
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLarge-area, flexible, and conductive porous films of interlinked carbon nanospheres for UV light filters and resistive heaters
Meng-Qi
Zhao†
,
Tian-Yi
Li†
,
Chuan-Bin
Li†
,
Hui-Qun
Huang
,
Yong-Sheng
Wang
,
Ling-Yu
Dong
,
Xiao-Dong
Shi
,
Yu-Tai
Wu
,
Guang-Ping
Hao
 * and
An-Hui
Lu
*
* and
An-Hui
Lu
*
State Key Laboratory of Fine Chemicals, Frontier Science Center for Smart Materials, Liaoning Key Laboratory for Catalytic Conversion of Carbon Resources, School of Chemical Engineering, Dalian University of Technology, Dalian 116024, P. R. China. E-mail: guangpinghao@dlut.edu.cn; anhuilu@dlut.edu.cn
First published on 20th September 2025
Abstract
Conductive and flexible thin films are promising for wearable electronic devices, but their fabrication is challenging due to their multiple requirements for structural and functional integration. Here, we present a chemical method for the preparation of large-area carbon films by in situ linking of uniform carbon nanospheres derived from stiff-shell polymeric crystals. The proof-of-concept interlinked carbon film features a dual-porous structure, consisting of intrinsic micropores within the nodes of carbon spheres and in-plane macropores within the interlinked carbon spheres patterned in mono-/few layers. The success in the preparation of such an interlinked carbon film relies on the unique combination of the liquid–solid configuration of the polymeric stiff-shell and soft-core, which induces a series of dynamic transformations from phase expansion, fusion, linking, and eventually forms large-area interlinked porous carbon nanospheres patterned in one layer. Such thin carbon films can be transferred onto various rigid or flexible substrates, and perform well as light filters that can block >90% UV light and as electrical-heating materials that delivered a heating efficiency up to 45.8 °C μm−1. This work provides a smart engineering platform and benefits the design of future electronic skin and intelligent wearable devices.
Introduction
Large-area, flexible, and conductive porous carbon films have attracted considerable research interest for optical and thermal management applications, driven by the ongoing trend toward miniaturization and integration in electronic devices.1–5 As optical functional materials, monolayer nanocarbon materials are one important option due to their light weight and tunable microstructures.6,7 However, conventional nanocarbon materials exhibit limited spectral tenability. For instance, high-quality single-layer graphene films show near-complete light transmission,8 while other nanocarbons, e.g., carbon nanospheres, display full-band absorption properties,9 making selective light-filtering highly difficult. For personal thermal management, it demands wearable Joule heaters, for which a thin conductive carbon film with high thermal conductivity is ideally required.10,11 Integrating both functions in one film material is highly challenging, which calls for targeted structural design of nanocarbons at both microscopic and macroscopic levels.From a microscopic aspect, the surface/interface structures of nanocarbons would improve selective light absorption or scattering.12,13 In particular, a surface structure with nanopatterns is one typical feature that could increase the accessible area, facilitate encoding surface properties,14,15 and thus tune optical characteristics.16–19 To date, various methods have been explored to engineer fine surface structures in carbon spheres, which include emulsion polymerization,20 surface coating,21 and other post-modifications.22 From the macroscopic aspect of a monolayer nanocarbon assembly, it needs to connect all the individual nanocarbons into a complete film, which not only ensures electron conductivity, but also preserves selective light transmission pathways. Although significant advances have been made in developing diverse nanocarbon materials,23–29 few studies can fulfil both of the aforementioned requirements.
Herein, we developed a chemical mimicry approach to construct a stiff-shell, soft-core polymeric structure, which spontaneously evolves into interlinked porous carbon nanospheres patterned in a large-area monolayer film. For the core material, thermotropic polymers such as liquid crystals were reasonably employed considering their soft crystalline features,30,31 which potentially exhibit favorable melting properties and thus create interactions between particles upon heating. Meanwhile, the highly crosslinked and stiff polymers were proposed as the shell material that can resist complete melting during thermal treatment.32 We showcased the fine control of the formation of the two components in spite of their fast and competitive reaction kinetics between nucleation and subsequent polymerization.33,34 The devised large-area monolayer film composed of interlinked nanocarbon spheres with controllable surface structures is well beyond the conventional nanocarbon films with multifunctionality that can effectively filter >90% UV light and enable thin and wearable Joule heating.
Results and discussion
The synthesis of quasi-liquid polymeric crystals (LPCs) is based on the rapid formation of Schiff-base liquid crystals as initial seeds, surrounding which highly crosslinked polymer shells in situ grow (Fig. 1a left). The heterogeneous stiff-shell and soft-core structure forms in one-step in the solution phase via cascading polymerization between Schiff-base seeds and benzoxazine monomers. First, Schiff-base condensation between terephthalaldehyde (TPA) and p-phenylenediamine (p-PDA) initiates the rapid chain propagation and thus fast nucleation at the early stage, owing to the highly reactive nucleophilic addition of aromatic amine to the carbonyl group.35 The π–π stacking and rigid C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds ensure the ordered arrangement of molecular chains, endowing the core with a soft polymeric structure.36 Subsequently, the unoccupied –NH2 in the outer layer of the Schiff-base core provides ample heterogeneous nucleation sites for the Mannich reaction upon the introduction of p-hydroquinone (HQ). During subsequent aging, the networks in the outer layer further crosslink, ending with the formation of a dense stiff shell. Upon heating, the highly uniform and monolayer LPCs sequentially expanded, fused, shrank, and transformed into an interlinked nanocarbon monolayer film (denoted as MLCF, Fig. 1a right).
N bonds ensure the ordered arrangement of molecular chains, endowing the core with a soft polymeric structure.36 Subsequently, the unoccupied –NH2 in the outer layer of the Schiff-base core provides ample heterogeneous nucleation sites for the Mannich reaction upon the introduction of p-hydroquinone (HQ). During subsequent aging, the networks in the outer layer further crosslink, ending with the formation of a dense stiff shell. Upon heating, the highly uniform and monolayer LPCs sequentially expanded, fused, shrank, and transformed into an interlinked nanocarbon monolayer film (denoted as MLCF, Fig. 1a right).
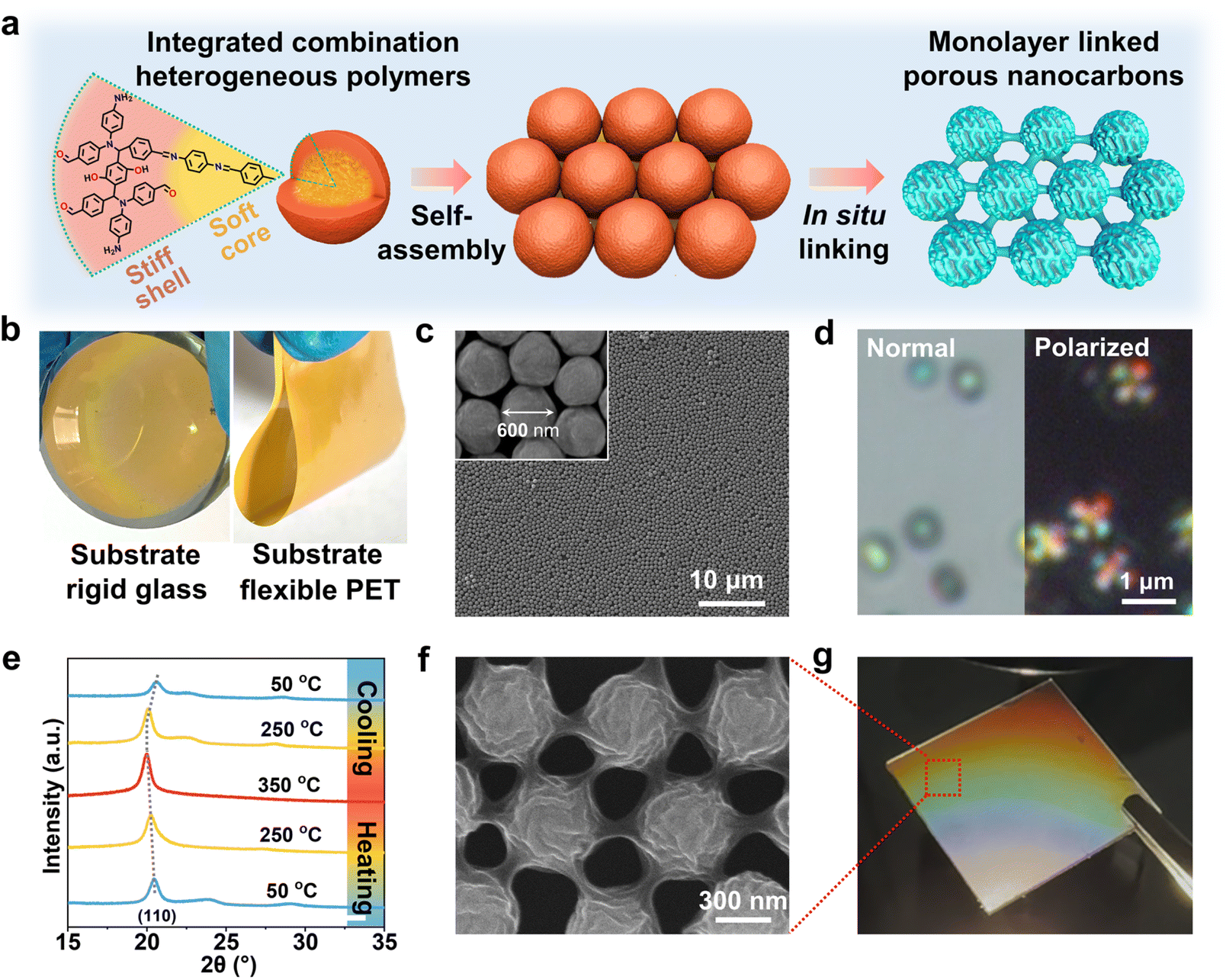 | ||
| Fig. 1 Illustration and morphological characterization of LPCs and MLCF. (a) Schematic of the synthesis of LPCs and the monolayer interlinked nanocarbon film. (b) Monolayer coating of the LPCs on substrates of glass and plastic of PET. (c) SEM image of the LPCs. Inset: high magnification image. (d) Optical microscopy images of LPCs with normal light and cross-polarized light. (e) In situ PXRD patterns during reversible thermal treatment. (f) SEM image of the monolayer film of interlinked nanocarbon spheres. (g) Optical image of the monolayer film of interlinked nanocarbon spheres under a light emitting diode. The SEM images in (c) and (f) and optical images in (d) and (g) were collected at room temperature. | ||
We also screened different kinds of amines and phenols for this synthesis (Fig. S1–S3), which failed to form such uniform nanospheres. This indicates that the reaction at the para-position of TPA, p-PDA, and HQ is unique for the three-component reactions and subsequent distribution of the soft and hard components. Notably, this self-assembly strategy exhibits exceptional substrate universality, which enables the formation of high-quality films over diverse substrates including silicon wafers, quartz glass, and flexible polyethylene terephthalate (PET) (Fig. 1b and S4). In addition, by controlling the thermal treatment conditions, nanocarbons in dispersed powder form can also be prepared, which we denote as DPC.
The scanning electron microscopy (SEM) images confirm that the resultant LPCs are highly uniform spheres with a size of ∼600 nm in diameter (Fig. 1c). The images of transmission electron microscopy (TEM) and scanning electron microscope-focused ion beam (SEM-FIB) reveal that the LPCs are solid structures (Fig. S5). Notably, the LPCs are not standard spheroids, but rather deformable due to the quasi-liquid nature of the soft-core. The crystalline feature of the LPCs was first evaluated by cross-polarized optical microscopy (Fig. 1d and S6). A dark cross (known as “Maltese cross”) was observed on LPCs, which is a typical feature of spherulites. Spherulites are spherical polymer crystals with radially aligned crystalline polymer lamellae.37 This observation suggests that the soft-core of LPCs is essentially the nanosized Schiff-base sheets assembled in spherical configurations.
Powder X-ray diffraction (PXRD) studies were conducted to further identify the crystal structure of LPCs. In situ XRD patterns were collected in the temperature range from 50 °C to 350 °C, and then back to 50 °C (Fig. 1e). The phase structures are consistent with the reported Schiff-base materials.38 The relatively sharp diffraction peak at 2θ of 20.4° coincides with the (110) plane of Schiff-base crystals. When the temperature increases to 350 °C, LPCs still largely preserve their crystalline structure, while the (110) plane at 20.4° moves to lower angles, indicating the thermally induced cell expansion. The structural change is highly reversible in the tested temperature range. We did not observe visible morphological changes of LPCs after temperature cycling, which confirmed their structural stability (Fig. S7).
We then assembled the LPCs into a monolayer film on a silicon (Si) wafer (Fig. S8). The thermal treatment of the LPCs patterned in a monolayer manner at 800 °C led to the formation of nanocarbon films. To our surprise, we observed two unique features of such nanocarbon films. One is their unique morphology with uniform surface folding with a depth of 20–40 nm determined by atomic force microscopy (Fig. S9 and 1f). This is distinct from common homogeneous polymer precursors, which resulted in smooth surfaces (Fig. S10). Previous simulation and experimental results at the macroscopic scale suggest that such gyrus-like nanofolds may arise from the joint effect of bending the hard shell and compressing the soft-core.39–41 We speculate that the non-synchronous structural transformation of LPCs followed a similar way due to the induced stress difference during heating. The structural evolution will be discussed in the next section of the formation mechanism. The other feature is that the nanocarbon spheres in the monolayer are interlinked with each other (Fig. S11). Each nanocarbon sphere functions as a node and connects with its neighbors by “nano-necks” (Fig. 1f). The formation of the nano-necks likely stemmed from the melting Schiff-base core, which seeped through the softening shell during heating. In this way, the monolayer carbon nanospheres were interlinked, forming an entire and complete film (MLCF). We transferred the MLCF to a flexible and transparent substrate of polydimethylsiloxane (PDMS). The obtained PDMS supported MLCF is rather transparent. Moreover, the digital camera image shows that the film displays vivid grating colors under light (Fig. 1g). This can be interpreted to mean that the wavelengths of light are selectively propagated upon interaction with periodically arranged micron or submicron structures, which also indicates the uniform size of the nanocarbon nodes. Notably, the large area film exhibited electron conductive properties (Fig. S12), suggesting its structural integrity due to the interlinked feature.
As mentioned above, the nanocarbon spheres with surface folds can be either in interlinked patterns or in dispersed powder form. When we thermally treat the dispersed polymer spheres in the powder state, the resultant carbon spheres are highly uniform in size and the gyrus-like surface folds are well maintained (Fig. 2a and b). Of note, our method is facile and versatile. The size and surface topography of the carbon nanospheres can be precisely controlled simply by adjusting the polymerization temperature. Specifically, as the temperature increases from 25 °C to 90 °C, the diameter of the obtained carbon nanospheres can be continuously tuned from 270 nm to 460 nm with high size uniformity. Concurrently, the folding depth of the carbon sphere surface could be synchronously modulated from 10 nm to 40 nm under corresponding preparation conditions (Fig. S13). This direct temperature-mediated regulation offers a simple pathway for the multiscale morphological design of functional carbon materials. In addition, we evaluated the porous structures and surface chemistry by gas adsorption using a series of molecular probes including N2 at 77 K, CO2 at 273 K, and H2O at 298 K (Fig. S14a–c). The N2 adsorption isotherm of DPC is typically type I, indicating its microporous feature. The specific surface area was determined to be around 529 m2 g−1 according to the Brunauer–Emmett–Teller theory. Furthermore, the dominant pore size of DPC is centered at 0.48 nm, which is determined based on the non-local density functional theory (Fig. S14a). However for H2O adsorption, it displays a sharp water uptake from the starting point to a relative humidity of 40% RH (Fig. S14c), indicating its relatively hydrophilic feature.
 | ||
| Fig. 2 Chemical structure of the inner core in LPCs. (a) and (b) Low magnification and high magnification SEM images of LPC derived nanocarbon spheres. (c) In situ FTIR spectra of LPCs collected at different reaction stages from 10 s to 2 h. (d) TEM image of LPCs after being leached with HCl solution. (e) MALDI-TOF mass spectrum of the composition released from the inner core of LPCs. (f) DSC curves of LPCs. The SEM images in (a) and (b) and the TEM image in (d) were collected at room temperature. | ||
To understand the formation pathway of the LPCs, we conducted an in situ Fourier transform infrared spectroscopy (FTIR) test (Fig. 2c). In 10 seconds, an absorption band at 1612 cm−1 was observed, which can be assigned to the CN stretching vibration, evidencing the fast nucleation of the Schiff-base core.42 Moreover, the C–N stretching band at 1195 cm−1 became obvious as the reaction continued, confirming the formation of crosslinked polybenzoxazine networks due to the co-polymerization of TPA, HQ, and p-PDA.32 The C1s and N1s X-ray photoelectron spectra of LPCs also show the coexistence of C–N bonds and CN bonds (Fig. S15). Considering the chemical properties of the Schiff-base core, we tried to extract it by dissolving it in acid or organic solvents. For instance, after soaking in 0.18 M HCl solution or 1-butanol for 24 h, the soft-core was indeed leached out (Fig. 2d, S16 and S17). We found that the leached nanospheres failed to form the fold structures after the identical thermal treatment, which evidenced the crucial role of the soft-core in the formation of the surface folding (Fig. S18). Subsequently, we analyzed the extracts by matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS). The mass distribution of the extracts is shown in Fig. 2e. The molecular mass of the adjacent peaks shows exactly a fixed interval of 206 dalton, which equals the molecular mass of a repeating unit of the Schiff-base. These results nicely verified the structure and composition of the soft Schiff-base core in the LPCs.
Next, we studied the thermotropic behavior of LPCs by thermogravimetric analysis (TGA) (Fig. S19) together with differential scanning calorimetry (DSC) (Fig. 2f). Based on the TG curve, we speculate that the conversion of LPCs experienced two stages. The weight loss in the first stage from RT to 450 °C is within 3%, which is relevant to the adsorbed water. Meanwhile, two endothermic peaks are observed at 170 °C and 261 °C in the DSC curve, which can be attributed to the glass transition of the stiff-shell and crystalline melting of the soft Schiff-base core. Similar observations were reported for other Schiff-base polymeric crystals.43,44 The sample experienced a significant weight loss of about 53% during the second stage of 450–1000 °C, which was due to the decomposition of polymeric molecular chains and subsequent evolution towards carbon structures. Notably, the formation of the nanofold structure started at around 500 °C, which coincides well with the TGA-DSC results.
Subsequently, we conducted a thorough investigation on the formation process of MLCF. The thermal transformation emerged as a pivotal stage in creating neck-like junctions among spheres. To directly observe this process, we employed in situ SEM and in situ XRD to provide a visual insight into the thermal transformation of LPCs as well as a comprehensive picture of the MLCF formation process (Fig. 3a and b). As shown, the monolayer LPCs were originally hexagonally assembled on the substrate (Fig. 3a bottom). Due to the non-standard spheroid shape of the polymer spheres, discernible gaps were left between them. Upon heating the sample to 450 °C, the LPCs exhibited volume expansion and thus squeezed each other, completely filling the interstitials (Fig. 3a middle). Further heating induced the fusion and connection of the thermotropic liquid crystals with adjacent spheres. However, the stiff shell of the LPCs prevented them from completely merging. Upon heating to 800 °C, the size of the obtained nanospheres shrank to 450 nm. However, each sphere was still interlinked with its neighboring spheres by the “nano-necks”; meanwhile, each sphere showed gyrus-like surface nanofolds (Fig. 3a top).
 | ||
| Fig. 3 In situ visualization of the dynamic structure evolution during thermal treatment and the proposed linking mechanism. (a) In situ SEM images collected at 20 °C, 50 °C, 450 °C, and 800 °C during the thermal transition of monolayer LPCs. Heating rate: 10 °C min−1. (b) In situ PXRD patterns collected during heating the LPCs from 20 to 450 °C, with pauses to take a set of XRD scans at an interval of 50 °C. The dashed line indicates the shift of the peak to lower angles. Inset: the model of the crystalline structure of the LPC inner core viewed in the direction perpendicular to the chains. (c) The proposed formation process of neck-like links in MLCF. | ||
Fig. 3b provides the results of in situ XRD measurements during the thermal treatment process. The dashed line shows the evolution of the most obvious features of the samples during the thermal transformation. In the heating process of 20–450 °C, the sharp peak at 20.4° (110) moves to lower angles, due to the expansion of the unit cell (Fig. 3b inset). Further increasing the temperature to 450 °C and above (Fig. S20), the XRD patterns changed obviously, and the peak at 20.4° disappeared and transformed into the (002) peak of graphitic carbons, indicating the formation of nanocarbon structures. Based on these data, we summarized the formation process in Fig. 3c. In brief, the nanolinks stem from the melting of the quasi-liquid crystal core. Upon heating, the melting core partly penetrated through the softening shell and fused with the neighboring spheres. Meanwhile, the heterogeneous polymeric spheres gradually shrank, which pulled the neighboring spheres apart. Such tension forces induced the nanowire drawing, ending with the neck-like junction among nanocarbon spheres. In contrast, we found that the carbon nanospheres derived from a pure stiff polymer failed to link with the surroundings (Fig. S21). These results reveal that the unique combination of the stiff-shell and soft-core structure is crucial for the formation of such interlinked nanocarbon arrays.
The monolayer assembly of LPCs can easily proceed on flexible substrates such as PDMS. The evident folding surface increases the adhesion to the substrate surface, thereby warranting a high success rate for device fabrication (Fig. S22). The large-area monolayer film of interlinked nanocarbon spheres motivated us to explore its application as light filters. The monolayer of LPCs underwent thermal treatment at 500 °C, yielding MLCF-500 (Fig. 4a and S23). Compared with the monolayer of LPCs without thermal treatment (Fig. S24), the formed voids in MLCF-500 due to the volume shrinkage of LPCs during thermal treatment allow light transmission freely (Fig. 4a inset).
 | ||
| Fig. 4 The UV light filtering and resistive heating properties of the MLCF. (a) SEM image of the MLCF prepared at 500 °C. Inset: the visible assessment of transparency of the monolayer film of MLCF-500. (b) Schematic illustration of UV light and HEBL filtering through the MLCF. (c) UV-vis transmission spectra of sample LPCs-RT and thermally treated MLCF at different temperatures. SEM images of Si supported CF-1L (d), CF-2L (e), and CF-3L (f). Inset: the cross-section of the CF-1L and its 2-layer (CF-2L) and 3-layer (CF-3L) counterparts. (g) Time-dependent temperatures of the MLCF with different layers with an applied potential of 11 V. (h) A performance comparison diagram of ΔT with thickness normalization for different film materials with an applied potential of 15 V.10,46–51 (i) Optical image of the flexible monolayer film of interlinked nanocarbon spheres on the PDMS substrate with an area of 126 cm2. The SEM images in (a), (d), (e) and (f) and the optical image in (i) were collected at room temperature. | ||
We then optimized the thermal treatment conditions (Fig. S25), and the typical films demonstrate high transmittance for visible light while effectively filtering most harmful UV light and high energy blue light (HEBL) (Fig. 4b). Among the tested samples, MLCF-450 and MLCF-500 demonstrated favorable filtration capabilities toward both UV and HEBL. In contrast, MLCF-50 was largely impermeable to these wavelengths, and MLCF-800 failed to selectively shield UV light and HEBL. The excellent performance of MLCF-500 is ascribed to its appropriate preparation temperature, which preserves the aromatic ring in the polymer as well as the C–N/CO bonds. These functionalities enable efficient absorption of UV light radiation and subsequent conversion of its energy into heat and fluorescence via π → π* transitions in the conjugated aromatic system and n → π* transitions related to carbon–heteroatom bonds (Fig. S26).45 As a result, MLCF-500 achieved high transmittance for visible light while blocking 90% and above UV light and HEBL (Fig. 4c), making it promising for optical applications.
In addition, the large area monolayer film exhibited consistent optical performance after exposure to high-power UV light irradiation for 72 h (Fig. S27). Even after immersing in water at different temperatures, the UV light transmittance of MLCF-500 only increased slightly by 1.2% (Fig. S28). The outstanding durability is ascribed to the inherent stability of carbon materials. Furthermore, we investigated the effect of the incident angle of light. As the light incident angle varied from 6 to 76°, the transmission of light in both UV light and visible spectra decreased to some extent. However, it retains over 90% absorption of UV light as well as considerable transparency under all the conditions (Fig. S29). To our knowledge, this is the first demonstration that large-area transparent monolayer films of interlinked nanocarbon spheres can be used for UV light protection. This might be useful for biological protection from UV light damage.
Due to the excellent conductivity and ultrathin properties of the MLCF, we further investigated its potential application as resistive heating materials. By repeating the single-layer assembly process of LPCs, carbon film resistance heaters with an area of 24 cm2 and different layers (Fig. 4d–f, S30 and S31) were successfully fabricated and named CF-1L, CF-2L, and CF-3L in turn. The electrothermal performance tests demonstrated that when a voltage of 11 V was applied across a 3.0 cm × 3.0 cm sample, the surface temperature of only 1.2 μm thick CF-3L rapidly reached 43 °C (Fig. 4g). Furthermore, we observed that as the number of MLCF layers increased, the electrical heating performance of the resistance heater increased regularly. Specifically, for every 0.4 μm increase in thickness, the surface temperature increased by about 9 °C (Fig. S32).
The repeatability over successive heating–cooling cycles is critical for the practical application of wearable heaters. The CF-3L heater consistently reached a maximum temperature of approximately 70 °C over 12 cycles under alternately switching on/off at 18 V, with no significant performance degradation (Fig. S33a). The corresponding SEM image confirmed the structural integrity after heating–cooling cycles (Fig. S33b). This demonstrates the electrothermal stability and mechanical robustness of the device.
Fig. S34 further showed that the surface temperature of CF-3L was proportional to the square of the applied voltage, following Joule's law (Q = U2R−1t), providing a theoretical basis for accurate temperature control of resistance heaters. It is important to note that CF-3L exhibited both efficient heat generation under low-voltage conditions (<36 V) and guaranteed operational safety (Fig. S35). Compared with the previously reported non-metallic ultrathin Joule heating materials, the MLCF demonstrated significant advantages in terms of thickness control and thermal conductivity (Fig. 4h and Table S1). In addition, the material preparation process exhibited good scalability, and its dimension was primarily constrained by the substrate area and heat treatment furnace capacity. For example, we have successfully prepared large area MLCF samples with dimensions up to 21.0 cm × 6.0 cm (Fig. 4i).
Conclusions
We demonstrated a facile fabrication of large-area conductive porous films of interlinked nanocarbon spheres derived from quasi-liquid polymeric crystals with a hybrid core–shell structure. Such a core–shell structure was prepared via a cascading polymerization strategy that allows the surface evolution toward nanofold morphology. The conductive and flexible films based on interlinked monolayer nanocarbons can serve as ideal candidates for optical materials such as UV light filtering glasses and electrically heated materials such as wearable heating devices. This scalable solution synthesis and the facile transformation represent a smart engineering platform at the nanoscale for the design of electronic skin and intelligent wearable devices.Author contributions
A.-H. L. and G.-P. H. conceived the presented idea and directed the project. M.-Q. Z., T.-Y. L. and C.-B. L. carried out the experiments with support from H.-Q. H., Y.-S. W., L.-Y. D., X.-D. S. and Y.-T. W., who helped in preparing and characterizing the samples with SEM, TEM and in situ XRD and analysed the data. All authors discussed the results and contributed to the final manuscript.Conflicts of interest
The authors declare no conflict of interest.Data availability
All the data can be found within the manuscript and SI files. Supplementary information: experimental procedures; materials characterization data including SEM, AFM, TEM, TG, XPS, XRD, gas adsorption; materials test data including UV light transmission test and electric heating test. See DOI: https://doi.org/10.1039/d5sc04466a.Acknowledgements
This work was supported by the National Natural Science Funds of China (No. 22275027) and the Fundamental Research Funds for the Central Universities (No. DUT22LAB607).Notes and references
- T. Cui, D. Li, T. Hirtz, J. Xu, Y. Qiao, H. Xu, H. Tian, H. Liu, Y. Yang and T. Ren, Carbon Future, 2024, 1, 9200005 CrossRef.
- Y. Meng, J. Feng, S. Han, Z. Xu, W. Mao, T. Zhang, J. S. Kim, I. Roh, Y. Zhao, D. Kim, Y. Yang, J. Lee, L. Yang, C. Qiu and S. Bae, Nat. Rev. Mater., 2023, 8, 498–517 CrossRef.
- A. T. Hoang, L. Hu, B. J. Kim, T. T. N. Van, K. D. Park, Y. Jeong, K. Lee, S. Ji, J. Hong, A. K. Katiyar, B. Shong, K. Kim, S. Im, W. J. Chung and J. Ahn, Nat. Nanotechnol., 2023, 18, 1439–1447 CrossRef PubMed.
- D. Ren, C. Zhao, S. Zhang, K. Zhang and F. Huang, Adv. Funct. Mater., 2023, 33, 2300517 CrossRef.
- X. Wang, T. Liu, F. Sun, J. Zhang, B. Yao, J. Xu and J. Fu, Smart Mol., 2024, 2, e20240008 CrossRef PubMed.
- C. Toh, H. Zhang, J. Lin, A. S. Mayorov, Y. Wang, C. M. Orofeo, D. B. Ferry, H. Andersen, N. Kakenov, Z. Guo, I. H. Abidi, H. Sims, K. Suenaga, S. T. Pantelides and B. Özyilmaz, Nature, 2020, 577, 199–203 CrossRef PubMed.
- L. Hou, X. Cui, B. Guan, S. Wang, R. Li, Y. Liu, D. Zhu and J. Zheng, Nature, 2022, 606, 507–510 CrossRef PubMed.
- M. Ramezani, J. Kim, X. Liu, C. Ren, A. Alothman, C. De-Eknamkul, M. N. Wilson, E. Cubukcu, V. Gilja, T. Komiyama and D. Kuzum, Nat. Nanotechnol., 2024, 19, 504–513 CrossRef.
- J. Liu, N. P. Wickramaratne, S. Z. Qiao and M. Jaroniec, Nat. Mater., 2015, 14, 763–774 CrossRef.
- D. Zhang, S. Xu, X. Zhao, W. Qian, C. R. Bowen and Y. Yang, Adv. Funct. Mater., 2020, 30, 1910809 CrossRef.
- Y. Chu, L. Sun, J. Wang, Z. Han, C. Wei, C. Han and H. Yan, Nanomaterials, 2024, 14, 911 CrossRef PubMed.
- M. Ghasemlou, N. Pn, K. Alexander, A. Zavabeti, P. C. Sherrell, E. P. Ivanova, B. Adhikari, M. Naebe and S. K. Bhargava, Adv. Mater., 2024, 36, 2312474 CrossRef.
- X. Ma, Q. Liu, N. Yu, D. Xu, S. Kim, Z. Liu, K. Jiang, B. M. Wong, R. Yan and M. Liu, Nat. Commun., 2021, 12, 6868 CrossRef.
- Z. Wang, X. Kong, Y. Huang, J. Li, L. Bao, K. Cao, Y. Hu, J. Cai, L. Wang, H. Chen, Y. Wu, Y. Zhang, F. Pang, Z. Cheng, P. Babor, M. Kolibal, Z. Liu, Y. Chen, Q. Zhang, Y. Cui, K. Liu, H. Yang, X. Bao, H. Gao, Z. Liu, W. Ji, F. Ding and M. Willinger, Nat. Mater., 2024, 23, 331–338 CrossRef.
- Y. Zhao, X. Yang, Z. Cheng, C. H. Lau, J. Ma and L. Shao, Nat. Commun., 2023, 14, 2679 CrossRef.
- S. Rahman, T. Yildirim, M. Tebyetekerwa, A. R. Khan and Y. Lu, ACS Nano, 2022, 16, 13959–13968 CrossRef.
- S. Lim, E. Jang, D. Yu, J. Koo, D. Kang, K. M. Lee, N. P. Godman, M. E. McConney, D. Kim and K. Jeong, Adv. Mater., 2023, 35, 2206764 CrossRef.
- Y. Liu, Z. Feng, C. Xu, A. Chatterjee and A. A. Gorodetsky, ACS Nano, 2021, 15, 17299–17309 CrossRef PubMed.
- T. Ma, T. Li, L. Zhou, X. Ma, J. Yin and X. Jiang, Nat. Commun., 2020, 11, 1811 CrossRef PubMed.
- L. Peng, C. Hung, S. Wang, X. Zhang, X. Zhu, Z. Zhao, C. Wang, Y. Tang, W. Li and D. Zhao, J. Am. Chem. Soc., 2019, 141, 7073–7080 CrossRef.
- L. Liu, S. Xu, Q. Yu, F. Wang, H. Zhu, R. Zhang and X. Liu, Chem. Commun., 2016, 52, 11693–11696 RSC.
- Z. Zhao, Y. Huang, J. Du and A. Chen, Chem.–Asian J., 2023, 18, e202300486 CrossRef PubMed.
- Y. Sun, C. Li, D. Liu, F. Zhang, J. Xue and Q. Zheng, ACS Nano, 2025, 19, 1944–1980 CrossRef PubMed.
- J. Jang, H. Choo, S. Lee, J. Song, K. Park, J. Yoon, D. Seong, S. An, H. Jung, J. Ju, J. Kang, J. Kang, I. S. Kim, M. Shin, J. Park and D. Son, Nat. Electron., 2025, 8, 474–484 CrossRef.
- Y. Yue, D. Zhang, P. Wang, X. Xia, X. Wu, Y. Zhang, J. Mei, S. Li, M. Li, Y. Wang, X. Zhang, X. Wei, H. Liu and W. Zhou, Adv. Mater., 2024, 36, 2313971 CrossRef PubMed.
- S. Arimura, I. Matsumoto, R. Sekiya and T. Haino, Angew. Chem., Int. Ed., 2024, 63, e202315508 CrossRef PubMed.
- L. Yu, L. Liang, I. Bajaj, K. Seabright, D. J. Keffer, I. N. Ivanov, H. Chen, S. Dai, A. J. Ragauskas, C. T. Maravelias and D. P. Harper, Carbon, 2023, 213, 118285 CrossRef.
- M. Kim, R. Xin, J. Earnshaw, J. Tang, J. P. Hill, A. Ashok, A. K. Nanjundan, J. Kim, C. Young, Y. Sugahara, J. Na and Y. Yamauchi, Nat. Protoc., 2022, 17, 2990–3027 CrossRef PubMed.
- R. Li, B. Ma, M. Li, D. Wang, P. Liu and P. An, Chem. Sci., 2024, 15, 11408–11417 RSC.
- J. Uchida, B. Soberats, M. Gupta and T. Kato, Adv. Mater., 2022, 34, 2109063 CrossRef.
- H. K. Bisoyi and Q. Li, Chem. Rev., 2022, 122, 4887–4926 CrossRef PubMed.
- S. Wang, W. Li, G. Hao, Y. Hao, Q. Sun, X. Zhang and A. Lu, J. Am. Chem. Soc., 2011, 133, 15304–15307 CrossRef.
- Z. Zheng, M. Wu, X. Zeng, X. Zhu, D. Luo, X. Chen, Y. Chen, G. Yang, D. Bin, X. Zhou and D. Li, Angew. Chem., Int. Ed., 2024, 63, e202400012 CrossRef.
- Y. Pi, L. Cui, W. Luo, H. Li, Y. Ma, N. Ta, X. Wang, R. Gao, D. Wang, Q. Yang and J. Liu, Angew. Chem., Int. Ed., 2023, 62, e202307096 CrossRef PubMed.
- J. L. Segura, M. J. Mancheño and F. Zamora, Chem. Soc. Rev., 2016, 45, 5635–5671 RSC.
- T. J. White and D. J. Broer, Nat. Mater., 2015, 14, 1087–1098 CrossRef CAS.
- B. Crist and J. M. Schultz, Prog. Polym. Sci., 2016, 56, 1–63 CAS.
- W. Łużny, E. Stochmal-Pomarzańska and A. Proń, Polymer, 1999, 40, 6611–6614 Search PubMed.
- T. Tallinen, J. Y. Chung, J. S. Biggins and L. Mahadevan, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 12667–12672 CrossRef.
- I. Tobasco, Y. Timounay, D. Todorova, G. C. Leggat, J. D. Paulsen and E. Katifori, Nat. Phys., 2022, 18, 1099–1104 Search PubMed.
- F. Xu, Y. Huang, S. Zhao and X. Feng, Nat. Comput. Sci., 2022, 2, 632–640 Search PubMed.
- F. Jin, T. Wang, H. Zheng, E. Lin, Y. Zheng, L. Hao, T. Wang, Y. Chen, P. Cheng, K. Yu and Z. Zhang, J. Am. Chem. Soc., 2023, 145, 6507–6515 CrossRef PubMed.
- G. Chen, B. Jin, Y. Shi, Q. Zhao, Y. Shen and T. Xie, Adv. Mater., 2022, 34, 2201679 CrossRef PubMed.
- S. T. Ha, L. K. Ong, S. T. Ong, G. Y. Yeap, J. P. W. Wong, T. M. Koh and H. C. Lin, Chin. Chem. Lett., 2009, 20, 767–770 CrossRef.
- B. K. Barman, Ø. Sele Handegård, A. Hashimoto and T. Nagao, ACS Sustain. Chem. Eng., 2021, 9, 9879–9890 CrossRef.
- Y. Wang, X. Ding, H. Wang, Q. Wang, M. Zhang and B. Yang, ACS Appl. Polym. Mater., 2024, 6, 7612–7620 CrossRef.
- J. Luo, H. Lu, Q. Zhang, Y. Yao, M. Chen and Q. Li, Carbon, 2016, 110, 343–349 CrossRef.
- H. Song, B. Nie, Y. Zhu, G. Qi, Y. Zhang, W. Peng, X. Li, J. Shao and R. Wei, Langmuir, 2024, 40, 6940–6948 CrossRef PubMed.
- Z. Shao, Y. Liu, P. Cai, Q. Wang, Z. Xiao, L. Zhang, B. Tong, B. Wang, Y. Zhao, W. Zhang and Y. Xia, J. Electron. Mater., 2024, 53, 4601–4612 CrossRef.
- H. Weerathunga, T. T. Do, H. D. Pham, R. Jones, J. MacLeod, T. Kim and D. Dubal, Adv. Mater. Technol., 2023, 8, 2201538 CrossRef.
- Y. Liu, Z. Xiao, W. Zhang, H. Huang, J. Zhang, Y. Gan, X. He, B. Wang, Y. Han and Y. Xia, J. Ind. Eng. Chem., 2022, 107, 401–409 CrossRef.
Footnote |
| † M.-Q. Z., T.-Y. L. and C.-B. L. contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |