entry 6). These findings are consistent with the earlier observation that, in chloroform solution, chlorinated phosphonium species tend to exist in the form of reactive ion-pairs.28a
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R | Solvent | t, h | T, °C | 7.1b, % | 8.1, % | 9.1,% |
| a 7.1 generated by using COCl2 or SOCl2 in sealed tube. b Determined by 31P NMR analysis of reaction mixture. c Using 1.5 eq. (COCl)2. d Using 1.2 eq. (COCl)2. e Using excess SOCl2. f Sealed tube. | |||||||
| 1 | Et | CDCl3 | 18 | 25 | 98 | 0 | 2 |
| 2c | Et | CDCl3 | 16 | 40 | 47 | 6 | (26) |
| 3 | Et | C6D6 | 96 | 60 | 4 | 61 | (6) |
| 4d | Et | C7H8 | 24 | 75 | 9 | 79 | (12) |
| 5d | Et | CDCl3 | 6 | 90f | 5 | 93 | (4) |
| 6d | Me | CDCl3 | 5 | 95f | 5 | 81 | (14) |
| 7e | Et | SOCl2 | 12 | 75 | 8 | 0 | (92) |
| 8e | Me | SOCl2 | 12 | 75 | 3 | 0 | (97) |
Remarkably, the unwanted formation of phosphinic chlorides 9.1 by oxidative MOM-deprotection, can become dominant: switching the source of chlorine to SOCl2 gave 9.1-b and 9.1-a virtually quantitatively (entries 7 and 8). This new unusual transformation corresponds to another interesting sub-class which can be termed “–E” (Scheme 1) because, it can be argued, it entails a conversion of a MOM-derived 5.1 into a phosphinic chloride 9.1i.e. formal replacement of an alkyl group (MOM) with a chlorine. Here, again, we are looking at a reversal of standard reactivity as phosphinic species 9.1 themselves are commonly used for accessing phosphine oxide 5.1. Accordingly, treatment of 9.1 with PhMgCl for example (Scheme 10) leads to mixed-group tertiary phosphine oxides 3.1-b and 3.1-c in high yields. Oxidative MOM-deprotection using SOCl2 as an alternative reagent further broadens the scope of this methodology. Depending on the chlorination reagent employed, either P(III) chlorinated phosphines 8 or the corresponding P(V) phosphinic chlorides 9 can be obtained from MOM-derived phosphine oxides 5 in high yields.
 | ||
| Scheme 10 Oxidative MOM-deprotection of 5.1 followed by introduction of a Ph-group. | ||
The synthetic versatility of MOM-derived intermediate 7.0-a was further examined in the Umpolung quaternization to prepare MOM-substituted phosphonium salts (Scheme 11A). When 7.0-a was treated with alkylmagnesium chlorides at 0 °C, the mixed-substituent structures 6.1a–d were obtained, mostly in high yields; for instance, the reaction of iso-BuMgCl leads to 6.1-d in 91% yield (86% isolated).
 | ||
| Scheme 11 Umpolung quaternization: (A) preparation of MOM-substituted 6.1; (B) preparation of MOM-substituted 10.0. | ||
Importantly, all quaternary phosphonium products were isolated by easy crystallization, including 6.1-e (62% isolated). Remarkably, the reaction of 7.0-a with tert-BuMgCl gave MOM-derived phosphine 10.0 (Scheme 11B) providing a new route to the poorly explored tertiary phosphines containing an alkoxyalkyl group at the phosphorus center. We believe this phosphine is produced via an attack28a of the Grignard reagent on the electron-deficient covalently bound chlorine of 7.0-a.
Turning now to new synthetic opportunities presented by MOM-protection, a variety of compounds can now be made from triphenylphosphine 1.0-a as the starting point. As summarized in Scheme 12, this basic structure can be converted to mixed-group tertiary phosphines Ph2RP, 1.1, via the stages of MOM-derived 5 and 6 followed by MOM-deprotection of the key chlorinated intermediate 7.0-a. Finally, standard replacement of Cl affords phosphines 1.1-b and 1.0-b in high yields of 95% and 93%, respectively, based on the original Ph3P. Crucially, using MOM-derived chlorophosphonium compound 7.0-a, a different reaction sequence (path II, Scheme 12) leads, via the stage of quaternary 6.1, to MOM-derived phosphine oxides 5.1.
