
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new generation of N-heterocyclic carbene (NHC) gold–selenolato complexes as potent anticancer agents: distinct synthetic routes and evaluation in 2D and 3D cancer models
Pierre
Arnaut†
a,
Nestor Bracho
Pozsoni†
 a,
Denys
Bondar
ab,
Petra
Lippmann
c,
Susanne
Boschuk
c,
Ivan
Semenyuta
d,
Subhrajyoti
Bhandary
e,
Kristof
Van Hecke
e,
Yevgen
Karpichev
b,
Enrico
Cavarzerani
f,
Vincenzo
Canzonieri
gh,
Flavio
Rizzolio
fg,
Thomas
Scattolin
i,
Georgios C.
Vougioukalakis
j,
Ingo
Ott
*c,
Nikolaos V.
Tzouras
*aj and
Steven P.
Nolan
*a
a,
Denys
Bondar
ab,
Petra
Lippmann
c,
Susanne
Boschuk
c,
Ivan
Semenyuta
d,
Subhrajyoti
Bhandary
e,
Kristof
Van Hecke
e,
Yevgen
Karpichev
b,
Enrico
Cavarzerani
f,
Vincenzo
Canzonieri
gh,
Flavio
Rizzolio
fg,
Thomas
Scattolin
i,
Georgios C.
Vougioukalakis
j,
Ingo
Ott
*c,
Nikolaos V.
Tzouras
*aj and
Steven P.
Nolan
*a
aDepartment of Chemistry and Centre for Sustainable Chemistry, Ghent University, Ghent, Belgium. E-mail: Nikolaos.Tzouras@ugent.be; Steven.Nolan@UGent.be
bDepartment of Chemistry and Biotechnology, Tallinn University of Technology, School of Science, Tallinn, Estonia
cInstitute of Medicinal and Pharmaceutical Chemistry, Technische Universität Braunschweig, Braunschweig, Germany. E-mail: ingo.ott@tu-bs.de
dDepartment of Chemistry Bioactive Nitrogen-containing Heterocyclic Bases, V. P. Kukhar Institute of Bioorganic Chemistry and Petrochemistry of the NASU, Kyiv, Ukraine
eXStruct, Department of Chemistry, Ghent University, Krijgslaan 281-S3, 9000 Ghent, Belgium
fDipartimento di Scienze Molecolari e Nanosistemi, Università Ca’ Foscari, Campus Scientifico Via Torino 155, 30174 Venezia-Mestre, Italy
gPathology Unit, Centro di Riferimento Oncologico di Aviano (C.R.O.) IRCCS, via Franco Gallini 2, Aviano, 33081, Italy
hDepartment of Medical, Surgical and Health Sciences, Università degli Studi di Trieste, Strada di Fiume 447, Trieste, Italy
iDipartimento di Scienze Chimiche, Università degli Studi di Padova, via Marzolo 1, 35131 Padova, Italy
jDepartment of Chemistry, National and Kapodistrian University of Athens, Panepistimiopolis, 15771 Athens, Greece
First published on 19th August 2025
Abstract
Two distinct synthetic pathways are disclosed that lead to new gold–selenolato complexes, stabilized by N-heterocyclic carbenes (NHCs). The weak base route can provide facile access to phenylselenolate complexes of gold, using both NHC and phopshine ligands. In addition, the pathway based on the carbometallation of elemental selenium enables the construction of a more diverse library of products, based on substituted aryl-selenide fragments whose selenol congeners are not commercially available. Biological studies performed on human cancer cell lines (A-549, H-T29, and MCF-7) and mammal healthy cell lines (Vero-E6) reveal that a selection of these complexes exhibit cytotoxic activity and are selective towards cancerous cells. In vitro experiments confirmed that our lead candidate is indeed a TrxR inhibitor. Finally, this complex showed strong cytotoxic activity even in advanced biological models, including patient-derived 3D tumor organoids. Noteworthily, it remained effective in both colon cancer and HGSOC organoids, even in patients resistant to standard chemotherapy agents.
Introduction
Selenium plays a role in numerous biological processes,1 one of which being the regulation of reactive oxygen species (ROS).2 Although these compounds are natural by-products of mitochondrial respiration and can be involved in cellular signaling,3,4 their accumulation has deleterious effects on DNA and proteins.5,6 Increased ROS levels are also characteristic of tumors, where they mediate both cell proliferation and angiogenesis. However, due to their high metabolism, tumors are particularly sensitive to oxidative stress, which explains why ROS can both serve as tumor-promoting and tumor-suppressing agents.7 This ambivalent role of ROS has led to different approaches for cancer treatment, by either increasing or lowering the production of ROS.8 This article will be focusing on the first strategy, through the inhibition of antioxidant systems in cancer cells.Thioredoxin reductases (TrxR) and glutathione peroxidases (GPx) are key antioxidative enzymes that depend directly on selenocysteine residues.9 Because of the instability of the Se–O bond, Se oxides can be rapidly reduced to selenols, which is why selenoenzymes tend to resist permanent inactivation by oxidation. TrxR exist in three forms: one located in the cytoplasm of cells, and the other two in the mitochondria; they represent a critical pharmacological target for apoptosis-inducing agents.10 The presence of a cysteine close to a selenocysteine enables TrxR to interact with organometallic complexes by coordinating to thiol and selenol groups.11–13 Conversely, GPx cannot form chelates with metals due to their distant selenol groups. Another aspect that makes TrxR an attractive therapeutic target is related to their incredible affinity for soft transition metals, especially gold(I) and platinum(II).14 For reference, only nanomolar (nM) concentrations of auranofin will reduce TrxR activity to 50%, whereas micromolar (μM) levels are needed for GPx.15
