
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Isolation of a phosphinidene sulfide and selenide†
Chenyang Hu ab,
Maren Pinkb and
Jose M. Goicoechea*a
ab,
Maren Pinkb and
Jose M. Goicoechea*a
aDepartment of Chemistry, Indiana University, 800 East Kirkwood Ave., Bloomington, Indiana 47405, USA. E-mail: jgoicoec@iu.edu
bDepartment of Chemistry, University of Oxford, Chemistry Research Laboratory, 12 Mansfield Rd., Oxford, OX1 3TA, UK
First published on 10th July 2025
Abstract
We describe the synthesis of an isolable phosphinidene sulfide and phosphinidene selenide (BnArNP![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Ch; Ch = S, Se) by reaction of a sterically protected phosphinidene oxide (BnArNPO) with Lawesson's or Woollins' reagents, respectively. These compounds reveal short PE bonds consistent with a high degree of pπ–pπ double bond character. The pendant amido group on the phosphorus atom plays an important role in their stabilization through nitrogen lone pair donation into the P–Ch π* orbital, and evidence for isomerism about the N–P bond was observed for both species. Dimeric intermediates featuring mixed phosphorus(III)/phosphorus(V) centers could be isolated and structurally authenticated during the course of these studies providing some insight into the mechanism of formation of the titular compounds.
Ch; Ch = S, Se) by reaction of a sterically protected phosphinidene oxide (BnArNPO) with Lawesson's or Woollins' reagents, respectively. These compounds reveal short PE bonds consistent with a high degree of pπ–pπ double bond character. The pendant amido group on the phosphorus atom plays an important role in their stabilization through nitrogen lone pair donation into the P–Ch π* orbital, and evidence for isomerism about the N–P bond was observed for both species. Dimeric intermediates featuring mixed phosphorus(III)/phosphorus(V) centers could be isolated and structurally authenticated during the course of these studies providing some insight into the mechanism of formation of the titular compounds.
Introduction
Phosphinidenes (R–P) are regarded as heavier analogues of carbenes (R2C) due to the diagonal relationship between phosphorus and carbon.1–13 Their oxidized phosphorus(III) counterparts, phosphinidene chalcogenides (R–PCh; where Ch = group 16 element), are also known to exhibit carbene-like reactivity.14,15 That being said, the chemical properties of phosphinidene chalcogenides remain poorly understood on account of their inherent instability under ambient conditions. The polarity of phosphorus–chalcogen bonds, and their weak π-bonding character, makes them highly reactive, which often leads to deleterious dimerization or oligomerization reactions. Given the difficulties in isolating heavier phosphinidene chalcogenides, they have thus far only been observed at extremely low temperatures (3–15 K) in inert gas matrices (Fig. 1a).16
 | ||
| Fig. 1 (a) Reported methods for the generation of phosphinidene sulfides and selenides; (b) an isolable phosphinidene sulfide and selenide supported by an ylidyl substituent; (c) a phosphinidene oxide and its phospha–Wittig reactivity; (d) current work: isolation of a phosphinidene sulfide and selenide. | ||
Two different approaches towards the chemical stabilization of phosphinidene chalcogenides have been previously explored in the chemical literature. The first of these, which we term the intermolecular approach, involves the trapping of in situ generated phosphinidene chalcogenides with chemical reagents (e.g. dienes).14,17–27 Using this approach, the formation of a transient phosphinidene chalcogenide can be inferred by the resulting products (often a heterocycle of some kind), however this does not allow for the isolation of the reactive PCh bond. The second strategy involves the intramolecular electronic stabilization of the PCh moiety by adjacent metal centres,28–31 or electron-donating R substituents.32–35 This latter strategy was successfully employed by Schmidpeter and co-workers to access the only structurally authenticated R–PS and R–PSe compounds reported to date (Fig. 1b). Both compounds are stabilized by electron-delocalization of the ylidyl substituent into the PCh π* orbital and were found to be moderately stable in solution for short periods of time.34,35
Recently, we reported the first example of an isolable phosphinidene oxide using a bulky amido supporting ligand for the stabilization of the highly reactive P(III)O bond (BnArNPO).36 During these studies, it became apparent that the nitrogen atom lone pair plays an important role in the stabilization of the reactive PO moiety. We have since gone on to show that this compound can act as an electrophilic phosphinidene precursor for the synthesis of phosphorus-main group element double bonds (Fig. 1c).37 This motivated us to explore the feasibility of employing the same ligand to support heavier phosphinidene chalcogenides. Herein, we expand this strategy and report the transformation of this phosphinidene oxide to its sulfide and selenide analogues using Lawesson's and Woollins' reagents, respectively (Fig. 1d). It is worth noting that recently Tan described the isolation of heavier antimony-containing analogues, the stibinidene chalcogenides (MsFluid*SbCh; Ch = S–Te; MsFluid* is a bulky hydrindacene substituent) in which the reactive SbCh bonds are protected by an extremely bulky hydrindacene ligand.38
Results and discussion
Inspired by the phospha–Wittig reaction, we initiated our studies by reacting phosphinidene oxide, A, with either triphenylphosphine sulfide (SPPh3) or trimethylphosphine sulfide (SPMe3). Given the similar electronic structures of SPR3 and phosphorus ylides, we reasoned that these reagents would allow chalcogen atom transfer to the phosphorus atom. To our surprise, unlike phosphorus ylides, which react with A instantly,37 no reaction was observed between SPPh3 or SPMe3 and A, even at elevated temperatures. This suggests that phosphine sulfides lack the requisite nucleophilicity to undergo reactions with phosphinidene oxides. Similarly, no Wittig-type reactions between phosphine sulfides and carbonyl compounds have been reported to date, further supporting the inertness of phosphine sulfides in such transformations.Given the structural and electronic similarity between A and carbonyl compounds, we trialled other strategies which have been shown to be effective in the chalcogenation of carbonyl-containing species. Lawesson's reagent (LR) is well established as a versatile thionation reagent. The mechanism which gives rise to its effective reactivity, involves the dissociation of the dimeric structure and the in situ generation of a dithioxo-phosphorane.39 We envisioned that the use of Lawesson's reagent could facilitate the conversion of our phosphinidene oxide, A, to its sulfide analogue, mirroring the thionation reactivity observed towards carbonyl compounds.