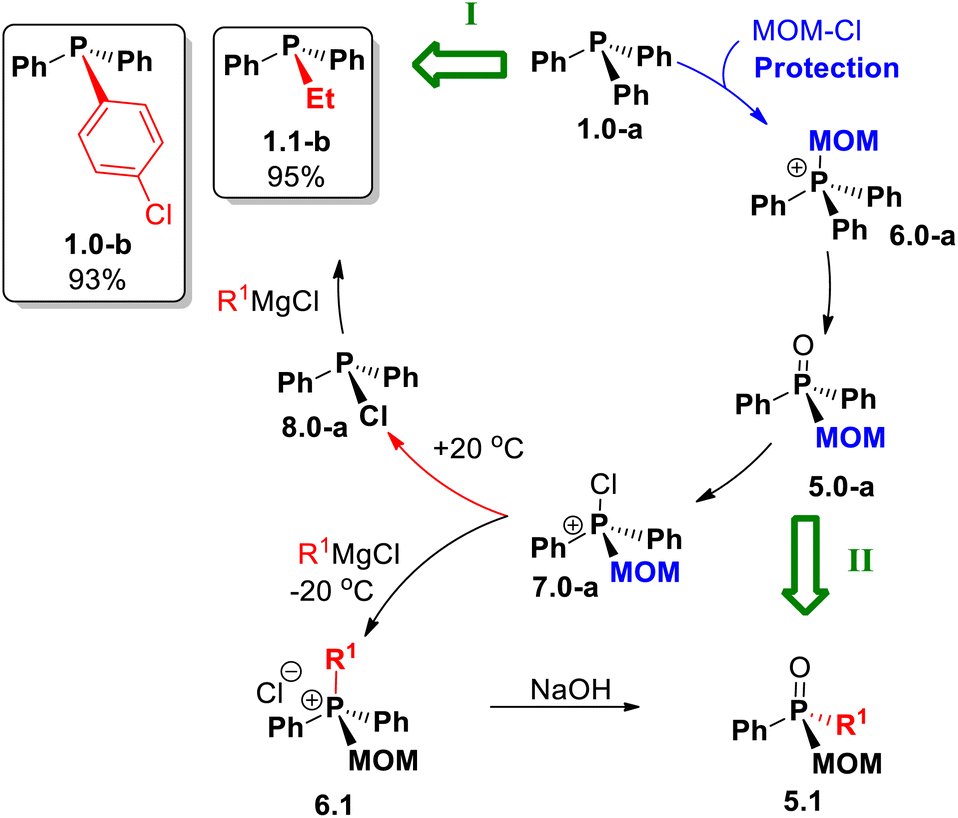 | ||
| Scheme 12 Efficient net transformations (green block arrows) of Ph3P enabled by MOM: (I) alkyl–aryl phosphines Ph2P–R1via the room temperature route. (II) mixed-group phosphine oxides 5.1 at low temperature. | ||
The mixed-group phosphine oxide 5.1, as a key intermediate, opens the access to even more structures shown in Scheme 13.
 | ||
| Scheme 13 Two-step synthesis of phosphines enabled by MOM-deprotection of 5.1 (see Scheme 12). | ||
As a prototypical example, the oxide 5.1-a, after chlorination–deprotection, leads to P(III) compound 8.1-a and, upon treatment with PhMgCl, gives the mixed-group phosphine 1.1-a in high overall yield (80%). Of course, in this example the re-introduction of a Ph group is degenerate and simply demonstrates the viability of a new MOM-protection approach. Significantly, preparation of the P-stereogenic 1.1-c from 5.1-b clearly demonstrates the synthetic value of the new MOM chemistry. Here, MOM-deprotection (via collapse of 7.1-b) leads to chlorophosphine 8.1-b (93% yield, Table 2) which, upon treatment with o-tolylmagnesium chloride, affords 1.1-c. Overall, this P-stereogenic phosphine has been prepared from symmetrical Ph3P in 56% yield over six steps (93% per step on average).
The results presented here are significant for phosphorus chemistry in two ways: firstly, the previously unknown pseudo-Arbuzov thermal collapse of MOM-derived chlorophosphonium species 7 leading to P-chlorophosphines is distinctly different from other organophosphorus chemistry reactions. It is highly complementary to the existing transformations forming the P-triangle (Scheme 1) as it belongs to the poorly explored reverse quaternization processes “–A”. Secondly, since the new route to P-chlorophosphines is selective and high yielding, it is very valuable in the design of new pathways to access a variety of asymmetric and P-stereogenic phosphines.
MOM-enabled sequential strategy
A broader synthetic framework was developed as an extension of the transformations shown in Scheme 12. It is represented in Scheme 14 as a sequence of interconnected reaction cycles. Starting with triphenylphosphine 1.0-a, through the first cycle (M1), the pathway reaches compound 7.0 – thermal branching point I. At elevated temperature, this route affords mixed tertiary phosphines 1.1, whereas maintaining the temperature below 0 °C leads, via inverse quaternization with Grignard reagents, to the MOM-derived phosphonium structure 6.1.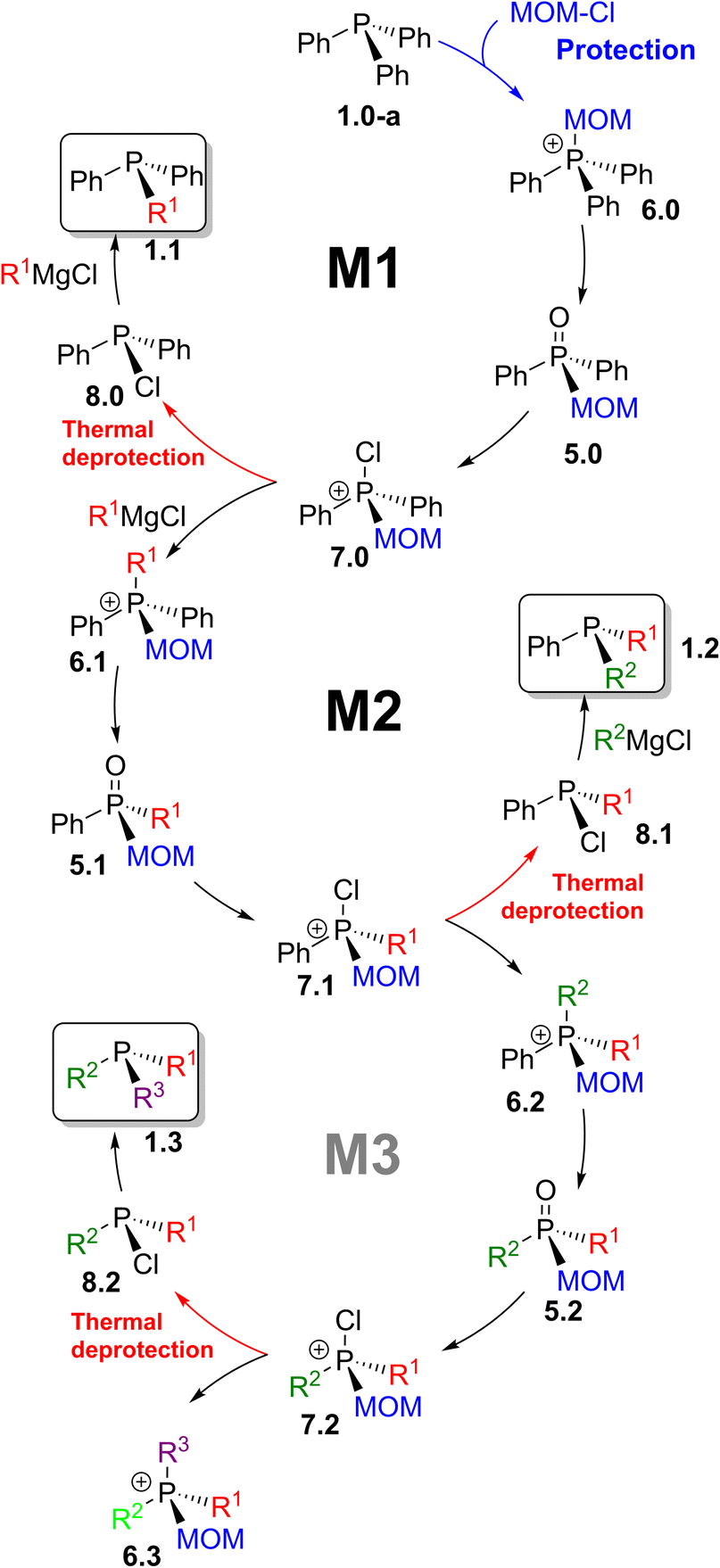 | ||
| Scheme 14 MOM-enabled chemistry: sequence M1 leads to phosphines 1.1; M2 gives stereogenic 1.2; M3 is a potential route to trialkylphosphines 1.3. | ||
This compound proceeds through the next cycle (M2), where a similar sequence of reactions leads to 7.1 – another thermally controlled branching point II. Upon heating, chlorinated intermediate 7.1 yields 8.1, which in turn produce mixed tertiary phosphines 1.2. Alternatively, at lower temperature, new mixed-group MOM-derived phosphonium species 6.2 are formed. These serve as entry point to another proposed cycle (M3), extending the methodology toward more extensively substituted phosphines 1.3 in which all three alkyl groups are introduced through successive replacement of phenyl substituents in Ph3P 1.0-a.
This synthetic approach relies on MOM-quaternization of phosphines 1, hydrolysis of phosphonium salts 6, and Grignard reagent metathesis with P-chlorophosphines 8. Fundamentally, by combining the Umpolung quaternization step (Scheme 2, A) with MOM-based protection–deprotection, symmetrical triphenylphosphine and its oxide can be efficiently transformed into a wide range of mixed-group and P-stereogenic phosphines 1.2 and 1.3 in a few steps.
Conclusions
The new systematic approach to the synthesis of P(V) compounds features high-yielding reverse transformations combined with established reactions of organophosphorus chemistry. Centred on the Umpolung quaternization process –C, this methodology enables the preparation of diverse mixed and P-stereogenic phosphine oxides and phosphonium salts from readily available and often discarded triphenylphosphine oxide using inexpensive and convenient reagent–condition combinations. The synthesis of the smallest P-stereogenic quaternary tetraalkylphosphonium structure from triphenylphosphine oxide exemplifies access to a fundamental chemical structure that was previously difficult or impossible to obtain.In addition, the key elements of this approach were combined with an unprecedented reverse quaternization of MOM-derived species –A. This process provides an efficient route from MOM-derived tertiary phosphine oxides to chlorinated P(III) compounds. For the first time, a multi-step protection–deprotection strategy was achieved at the nucleophilic phosphorus centre using standard MOM–Cl as the protecting reagent which can be recycled.
The missing connections, –A and –C, between the three key classes of organophosphorus compounds 1, 2 and 3 have been added to the P-triangle (compare to Scheme 1) as follows:
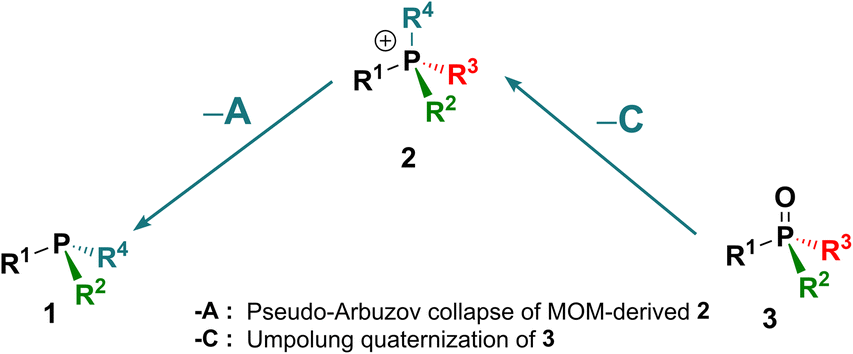
This simple diagram illustrates how the new findings complete the existing network of organophosphorus transformations by introducing two reverse processes. The reactions are complementary: path –A is a P–C bond-breaking process, while path –C is a P–C bond-forming process. Moreover, the P–C bond created in process –C is typically not the same bond cleaved in reaction –A, making their combination a powerful synthetic tool for constructing diverse organophosphorus architectures.
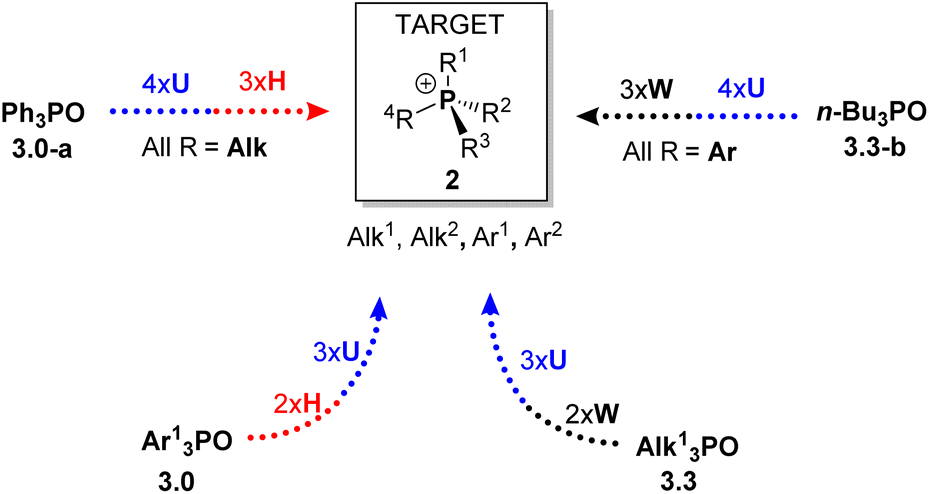
For instance, new structures R4P (where R represents alkyl or aryl groups, or O for phosphine oxides 3) can be synthesized through iterative application of U and H steps. All-alkyl species 2 can be obtained from Ph3PO (3.0-a) via four U and three H steps, while all-aryl structures can be accessed from symmetrical n-Bu3PO (3.3-b) using U and W steps. For mixed structures lacking Ph or n-Bu substituents, the corresponding symmetrical triaryl 3.0 or trialkyl 3.3 derivative can serve as the starting point, following then the same iterative approach.
In summary, two distinct and complementary missing links have been established between the principal classes of organophosphorus compounds. Through these transformations, many mixed aryl–alkyl phosphines can be prepared from Ph3P, and asymmetric (P-stereogenic) quaternary phosphonium salts can be synthesized from Ph3PO and other phosphine oxides in only a few steps. These new synthetic tools provide straightforward access to a wide range of mixed and P-stereogenic phosphines, their oxides, and phosphonium salts from inexpensive and widely available phosphorus precursors. The results presented here, together with earlier findings, indicate that these transformations open extensive new possibilities in organophosphorus chemistry, with broad potential for the synthesis of valuable P(III) and P(V) target compounds.
Author contributions
Conceptualization and design: A. C. V., K. N., D. G. G. Data collection: A. C. V., Y. O. (supporting), K. N.; Writing: A. C. V., K. N., DGG (supporting); supervision and funding acquisition: D. G. G.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: details of experimental methods, characterisation data. See DOI: https://doi.org/10.1039/d5sc04496k.Acknowledgements
This publication has emanated primarily from research supported by Science Foundation Ireland through the SFI-funded Synthesis & Solid-State Pharmaceutical Centre 12/RC/2275 Grant to D. G. G. The authors are grateful to UCD School of Chemistry for use of their analysis facilities.Notes and references
- (a) F. H. Westheimer, Science, 1987, 235, 1173–1178 CrossRef CAS; (b) D. E. C. Corbridge, Phosphorus Chemistry, Biochemistry and Technology, CRC Press, 6th edn, 2013 Search PubMed; (c) Y. Zhao, Y. Liu, X. Gao and P. Xu, Phosphorus Chemistry. The Role of Phosphorus in Prebiotic Chemistry, DeGruyter, 2018 CrossRef; (d) C. T. Walsh, The Chemical Biology of Phosphorus, Royal Society of Chemistry, 2020 RSC; (e) E. Macia-Barber, The Chemical Evolution of Phosphorus: An Interdisciplinary Approach to Astrobiology, Apple Academic Press, 2021 Search PubMed; (f) D. Egan, The Devil's Element: Phosphorus and a World Out of Balance, W. W. Norton & Company, 2023 Search PubMed.
- (a) H. Goldwhite, Introduction to Phosphorus Chemistry, Cambridge University Press, 1981 Search PubMed; (b) L. D. Quin, A Guide to Organophosphorus Chemistry, Wiley, 2000 Search PubMed; (c) L. D. Quin, The Development of Organic Phosphorus Chemistry: Discoveries of the Nineteenth Century, Nova Science Publishers, 2014 Search PubMed; (d) L. D. Quin, Heteroat. Chem., 2013, 24, 243–251 CrossRef CAS.
- (a) Organic Phosphorus Compounds, ed. G. M. Kosolapoff and L. Maier, Wiley, 1972–1976, vol. 1–7 Search PubMed; (b) Organophosphorus Compounds, ed. F. R. Hartley, Wiley, 1990–1996, vol. 1–4 Search PubMed; (c) K. B. Dillon, F. Mathey and J. F. Nixon, Phosphorus: The Carbon Copy: From Organophosphorus to Phospha-organic Chemistry, Wiley, 1998 Search PubMed; (d) R. Engel and J. I. Cohen, Synthesis of Carbon–Phosphorus Bonds, CRC Press, 2nd edn, 2004 Search PubMed; (e) Phosphorus Chemistry I: Asymmetric Synthesis and Bioactive Compounds, ed. J.-L. Montchamp, Springer, 2015 Search PubMed; (f) V. Iaroshenko, Organophosphorus Chemistry: From Molecules to Applications, Wiley-VCH Verlag, 2019 CrossRef.
- (a) J. I. G. Cadogan, Organophosphorus Reagents in Organic Synthesis, Academic Press, 1979 Search PubMed; (b) F. Karanam, G. M. Reddy, S. R. Koppolu and W. Lin, Tetrahedron Lett., 2018, 59, 59–76 CrossRef; (c) D. H. Valentine and J. H. Hillhouse, Synthesis, 2003, 35, 317–334 Search PubMed; (d) S. L. Xu and Z. J. He, RSC Adv., 2013, 3, 16885–16904 RSC.
- (a) R. F. Heck, Org. React., 1982, 27, 345–390 CAS; (b) S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1439 CrossRef CAS; (c) C. A. Fleckenstein and H. Plenio, Chem. Soc. Rev., 2010, 39, 694–711 RSC; (d) J. Margalef, M. Biosca, P. de la Cruz Sánchez, J. Faiges, O. Pàmies and M. Diéguez, Coord. Chem. Rev., 2021, 446, 214120 CrossRef CAS.
- (a) Y. M. Xiao, Z. H. Sun, H. C. Guo and O. Kwon, Beilstein J. Org. Chem., 2014, 10, 2089–2121 CrossRef; (b) S. Y. Liu, Y. Kumatabara and S. Shirakawa, Green Chem., 2016, 18, 331–341 Search PubMed; (c) D. Enders and T. V. Nguyen, Org. Biomol. Chem., 2012, 10, 5327–5331 RSC; (d) J. L. Methot and W. R. Roush, Adv. Synth. Catal., 2004, 346, 1035–1050 CrossRef CAS; (e) H. C. Guo, Y. C. Fan, Z. H. Sun, Y. Wu and O. Kwon, Chem. Rev., 2018, 118, 10049–10293 Search PubMed; (f) T. Werner, Adv. Synth. Catal., 2009, 351, 1469–1481 CrossRef CAS; (g) A. Golandaj, A. Ahmad and D. Ramjugernath, Adv. Synth. Catal., 2017, 359, 3676–3706 CrossRef CAS.
- (a) D. J. Scott, J. Cammarata, M. Schimpf and R. Wolf, Nat. Chem., 2021, 13, 458–464 CrossRef CAS PubMed; (b) M. B. Geeson and C. C. Cummins, Science, 2018, 359, 1383–1385 CrossRef CAS.
- (a) D. G. Gilheany and C. M. Mitchell, Preparation of Phosphines, in ref. 3b, 1990, ch. 7, vol. 1, pp. 151–190 Search PubMed; (b) X. Chen, H. Wu, R. Yu, H. Zhu and Z. Wang, J. Org. Chem., 2021, 86, 8987–8996 CrossRef CAS PubMed; (c) C. Baillie and J. L. Xiao, Curr. Org. Chem., 2003, 7, 477–514 CrossRef CAS.
- (a) H.-J. Cristau and F. Plenat, Preparation, Properties and Reactions of Phosphonium Salts, in ref. 3b, 1994, vol. 3, pp. 45–183 Search PubMed; (b) A. C. Shaikh, J. M. Veleta, J. Bloch, H. J. Goodman and T. L. Gianetti, Eur. J. Org Chem., 2020, 2020, 2553–2559 Search PubMed; (c) C. C. Xu, T. Li, P. K. Jiang and Y. J. Zhang, Tetrahedron, 2020, 76, 131107 Search PubMed; (d) A. K. Bhattacharya and N. K. Roy, Methods of Preparation of Phosphine Chalcogenides, in ref. 3b, 1992, vol. 2, pp. 195–285 Search PubMed.
- (a) P. A. Byrne and D. G. Gilheany, Chem.–Eur. J., 2016, 22, 9140–9154 CrossRef CAS PubMed; (b) P. A. Byrne and D. G. Gilheany, Chem. Soc. Rev., 2013, 42, 6670–6696 RSC; (c) O. Korpiun, R. A. Lewis, J. Chickos and K. Mislow, J. Am. Chem. Soc., 1968, 90, 4842–4846 CrossRef CAS; (d) D. Valentine, J. F. Blount and K. Toth, J. Org. Chem., 1980, 45, 3691–3698 CrossRef CAS; (e) C.-H. Zhong and W. Huang, ACS Omega, 2020, 5, 16010–16020 CrossRef CAS; (f) C.-H. Zhong and W. Huang, Synth. Commun., 2021, 51, 1537–1546 CAS; (g) K. Nikitin, Y. Ortin, H. Müller-Bunz, D. G. Gilheany and M. J. McGlinchey, Eur. J. Org Chem., 2018, 2018, 5260–5267 CrossRef CAS.
- (a) R. S. Edmundson, The Synthesis of Phosphonic and Phosphinic Acids and their Derivatives, in ref. 3b, 1996, ch 2, vol. 4, pp. 47–144 Search PubMed; (b) J. Ash, H. Huang, P. Cordero and J. Y. Kang, Org. Biomol. Chem., 2021, 19, 6007–6014 RSC; (c) K. C. Forbes and E. N. Jacobsen, Science, 2022, 376, 1230–1236 Search PubMed; (d) M. Stankevic, J. Pisklak and K. Wlodarczyk, Tetrahedron, 2016, 72, 810–824 Search PubMed.
- (a) E. V. Jennings, K. Nikitin, Y. Ortin and D. G. Gilheany, J. Am. Chem. Soc., 2014, 136, 16217–16226 CrossRef CAS PubMed; (b) K. Nikitin, E. V. Jennings, S. S. Al Sulaimi, Y. Ortin and D. G. Gilheany, Angew. Chem., Int. Ed., 2018, 57, 1480–1484 CrossRef CAS; (c) K. V. Rajendran and D. G. Gilheany, Chem. Commun., 2012, 48, 10040–10042 RSC; (d) K. Nikitin, K. V. Rajendran, H. Müller-Bunz and D. G. Gilheany, Angew. Chem., Int. Ed., 2014, 53, 1906–1909 CrossRef CAS PubMed; (e) D. A. Tatarinov, D. M. Kuznetsov, A. D. Voloshina, A. P. Lyubina, A. S. Strobykina, F. K. Mukhitova, F. M. Polyancev and V. F. Mironov, Tetrahedron, 2016, 72, 8493–8501 CrossRef CAS.
- (a) Z. Xu, P. Wang, Q. Chen and M. Cai, J. Organomet. Chem., 2018, 866, 50–58 Search PubMed; (b) C. J. Li, J. Lu, Z. X. Zhang, K. Zhou, Y. Li and G. H. Qi, Res. Chem. Intermed., 2018, 44, 4547–4562 CrossRef CAS; (c) R. R. Yu, X. Y. Chen, S. F. Martin and Z. Q. Wang, Org. Lett., 2017, 19, 1808–1811 CrossRef CAS PubMed; (d) D. S. Glueck, Dalton Trans., 2008, 2008, 5276–5286 RSC; (e) J. Ye, J.-Q. Zhang, Y. Saga, S.-y. Onozawa, S. Kobayashi, K. Sato, N. Fukaya and L.-B. Han, Organometallics, 2020, 39, 2682–2694 CrossRef CAS; (f) P. Nandi, J. L. Dye, P. Bentley and J. E. Jackson, Org. Lett., 2009, 11, 1689–1692 Search PubMed.
- (a) S.-G. Li, M. Yuan, F. Topic, Z. S. Han, C. H. Senanayake and Y. S. Tsantrizos, J. Org. Chem., 2019, 84, 7291–7302 CrossRef CAS; (b) Y. Nishiyama, Y. Hazama, S. Yoshida and T. Hosoya, Org. Lett., 2017, 19, 3899–3902 Search PubMed; (c) M. B. Kurosawa, R. Isshiki, K. Muto and J. Yamaguchi, J. Am. Chem. Soc., 2020, 142, 7386–7392 CrossRef CAS PubMed; (d) R. Isshiki, K. Muto and J. Yamaguchi, Org. Lett., 2018, 20, 1150–1153 CrossRef CAS; (e) J. Yang, J. Xiao, T. Chen, D.-F. Yin and L.-B. Han, Chem. Commun., 2016, 52, 12233–12236 Search PubMed; (f) Q. Dai, W. B. Li, Z. Lo and J. L. Zhang, J. Am. Chem. Soc., 2019, 141, 20556–20564 Search PubMed; (g) H. Ohta, Q. Xue and M. Hayashi, Eur. J. Org Chem., 2018, 2018, 735–738 CrossRef CAS; (h) J.-Q. Zhang, J. Ye, T. Huang, H. Shinohara, H. Fujino and L.-B. Han, Commun. Chem., 2020, 3, 1 CrossRef CAS; (i) S. Lemouzy, R. Membrat, E. Olivieri, M. Jean, M. Albalat, D. Nuel, L. Giordano, D. Hérault and G. Buono, J. Org. Chem., 2019, 84, 4551–4557 CrossRef CAS; (j) Y. Zhang, H. He, Q. Wang and Q. Cai, Tetrahedron Lett., 2016, 57, 5308–5311 CrossRef CAS; (k) B. Verbelen, W. Dehaen and K. Binnemans, Synthesis, 2018, 50, 2019–2026 CrossRef CAS.
- (a) W. A. Henderson and S. A. Buckler, J. Am. Chem. Soc., 1960, 82, 5794–5800 CrossRef CAS; (b) M. T. Honaker, J. M. Hovland and R. N. Salvatore, Curr. Org. Synth., 2007, 4, 31–45 CrossRef CAS; (c) F. M. J. Tappe, V. T. Trepohl and M. Oestreich, Synthesis, 2010, 42, 3037–3062 Search PubMed; (d) D. I. Bugaenko, A. A. Volkov, M. V. Livantsov, M. A. Yurovskaya and A. V. Karchava, Chem.–Eur. J., 2019, 25, 12502–12506 Search PubMed; (e) P. N. Chalikidi, T. T. Magkoev, A. V. Gutnov, O. P. Demidov, M. G. Uchuskin, I. V. Trushkov and V. T. Abaev, J. Org. Chem., 2021, 86, 9838–9846 CrossRef CAS PubMed.
- C. L. Chernick and H. A. Skinner, J. Chem. Soc., 1956, 1401–1405 RSC.
- (a) D. Herault, D. H. Nguyen, D. Nuel and G. Buono, Chem. Soc. Rev., 2015, 44, 2508–2528 RSC; (b) T. Kovacs and G. Keglevich, Curr. Org. Chem., 2017, 21, 569–585 CrossRef CAS; (c) E. Podyacheva, E. Kuchuk and D. Chusov, Tetrahedron Lett., 2019, 60, 575–582 Search PubMed; (d) A. J. Stepen, M. Bursch, S. Grimme, D. W. Stephan and J. Paradies, Angew. Chem., Int. Ed., 2018, 57, 15253–15256 CrossRef CAS; (e) Y. Li, S. Das, S. Zhou, K. Junge and M. Beller, J. Am. Chem. Soc., 2012, 134, 9727–9732 Search PubMed; (f) Y. Li, L.-Q. Lu, S. Das, S. Pisiewicz, K. Junge and M. Beller, J. Am. Chem. Soc., 2012, 134, 18325–18329 Search PubMed; (g) P. Li, R. Wischert and P. Metivier, Angew. Chem., Int. Ed., 2017, 56, 15989–15992 Search PubMed; (h) Ł. Kapuśniak, P. N. Plessow, D. Trzybiński, K. Woźniak, P. Hofmann and P. I. Jolly, Organometallics, 2021, 40, 693–701 CrossRef; (i) J. Xue, Y. S. Zhang, Z. Huan, J. D. Yang and J. P. Cheng, J. Am. Chem. Soc., 2023, 145, 15589–15599 CrossRef CAS PubMed.
- (a) K. V. Rajendran and D. G. Gilheany, Chem. Commun., 2012, 48, 817–819 Search PubMed; (b) K. V. Rajendran, J. S. Kudavalli, K. S. Dunne and D. G. Gilheany, Eur. J. Org Chem., 2020, 2012, 2720–2723 Search PubMed; (c) S. S. Al Sulaimi, K. V. Rajendran and D. G. Gilheany, Eur. J. Org Chem., 2015, 2015, 5959–5965 CrossRef CAS.
- Note that reduction generating 1 can further be telescoped to the formation of the corresponding QPS 2, a principle which is utilized for a catalytic version of Wittig olefination: C. J. O'Brien, J. L. Tellez, Z. S. Nixon, L. J. Kang, A. L. Carter, S. R. Kunkel, K. C. Przeworski and G. A. Chass, Angew. Chem., Int. Ed., 2009, 48, 6836–6839 Search PubMed.
- (a) I. J. S. Fairlamb, ChemSusChem, 2009, 2, 1021–1024 CrossRef CAS PubMed; (b) E. E. Coyle, B. J. Doonan, A. J. Holohan, K. A. Walsh, F. Lavigne, E. H. Krenske and C. J. O'Brien, Angew. Chem., Int. Ed., 2014, 53, 12907–12911 CrossRef CAS PubMed; (c) J. Tönjes, L. Longwitz and T. Werner, Green Chem., 2021, 23, 4852–4857 Search PubMed.
- The reduction could also be combined with C–P bond formation: e.g.: L. Zhang, C. Liu, L. Yang, L. Cao, C. Liang, M. Sun, Y. Ma, R. Cheng and J. Ye, Org. Biomol. Chem., 2022, 20, 3897–3901 RSC.
- (a) D. Virieux, J.-N. Volle and J.-L. Pirat, Alkylphosphonium Salts in Science of Synthesis, ed. F. Mathey and B. M. Trost, Thieme, 2009, vol. 42, pp. 503–594 Search PubMed; (b) J. C. Tebby and D. W. Allen, Arylphosphonium Salts and Derivatives in Science of Synthesis, ed. C. A. Ramsden, Thieme, 2007, vol. 31, pp. 2083–2104 Search PubMed; (c) I. Wauters, W. Debrouwer and C. V. Stevens, Beilstein J. Org. Chem., 2014, 10, 1064–1096 CrossRef PubMed.
- (a) L. Horner and A. Mentrup, Annalen, 1961, 646, 65–77 Search PubMed; (b) L. Horner, H. Fuchs and F. Rottger, Chem. Ber., 1963, 96, 3141–3147 CrossRef CAS; (c) J. Grimshaw and J. S. Ramsey, J. Chem. Soc. B, 1968, 63–64 Search PubMed; (d) K. S. V. Santhanam, Electrochemistry of Ylides, Phosphoranes and Phosphonium Salts, in ref 3b, 1994, vol. 3, pp. 303–323 Search PubMed.
- Hydrolytic dealkylation can occur under some circumstances e.g.: J. J. Brophy and M. J. Gallagher, Aust. J. Chem., 1969, 22, 1385–1398 CrossRef CAS.
- (a) N. Donoghue and M. J. Gallagher, Chem. Commun., 1998, 1973–1974 RSC; (b) S. Roediger, S. U. Leutenegger and B. Morandi, Chem. Sci., 2022, 13, 7914–7919 RSC; (c) M. Lei, X. Chen, Y. Wang, L. Zhang, H. Zhu and Z. Wang, Org. Lett., 2022, 24, 2868–2872 Search PubMed; (d) X. Zhang and A. McNally, Angew. Chem., Int. Ed., 2017, 56, 9833–9836 Search PubMed.
- (a) J. B. Hendrickson and S. M. Schwartzman, Tetrahedron Lett., 1975, 16, 277–280 CrossRef; (b) M. Masaki and K. Fukui, Chem. Lett., 1977, 1977, 151–152 CrossRef; (c) L. D. Quin, J. C. Kisalus, J. J. Skolimowski and N. S. Rao, Phosphorus, Sulfur Silicon Relat. Elem., 1990, 54, 1–7 CrossRef CAS; (d) N. P. Kenny, K. V. Rajendran, E. V. Jennings and D. G. Gilheany, Chem.–Eur. J., 2013, 19, 14210–14214 CrossRef CAS PubMed; (e) N. P. Kenny, K. V. Rajendran and D. G. Gilheany, Chem. Commun., 2015, 51, 16561–16564 RSC; (f) K. Nikitin, H. Müller-Bunz and D. G. Gilheany, Chem. Commun., 2013, 49, 1434–1436 RSC.
- (a) R. M. Denton, J. An, B. Adeniran, A. J. Blake, W. Lewis and A. M. Poulton, J. Org. Chem., 2011, 76, 6749–6767 CrossRef CAS PubMed; (b) R. H. Beddoe, K. G. Andrews, V. Magné, J. D. Cuthbertson, J. Saska, A. L. Shannon-Little, S. E. Shanahan, H. F. Sneddon and R. M. Denton, Science, 2019, 365, 910–914 Search PubMed; (c) Y. Zou, J. J. Wong and K. N. Houk, J. Am. Chem. Soc., 2020, 142, 16403–16408 CrossRef CAS; (d) C. M. Xie, A. J. Smaligo, X. R. Song and O. Kwon, ACS Cent. Sci., 2021, 7, 536–558 Search PubMed.
- (a) A. C. Vetter, K. Nikitin and D. G. Gilheany, Chem. Commun., 2018, 54, 5843–5846 RSC; (b) presented at the 22nd International Conference on Phosphorus Chemistry (ICPC) held in Budapest, Hungary, July 2018: A. C. Vetter, K. Nikitin and D. G. Gilheany, Phosphorus, Sulfur Silicon Relat. Elem., 2019, 194, 339–342 CrossRef CAS.
- (a) H. A. van Kalkeren, F. L. van Delft and F. Rutjes, ChemSusChem, 2013, 6, 1615–1624 CrossRef CAS; (b) M. Y. Pei, A. Q. Tian, Q. Q. Yang, N. Y. Huang, L. Wang and D. S. Li, Green Synth. Catal., 2023, 4, 135–149 Search PubMed.
- Oxide 3.2-b is not in the literature but its precursor phosphine has been made by multiple alkylation of phenylphosphide anion, iself prepared from primary (and spontaneously flammable) phenylphosphine: E. N. Tsvetkov, N. A. Bondarenko, I. G. Malakhova and M. I. Kabachnik, Synthesis, 1986, 18, 198–208 CrossRef.
- A. Kuźnik, R. Mazurkiewicz and B. Fryczkowska, Beilstein J. Org. Chem., 2017, 13, 2710–2738 CrossRef.
- There is a single report of vinylphosphonium chloride 2.3-e made by dehydrobromination of the 2-bromoethyl salt resultant from the reaction of tributylphosphine and dibromoethane and subsequent chloride ion exchange: R. Rabinowitz, A. C. Henry and R. Marcus, J. Polym. Sci., Part A: Gen. Pap., 1965, 3, 2055–2061 CrossRef CAS.
- At least on laboratory scale, Ph3PO (Sigma-Aldrich 100 g, $98) and Ph2PCl (100 g, $155) have similar costs.
- The synthesis of this QPS has been claimed but with no yield or characterisation data: M. Schmidt and W. R. Neeff, Angew. Chem., Int. Ed. Engl., 1970, 9, 807–808 CrossRef CAS.
- B. Streicher and B. Wünsch, Carbohydr. Res., 2003, 338, 2375–2385 Search PubMed.
- P. Xing, Z.-g. Huang, Y. Jin and B. A. Jiang, Synthesis, 2013, 45, 596–600 Search PubMed.
- Analogous spontaneous collapse of mixed group P-chloroacetylphosphines has been documented: E. Lindner, R. D. Merkle and H. A. Mayer, Chem. Ber., 1986, 119, 645–658 CrossRef CAS.
- K. Nikitin, H. Müller-Bunz, J. Muldoon and D. G. Gilheany, Chem.–Eur. J., 2017, 23, 4794–4802 CrossRef CAS.
- (a) J. M. Mattalia, B. Vacher, A. Samat and M. Chanon, J. Am. Chem. Soc., 1992, 114, 4111–4119 CrossRef CAS; (b) J. P. Snyder, L. Lee, V. T. Bandurco, C. Y. Yu and R. J. Boyd, J. Am. Chem. Soc., 1972, 94, 3260–3261 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2026 |