Cancer cells exhibit elevated mitochondrial membrane potential compared to healthy cells,16 which causes the accumulation of lipophilic cations within the mitochondria. Therefore, following the design of auranofin in the late 1970s,17 one of the earlier examples of analogue structures was a bis-chelated cationic gold–phosphine complex.18,19 Changing from phosphines to triazaphosphaadamantane ligands increased the water-solubility of the complex, with limited results against cancer cells.20,21 It is worth noting that even though the thiosugar ligand is not essential for the cytotoxic activity of auranofin,22,23 the presence of a glycosidic unit is still an important factor in the design of chemotherapeutic agents, since carbohydrates can act as active sites for molecular recognition, in accordance with the Warburg effect.24 As exemplified with cisplatin,25 ligand exchange is a critical step for metallodrugs before the actual binding process.26,27 The use of N-heterocyclic carbene (NHC) ligands paved the way to more selective compounds thanks to a more refined balance of hydrophilic and lipophilic character for membrane permeation.28 For instance, shorter alkyl chains (IMe = 1,3-dimethylimidazol-2-ylidene) display lower lipophilicity than longer ones (such as nBu = 1,3-di(tert-butyl)imidazol-2-ylidene or ICy = 1,3-di(cyclohexyl)imidazol-2-ylidene), as is to be expected.29 Another benefit of NHCs, besides substituent versatility, is the ease of synthesis of their transition metal complexes via the weak base route, which can be conducted under air with green solvents (including acetone, ethanol, and ethyl acetate).30–32 Gold–NHC complexes can have antioxidant,33 antibacterial,34 and anticancer35–37 properties and are capable of binding to cysteine and selenocysteine residues in proteins.38–40 Bis-NHC gold complexes have also shown cytotoxic activity against acute myeloid leukemia cells (HL60) and cisplatin resistant cell lines, notably ovarian cancer (A2780).38 [(NHC)2Au]+ cationic complexes were reported to selectively target mitochondrial TrxR to induce the apoptosis of cancer cells without affecting the corresponding healthy cells. In this case, it was shown that the gold center forms bridges between cysteine and selenocysteine residues, and that selenols are preferred over thiols by several orders of magnitude.41
The first characterized gold–selenolato complex dates to the 1990s.42 The reason behind the incorporation of a selenium atom, as a substitute to sulfur, was to reduce toxicity and adverse side effects.43–45 A common synthetic pathway was to mix phosphine gold chlorides with a selenolate.46–49 These protocols, however, require strict inert conditions, due to selenols being typically air sensitive. Alternatively, it is possible to protect the selenol by silylation to obtain gold–selenolates in air, as it was performed in the specific case of dinuclear complexes with ferrocene (Scheme 1).50 Since then, even though numerous gold derivatives have been developed, a larger emphasis has been given to Au–S bond-containing complexes.51–54 Besides gold complexes bearing selenourea ligands, examples of Au–Se bond-containing complexes remain scarce.43,55–57 Therefore, it is in this context that we aimed to synthesize M–selenolato derivatives (M = Au, Cu, Ag), in an attempt to gain a better understanding of the mechanism behind the biological activity of these complexes, as well as the structural parameters which affect this activity. To this end, we considered two possible synthetic pathways: the weak base route, to access M–arylselenolato complexes from the corresponding [M(NHC)Cl] complexes and selenophenol, and a second method – the formal Se insertion into the Au–C bond which occurs when a Au–aryl complex is heated in the presence of elemental selenium. The use of the weak base route for selenol metalation represents a new synthetic approach, although being a logical extension of the analogous thiol metalation.58 The limited number of commercially available/low cost selenols may certainly restrict the diversity of such a library of complexes intended for biological studies. Therefore, we also developed the second synthetic approach in order to establish the potential of gaining rapid access to arylselenolato complexes which vary with respect to the arylselenolato fragment, benefiting from the variety and facile synthesis of Au–aryl complexes.59,60 The formal insertion of Se into Au–aryl bonds to form a Au–Se–C bonding arrangement is without precedent in the literature (Scheme 1). After gaining synthetic access to a new library of gold–selenolato complexes, their anticancer activity was evaluated in vitro. For the most promising complex, further in vitro experiments were performed, revealing the role of this compound as a TrxR inhibitor. Then, the performance of our lead compound was tested ex vivo, using patient-derived tumor organoids as an advanced 3D cancer model.
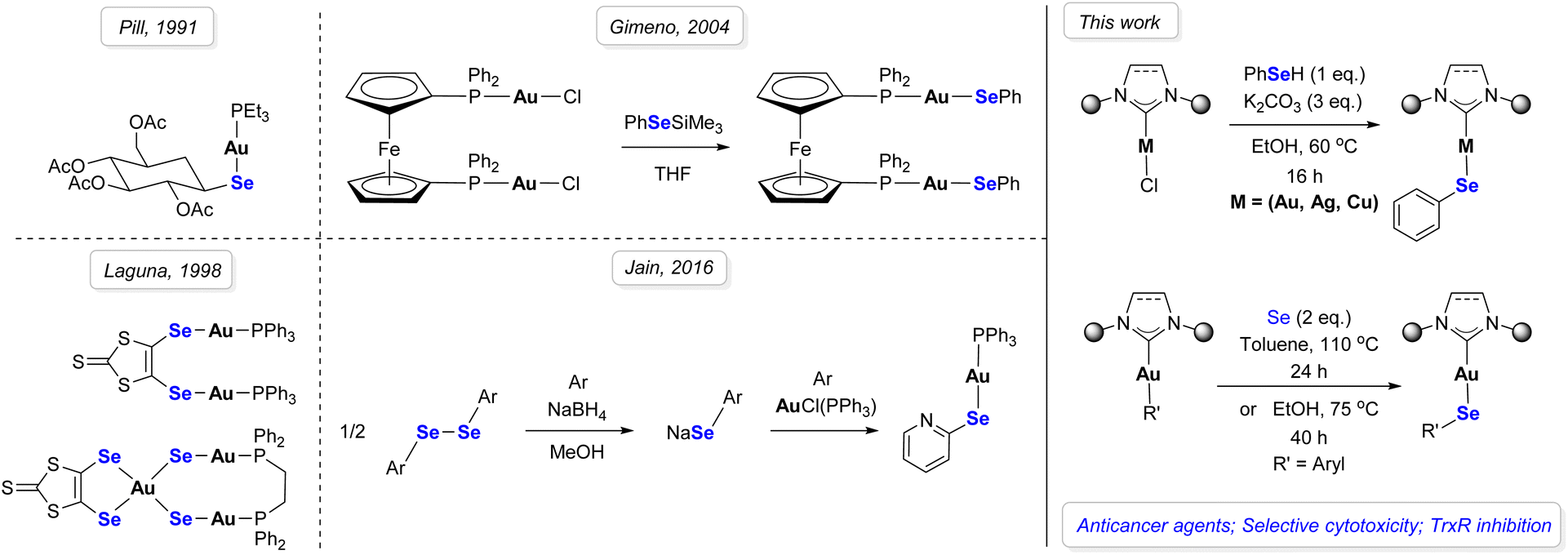 | ||
| Scheme 1 Synthetic background of selenium-based gold complexes. | ||
Results and discussion
Synthesis of metal–selenolato complexes: weak base route and Se insertion
The weak base route is applicable to a wide variety of substrates with moderate to high yields (Scheme 2), in green solvents, such as ethanol and acetone. While the standard procedure was conducted for 16 hours overnight, the reaction time can be shorter in some cases, such as for monoIPent (1m), which can reach full conversion in one hour. Ethyl acetate can also be used as an alternative to dichloromethane for product purification. | ||
| Scheme 2 Scope of ligands for the weak base route with selenophenol. The X-ray molecular structures of complexes 1b, 1e and 1l are presented, showing thermal displacement ellipsoids at the 50% probability level and hydrogen atoms omitted for clarity. CCDC: 1b 2379975, 1e 2379977, 1l 2379976. | ||
The isolated yields are highly dependent on the ligand and the metal center. To begin with the effect of the ancillary ligand of the gold complexes, IPr performed best in acetone with up to 90% yield, compared to 62% in ethanol (1a). The same trend was observed on SIPr (1b) and BIAN-IPr (1e), where the yield increased to 84% and 88%, respectively, by performing the reaction in acetone. Complexes bearing unsymmetrical ligands also reacted favorably with 88% yield for MeIPr (1g), 72% for MeIMes (1h), and 91% for ImPyIPr (1i) in ethanol.
In addition to NHCs, a triphenylphosphine-based complex was successfully synthesized with 61% yield (1o, Scheme 3). Lastly, this method was extended to both copper (1p) and silver (1q) complexes, which were isolated with 75% and 64% yield, respectively. Special care should be taken with silver derivatives, due to their light sensitivity. More details on reaction conditions can be found in the SI (Table S1).
 | ||
| Scheme 3 Additional entries for non-NHC gold complexes. | ||
To further increase the scope of products, we investigated an alternative synthetic pathway, based on the formal insertion of selenium into the Au–C bond of Au–aryl derivatives. In this method, the gold center is functionalized pre-emptively through base-assisted transmetallation of an aryl group from arylboronic acids, leading to the corresponding Au–aryl complex. Then, this complex is heated in the presence of elemental Se, resulting in the desired gold–selenolato complex via carboauration. All synthetic procedures towards substrates for the Se insertion can be found in the SI (Table S2). Because the starting material is fully tunable before the insertion, this two-step process enables more elaborate products, such as substituted aryl complexes (Scheme 4). Among Au–aryls, the insertion performed on the 4-trifluoromethylphenyl derivative resulted in complex 2a with 92% yield in toluene. The meta-equivalent 2b can be prepared under the same conditions with 71% yield. 2a is also accessible through a one pot reaction from the Au–Cl complex with 87% yield. The synthesis of 1a was cleaner in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of dioxane/water, compared to toluene, which is why protonated solvents were subsequently investigated. 1a can be isolated with 83% yield in ethanol at 75 °C after 40 hours. Unfortunately, neither 2a nor 2b was obtained when the reaction was performed in ethanol. Based on our studies, the outcome of this reaction appears to be highly dependent on the electron density of the aryl group. Even though the reaction is possible in both cases, it is more selective when the aryl fragment bears electron-withdrawing groups than it is when electron-donating groups are present.
:1 mixture of dioxane/water, compared to toluene, which is why protonated solvents were subsequently investigated. 1a can be isolated with 83% yield in ethanol at 75 °C after 40 hours. Unfortunately, neither 2a nor 2b was obtained when the reaction was performed in ethanol. Based on our studies, the outcome of this reaction appears to be highly dependent on the electron density of the aryl group. Even though the reaction is possible in both cases, it is more selective when the aryl fragment bears electron-withdrawing groups than it is when electron-donating groups are present.
 | ||
| Scheme 4 Scope of the formal Se insertion. The X-ray molecular structure of complex 2a is presented, showing thermal displacement ellipsoids at the 50% probability level and hydrogen atoms omitted for clarity. CCDC: 2a 2379974. | ||
The electronic environment of the Se-based complex is naturally influenced by the ancillary ligand. The chemical shift of 77Se in NMR indicates a correlation of 83% with the Tolman electronic parameter (TEP) of the NHC in NHC–Au–Se–aryl complexes, as shown in Fig. 1. It is known that the reactivity of NHC gold complexes is modulated by the σ-donor character of the ancillary ligand.61 Here, the metal center acts as a mediator that facilitates the transfer of electron density from the NHC ligand to selenium, hence the correlation observed between the TEP and the Se chemical shift. Changes in electron density at gold and electronic communication between ancillary ligands and other gold-bound fragments are well-documented in the literature.62 The use of selenium as a probe in the context of L-Au → donating ability (L = ancillary ligand), is unprecedented,63 to the best of our knowledge. Combined with the facile synthesis and stability of the compounds in question, this correlation suggests that NHC–Au–selenolato complexes could serve as a new type of probe for the electron-donating ability of ancillary ligands.
 | ||
| Fig. 1 Plot of TEP of NHC ligands vs. Se shift of complexes 1a, 1b, 1c, 1j, 1k, and 1l (TEP determined by IR of the corresponding [Ni(CO)3(NHC)] complexes).64 | ||
Furthermore, the stability of the complexes 1a and 1m in water was studied by dissolving a known quantity of the corresponding complex (4 mg) in DMSO-d6 (0.4 mL), followed by diluting the solution with D2O to obtain a total volume of 0.5 mL. No change was observed after 48 hours. This indicates that gold–selenolato complexes are stable in aqueous solution.
Caution should be exercised when using complexes with potential minor impurities suggested by discrepancies between theoretical and experimental values in the contents of some elements (between ca. 0.6–0.9%) in elemental analysis, such as 1e, 1i and 1j. Despite revealing no sign of impurities via1H NMR, complexes 1e and 1i could not be validated by elemental analysis successfully. We suspect 1e to slowly decompose at ambient temperature. No organic impurity was detected via1H NMR for 1i, therefore the failure of elemental analysis must have been due to minor inorganic impurities. Among these cases, only 1i was used for biological analysis along with the other candidates.