Reaction of 0.5 equivalents of LR with 1 equivalent of compound A (Scheme 1) gave rise to multiple products, as evidenced by in situ 31P{1H} NMR spectroscopy. Addition of excess LR drove conversion to a major product which features a set of doublets at 42.5 and 47.8 ppm with a 2JP–P coupling constant of 93.2 Hz. The proton-coupled 31P NMR spectrum revealed that the resonance at 42.5 ppm splits into a doublet of triplets, with a 3JP–H coupling constant of 15.2 Hz, indicating that it originates from the LR fragment (i.e. coupling to a p-methoxy-phenyl group is observed). These data suggests that this product, 1, is a dimeric phosphinidene sulfide, incorporating a dithio-phosphorane unit derived from LR dissociation. Similar dimeric structures have been reported previously for germanium and tin, formed via the reaction of germylenes, stannylenes or stannylene sulfides with LR.40 The reaction also affords an intractable mixture of side-products which we believe to be oligomers of (MeO)C6H4P(O)(S) and (MeO)C6H4P(S)2, as previously reported in the literature.41
 | ||
| Scheme 1 Synthesis of compounds 1 and 2. | ||
The structure of compound 1 was confirmed by single crystal X-ray diffraction.† As expected, the structure revealed a four-membered P2S2 ring containing one P(III) centre and one P(V) centre. Given the poor quality of the crystal and the presence of extensive positional disorder, the discussion of bond metric data is not viable, however the connectivity is consistent with a dimeric compound (see ESI, Fig. S27†). It is worth noting that a related dimeric compound DippTerP(μ-S)2P(S)DippTer, was recently reported by Hering-Junghans and co-workers (DippTer 2,6-(2,6-iPr2–C6H3)2–C6H3).42
During the purification of 1, a minor product caught our attention. Upon removal of the excess LR from the reaction mixture by washing with MeCN, two new singlet resonances appeared at 477.0 and 472.1 ppm in the 31P{1H} NMR spectrum in a 4.28![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, the chemical shifts of these resonances comparable favourably with the R–PS compound reported by Schmidpeter.34 These two peaks are also similar to those observed for the phosphinidene oxide A, which also displays two 31P{1H} NMR singlet resonances arising from cis–trans isomers. Based on this spectral similarity, we assigned these peaks to phosphinidene sulfide 2. A dynamic equilibrium exists between compounds 1, 2 and LR, with the equilibrium favouring the formation of compound 1. Notably, while the removal of excess LR shifts the equilibrium toward compound 2, washing with MeCN alone was insufficient to eliminate all the residual LR.
:1 ratio, the chemical shifts of these resonances comparable favourably with the R–PS compound reported by Schmidpeter.34 These two peaks are also similar to those observed for the phosphinidene oxide A, which also displays two 31P{1H} NMR singlet resonances arising from cis–trans isomers. Based on this spectral similarity, we assigned these peaks to phosphinidene sulfide 2. A dynamic equilibrium exists between compounds 1, 2 and LR, with the equilibrium favouring the formation of compound 1. Notably, while the removal of excess LR shifts the equilibrium toward compound 2, washing with MeCN alone was insufficient to eliminate all the residual LR.
We hypothesized that introducing an additional reagent capable of consuming LR while leaving compound 2 intact would provide the additional thermodynamic driving force needed to generate 2. LR and its analogues are known to react with Lewis bases, forming four-coordinate monomeric adducts.43,44 For this purpose, 4-dimethylaminopyridine (DMAP) was selected as an external Lewis base to neutralize LR. Satisfactorily, upon the addition of excess DMAP to compound 1, in situ 31P{1H} NMR spectroscopy revealed the complete consumption of 1, and increased signal intensity for compound 2, together with a new singlet at 109.4 ppm. Independent reaction of LR with DMAP in THF confirmed that the new 31P{1H} resonance corresponds to the monomeric LR-DMAP adduct. These findings provide strong support for the success of our synthetic strategy. Subsequently, a one-pot synthesis of compound 2 was performed by sequential addition of LR and DMAP to compound A, affording 2 in excellent yields (83%) on a 50 mg scale (Scheme 1). Addition of excess LR to 2 leads to the reformation of 1 again, further confirming the equilibrium nature of this transformation.
The solid-state molecular structure of compound 2 (Fig. 2) was determined by single crystal X-ray diffraction. The crystal structure reveals a trans-arrangement of the BnArN–PS moiety about the N–P bond, consistent with the solid-state structure of A. The PS bond length in 2, 1.9166(14) Å, is significantly shorter than the P–S bond in Schmidpeter's ylide-substituted phosphinidene sulfide (1.981(2) Å), which the authors attribute to a significant contribution from the “dipolar resonance formula”.34 Base-stabilized phosphinidene sulfides, such as an intramolecularly stabilized species recently reported by Nikonov, also exhibit longer P–S bonds (e.g. 1.998(2) Å).45 Notably, the PS bond in 2 is also shorter than the sum of double bond covalent radii (1.96 Å)46 and the P(V)–S bond in SPPh3 (1.9554 Å).47 It is worth noting that the short P–S bond in SPPh3 arises from hyperconjugation of the sulphur atom lone pairs into a doubly degenerate set of P–C σ* antibonding orbitals and that, consequently, such bonds cannot be referred to as double bonds. In contrast, in compound 2, the PS bond length is attributed to a strong π-interaction between phosphorus and sulphur, reinforcing the PS double bond character (vide infra). Notably, the nitrogen atom still retains its planar geometry  , consistent with A and previously reported amido phosphaalkenes and phosphaimines.48–50 The N–P bond length (1.674(3) Å) falls between that of single and double bond, closely resembling previously reported N–P bond length in amido phosphaalkenes and phosphaimines. These structural features indicate that the nitrogen atom lone pair is delocalized into P–S π* orbital, which aligns with the restricted N–P bond rotation observed in the aforementioned NMR studies.
, consistent with A and previously reported amido phosphaalkenes and phosphaimines.48–50 The N–P bond length (1.674(3) Å) falls between that of single and double bond, closely resembling previously reported N–P bond length in amido phosphaalkenes and phosphaimines. These structural features indicate that the nitrogen atom lone pair is delocalized into P–S π* orbital, which aligns with the restricted N–P bond rotation observed in the aforementioned NMR studies.
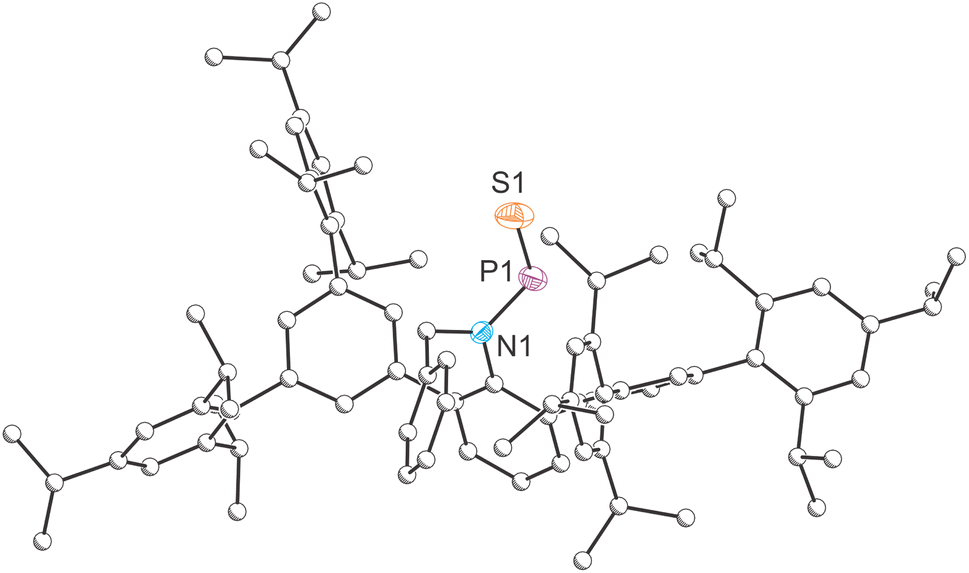 | ||
| Fig. 2 Single-crystal X-ray structures of 2. Carbon atoms depicted with an arbitrary radius. Thermal ellipsoids pictured at 50% probability level. Positional disorder and hydrogen atoms are removed for clarity. | ||
To gain deeper insight into the electronic structure of 2, density functional theory (DFT) calculations were performed at the M06-2X/Def2-SVP level of theory. The optimized geometry is in good agreement with that obtained by X-ray crystallography (e.g. P–S: 1.941 Å; N–P: 1.689 Å;  ). While the LUMO orbital reveals a highly localized P–S π* orbital, the P–S π orbital is diffuse and low in energy, observed as the HOMO−12. Additionally, one of the lone pairs on sulphur is distinctly characterized in the HOMO−1 (Fig. 3a). Natural bond orbital (NBO) analysis further confirmed the presence of a well-defined σ-bond and π-bond within the PS moiety (Fig. S29 in ESI†). The π-interaction is predominantly composed of p-orbital overlap between phosphorus (96.97% p-character) and sulphur (98.14% p-character), with a significant polarization toward sulphur (70.47%). Second-order perturbation theory analysis revealed a back-donation energy of 36.05 kcal mol−1 from the nitrogen atom lone pair to the P–S π* orbital, which is comparable to that of A (41.12 kcal mol−1). The back-donation was also evidenced by the low occupancy of nitrogen lone pair (1.699 e−) and the partially filled P–S π* orbital (0.229 e−). Mayer bond indices corroborate these findings, indicating a bond order of 1.09 for N–P and 1.81 for PS. Since the electronic structure of 2 closely resembles that of A, a similar energy barrier for N–P bond rotation should also be expected. Energy calculations at SMD-M06-2X/def2-TZVP//M06-2X/def2-SVP level revealed a rotation barrier of 20.7 kcal mol−1, closely matching that of A (21.8 kcal mol−1). The trans-isomer of 2 is thermodynamically favoured by 0.6 kcal mol−1, agreeing with the crystallographic observed trans-geometry for the N–P bond (Fig. 3b).
). While the LUMO orbital reveals a highly localized P–S π* orbital, the P–S π orbital is diffuse and low in energy, observed as the HOMO−12. Additionally, one of the lone pairs on sulphur is distinctly characterized in the HOMO−1 (Fig. 3a). Natural bond orbital (NBO) analysis further confirmed the presence of a well-defined σ-bond and π-bond within the PS moiety (Fig. S29 in ESI†). The π-interaction is predominantly composed of p-orbital overlap between phosphorus (96.97% p-character) and sulphur (98.14% p-character), with a significant polarization toward sulphur (70.47%). Second-order perturbation theory analysis revealed a back-donation energy of 36.05 kcal mol−1 from the nitrogen atom lone pair to the P–S π* orbital, which is comparable to that of A (41.12 kcal mol−1). The back-donation was also evidenced by the low occupancy of nitrogen lone pair (1.699 e−) and the partially filled P–S π* orbital (0.229 e−). Mayer bond indices corroborate these findings, indicating a bond order of 1.09 for N–P and 1.81 for PS. Since the electronic structure of 2 closely resembles that of A, a similar energy barrier for N–P bond rotation should also be expected. Energy calculations at SMD-M06-2X/def2-TZVP//M06-2X/def2-SVP level revealed a rotation barrier of 20.7 kcal mol−1, closely matching that of A (21.8 kcal mol−1). The trans-isomer of 2 is thermodynamically favoured by 0.6 kcal mol−1, agreeing with the crystallographic observed trans-geometry for the N–P bond (Fig. 3b).
 | ||
| Fig. 3 (a) Selected Kohn–Sham molecular orbital depictions (isovalue = 0.03) for compound 2; (b) calculated N–P bond rotation energy profiles for 2 and 4. | ||
The thermodynamic profile for the equilibrium between 1, 2 and LR was further investigated using DFT calculations (see Scheme S8 in ESI† for full details). Reaction of 2 with 0.5 equivalents of LR to generate compound 1 was found to be spontaneous and thermodynamically favoured by 2.9 kcal mol−1, which aligns well with experimental observations, as compound 1 is the predominant species in the reaction mixture. DMAP-induced LR cleavage is determined to be an exothermic reaction (−6.5 kcal mol−1). By coupling these reactions, the overall transformation (formation of 2 from reaction of 1 and DMAP) favours the formation of 2, with an overall free energy change of −3.6 kcal mol−1. These computational results are in strong agreement with experimental observations, where the addition of DMAP effectively shifts the equilibrium between compounds 1, 2 and LR, to afford 2.
The successful thionation of A prompted us to explore the use of Woollins' Reagent (WR) to access a phosphinidene selenide. Woollins' reagent is the selenium analogue of Lawesson's reagent and is widely utilized in organic synthesis for the selenation of carbonyl compounds.51 Building on insights gained from the reaction between compound A and LR, we treated A with excess WR in benzene (Scheme 2). After stirring overnight, a bright-yellow solution was obtained, and in situ 31P NMR spectroscopy revealed a doublet at 46.0 ppm with a 2JP–P coupling constant of 80.6 Hz, and a doublet of triplets at −46.0 ppm (2JP–P = 80.6 Hz and 3JP–H = 17.0 Hz). The similarity of this 31P NMR spectrum to that of compound 1 suggests the formation of the dimeric structure 3. Since Woollins' reagent can also be cleaved using a Lewis base,52 we envisioned that addition of DMAP can promote the formation of phosphinidene selenide 4, following an analogous mechanism to that proposed for the formation of 2. Indeed, upon the addition of excess DMAP to the reaction mixture (Scheme 2), the solution rapidly changed colour to light red. In situ 31P{1H} NMR spectroscopy revealed two singlet resonances at 535.4 and 528.6 ppm in a 4.81:1 ratio, closely resembling the 31P{1H} NMR spectrum of compounds A and 2. Extended 31P{1H} NMR scans further revealed the presence of selenium satellites, with a 31P–77Se coupling constant of 788.1 Hz, confirming the formation of phosphinidene selenide 4. While no signals were observed in the 77Se NMR spectrum at room temperature, a broad doublet appeared at 1081.6 ppm when the NMR spectrum was recorded at −45 °C. These observations are tentatively attributed to the chemical exchange process between the cis–trans isomers at room temperature, leading to broad and weak signals. Upon cooling, the chemical exchange process is effectively frozen, resulting in enhanced spectral resolution and signal intensity in the 77Se NMR spectrum. Upon re-addition of excess Woollins' reagent, 4 can cleanly convert back to 3.
 | ||
| Scheme 2 Synthesis of compounds 3 and 4. | ||
The structures of 3 and 4 were authenticated by single crystal X-ray diffraction and are comparable to those of 1 and 2, demonstrating structural consistency across the series (Fig. 4). As with its sulphur-containing analogue, phosphinidene selenide 4 adopts a trans-geometry in solid state. The PSe bond length in 4 was determined to be 2.0662(15) Å, which again is notably shorter that the ylidyl-functionalised analogue (2.129(2) Å). This bond is also shorter than the phosphorus(V)–Se bond (2.106(1) Å) in SePPh3,53 due to presence of a strong P–Se π interaction. The planar geometry of the nitrogen atom  and the short N–P bond length (1.669(4) Å) are consistent with those observed for A and 2, indicating the back-donation from the nitrogen lone pair to P–Se σ* antibonding orbital.
and the short N–P bond length (1.669(4) Å) are consistent with those observed for A and 2, indicating the back-donation from the nitrogen lone pair to P–Se σ* antibonding orbital.
 | ||
| Fig. 4 Single-crystal X-ray structure of 4. Carbon atoms depicted with an arbitrary radius. Thermal ellipsoids pictured at 50% probability level. Positional disorder and hydrogen atoms are removed for clarity. | ||
DFT calculations were conducted to elucidate the electronic structure of 4. While the LUMO of 4 is predominantly composed of the P–Se π* interaction, the HOMO represents the lone pair of the selenium atom (Fig. 5). The P–Se π bond is highly delocalized and is observed in the HOMO−3 and HOMO−12. Natural bond orbital (NBO) analysis confirmed the presence of both σ- and π-bonding orbital between phosphorus and selenium (Fig. S29 in ESI†). Due to the similar electronegativity of Se (2.55) and S (2.58), the p-type π orbital remains polarized, with Se contributing 64.72% and P contributing 35.28%, which is comparable to the values determined for 2.
 | ||
| Fig. 5 Selected Kohn–Sham molecular orbital depictions (isovalue = 0.03) for compound 4. | ||
Second-Order Perturbation Theory analysis revealed a back-donation energy of 19.04 kcal mol−1 for nitrogen lone pair donating to the P–Se π* bond and 12.49 kcal mol−1 to σ* bond, in agreement with the crystal structure of 4. Notably, while the back-donation to π* bond is weaker than calculated for 2 (36.05 kcal mol−1), the σ* back-donation is much stronger in 4 (12.49 kcal mol−1 versus 3.52 kcal mol−1 in 2). We attribute this difference to the lower electronegativity and larger atomic orbital size of Se. The P–Se π-bond in 4 is less polar, which disfavours the π* back-donation, whereas the orbital mismatch between phosphorus and selenium weakens the σ-bond, thereby enhancing the σ* back-donation. Energy calculations determined a cis–trans isomerization barrier of 21.8 kcal mol−1, with the trans-isomer being energetically favoured by 0.2 kcal mol−1, consistent with the crystallographic structure of 4. While the back-donation in 4 is weaker than that in 2, it may seem counterintuitive that the rotation barrier is higher in 4 (21.8 kcal mol−1 versus 20.7 kcal mol−1 in 2). This can be rationalized by the longer P–Se bond length in 4 (2.0662(15) Å) compared to the P–S bond in 2 (1.9166(14) Å), which introduces additional steric hindrance in the transition state. This energy barrier also matches the observed 31P{1H} NMR spectrum of 4, which is similar to those observed for A and 2. Mayer bond indices suggest a bond order of 1.11 for the N–P bond and 1.75 for the PSe bond, reinforcing the proposed bonding model.
Conclusions
Following the successful isolation of a free phosphinidene oxide, we herein report the isolation of its heavier analogues, a phosphinidene sulfide 2 and a phosphinidene selenide 4, further expanding the chemistry of phosphorus(III)–chalcogen double bonds. Lawesson's reagent (LR) and Woollins' reagent (WR) are widely employed in organic synthesis for the transformation of carbonyl compounds into corresponding thio- and seleno-carbonyl derivatives. Given the similarity between carbonyl compounds and phosphinidene oxide A, we have shown that these reagents are also effective in converting phosphinidene oxide A to the corresponding sulfide and selenide. Upon reaction of phosphinidene oxide A with excess LR or WR, the in situ generated phosphinidene chalcogenide overreacted with LR or WR, forming dimeric structures 1 and 3, which features a P2Ch2 four-membered ring motif. Addition of excess DMAP to 1 or 3 introduces an external thermodynamic driving force, facilitating the generation of monomeric phosphinidene sulfide 2 and selenide 4. Crystal structures of both 2 and 4 revealed a trans-geometry within the ArBnN–PCh moiety and back-donation from the nitrogen atom lone pair to P–Ch π* orbital, which is also observed in A. DFT calculations further support the presence of significant back-donation, and an energetically significant rotation barrier was determined for 2 (20.7 kcal mol−1) and 4 (21.8 kcal mol−1), which is comparable to that observed in A.
Experimental section
General synthetic methods
All reactions and product manipulations were carried out under an inert atmosphere of argon or dinitrogen using standard Schlenk-line or glovebox techniques (MBraun UNIlab glovebox maintained at <0.1 ppm H2O and <0.1 ppm O2). Benzene (anhydrous, Sigma-Aldrich), toluene (Fisher Chemical, HPLC grade), hexane (Fisher Chemical, HPLC grade) and acetonitrile (Fisher Chemical, HPLC grade) were purified using a Pure Process Technology (PPT) solvent purification system (SPS). C6D6 (Aldrich, 99.5%) and d8-toluene (Aldrich, 99%) were distilled over sodium/benzophenone. CDCl3 was distilled over CaH2. All dry solvents were stored under argon in gas-tight ampoules over activated 3 Å molecular sieves. Compound A was synthesized following the procedure described in the literature.36 Lawesson's reagent, Woollins' reagent and DMAP were purchased from TCI and Sigma-Aldrich, respectively, and used as received.Analytical techniques
NMR spectra were acquired on a Bruker 500 MHz Avance Neo, a Varian 500 MHz Inova, or a Varian 400 MHz Inova NMR spectrometer. Chemical shifts (δ) are reported in parts per million (ppm). 1H and 13C NMR spectra are referenced to TMS using residual protio-solvent resonance (1H NMR C6D6: δ = 7.16 ppm, 13C NMR C6D6: δ = 128.06 ppm; 1H NMR CDCl3: δ = 7.26 ppm, 13C NMR CDCl3: δ = 77.16 ppm, 1H NMR d8-toluene: δ = 2.08 ppm). 31P NMR spectra were externally referenced to an 85% solution of H3PO4 in H2O. 77Se NMR spectra were externally referenced to a solution of SeMe2 in C6D6. High-resolution mass spectra were recorded on a Thermo Q-Exactive Plus (ESI-TOF, positive ion mode) instrument at the Mass Spectrometry Facility of the Department of Chemistry of Indiana University.Synthesis of 1
To a solid mixture of A (30 mg, 0.025 mmol, 1.00 eq.) and Lawesson's reagent (20 mg, 0.049 mmol, 2 eq.), benzene (0.5 mL) was added. The mixture was stirred overnight at room temperature and monitored by 31P{1H} NMR spectroscopy. Once compound A was fully consumed, the mixture was filtered, and MeCN (20 mL) was added to the filtrate. The mixture was left in a −35 °C freezer overnight to crystalize. The supernatant was removed, and the resulting solid was dried in vacuo to afford 1 (28 mg, 0.020 mmol; 79% yield) as a colourless powder. Single crystals of 1 suitable for X-ray diffraction were obtained by slow evaporation of a concentrated hexane solution. 1H NMR (500 MHz, C6D6): δ (ppm) 1.12 (d, 3JH–H = 6.8 Hz, 12H; CH(CH3)2), 1.15 (d, 3JH–H = 6.8 Hz, 18H; CH(CH3)2), 1.24–1.29 (m, 42H; CH(CH3)2), 2.85 (sept, 3JH–H = 6.8 Hz, 4H; CH(CH3)2), 3.04–3.10 (m, 8H; CH(CH3)2), 3.12 (s, 3H; OCH3), 5.44 (s, 2H; NCH2Ph), 6.59–6.62 (m, 2H; ArH), 6.68–6.75 (m, 3H; ArH), 7.00 (t, 3JH–H = 7.8 Hz, 1H; ArH), 7.16 (s, overlapping with C6D6, 8H, ArH), 7.17 (d, 4JH–H = 1.7 Hz, 2H; ArH), 7.19 (d, 4JP–H = 1.7 Hz, 2H; ArH), 7.22 (d, 4JH–H = 1.7 Hz, 2H; ArH), 7.30 (d, 3JH–H = 7.8 Hz, 2H; ArH), 7.56 (d, 3JH–H = 8.6 Hz, 2H; ArH), 8.26 (dd, 3JH–P = 15.2 Hz, 3JH–H = 8.6 Hz, 2H; ArH). (Accurate integration was not possible as, in solution, 1 is in equilibrium with 2 and LR). 13C{1H} NMR (125 MHz, C6D6): δ (ppm) 24.40, 24.43, 24.68, 25.04, 25.23, (s; CH(CH3)2) 30.71, 30.99, 31.11, 34.84, 34.93 (s; CH(CH3)2), 52.80 (br; NCH2Ph) 54.88 (s; OCH3), 113.81 (d, 1JP–C = 16.7 Hz), 120.89, 128.59, 129.63, 131.53, 131.72, 132.39, 133.49, 133.54, 133.61, 135.20 (d, 1JP–C = 17.1 Hz), 136.39, 137.07, 137.38, 140.28, 141.39, 141.90, 142.39 (d, 2JP–C = 6.6 Hz), 147.00, 148.53, 163.07 (s; ArC). 31P NMR (162 MHz, C6D6): δ (ppm) 42.5 (dt, 2JP–P = 93.2 Hz 3JP–H = 15.2 Hz; PSP(S)C6H4OMe), 47.8 (d, 3JP–P = 93.2 Hz; PSP(S)C6H4OMe). 31P{1H} NMR (162 MHz, C6D6): δ (ppm) 42.48 (d, 3JP–P = 93.2 Hz; PSP(S)C6H4OMe), 47.84 (d, 3JP–P = 93.2 Hz; PSP(S)C6H4OMe). HRMS (m/z): [M + H]+ calcd. For C92H116NOP2S3, 1408.7689; found 1408.7670.Synthesis of 2
To a solid mixture of A (50 mg, 0.042 mmol, 1.00 eq.) and Lawesson's reagent (40 mg, 0.099 mmol, 2.3 eq.), benzene (0.7 mL) was added. The mixture was stirred overnight at room temperature and monitored by 31P{1H} NMR spectroscopy. Once compound A was fully consumed, the mixture was filtered, and DMAP (20 mg, 0.164 mmol, 3.9 eq.) was added to the filtrate. After stirring the mixture for 30 min at room temperature, the formation of a precipitate was observed. The mixture was filtered again, and MeCN (20 mL) was added to the filtrate. The mixture was left in a −35 °C freezer overnight to crystalize. The supernatant was removed, and the resulting solid was dried in vacuo to afford 2 (42 mg, 0.035 mmol; 83% yield) as a light-green powder. Single crystals of 2 suitable for X-ray diffraction were obtained by slow evaporation of a concentrated hexane solution. 1H NMR (500 MHz, C6D6): δ (ppm) 1.16 (d, 3JH–H = 6.8 Hz, 12H; CH(CH3)2), 1.20–1.22 (m, 24H; CH(CH3)2), 1.28–1.32 (m, 36H; CH(CH3)2), 2.87 (sept, 3JH–H = 6.8 Hz, 4H; CH(CH3)2), 2.93–3.06 (m, 8H; CH(CH3)2), 5.46 (s, 2H; NCH2Ph), 6.61 (t, 3JH–H = 7.6 Hz, 2H; ArH), 6.74 (t, 3JH–H = 7.6 Hz, 2H; ArH), 6.79–6.83 (m, 1H; ArH), 6.95 (t, 3JH–H = 7.8 Hz, 1H; ArH), 7.10 (t, 4JH–H = 1.6 Hz, 2H; ArH), 7.16–7.17 (m, overlapping with C6D6, 8H; ArH), 7.19 (t, 4JH–H = 1.6 Hz, 2H; ArH), 7.21 (t, 4JH–H = 1.6 Hz, 2H; ArH), 7.31 (t, 3JH–H = 7.8 Hz, 2H; ArH). (Accurate integration was not possible due to the presence of the minor isomer, 2-cis). 13C{1H} NMR (125 MHz, C6D6): δ (ppm) 24.17, 24.37, 24.41, 24.45, 24.70, 24.81, 25.02, 25.09 (s; CH(CH3)2), 30.86, 30.89, 30.99, 31.00, 31.11, 34.88 (s; CH(CH3)2), 51.02 (br; NCH2Ph), 120.72, 120.78, 128.59, 129.37, 130.53, 131.19, 131.37, 131.81, 131.91, 132.10, 135.78, 137.06, 137.24, 139.86, 140.61 (d, JP–C = 5.2 Hz), 140.94, 141.57, 146.78, 146.90, 148.60 (s; ArC). 31P{1H} NMR (202 MHz, C6D6): δ (ppm) 477.0 (s; 2-trans), 472.1 (s; 2-cis). Ratio 2-trans:2-cis = 4.28:1. HRMS (m/z): [M + H]+ calcd. For C85H109NPS, 1206.8013; found 1206.8017.
Synthesis of 3
To a solid mixture of A (30 mg, 0.025 mmol, 1.00 eq.) and Woollins' reagent (26 mg, 0.049 mmol, 2 eq.), benzene (0.5 mL) was added. The mixture was stirred overnight at room temperature and monitored by 31P{1H} NMR spectroscopy. Once compound A was fully consumed, the mixture was filtered, and MeCN (20 mL) was added to the filtrate. The mixture was left in a −35 °C freezer overnight to crystalize. The supernatant was removed, and the resulting solid was dried in vacuo to afford 3 (31 mg, 0.020 mmol; 82% yield) as a bright-yellow powder. Single crystals of 3 suitable for X-ray diffraction were obtained by slow evaporation of a concentrated hexane solution. 1H NMR (500 MHz, C6D6): δ (ppm) 1.11 (d, 3JH–H = 6.9 Hz, 18H; CH(CH3)2), 1.17 (d, 3JH–H = 6.9 Hz, 18H; CH(CH3)2), 1.24–1.30 (m, 36H; CH(CH3)2), 2.85 (sept, 3JH–H = 6.9 Hz, 4H; CH(CH3)2), 3.01–3.14 (m, 8H; C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) (CH3)2), 5.53 (s, 2H; NC2Ph), 6.69–6.75 (m, 3H; ArH), 6.97–7.02 (m, 3H; ArH), 7.16 (s, overlapping with C6D6, 8H; ArH), 7.18 (d, 4JH–H = 1.7 Hz, 2H; ArH), 7.20 (d, 4JH–H = 1.7 Hz, 2H; ArH), 7.25 (t, 4JH–H = 1.7 Hz, 2H; ArH), 7.29 (d, 3JH–H = 7.7 Hz, 2H; ArH), 7.53–7.58 (m, 3H; ArH), 8.42 (dd, 3JH–P = 17.0 Hz, 3JH–H = 7.2 Hz, 2H; ArH). (Accurate integration was not possible as, in solution, 3 is in equilibrium with 4 and WR). 13C{1H} NMR (125 MHz, C6D6): δ (ppm) 24.40, 24.44, 24.68, 25.04, 25.32 (s; CH(CH3)2), 30.77, 30.84, 30.90, 31.05, 31.11, 34.84, 34.88 (s; CH(CH3)2), 55.24 (br; NCH2Ph), 120.71, 120.82, 120.96, 128.59, 129.63, 131.46, 131.56, 131.62, 131.82, 132.39, 136.26, 136.95, 137.05, 137.17, 137.38, 138.87 (d, 1JC–P = 25.3 Hz), 140.13, 140.88, 141.40, 141.54, 141.90, 142.40 (d, 2JC–P = 6.2 Hz) 143.84 (br), 146.79, 146.91, 146.96, 147.05, 147.10, 148.56, 148.63 (s; ArC). 31P NMR (202 MHz, C6D6): δ (ppm) −46.0 (dt, 2JP–P = 80.6 Hz, 3JP–H = 17.0 Hz; PSeP(Se)Ph), 46.0 (d, 2JP–P = 80.6 Hz, PSeP(Se)Ph). 31P{1H} NMR (202 MHz, C6D6): δ (ppm) −45.95 (d, 2JP–P = 80.6 Hz; PSeP(Se)Ph), 46.04 (d, 2JP–P = 80.6 Hz; PSeP(Se)Ph). HRMS (m/z): [M + H]+ calcd. For C91H114NP2Se3, 1520.5925; found 1520.5954.
(CH3)2), 5.53 (s, 2H; NC2Ph), 6.69–6.75 (m, 3H; ArH), 6.97–7.02 (m, 3H; ArH), 7.16 (s, overlapping with C6D6, 8H; ArH), 7.18 (d, 4JH–H = 1.7 Hz, 2H; ArH), 7.20 (d, 4JH–H = 1.7 Hz, 2H; ArH), 7.25 (t, 4JH–H = 1.7 Hz, 2H; ArH), 7.29 (d, 3JH–H = 7.7 Hz, 2H; ArH), 7.53–7.58 (m, 3H; ArH), 8.42 (dd, 3JH–P = 17.0 Hz, 3JH–H = 7.2 Hz, 2H; ArH). (Accurate integration was not possible as, in solution, 3 is in equilibrium with 4 and WR). 13C{1H} NMR (125 MHz, C6D6): δ (ppm) 24.40, 24.44, 24.68, 25.04, 25.32 (s; CH(CH3)2), 30.77, 30.84, 30.90, 31.05, 31.11, 34.84, 34.88 (s; CH(CH3)2), 55.24 (br; NCH2Ph), 120.71, 120.82, 120.96, 128.59, 129.63, 131.46, 131.56, 131.62, 131.82, 132.39, 136.26, 136.95, 137.05, 137.17, 137.38, 138.87 (d, 1JC–P = 25.3 Hz), 140.13, 140.88, 141.40, 141.54, 141.90, 142.40 (d, 2JC–P = 6.2 Hz) 143.84 (br), 146.79, 146.91, 146.96, 147.05, 147.10, 148.56, 148.63 (s; ArC). 31P NMR (202 MHz, C6D6): δ (ppm) −46.0 (dt, 2JP–P = 80.6 Hz, 3JP–H = 17.0 Hz; PSeP(Se)Ph), 46.0 (d, 2JP–P = 80.6 Hz, PSeP(Se)Ph). 31P{1H} NMR (202 MHz, C6D6): δ (ppm) −45.95 (d, 2JP–P = 80.6 Hz; PSeP(Se)Ph), 46.04 (d, 2JP–P = 80.6 Hz; PSeP(Se)Ph). HRMS (m/z): [M + H]+ calcd. For C91H114NP2Se3, 1520.5925; found 1520.5954.
Synthesis of 4
To a solid mixture of A (50 mg, 0.042 mmol, 1.00 eq.) and Woollin's reagent (40 mg, 0.075 mmol, 1.8 eq.), benzene (0.7 mL) was added. The mixture was stirred overnight at room temperature and monitored by 31P{1H} NMR spectroscopy. Once compound A was fully consumed, the mixture was filtered, and DMAP (20 mg, 0.164 mmol, 3.9 eq.) was added to the filtrate. After stirring the mixture for 30 min at room temperature, the formation of a precipitate was observed. The mixture was filtered again, and MeCN (20 mL) was added to the filtrate. The mixture was left in a −35 °C freezer overnight to crystalize. The supernatant was removed, and the resulting solid was dried in vacuo to afford 4 (42 mg, 0.033 mmol; 80% yield) as a light-red powder. Single crystals of 4 suitable for X-ray diffraction were obtained by slow evaporation of a concentrated hexane solution at room temperature. 1H NMR (500 MHz, C6D6): δ (ppm) 1.15 (d, 3JH–H = 6.8 Hz, 12H; CH(CH3)2), 1.18 (d, 3JH–H = 6.8 Hz, 12H; CH(CH3)2), 1.21 (d, 3JH–H = 6.8 Hz, 12H; CH(CH3)2), 1.28 (d, 3JH–H = 6.8 Hz, 24H; CH(CH3)2), 1.30–1.32 (m, 12H; CH(CH3)2), 2.86 (sept, 3JH–H = 6.8 Hz, 4H; CH(CH3)2), 2.92–3.07 (m, 8H; CH(CH3)2), 5.49 (s, 2H; NCH2Ph), 6.61 (t, 3JH–H = 7.5 Hz, 2H; ArH), 6.72–6.75 (m, 2H; ArH), 6.79–6.81 (m, 1H; ArH), 6.94 (t, 3JH–H = 7.7 Hz, 1H; ArH), 7.09 (t, 4JH–H = 1.5 Hz, 2H; ArH), 7.16–7.17 (m, overlapping with C6D6, 8H; ArH), 7.19 (d, 4JH–H = 1.5 Hz, 4H; ArH), 7.29 (d, 3JH–H = 7.7 Hz, 2H; ArH). (Accurate integration was not possible due to the presence of the minor isomer, 4-cis). 13C{1H} NMR (125 MHz, C6D6): δ (ppm) 24.17, 24.41, 24.45, 24.71, 24.81, 25.05, 25.10 (s; CH(CH3)2), 30.84, 30.86, 30.95, 31.01, 31.11, 34.88 (s; CH(CH3)2), 54.14 (br; NCH2Ph), 120.71, 120.80, 128.77, 129.01, 129.35, 130.55, 131.23, 131.38, 131.83, 132.05, 135.47, 137.05, 137.23, 139.80, 140.15, 140.54, 140.71, 141.54, 141.90, 146.77, 146.80, 146.86, 146.91, 148.49, 148.59 (s; ArC). 31P{1H} NMR (202 MHz, C6D6): δ (ppm) 535.4 (s, 1JP–Se = 788.1 Hz; 4-trans), 528.6 (s; 4-cis). Ratio 4-trans:4-cis = 4.81:1. 77Se NMR (95.4 MHz, d8-tol, −45 °C): δ (ppm) 1081.62 (d, 1JP–Se = 875.0 Hz; 4-trans). HRMS (m/z): [M + H]+ calcd. For C85H109NPSe, 1254.7457; found 1254.7473.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Conceptualization: C. H. and J. M. G.; experimental work: C. H.; X-ray crystallography: C. H. and M. P.; computational work: C. H.; writing – original draft: C. H. and J. M. G.; writing & editing: all authors; supervision: J. M. G.; funding acquisition: J. M. G.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based upon work supported by the National Science Foundation (Grant No. 2348777) and by Indiana University. Dr Arifuzzaman Tapash is acknowledged for help with collecting 77Se NMR data.Notes and references
- G. Fritz, T. Vaahs, H. Fleischer and E. Matern, Angew Chem. Int. Ed. Engl., 1989, 28, 315–316 CrossRef.
- F. Mathey, Angew Chem. Int. Ed. Engl., 1987, 26, 275–286 CrossRef.
- X. Li, S. I. Weissman, T.-S. Lin, P. P. Gaspar, A. H. Cowley and A. I. Smirnov, J. Am. Chem. Soc., 1994, 116, 7899–7900 CrossRef CAS.
- A. V. Akimov, Y. S. Ganushevich, D. V. Korchagin, V. A. Miluykov and E. Y. Misochko, Angew. Chem., Int. Ed., 2017, 56, 7944–7947 CrossRef CAS PubMed.
- S. Shah, M. C. Simpson, R. C. Smith and J. D. Protasiewicz, J. Am. Chem. Soc., 2001, 123, 6925–6926 CrossRef CAS PubMed.
- A. H. Cowley, Acc. Chem. Res., 1997, 30, 445–451 CrossRef CAS.
- K. Lammertsma and M. J. M. Vlaar, Eur. J. Org Chem., 2002, 2002, 1127–1138 CrossRef.
- K. Lammertsma, Phosphinidenes, in New Aspects in Phosphorus Chemistry III, ed. J.-P. Majoral, Springer, Berlin-Heidelberg, 2003, pp. 95–119 Search PubMed.
- F. Mathey, Dalton Trans., 2007, 1861–1868 RSC.
- H. Aktaş, J. C. Slootweg and K. Lammertsma, Angew. Chem., Int. Ed., 2010, 49, 2102–2113 CrossRef PubMed.
- L. Liu, D. A. Ruiz, D. Munz and G. Bertrand, Chem, 2016, 1, 147–153 CAS.
- M. C. Neben, N. Wegerich, T. A. Al Said, R. R. Thompson, S. Demeshko, K. Dollberg, I. Tkach, G. P. Van Trieste III, H. Verplancke, C. von Hänisch, M. C. Holthausen, D. C. Powers and S. Schenider, J. Am. Chem. Soc., 2025, 147, 5330–5339 CrossRef CAS PubMed.
- Q. Luo, T. Liu, L. Huang, C. Yang and W. Lu, Angew. Chem., Int. Ed., 2024, 63, e202405122 CrossRef CAS PubMed.
- P. P. Gaspar, H. Qian, A. M. Beatty, D. A. d'Avignon, J. L. F. Kao, J. C. Watt and N. P. Rath, Tetrahedron, 2000, 56, 105–119 CrossRef CAS.
- C. M. E. Graham, C. L. B. Macdonald, P. D. Boyle, J. A. Wisner and P. J. Ragogna, Chem.–Eur. J., 2018, 24, 743–749 CrossRef CAS PubMed.
- A. Mardyukov, F. Keul and P. R. Schreiner, Angew. Chem., Int. Ed., 2020, 59, 12445–12449 CrossRef CAS PubMed.
- P. P. Gaspar, A. M. Beatty, X. Li, H. Qian, N. P. Rath and J. C. Watt, Phosphorus Sulfur Silicon Relat. Elem., 1999, 144, 277–280 CrossRef.
- C. M. E. Graham, T. E. Pritchard, P. D. Boyle, J. Valjus, H. M. Tuononen and P. J. Ragogna, Angew. Chem., Int. Ed., 2017, 56, 6236–6240 CrossRef CAS PubMed.
- W. J. Transue, M. Nava, M. W. Terban, J. Yang, M. W. Greenberg, G. Wu, E. S. Foreman, C. L. Mustoe, P. Kennepohl, J. S. Owen, S. L. Billinge, H. J. Kulik and C. C. Cummins, J. Am. Chem. Soc., 2019, 141, 431–440 CrossRef CAS PubMed.
- L. Wang, R. Ganguly and F. Mathey, Organometallics, 2014, 33, 5614–5617 CrossRef CAS.
- S. Nakayama, M. Yoshifuji, R. Okazaki and N. Inamoto, J. Chem. Soc. D, 1971, 1186–1187 RSC.
- H. Tomioka, S. Takata, Y. Kato and Y. Izawa, J. Chem. Soc., Perkin Trans. 2, 1980, 1017–1021 RSC.
- S. Nakayama, M. Yoshifuji, R. Okazaki and N. Inamoto, Bull. Chem. Soc. Jpn., 2006, 48, 546–548 CrossRef.
- S. Holand and F. Mathey, J. Org. Chem., 1981, 46, 4386–4389 CrossRef CAS.
- H. Tomioka, S. Miura and Y. Izawa, Tetrahedron Lett., 1983, 24, 3353–3356 CrossRef CAS.
- C. C. Santini, J. Fischer, F. Mathey and A. Mitschler, J. Am. Chem. Soc., 1980, 102, 5809–5815 CrossRef CAS.
- M. Yoshifuji, S. Nakayama, R. Okazaki and N. Inamoto, J. Chem. Soc., Perkin Trans. 1, 1973, 2065–2068 RSC.
- G. Balázs, J. C. Green and M. Scheer, Chem.–Eur. J., 2006, 12, 8603–8608 CrossRef PubMed.
- I. G. Albuerne, M. A. Alvarez, M. E. García, D. García-Vivó and M. A. Ruiz, Inorg. Chem., 2015, 54, 9810–9820 CrossRef CAS PubMed.
- M. A. Alvarez, M. E. García, R. González and M. A. Ruiz, Dalton Trans., 2012, 41, 14498–14513 RSC.
- M. A. Alvarez, E. García, D. García-Vivó, M. A. Ruiz and P. Vega, Inorg. Chem., 2023, 62, 5677–5689 CrossRef CAS PubMed.
- M. Yoshifuji, S. Sangu, M. Hirano and K. Toyota, Chem. Lett., 1993, 22, 1715–1718 CrossRef.
- M. Yoshifuji, M. Hirano and K. Toyota, Tet. Lett., 1993, 34, 1043–1046 CrossRef CAS.
- G. Jochem, H. Noth and A. Schmidpeter, Angew Chem. Int. Ed. Engl., 1993, 32, 1089–1091 CrossRef.
- G. Jochem, K. Karaghiosoff, S. Plank, S. Dick and A. Schmidpeter, Chem. Ber., 1995, 128, 1207–1219 CrossRef CAS.
- C. Hu, N. H. Rees, M. Pink and J. M. Goicoechea, Nat. Chem., 2024, 16, 1855–1860 CrossRef CAS PubMed.
- C. Hu, M. Pink and J. M. Goicoechea, J. Am. Chem. Soc., 2025, 147, 12331–12337 CrossRef CAS PubMed.
- X. Li, Y. Chen, S. Dong, D. Wang, L. Xu, J. Zhu and G. Tan, J. Am. Chem. Soc., 2025, 147, 9858–9864 CrossRef CAS PubMed.
- T. Ozturk, E. Ertas and O. Mert, Chem. Rev., 2007, 107, 5210–5278 CrossRef CAS PubMed.
- C. J. Carmalt, Chem. Commun., 1998, 243–244 RSC.
- T. Wen, R. Bau and C. E. McKenna, J. Chem. Soc., Chem. Commun., 1991, 1223–1224 RSC.
- F. Dankert, P. Gupta, T. Wellnitz, W. Baumann and C. Hering-Junghans, Dalton Trans., 2022, 51, 18642–18651 RSC.
- E. Soleimani, K. N. Robertson, C. C. Pye and J. A. C. Clyburne, Dalton Trans., 2018, 47, 6299–6303 RSC.
- L. E. English, A. Pajak, C. L. McMullin, J. P. Lowe, M. F. Mahon and D. J. Liptrot, Chem.–Eur. J., 2022, 28, e202200376 CrossRef CAS PubMed.
- A. G. Baradzenka, S. F. Vyboishchikov, M. Pilkington and G. I. Nikonov, Chem.–Eur. J., 2023, 29, e202301842 CrossRef CAS PubMed.
- P. Pyykkö and M. Atsumi, Chem.–Eur. J., 2009, 15, 12770–12779 CrossRef PubMed.
- C. Foces-Foces and A. L. Llamas-Saiz, Acta Crystallogr., 1998, C54, IUC9800013 Search PubMed.
- P. Royla, K. Schwedtmann, Z. Han, J. Fidelius, D. P. Gates, R. M. Gomila, A. Frontera and J. J. Weigand, J. Am. Chem. Soc., 2023, 145, 10364–10375 CrossRef CAS PubMed.
- R. Appel, U. Kündgen and F. Knoch, Chem. Ber., 1985, 118, 1352–1370 CrossRef CAS.
- E. Niecke and D. Gudat, Angew. Chem., Int. Ed. Engl., 1991, 30, 217–237 CrossRef.
- J. V. X. da Silva, I. C. Chipoline and S. B. Ferreira, SynOpen, 2024, 08, 223–231 CrossRef CAS.
- L. Ascherl, A. Nordheider, K. S. A. Arachchige, D. B. Cordes, K. Karaghiosoff, M. Bühl, A. M. Z. Slawin and J. D. Woollins, Chem. Commun., 2014, 50, 6214–6216 RSC.
- P. W. Codding and K. A. Kerr, Acta Crystallogr. B, 1979, 35, 1261–1263 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental data, spectra, computational information, and crystallographic data can be found in the ESI. Computationally optimized structures (.xyz) are also provided. CCDC 2444805 (for 1), 2444806 (for 2), 2444807 (for 3) and 2444808 (for 4). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc04352b |
| This journal is © The Royal Society of Chemistry 2025 |