Biological analyses of metal–selenolato complexes
| Complex | IC50 (μM) | |||
|---|---|---|---|---|
| (Entry) | A-549 | HT-29 | MCF-7 | Vero-E6 |
| Auranofin | 3.7 ± 0.4 | 4.9 ± 0.5 | 1.5 ± 0.2 | 2.6 ± 0.1 |
| Cisplatin | 3.0 ± 0.4 | 7.0 ± 0.5 | 5.3 ± 0.8 | 2.06 ± 0.02 |
| [Au(IPr)Se(C6H5)] (1a) | 4.5 ± 0.9 | 3.1 ± 0.5 | 3.6 ± 0.5 | 7.1 ± 0.6 |
| [Au(IPr)Se(C6H4CF3)] (2a) | 55 ± 10 | 88 ± 8 | 48 ± 5 | >100 |
| [Au(SIPr)Se(C6H5)] (1b) | >100 | >100 | >100 | >100 |
| [Au(IPr*)Se(C6H5)] (1c) | >100 | >100 | >100 | >100 |
| [Au(IPrMe)Se(C6H5)] (1d) | >100 | >100 | 6.1 ± 0.3 | >100 |
| [Au(BzIPr)Se(C6H5)] (1f) | 0.32 ± 0.04 | 1.08 ± 0.05 | 1.9 ± 0.4 | 2.9 ± 0.2 |
| [Au(MeIPr)Se(C6H5)] (1g) | 1.5 ± 0.3 | 2.2 ± 0.5 | 2.4 ± 0.5 | 3.8 ± 0.1 |
| [Au(MeIMes)Se(C6H5)] (1h) | 4.0 ± 0.5 | 4 ± 1 | 3.4 ± 0.6 | 3.5 ± 0.2 |
| [Au(ImPyIPr)Se(C6H5)] (1i) | 5.97 ± 0.08 | 12.6 ± 0.8 | 4.3 ± 0.4 | 6.3 ± 0.1 |
| [Au(SIMes)Se(C6H5)] (1k) | 6.3 ± 0.6 | 9.9 ± 0.3 | 4.4 ± 0.1 | 6.0 ± 0.3 |
| [Au(IPent)Se(C6H5)] (1l) | >100 | >100 | >100 | >100 |
| [Au(MonoIPent)Se(C6H5)] (1m) | 0.27 ±0.03 | 0.35 ±0.05 | 0.27 ±0.04 | 4.5 ±0.2 |
| MonoIPent·HCl (1m′) | 0.76 ± 0.08 | 0.8 ± 0.2 | 0.79 ± 0.09 | 12.3 ± 0.8 |
| [Au(PPh3)Se(C6H5)] (1o) | 2.9 ± 0.4 | 2.8 ± 0.2 | 1.70 ± 0.08 | 1.9 ± 0.3 |
| [Cu(IPr)Se(C6H5)] (1p) | 0.7 ± 0.1 | 1.0 ± 0.3 | 0.4 ± 0.3 | 1.1 ± 0.2 |
| [Ag(IPr)Se(C6H5)] (1q) | 0.8 ± 0.1 | 1.2 ± 0.3 | 0.9 ± 0.2 | 6.0 ± 0.7 |
In preparation for testing on patient-derived colon and ovarian cancer organoids, we evaluated our lead compound (complex 1m) on the widely used ovarian cancer cell line A2780, obtaining an IC50 of 0.4 ± 0.1 μM (compared to 1 μM for cisplatin). This result is consistent with the IC50 values previously determined for complex 1m in A-549, HT-29, and MCF-7 cell lines. Moreover, since oxaliplatin and carboplatin will be employed as positive controls in colon and ovarian cancer tumoroid assays, respectively—being the standard treatments for these cancer types—we find it relevant to report the average IC50 values for these second- and third-generation platinum drugs as described in the literature: (i) 7 μM for oxaliplatin in HT-29 colon cancer cells,65 and (ii) 8 μM for carboplatin in A2780 ovarian cancer cells.66
Ex vivo experiments on 3D patient-derived organoids
Encouraged by the promising in vitro results obtained in a wide range of 2D cancer cell lines, we sought to assess the efficacy of our synthesized compounds in more advanced biological models. Among these models, patient-derived tumor organoids (PDTOs) and spheroids derived from commercially available tumor cell lines are two key approaches. However, PDTOs offer significant advantages over spheroids in drug screening, particularly in the context of metallodrugs.PDTOs are established directly from patient tumors, preserving the genetic, epigenetic, and histological heterogeneity of the original cancer.67 This is in stark contrast to spheroids from commercial cell lines, which often undergo extensive adaptation in culture, leading to reduced genetic diversity and loss of key tumor characteristics. The ability of PDTOs to reflect real patient tumor variability is crucial for assessing the efficacy of metallodrugs across different cancer subtypes.68 Unlike conventional spheroids, PDTOs better recapitulate the three-dimensional (3D) architecture and microenvironment of tumors, including cellular interactions and extracellular matrix components. Since PDTOs are directly derived from patient tumors, they allow for personalized drug screening, enabling clinicians to test metallodrugs on a patient's specific cancer before treatment.69 This personalized approach increases the likelihood of selecting the most effective therapy while reducing unnecessary exposure to ineffective drugs, an advantage that commercial spheroids cannot offer.
Cancer cell lines used for spheroids often lack the ability to adapt and evolve under drug pressure in the same way real tumors do. PDTOs, however, can capture mechanisms of drug resistance and tumor evolution more effectively, making them superior models for studying long-term responses and resistance development to metallodrugs. Moreover, metallodrugs often require specific metabolic conditions to become activated or exert their effects. PDTOs, derived from primary tissues, better reflect patient-specific drug metabolism, helping to predict cytotoxicity profiles more accurately than cell line-derived spheroids. This is particularly important for metallodrugs, whose activity can be influenced by metal-binding proteins, enzymatic modifications, and redox conditions that may not be properly maintained in conventional spheroid models.70
Taking advantage of organoid biotechnology, four organoids derived from tumor tissues of patients affected by colon cancer (CRC-PDTO1-4) were selected to test the efficacy of the most promising gold(I)–selenolato complex 1m.
The IC50 values reported in Table 2 clearly indicate that complex 1m retains remarkable cytotoxicity even in these more complex models of colon cancer, in addition to the promising results obtained with HT-29 2D cells. In this case, oxaliplatin was used as a reference, as it represents the standard chemotherapy agent employed in the clinical treatment of colon cancer. Interestingly, in the two patients for whom oxaliplatin proved to be ineffective (CRC-PDTO3 and CRC-PDTO4), our complex exhibits cytotoxicity comparable to that observed in patients sensitive to oxaliplatin (CRC-PDTO1 and CRC-PDTO2). This finding suggests that complex 1m warrants further investigation as a potential metallodrug for the treatment of aggressive forms of colon cancer that are resistant to conventional first-line oxaliplatin-based therapy.
| Organoid | IC50 (μM) | |
|---|---|---|
| Oxaliplatin | 1m | |
| CRC-PDTO1 | 1.4 ± 0.9 | 13 ± 9 |
| CRC-PDTO2 | 4 ± 1 | 6 ± 2 |
| CRC-PDTO3 | >100 | 3.4 ± 0.5 |
| CRC-PDTO4 | >100 | 9 ± 5 |
These promising results prompted us to assess the efficacy of complex 1m in a highly aggressive and lethal type of cancer affecting the female population, namely High-Grade Serous Ovarian Cancer (HGSOC). Among the various subtypes of ovarian cancer, HGSOC is the most prevalent and is associated with the poorest five-year survival rate.71 Moreover, nearly 30% of HGSOC patients develop ascites, a condition characterized by the accumulation of free-floating malignant cells that facilitate intraperitoneal metastasis.72 This clinical complication not only hampers the efficacy of standard chemotherapy but also necessitates frequent paracentesis for symptomatic relief.73
Given these therapeutic limitations, current treatment strategies often fail to achieve lasting efficacy. This highlights the urgent need for innovative drugs capable of overcoming both intrinsic and acquired resistance, which compromise treatment outcomes while simultaneously increasing toxicity. With the aim of contributing to research against ovarian cancer, we tested complex 1m and carboplatin (standard therapy) towards four patient-derived tumoroids recently prepared in our laboratory from HGSOC samples (HGSOC-PDTO-1-4).74–78 In previous studies, immunohistochemistry (IHC) analysis confirmed that these organoids retained key histological features of their original tumors, including the expression of markers such as PAX8, WT-1, and CA-125.
Even in this type of cancer, complex 1m demonstrates high efficacy, with IC50 values in the micromolar range (see Table 3). Similar to the results observed in organoids derived from colon cancer tissues, complex 1m exhibits potent cytotoxicity across all tested organoids (Fig. 2). This finding is particularly noteworthy when examining the HGSOC organoids HGSOC-PDTO3 and HGSOC-PDTO4, where conventional carboplatin-based therapy is ineffective.
| Organoid | IC50 (μM) | |
|---|---|---|
| Carboplatin | 1m | |
| HGSOC-PDTO1 | 3.5 ± 0.7 | 17 ± 8 |
| HGSOC-PDTO2 | 3 ± 1 | 4 ± 2 |
| HGSOC-PDTO3 | >100 | 6 ± 2 |
| HGSOC-PDTO4 | >100 | 7 ± 5 |
 | ||
| Fig. 2 Representative brightfield images of PDTOs from HGSOC (A) and CRC (B) following treatment with complex 1m at 50 μM for 96 hours, compared to non-treated controls. Complex 1m treatment resulted in visible reductions in organoid size, density, and structural integrity in both HGSOC and CRC models. Scale bar = 200 μm. | ||
TrxR1 inhibition
Based on the obtained in vitro results, three compounds were then tested for TrxR1 inhibition: the most active complex (1m), the corresponding imidazolium (1m′) and one featuring the most common NHC-ligand in gold chemistry (1a). These experiments were focused on gold complexes, since TrxR is not the most common target for copper and silver complexes. The inhibitory activity on TrxR by our lead candidate 1m was then confirmed in vitro with an average IC50 value of 0.18 ± 0.05 μM. The imidazolium salt 1m′ and complex 1a did not show any inhibition of the enzyme (i.e. no reduction of the enzymatic activity was notable at the highest applied dosage of 100 μM), which suggests that their cytotoxic potency is related to a separate mechanism.Molecular docking of complexes 1m, 1m′ and 1a into the TrxR1 redox site
Next, molecular docking studies on these three candidates (1m, 1m′, and 1a) were conducted to compare both active and inactive complexes and to understand the potential interactions between 1m and TrxR1 redox sites. Even though there is a possibility of ligand exchange upon binding to the active site, we chose to conduct this study with the intact gold–selenolato complexes to gauge interactions that precede ligand exchange and this may explain the inhibitory activity observed for 1m. The RSCB Protein Data Bank file PDB: 3EAN of the crystal structure of thioredoxin reductase I, containing selenocysteine (Sec) amino acid residues, was used for the docking research and the amino acid residue SEC498 (subunit A) was set as the docking box center. Fig. 3 shows the comparative localization of the docking positions of 1m in the TrxR1 redox site (top), as well as the key interactions between various sites of 1m and specific amino acid residues (bottom). For the investigation of the proposed mechanism of the inhibition of TrxR, molecular docking was performed on compounds 1m, 1m′ and 1a into the thioredoxin reductase redox site (subunit A), as illustrated in Fig. S1 (see SI). Auranofin was docked as a control at the redox site of the TrxR1 with the formation of a protein–auranofin complex (−9.5 kcal mol−1). The molecular docking method has established the complexation of 1m, 1m′ and 1a with the TrxR1 C-terminal redox center with a binding energy (ΔG) of −9.3, −7.8 and −7.4 kcal mol−1 respectively. A key aspect of this process is the binding of the gold atom of 1m with the selenium atom of SEC498 through metal acceptor bonds (2.77–2.91 Å) (Fig. 3). Secondly, complexation is assisted by hydrogen bonds (2.46–2.95 Å) between the selenium atom of the organometallic complex with the amino acids LYS29 and LYS123 of TrxR1. These interactions may represent the onset of a protodeauration/gold binding sequence after which gold would be bound to the selenium atom of SEC498. | ||
| Fig. 3 Localization of 1m at the TrxR1 redox site (top), and molecular docking of 1m into the redox center of TrxR1 (bottom); grey – complex. | ||
The important factors in the stabilization of the protein–1m complex are hydrogen bonds between the imidazolylidene ring of 1m and amino acid LEU493. It should be noted that the formation of electrostatic interactions occurs between the phenyl rings of 1m and amino acids LYS29 and SEC498. For 1m′, the imidazolium is stabilized by one hydrogen bond (3.42 Å) between the heterocyclic ring and amino acid GLN494; two electrostatic interactions, SEC498 (4.85 Å) and LYS29 (4.45 Å) with the phenyl ring; and two hydrophobic interactions, the isopentyl group with VAL484 (3.92, 4.89 Å) (Fig. S1b). Molecular docking of non-inhibitor 1a demonstrates a lack of bond between the gold atom and the SEC498 selenium atom (Fig. S1c). The stabilization of the protein–1a complex only occurs through two electrostatic interactions (2.98–5.47 Å) between the phenyl rings and amino acids LYS123 and SEC498, and four hydrophobic interactions (3.22–5.46 Å) between phenyl and isopropyl groups and amino acids ALA119 and VAL484. The localization and certain interactions observed at the TrxR1 redox center in the case of 1m are comparable to those found in cases of NHC–gold–selenone complexes described in the literature.79,80 Considering all the above results, the candidate molecule can be considered an inhibitor of TrxR1, affirming the proposed theory on the anticancer mechanism of action of such complexes.
Conclusion
We have developed two simple pathways for the synthesis of metal–selenolato complexes under air, with gold, copper, or silver. A large number of ligands are compatible with the weak base route, while the Se insertion route enables the synthesis of gold–selenolato complexes from gold–aryl derivatives, bypassing the use of selenol. The importance of this newly discovered reactivity of coinage metal–aryl complexes lies in the capacity of converting any (aryl)boron-containing substrate to the corresponding coinage metal–selenolato complex. Additionally, this reaction reveals the potential of converting other types of coinage metal organometallics into their organoselenolato congeners, thus providing access to a significantly larger pool of such complexes for biological or other applications. Both routes make use of very simple, atom-economical procedures, with desirable solvents such as ethanol and acetone with straightforward purification, leading to high yields. In vitro analysis provided clear evidence of selective cytotoxicity for a selection of complexes on human cancer cells and confirmed the inhibitory activity on TrxR with our most promising candidate, complex 1m. Molecular docking has demonstrated the complexation of 1m with the TrxR1 C-terminal redox center with a binding energy (ΔG) of −9.3 kcal mol−1. Based on the computational studies and biological tests, it is plausible to infer that the binding of the studied compounds on the redox centers of TrxR1 causes oxidative stress followed by the apoptosis of cancer cells.Finally, complex 1m exhibited promising cytotoxic effects even in advanced and physiologically relevant biological models, such as 3D organoids derived from tumor tissues of multiple patients. In both colon cancer and HGSOC organoids, our compound demonstrates high efficacy even in patients resistant to the standard chemotherapy agents used in clinical practice (oxaliplatin and carboplatin, respectively).
The strong activity observed in vitro, combined with its effectiveness in ex vivo models, provides a solid foundation for the potential clinical development of the compounds discussed in this study. However, additional research is currently underway in our laboratories, with a primary focus on assessing their efficacy in in vivo models as well as in better defining their mode of action.
Ethical statement
All experiments were performed in accordance with the relevant laws and institutional guidelines (European Union Directive 2010/63/EU for animal and in vitro research) and were approved by the Ethics Committee of the Centro di Riferimento Oncologico di Aviano (CRO) IRCCS, Italy. Written informed consent was obtained from all human participants or their legal guardians prior to sample collection.Specifically, patient-derived tumour organoids (PDTOs) were obtained for research purposes from completely anonymous specimens collected under a signed biobank informed consent at the National Cancer Institute (CRO) of Aviano, Italy and approved by the local ethical committee.
Author contributions
IO, NVT and SPN conceived and designed the project. PA, NBP and DB synthesized all metal-selenolate complexes through the weak base route. PA performed the synthesis of substrates and metal-selenolate complexes for the Se insertion. SB and KVH solved all XRD crystal structures for characterization of metal-selenolate complexes. PL, SB and IO conducted in vitro biological analyses for cytotoxicity assessment as well as inhibition tests. IS performed computational studies on molecular docking. EC and TS conducted ex vivo experiments, while VC and FR provided organoid samples from the C.R.O. biobank. PA, NBP, TS, IO, NVT and SPN wrote and corrected the manuscript with input from all authors.Conflicts of interest
The authors declare no conflict of interest.Data availability
The primary data associated with this work can be found in the SI.CCDC 2379974–2379977 contain the supplementary crystallographic data for this paper.81–84
Supplementary information: characterization data, Spectroscopy and experimental details. See DOI: https://doi.org/10.1039/d5sc04490a.
Acknowledgements
The Research Foundation – Flanders (FWO) is acknowledged for a Fundamental Research PhD fellowship to NVT (11I6921N) and research grant G0A6823N to SPN. We are also grateful to the SBO (D2M to SPN) and the BOF (starting and senior grants to SPN) as well as the iBOF C3 project for financial support. NVT is the recipient of the 2023 Incentive Award from C. G. B.-C. B. B., the Management Committee of the Bulletin of the Belgian Chemical Societies: Koninklijke Vlaamse Chemische Vereniging (KVCV) and Société Royale de Chimie (SRC). KVH and SB thank the Special Research Fund (BOF) – UGent (Project: BOF/24J/2023/084). FR was financially supported by Fondazione AIRC per la Ricerca sul Cancro (Grant AIRC IG23566) and VC from Ministero della Salute – Ricerca Corrente. UGent is gratefully acknowledged for the doctoral assistant fellowship awarded to NBP.Notes and references
- K. M. Brown and J. R. Arthur, Public Health Nutr., 2007, 4, 5933–5993 Search PubMed.
- J. K. Wrobel, R. Power and M. Toborek, IUBMB Life, 2016, 68, 97–105 Search PubMed.
- T. S. Gechev and J. Hille, J. Cell Biol., 2005, 168, 17–20 Search PubMed.
- D. R. Gough and T. G. Cotter, Cell Death Dis., 2011, 2, e213 Search PubMed.
- E. Lubos, J. Loscalzo and D. E. Handy, Antioxid. Redox Signaling, 2011, 15, 1957–1997 Search PubMed.
- H.-U. Simon, A. Haj-Yehia and F. Levi-Schaffer, Apoptosis, 2000, 5, 415–418 Search PubMed.
- N. Diwanji and A. Bergmann, Semin. Cell Biol., 2018, 80, 74–82 Search PubMed.
- M. H. Raza, S. Siraj, A. Arshad, U. Waheed, F. Aldakheel, S. Alduraywish and M. Arshad, J. Cancer Res. Clin. Oncol., 2017, 143, 1789–1809 Search PubMed.
- A. Borchert, J. Kalms, S. R. Roth, M. Rademacher, A. Schmidt, H.-G. Holzhutter, H. Kuhn and P. Scheerer, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2018, 1863, 1095–1107 Search PubMed.
- P. Singh and B. Lim, Curr. Oncol. Rep., 2022, 24, 273–284 Search PubMed.
- Q. Cheng, T. Sandalova, Y. Lindqvist and E. S. J. Arnér, J. Biol. Chem., 2009, 284, 3998–4008 Search PubMed.
- J. Lu, A. Vlamis-Gardikas, K. Kandasamy, R. Zhao, T. N. Gustafsson, L. Engstrand, S. Hoffner, L. Engman and A. Holmgren, FASEB J., 2013, 27, 1394–1403 Search PubMed.
- R. Rubbiani, S. Can, I. Kitanovic, H. Alborzinia, M. Stefanopoulou, M. Kokoschka, S. Mönchgesang, W. S. Sheldrick, S. Wölfl and I. Ott, J. Med. Chem., 2011, 54, 8646–8657 Search PubMed.
- F. Saccoccia, F. Angelucci, G. Boumis, D. Carotti, G. Desiato, A. E. Miele and A. Bellelli, Curr. Protein Pept. Sci., 2014, 15, 621–646 Search PubMed.
- G. Bjørklund, L. Zou, J. Wang, C. T. Chasapis and M. Peana, Pharmacol. Res., 2021, 174, 105854 Search PubMed.
- L. B. Chen, Annu. Rev. Cell Biol., 1988, 4, 155–181 Search PubMed.
- A. E. Finkelstein, D. T. Walz, V. Batista, M. Mizraji, F. Roisman and A. Misher, Ann. Rheum. Dis., 1976, 35, 251–257 Search PubMed.
- S. J. Berners-Price, C. K. Mirabelli, R. K. Johnson, M. R. Mattern, F. L. McCabe, L. F. Faucette, C.-M. Sung, S.-M. Mong, P. J. Sadler and S. T. Crooke, Cancer Res., 1986, 46, 5486–5493 Search PubMed.
- M. J. McKeage, L. Maharaj and S. J. Berners-Price, Coord. Chem. Rev., 2002, 232, 127–135 Search PubMed.
- E. Vergara, S. Miranda, F. Mohr, E. Cerrada, E. R. T. Tiekink, P. Romero, A. Mendía and M. Laguna, Eur. J. Inorg. Chem., 2007, 2007, 2926–2933 Search PubMed.
- S. Miranda, E. Vergara, F. Mohr, D. de Vos, E. Cerrada, A. Mendía and M. Laguna, Inorg. Chem., 2008, 47, 5641–5648 Search PubMed.
- I. Landini, L. Massai, D. Cirri, T. Gamberi, P. Paoli, L. Messori, E. Mini and S. Nobili, J. Inorg. Biochem., 2020, 208, 111079 Search PubMed.
- T. Marzo, D. Cirri, C. Gabbiani, T. Gamberi, F. Magherini, A. Pratesi, A. Guerri, T. Biver, F. Binacchi, M. Stefanini, A. Arcangeli and L. Messori, ACS Med. Chem. Lett., 2017, 8, 997–1001 Search PubMed.
- T. Scattolin, E. Bortolamiol, F. Rizzolio, N. Demitri and F. Visentin, Appl. Organomet. Chem., 2020, 34, e5876 Search PubMed.
- S. K. Goetzfried, S. M. C. Koenig, C. M. Gallati and R. Gust, Inorg. Chem., 2021, 60, 8546–8553 Search PubMed.
- H. F. Dos Santos, M. A. Vieira, G. Y. Sánchez Delgado and D. Paschoal, J. Phys. Chem. A, 2016, 120, 2250–2259 Search PubMed.
- P. J. Barnard, M. V. Baker, S. J. Berners-Price and D. A. Day, J. Inorg. Biochem., 2004, 98, 1642–1647 Search PubMed.
- M. V. Baker, P. J. Barnard, S. J. Berners-Price, S. K. Brayshaw, J. L. Hickey, B. W. Skelton and A. H. White, J. Organomet. Chem., 2005, 690, 5625–5635 Search PubMed.
- M. V. Baker, P. J. Barnard, S. J. Berners-Price, S. K. Brayshaw, J. L. Hickey, B. W. Skelton and A. H. White, Dalton Trans., 2006, 3708 Search PubMed.
- A. Collado, A. Gómez-Suárez, A. R. Martin, A. M. Z. Slawin and S. P. Nolan, Chem. Commun., 2013, 49, 5541 Search PubMed.
- N. V. Tzouras, F. Nahra, L. Falivene, L. Cavallo, M. Saab, K. Van Hecke, A. Collado, C. J. Collett, A. D. Smith, C. S. J. Cazin and S. P. Nolan, Chem.–Eur. J., 2020, 26, 4515–4519 Search PubMed.
- E. A. Martynova, N. V. Tzouras, G. Pisano, C. S. J. Cazin and S. P. Nolan, Chem. Commun., 2021, 57, 3836–3856 Search PubMed.
- A. M. Al-Majid, S. Yousuf, M. I. Choudhary, F. Nahra and S. P. Nolan, ChemistrySelect, 2016, 1, 76–80 Search PubMed.
- R. Büssing, B. Karge, P. Lippmann, P. G. Jones, M. Brönstrup and I. Ott, ChemMedChem, 2021, 16, 3402–3409 Search PubMed.
- J. Zhang, M. A. Abu el Maaty, H. Hoffmeister, C. Schmidt, J. K. Muenzner, R. Schobert, S. Wölfl and I. Ott, Angew. Chem., Int. Ed., 2020, 59, 16795–16800 Search PubMed.
- B. Bertrand and A. Casini, Dalton Trans., 2014, 43, 4209–4219 Search PubMed.
- M. C. Dietl, M. Maag, S. Ber, F. Rominger, M. Rudolph, I. Caligiuri, P. K. Andele, I. A. I. Mkhalid, F. Rizzolio, P. A. Nogara, L. Orian, T. Scattolin and A. S. K. Hashmi, Chem. Sci., 2024, 15, 15291–15298 Search PubMed.
- S. K. Goetzfried, P. Kapitza, C. M. Gallati, A. Nindl, M. Cziferszky, M. Hermann, K. Wurst, B. Kircher and R. Gust, Dalton Trans., 2022, 51, 1395–1406 Search PubMed.
- L. Cosottini, A. Giachetti, A. Guerri, A. Martinez-Castillo, A. Geri, S. Zineddu, N. G. A. Abrescia, L. Messori, P. Turano and A. Rosato, Angew. Chem., Int. Ed., 2025, 137(30), e202503778 Search PubMed.
- C. Schmidt, L. Albrecht, S. Balasupramaniam, R. Misgeld, B. Karge, M. Brönstrup, A. Prokop, K. Baumann, S. Reichl and I. Ott, Metallomics, 2019, 11, 533–545 Search PubMed.
- J. L. Hickey, R. A. Ruhayel, P. J. Barnard, M. V. Baker, S. J. Berners-Price and A. Filipovska, J. Am. Chem. Soc., 2008, 130, 12570–12571 Search PubMed.
- W. Eikens, C. Kienitz, P. G. Jones and C. Thöne, J. Chem. Soc., Dalton Trans., 1994, 83–90 Search PubMed.
- S. Ahmad and A. A. Isab, Transition Met. Chem., 2003, 28, 540–543 Search PubMed.
- N. Sato, S. Iwata, K. Nakamura, T. Hori, K. Mori and J. Yodoi, J. Immunol., 1995, 154, 3194–3203 Search PubMed.
- K. D. Held, F. C. Sylvester, K. L. Hopcia and J. E. Biaglow, Radiat. Res., 1996, 145, 542 Search PubMed.
- K. P. Bhabak and G. Mugesh, J. Chem. Sci., 2011, 123, 783–789 Search PubMed.
- D. Menia, H. Kopacka, K. Wurst, T. Müller, P. Lippmann, I. Ott and B. Bildstein, Eur. J. Inorg. Chem., 2021, 2021, 2784–2786 Search PubMed.
- G. K. Kole, A. P. Wadawale, S. Nigam, C. Majumder and V. K. Jain, ChemistrySelect, 2016, 1, 4131–4136 Search PubMed.
- O. Crespo, M. C. Gimeno, A. Laguna, M. Kulcsar and C. Silvestru, Inorg. Chem., 2009, 48, 4134–4142 Search PubMed.
- S. Canales, O. Crespo, M. C. Gimeno, P. G. Jones and A. Laguna, Inorg. Chem., 2004, 43, 7234–7238 Search PubMed.
- M. Mora, M. C. Gimeno and R. Visbal, Chem. Soc. Rev., 2019, 48, 447–462 Search PubMed.
- E. Schuh, C. Pflüger, A. Citta, A. Folda, M. P. Rigobello, A. Bindoli, A. Casini and F. Mohr, J. Med. Chem., 2012, 55, 5518–5528 Search PubMed.
- O. Dada, D. Curran, C. O'Beirne, H. Müller-Bunz, X. Zhu and M. Tacke, J. Organomet. Chem., 2017, 840, 30–37 Search PubMed.
- S. W. Schleser, L. H. F. Köhler, F. Riethmüller, S. Reich, R. Fertig, L. Schlotte, J. Seib, A. Goller, G. Begemann, R. Kempe and R. Schobert, ChemPlusChem, 2023, 88, e202300167 Search PubMed.
- A. A. A. Seliman, M. Altaf, N. A. Odewunmi, A.-N. Kawde, W. Zierkiewicz, S. Ahmad, S. Altuwaijri and A. A. Isab, Polyhedron, 2017, 137, 197–206 Search PubMed.
- M. De Franco, M. Saab, M. Porchia, C. Marzano, S. P. Nolan, F. Nahra, K. Van Hecke and V. Gandin, Chem.–Eur. J., 2022, 28, e202201898 Search PubMed.
- A. Molter and F. Mohr, Coord. Chem. Rev., 2010, 254, 19–45 Search PubMed.
- M. Safir Filho, T. Scattolin, P. Dao, N. V. Tzouras, R. Benhida, M. Saab, K. Van Hecke, P. Lippmann, A. R. Martin, I. Ott and S. P. Nolan, New J. Chem., 2021, 45, 9995–10001 Search PubMed.
- N. V. Tzouras, M. Saab, W. Janssens, T. Cauwenbergh, K. Van Hecke, F. Nahra and S. P. Nolan, Chem.–Eur. J., 2020, 26, 5541–5551 Search PubMed.
- T. Cauwenbergh, N. V. Tzouras, T. Scattolin, S. Bhandary, A. Simoens, K. Van Hecke, C. V. Stevens and S. P. Nolan, Chem.–Eur. J., 2021, 27, 13342–13345 Search PubMed.
- R. Döpp, C. Lothschütz, T. Wurm, M. Pernpointner, S. Keller, F. Rominger and A. S. K. Hashmi, Organometallics, 2011, 30, 5894–5903 Search PubMed.
- R. G. Carden, N. Lam and R. A. Widenhoefer, Chem.–Eur. J., 2017, 23, 17992–18001 Search PubMed.
- G. Marrazzini, C. Gabbiani and G. Ciancaleoni, ACS Omega, 2019, 4, 1344–1353 Search PubMed.
- D. J. Nelson and S. P. Nolan, Chem. Soc. Rev., 2013, 42, 6723 Search PubMed.
- S. William-Faltaos, D. Rouillard, P. Lechat and G. Bastian, Fundam. Clin. Pharmacol., 2007, 21, 165–172 Search PubMed.
- U. Banerji, N. Sain, S. Y. Sharp, M. Valenti, Y. Asad, R. Ruddle, F. Raynaud, M. Walton, S. A. Eccles, I. Judson, A. L. Jackman and P. Workman, Cancer Chemother. Pharmacol., 2008, 62, 769–778 Search PubMed.
- J. Drost and H. Clevers, Nat. Rev. Cancer, 2018, 18, 407–418 Search PubMed.
- Y.-H. Lo, K. Karlsson and C. J. Kuo, Nat. Cancer, 2020, 1, 761–773 Search PubMed.
- T. Takahashi, Annu. Rev. Pharmacol. Toxicol., 2019, 59, 447–462 Search PubMed.
- E. J. Anthony, E. M. Bolitho, H. E. Bridgewater, O. W. L. Carter, J. M. Donnelly, C. Imberti, E. C. Lant, F. Lermyte, R. J. Needham, M. Palau, P. J. Sadler, H. Shi, F.-X. Wang, W.-Y. Zhang and Z. Zhang, Chem. Sci., 2020, 11, 12888–12917 Search PubMed.
- D. D. Bowtell, S. Böhm, A. A. Ahmed, P.-J. Aspuria, R. C. Bast, V. Beral, J. S. Berek, M. J. Birrer, S. Blagden, M. A. Bookman, J. D. Brenton, K. B. Chiappinelli, F. C. Martins, G. Coukos, R. Drapkin, R. Edmondson, C. Fotopoulou, H. Gabra, J. Galon, C. Gourley, V. Heong, D. G. Huntsman, M. Iwanicki, B. Y. Karlan, A. Kaye, E. Lengyel, D. A. Levine, K. H. Lu, I. A. McNeish, U. Menon, S. A. Narod, B. H. Nelson, K. P. Nephew, P. Pharoah, D. J. Powell, P. Ramos, I. L. Romero, C. L. Scott, A. K. Sood, E. A. Stronach and F. R. Balkwill, Nat. Rev. Cancer, 2015, 15, 668–679 Search PubMed.
- E. Smolle, V. Taucher and J. Haybaeck, Anticancer Res., 2014, 34, 1553–1561 Search PubMed.
- E. Kipps, D. S. P. Tan and S. B. Kaye, Nat. Rev. Cancer, 2013, 13, 273–282 Search PubMed.
- E. Cavarzerani, I. Caligiuri, M. Bartoletti, V. Canzonieri and F. Rizzolio, Front. Bioeng. Biotechnol., 2023, 11, 1135374 Search PubMed.
- E. Cavarzerani, I. Caligiuri, M. Bartoletti, G. Corona, T. Perin, A. Palumbo, V. Canzonieri and F. Rizzolio, BiorXiv, 2024, preprint, DOI:10.1101/2024.05.09.593311.
- G. Saorin, M. Mauceri, E. Cavarzerani, I. Caligiuri, G. Bononi, C. Granchi, M. Bartoletti, T. Perin, T. Tuccinardi, V. Canzonieri, M. Adeel and F. Rizzolio, J. Drug Deliv. Sci. Technol., 2023, 87, 104718 Search PubMed.
- T. Scattolin, I. Pessotto, E. Cavarzerani, V. Canzonieri, L. Orian, N. Demitri, C. Schmidt, A. Casini, E. Bortolamiol, F. Visentin, F. Rizzolio and S. P. Nolan, Eur. J. Inorg. Chem., 2022, 2022, e202200103 Search PubMed.
- G. Tonon, M. Mauceri, E. Cavarzerani, R. Piccolo, C. Santo, N. Demitri, L. Orian, P. A. Nogara, J. B. T. Rocha, V. Canzonieri, F. Rizzolio, F. Visentin and T. Scattolin, Dalton Trans., 2024, 53, 8463–8477 Search PubMed.
- A. A. A. Seliman, M. Altaf, A. T. Onawole, S. Ahmad, M. Y. Ahmed, A. A. Al-Saadi, S. Altuwaijri, G. Bhatia, J. Singh and A. A. Isab, J. Organomet. Chem., 2017, 848, 175–183 Search PubMed.
- T. Shoeib, D. W. Atkinson and B. L. Sharp, Inorg. Chim. Acta, 2010, 363, 184–192 Search PubMed.
- P. Arnaut, N. B. Pozsoni, D. Bondar, P. Lippmann, S. Boschuk, I. Semenyuta, S. Bhandary, K. van Hecke, Y. Karpichev, E. Cavarzerani, V. Canzonieri, F. Rizzolio, T. Scattolin, G. C. Vougioukalakis, I. Ott, N. V. Tzouras and S. P. Nolan, CCDC 2379974: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2kwkcd.
- P. Arnaut, N. B. Pozsoni, D. Bondar, P. Lippmann, S. Boschuk, I. Semenyuta, S. Bhandary, K. van Hecke, Y. Karpichev, E. Cavarzerani, V. Canzonieri, F. Rizzolio, T. Scattolin, G. C. Vougioukalakis, I. Ott, N. V. Tzouras and S. P. Nolan, CCDC 2379975: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2kwkdf.
- P. Arnaut, N. B. Pozsoni, D. Bondar, P. Lippmann, S. Boschuk, I. Semenyuta, S. Bhandary, K. van Hecke, Y. Karpichev, E. Cavarzerani, V. Canzonieri, F. Rizzolio, T. Scattolin, G. C. Vougioukalakis, I. Ott, N. V. Tzouras and S. P. Nolan, CCDC 2379976: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2kwkfg.
- P. Arnaut, N. B. Pozsoni, D. Bondar, P. Lippmann, S. Boschuk, I. Semenyuta, S. Bhandary, K. van Hecke, Y. Karpichev, E. Cavarzerani, V. Canzonieri, F. Rizzolio, T. Scattolin, G. C. Vougioukalakis, I. Ott, N. V. Tzouras and S. P. Nolan, CCDC 2379977: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2kwkgh.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |