
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnraveling disorder-to-order transitions and chemical ordering in PtCoM ternary alloys using machine learning potential†
Xiangfu
Niu
abcd,
Shiyu
Zhen
 abcd,
Rui
Zhang
d,
Jianqiu
Li
bcd and
Liang
Zhang
*abcd
abcd,
Rui
Zhang
d,
Jianqiu
Li
bcd and
Liang
Zhang
*abcd
aCenter for Combustion Energy, Tsinghua University, Beijing, 100084, China. E-mail: zhangbright@tsinghua.edu.cn
bSchool of Vehicle and Mobility, Tsinghua University, Beijing, 100084, China
cState Key Laboratory of Intelligent Green Vehicle and Mobility, China
dBeijing Huairou Laboratory, Beijing, 101400, China
First published on 17th July 2025
Abstract
PtCo intermetallic alloy nanoparticles are highly active and stable catalysts for the oxygen reduction reaction (ORR), making them key materials for proton-exchange membrane fuel cells. However, the high-temperature annealing required for ordering into the intermetallic phase often leads to particle growth. In this work, we developed a machine learning interatomic potential to model the disorder-to-order transition in PtCo-based ternary alloys with high accuracy and computational efficiency. Monte Carlo simulations reveal that introducing a third element significantly affects both the ordering process and the critical temperature for the disorder-to-order transition. The thermodynamic driving forces for ordering in various PtCoM alloys were systematically investigated to identify potential high-performance PtCoM catalysts. Kinetic analysis further indicates that the accelerated ordering transition in PtCo alloys is primarily driven by lower migration energy barriers and enhanced directional diffusion. These findings provide valuable atomic-scale insights into the chemical ordering mechanisms and suggest a pathway for designing highly ordered PtCo-based nanoparticles for energy conversion and storage applications.
Introduction
Proton-exchange membrane fuel cells (PEMFCs) are promising clean energy technologies with significant potential in transportation and power generation applications.1–5 However, the sluggish kinetics of the oxygen reduction reaction (ORR) at the cathode significantly limit their efficiency, necessitating the use of platinum (Pt)-based catalysts for optimal performance.6–8 The high cost and scarcity of Pt remain key barriers to the large-scale adoption of PEMFCs.4 Alloy catalysts, which combine Pt with other metals such as Fe, Co, and Ni, present a feasible solution by enhancing catalytic activity through electronic and ligand effects while reducing Pt content.9–13 This approach is crucial for achieving high performance at a lower cost, which is essential for the broader commercialization of PEMFCs.Intermetallic alloys refer to crystalline structures in which the constituent elements are arranged in an ordered and uniform manner. It is widely reported in the literature that ordered intermetallic compounds exhibit superior activity and stability compared to their disordered counterparts.14–17 The enhanced activity and stability of intermetallic compounds are attributed to stronger electronic interactions between Pt and the doped metals, greater compressive strain, and lower formation enthalpy.15,18 Although ordered intermetallic compounds are thermodynamically more stable, the synthesis of multicomponent metallic nanocrystals through wet-chemical methods often results in disordered solid-solution alloys or phase separation due to the varying reduction potentials of the constituent metals.19 Therefore, annealing at elevated temperatures is commonly employed to facilitate the disorder-to-order transition. However, this process frequently causes catalyst sintering, leading to particle growth and a consequent reduction in the atomic utilization of noble metals.20,21
Several strategies have been explored to mitigate particle sintering during the disorder-to-order transition, including enhancing the interaction between nanoparticles and the support through sulfur atoms,15 forming protective shells around the particles using molecular additives,22,23 and elaborately controlling the heating and cooling rates.24 Additionally, introducing a third element, such as Ga, Cu, or Ni, into bimetallic alloys has proven effective in promoting the ordered transition of the alloy.25–28 Previous studies have primarily focused on representative ordered and disordered structures, identifying trends through key indicators such as formation enthalpy and migration energy barriers derived from first-principles calculations.24,26,29 The use of empirical potentials and global optimization algorithms has further expanded the scope of investigations, offering alternative approaches to studying the transition process.30–33 Despite these advancements, microscopic insights into the processes governing disorder-to-order transitions and the chemical ordering of ternary alloys remain limited. These challenges are further complicated by the wide range of alloy configurations and the substantial computational resources needed to achieve statistically significant results with first-principles accuracy.
Recent advances in machine learning potentials have introduced a novel approach for modeling interatomic interactions that combines the precision of first-principles calculations with the computational efficiency of empirical potentials.34,35 In this work, we developed a machine learning interatomic potential using transfer learning, based on a pre-trained large atomic model.36 This machine learning potential enables efficient Monte Carlo simulations of the PtCoM disorder-to-order transition process with first-principles accuracy, providing atomic-scale insights into the ordering process. Our work not only advances the understanding of the mechanisms driving chemical ordering but also provides a scalable framework for investigating other alloy systems, facilitating the rational design and optimization of advanced materials.
Results and discussion
Development of machine learning potential
The machine learning potential were trained by fine-tuning the Deep Potential model with a gated attention mechanism (DPA-1), as illustrated in Fig. 1a.36 The dataset for fine tuning includes alloy structures (PtCo and PtCoM) with various degrees of chemical ordering (Fig. S1–S3 and Table S1†). The final dataset contains a total of 36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 331 frames from the optimization calculations, which were divided into a training set and a validation set in a 9:1 ratio. The convergence process of the model's accuracy is shown in Fig. 1b. A total of 5 million training steps were performed. After these steps, the errors for both the training and validation sets became nearly identical, indicating that no overfitting occurred. Additionally, the model's accuracy plateaued, suggesting that 5 million training steps were sufficient. We evaluated different training strategies using the same training data and the same number of steps but different parameter fine-tuning schemes. The results demonstrate that directly applying the pre-trained DPA-1 model without any modification leads to substantial prediction errors (Fig. 1c). In contrast, fine-tuning all parameters of DPA-1 yields the highest accuracy, outperforming models trained from scratch (Fig. S4†). This performance gain can be attributed to the prior knowledge encoded in the pre-trained model, which provides a better initialization and facilitates more efficient parameter optimization compared to training from scratch. These findings underscore the efficiency and effectiveness of the pre-training and fine-tuning paradigm in developing accurate machine learning potentials.
331 frames from the optimization calculations, which were divided into a training set and a validation set in a 9:1 ratio. The convergence process of the model's accuracy is shown in Fig. 1b. A total of 5 million training steps were performed. After these steps, the errors for both the training and validation sets became nearly identical, indicating that no overfitting occurred. Additionally, the model's accuracy plateaued, suggesting that 5 million training steps were sufficient. We evaluated different training strategies using the same training data and the same number of steps but different parameter fine-tuning schemes. The results demonstrate that directly applying the pre-trained DPA-1 model without any modification leads to substantial prediction errors (Fig. 1c). In contrast, fine-tuning all parameters of DPA-1 yields the highest accuracy, outperforming models trained from scratch (Fig. S4†). This performance gain can be attributed to the prior knowledge encoded in the pre-trained model, which provides a better initialization and facilitates more efficient parameter optimization compared to training from scratch. These findings underscore the efficiency and effectiveness of the pre-training and fine-tuning paradigm in developing accurate machine learning potentials.
 | ||
| Fig. 1 Development and accuracy of the machine learning interatomic potential. (a) Schematic of the workflow for developing the machine learning potential for PtCoM. (b) Convergence curve of the fine-tuning process. (c) Comparison of accuracy for different machine learning potential models: ‘Pure DPA1’ refers to the DPA1 model whose parameters are not adjusted; ‘From scratch’ refers to training a new model with randomly initialized parameters, without inheriting any from the pre-trained DPA1 model; ‘Finetuned’ refers to training a new model that inherits its initial parameters from the pre-trained DPA1 model. (d) Energy error and (e) force error of the fine-tuned machine learning potential on the validation test. | ||
Through pre-training and fine-tuning, a machine learning potential was developed, achieving an energy error of 0.0022 eV per atom and a force error of 0.04 eV Å−1 (Fig. 1d and e). We are confident that the model is sufficiently accurate to replace first-principles calculations, as its energy error surpasses those reported in existing literature for machine learning models applied to alloy systems.37–40 Additionally, its force error is comparable to the force convergence criterion of 0.03 eV Å−1 commonly used in DFT structural optimizations. We also performed additional benchmarking against several representative interatomic modeling approaches for PtCoM ternary alloys. Specifically, we compared the energy prediction accuracy of the fine-tuned DPA-1 model with that of a fine-tuned MACE model, as well as more traditional approaches including cluster expansion and descriptor-based regression using Gaussian process regression. As summarized in Table S2,† the fine-tuned DPA-1 model achieves a lower energy MAE (0.0022 eV per atom) compared to the MACE model (0.0029 eV per atom), and outperforms both the cluster expansion (0.0035–0.0082 eV per atom) and the descriptor-based GPR model (0.0036–0.0058 eV per atom), which were trained on representative PtCoCu and PtCoNi subsets. These results highlight the accuracy and transferability of the fine-tuned DPA-1 model across a broader compositional space and support its use in our study.
Thermodynamics of disorder-to-order transition of PtCoM
Using the machine learning potential, we conducted virtual experiments via Monte Carlo simulations to investigate the disorder-to-order transition process of PtCoM at different temperatures. To evaluate the temperature dependence, we adopted a simplified thermodynamic model, considering the high computational cost of rigorous phase space sampling methods. Specifically, the free energy change ΔG was approximated as ΔG = ΔH − TΔSconfig, where the enthalpy change ΔH was assumed to be temperature-independent, and the entropy contribution was limited to the configurational entropy ΔSconfig. This approximation neglects vibrational and electronic entropy contributions, which are computationally demanding to compute via first-principles methods. Although more accurate evaluations could be achieved using molecular dynamics or metadynamics, these techniques remain impractical for high-throughput alloy screening due to their substantial resource requirements. Despite these simplifications, our approach captures the dominant thermodynamic driving forces and aligns with widely adopted methodologies in prior studies,24,41 offering a reasonable approximation for analyzing disorder-to-order transition trends in PtCoM systems. The enthalpy was calculated using the trained machine learning potentials, and the configurational entropy was determined based on chemical ordering using the method proposed by Yang.42 The initial atomic occupations in PtCoM were randomly assigned; after that, Monte Carlo simulations were performed to iteratively swap the positions of two atoms, allowing the system to evolve toward the configuration with the lowest free energy. The Warren–Cowley short-range order (SRO) was used to quantify the ordering degree, which is defined as: | (1) |
We monitored the evolution of chemical ordering during the disorder-to-order transition process of Pt2CoCu at 600 K (Fig. 2a). Initially, Pt and non-Pt atoms, as well as Co and Cu atoms, are randomly distributed. As the simulation progresses, Pt and non-Pt atoms arrange in an increasingly ordered manner, while Co and Cu atoms tend to segregate. This phenomenon can be attributed to the differing interactions between various atomic pairs. Notably, this behavior provides insights into multi-component intermetallic compounds, where certain element pairs still exhibit local disorder despite the overall trend towards ordering. Machine-learning-assisted Monte Carlo simulations provide atomic-level insights into chemical ordering since local short-range order is particularly challenging to observe directly in experiments—especially for elements like Co and Cu, which have similar physicochemical properties.43
 | ||
| Fig. 2 Thermodynamic insights into the disorder-to-order transition of PtCoM. (a) The 3D distribution of αPt–CoCu and αCo–Cu of PtCoCu at the beginning and after 400 steps, 800 steps, and 1200 steps of the Monte Carlo simulation at 600 K. (b) The final chemical ordering αPt–CoM of PtCo, PtCoNi, PtCoCu and PtCoZn as a function of simulated temperature in Monte Carlo simulation. A critical temperature that separates the map into two distinct regimes: fully ordered regime and not fully ordered regime. (c) The final chemical ordering αPt–CoM of PtCoCr, PtCoMn and PtCoFe as a function of simulated temperature in Monte Carlo simulations. (d) Schematic diagram of the relationship between Gibbs free energy change ΔG, enthalpy change ΔH, and temperature T during the disorder-to-order transition. | ||
Monte Carlo simulations were also performed at various temperatures for different Pt2CoM systems, and the degree of ordering corresponding to the configuration with the lowest free energy is shown in Fig. 2b and c. These simulations indicate that for PtCo, Pt2CoNi, Pt2CoCu and Pt2CoZn, there is a critical transition temperature. Below this temperature, complete ordering is attainable for Pt and Co/M (αPt–CoM = −1/3), but above it, such ordering is not feasible (αPt–CoM > −1/3). This critical temperature, which aligns with findings from other studies, is influenced by two primary factors.30,31,44 Firstly, at elevated temperatures, the increased randomness in the Monte Carlo simulation trajectories hampers convergence to the global minimum energy state. Secondly, based on the free energy relation ΔG = ΔH − TΔSconfig, the enthalpy change ΔH during the ordering process is assumed to be temperature-independent. The configurational entropy change ΔSconfig is negative during the disorder-to-order transition, as disordered states have higher entropy than ordered ones. Consequently, the entropic term −TΔSconfig is positive and increases with temperature, which raises the free energy ΔG and destabilizes the ordered phase at higher temperatures. For Pt2CoCr, Pt2CoMn and Pt2CoFe, complete ordering is not achieved at any temperature. As shown in Fig. 2d, theoretically, a higher critical transition temperature  is generally associated with a larger thermodynamic driving force for ordering – namely, a more negative enthalpy change ΔH, which facilitates the disorder-to-order transition. The enhanced ordering observed upon the addition of Ni, Cu, and Zn can thus be attributed to their ability to increase this thermodynamic driving force relative to PtCo. In contrast, the incorporation of Cr, Mn, and Fe reduces the driving force, thereby suppressing the ordering tendency.
is generally associated with a larger thermodynamic driving force for ordering – namely, a more negative enthalpy change ΔH, which facilitates the disorder-to-order transition. The enhanced ordering observed upon the addition of Ni, Cu, and Zn can thus be attributed to their ability to increase this thermodynamic driving force relative to PtCo. In contrast, the incorporation of Cr, Mn, and Fe reduces the driving force, thereby suppressing the ordering tendency.
Element screening
In addition to the six elements discussed above, we conducted a systematic investigation on the effects of various other elements on the ordering process of PtCo, covering most transition metals from the fourth, fifth, and sixth periods. Lanthanide elements and technetium (Tc) were not included in this study. Tc is extremely scarce in the Earth's crust, and accurately modeling it using DFT presents significant challenges.45 In addition, Sc, Y, Zr, and Hf were also excluded, as DFT optimizations for the corresponding Pt2CoM structures failed to converge. Additionally, a general trend between the critical transition temperature and the thermodynamic driving force for ordering suggests that Eordering can serve as a qualitative indicator for screening alloying elements. A higher Eordering than that of PtCo indicates that incorporation of element M thermodynamically promotes ordering between Pt and Co/M. Even in cases where a well-defined transition temperature is absent—such as for Cr, Mn, and Fe—the ordering energy remains a meaningful measure of chemical ordering tendencies. This Eordering based screening approach enables us to efficiently identify promising alloying elements without relying on time-consuming Monte Carlo simulations.A total of 180 structures with Pt and Co/M arranged in a random pattern (αPt–CoM = 0) were generated. Simultaneously, 180 structures with Pt and Co/M arranged in an ordered pattern (αPt–CoM = −1/3), featuring varying degrees of ordering between Co and M (αCo–M ranging from −3/27 to 25/27), were also created. Their energies were calculated and compared to that of PtCo. We tested the effect of sampling size on the results, as shown in Table S3,† and found that increasing the number of sampled structures beyond 180 has a negligible impact on the calculated energies. As shown in Fig. 3a, compared to the ordered Pt–Co structure, which has a single energy value, the energies of the ordered Pt–CoM structures exhibit a broader distribution due to the varying degree of Co–M ordering. This spread provides an additional thermodynamic driving force that promotes the disorder-to-order transition. As shown in Fig. 3a, for the binary PtCo alloy, the ordering energy Eordering is defined as the energy difference between the ordered structure with αPt–Co = −1/3 and the average energy of randomly generated configurations with αPt–Co = 0. For the ternary Pt2CoM alloy, the concept is extended due to the presence of a third element. The total ordering energy can be expressed as Etotal = Eordering + Eextra, where the first term accounts for the energy difference between the average energy of randomly generated Pt–Co/M disordered configurations (αPt–CoM = 0) and the average energy of randomly generated Pt–Co/M ordered structures (αPt–CoM = −1/3). The second term represents the additional stabilization associated with Co–M interactions, which is calculated as the energy difference between the average energy of Pt/CoM ordered structures, with Co/M ordering or disordering, and the average energy of the ordered structures (αPt–CoM = −1/3).
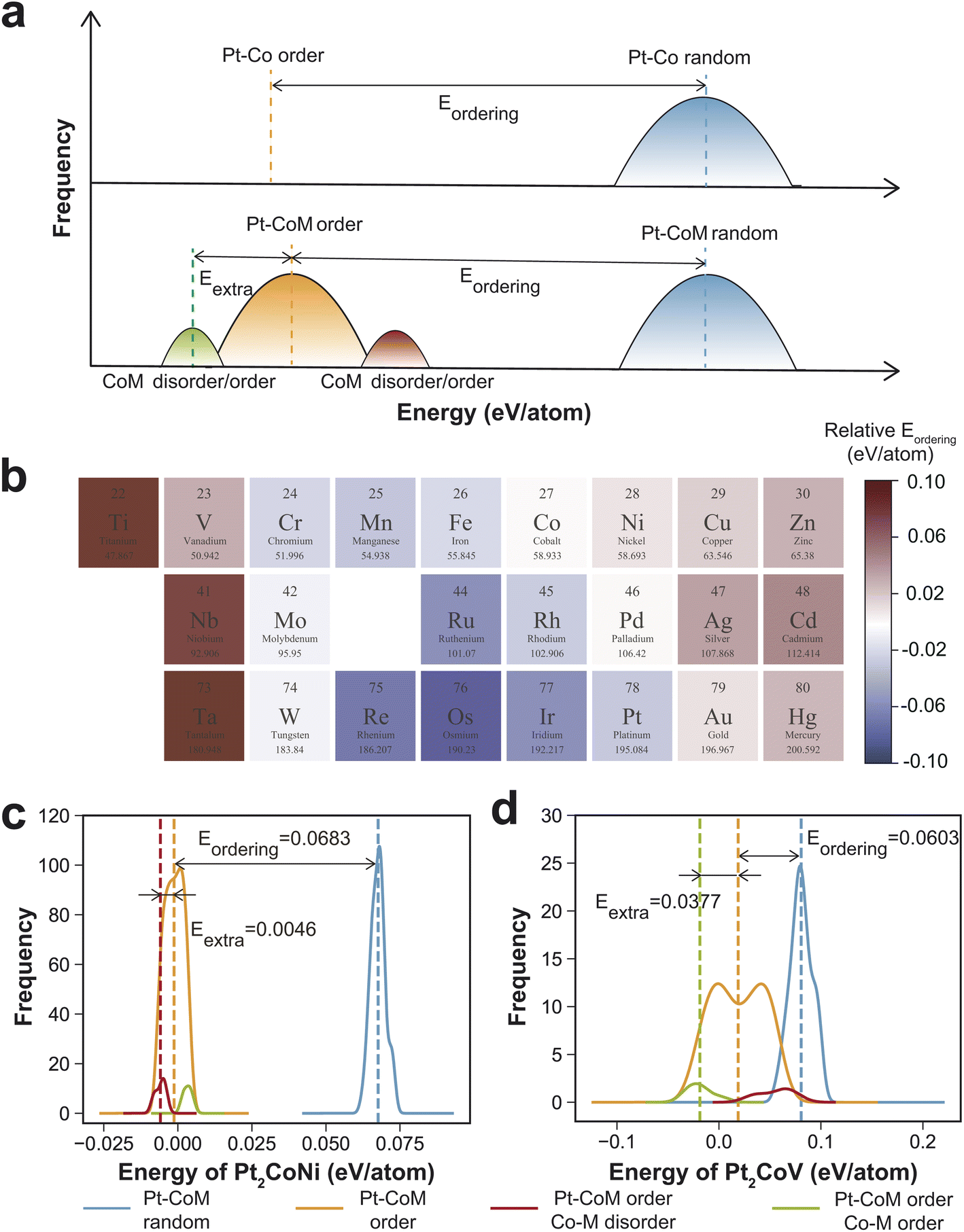 | ||
| Fig. 3 Element screening in Pt2CoM systems. (a) Schematic diagram of disorder-to-order driving energy Eordering for Pt2CoM and PtCo. (b) Comparative energy analysis of ordered (αPt–CoM = −1/3) and randomly arranged (αPt–CoM = 0) Pt and Co/M atom configurations, with energy values benchmarked to PtCo. (c) Disorder-to-order driving energy Eordering for Pt2CoNi. (d) Disorder-to-order driving energy Eordering for Pt2CoV. | ||
We first systematically investigated the case where Pt is ordered with Co/M, examining how the chemical ordering of Co and M affects the energy of PtCoM (Fig. S5 and S6†). As shown in Fig. S5,† for certain elements such as Ti, V, and Nb, the energy increases linearly with increasing αCo–M, indicating a thermodynamic preference for these elements to adopt an ordered arrangement with Co. In contrast, for elements such as Ni, Cu, and Zn, the energy decreases linearly with increasing αCo–M, suggesting a tendency toward phase separation from Co. For a few elements, such as Ru and Re, no significant linear correlation is observed between energy and αCo–M. Fig. S6† summarizes the additional thermodynamic driving force arising from either ordering (Fig. S6b†) or segregation (Fig. S6c†) between Co and M. In Fig. S6b,† elements highlighted in red exhibit lower energy when ordered with Co compared to the average energy across different Co–M orderings, indicating a preference for ordering. Conversely, in Fig. S6c,† red-highlighted elements show lower energy in a segregated configuration, suggesting a tendency to phase-separate from Co. Overall, early transition metals with fewer d-electrons tend to order with Co, while late transition metals with more d-electrons tend to segregate. This behavior can be attributed to the relative bond strengths among Co–Co, M–M, and Co–M. Specifically, if the sum of the Co–Co and M–M bond energies (ECo–Co + EM–M) is smaller than twice the Co–M bond energies (2ECo–M), an ordered arrangement is energetically favored. Otherwise, Co and M tend to segregate (Fig. S7†).
Fig. 3b presents the total ordering energy of Pt2CoM relative to PtCo. Elements highlighted in red indicate an enhancement in the thermodynamic driving force for ordering, while those in blue suggest a reduction. Among the investigated elements, doping with Ti, V, Cr, Ni, Cu, Zn, Nb, Ag, Cd, Ta, Au, and Hg was found to increase the thermodynamic driving force for ordering between Pt and non-Pt components, suggesting their potential as favorable alloying elements. Notably, ordered phases incorporating Ti, V, Ni, Cu, Zn, Ag, and Au have already been experimentally synthesized, as summarized in Table S4.† For example, Liao et al. synthesized PtCoTi ternary nanoparticles (∼3 nm) consisting of a Pt3Co0.6Ti0.4 intermetallic core and three atomic layers of Pt shells using a modified impregnation–reduction approach. This catalyst exhibits superior activity and durability compared with Pt3Co and Pt in rotating disk electrode (RDE) and single fuel cell tests.46 Zhong et al. demonstrated the synthesis of highly ordered PtCoV nanoalloys via oxidative–reductive thermochemical treatments. At elevated temperatures, enhanced atomic-scale chemical ordering and reduced interatomic distances significantly improve ORR activity, achieving up to 1 A mg−1 of Pt in RDE measurements.47 Shen et al. reported the synthesis of highly ordered PtCoZn nanoparticles (∼5.3 nm) through a controlled two-stage confinement strategy that enables both small particle size and high atomic ordering by resolving the conflict between atomic migration and spatial confinement. The resulting intermetallic PtCoZn catalyst exhibits exceptional ORR performance and durability in PEMFCs, attributed to enhanced Pt–Co coupling, high Pt surface exposure, and agglomeration prevention within mesoporous carbon walls.48
Considering that the average energy difference between the ordered and random phases of Pt2CoM relative to PtCo ranges from −0.11 to 0.03 eV per atom (Table S5†), the ordering or segregation of Co and M provides additional thermodynamic driving force, leading to a maximum energy decrease of 0.12 eV per atom for the ordered phase (Table S6†). This highlights the critical role of Co–M chemical ordering in facilitating the disorder-to-order transition of Pt and non-Pt elements. As illustrated in Fig. 3b and c, Co–Ni segregation contributes a thermodynamic driving force of 0.0046 eV per atom in Pt2CoNi, while Co–V ordering contributes a notably higher value of 0.0377 eV per atom in Pt2CoV.
Kinetics of disorder-to-order transition of PtCoCu
The previous section investigated chemical ordering in thermodynamic equilibrium using Monte Carlo simulations based on atomic exchanges. However, atomic exchanges are primarily a computational tool for identifying stable configurations and do not directly reflect physical atomic migration mechanisms. In reality, atomic movement occurs via vacancy-mediated diffusion, and the presence of vacancies plays a key role in facilitating the ordering process.49 To enhance the accuracy of migration barrier predictions, we fine-tuned the machine learning potential by incorporating a small dataset of transition-state configurations obtained from climbing-image nudged elastic band (CI-NEB) calculations of vacancy migration in PtCo. As shown in Fig. 4a, the fine-tuned model achieves a prediction error of just 0.07 eV for the vacancy migration energy barrier in PtCo.In FCC crystals, each atom has 12 nearest neighbors, giving rise to 12 possible vacancy migration pathways (Fig. 4b). By combining the fine-tuned machine learning potentials with the CI-NEB method, we calculated 5000 migration energy barriers for Pt, Co, and Cu atoms migrating to neighboring vacancies in Pt2CoCu and PtCo alloys, thereby investigating the kinetics of the ordering process. As shown in Fig. 4c, Cu atoms exhibit, on average, lower migration energy barriers compared to Pt and Co atoms in Pt2CoCu, suggesting that the incorporation of Cu can accelerate the disorder-to-order transition. Furthermore, the average migration barriers of Pt and Co atoms in Pt2CoCu (Pt: 1.07 eV; Co: 1.02 eV) are notably reduced compared to those in PtCo (Pt: 1.33 eV; Co: 1.17 eV), indicating that the presence of Cu not only enhances the mobility of Cu atoms themselves but also facilitates the diffusion of Pt and Co atoms, further promoting the ordering kinetics. To assess whether this kinetic promotion effect extends to other dopants, we performed additional vacancy-mediated diffusion barrier calculations for Pt2CoNi and Pt2CoZn. The results show that Ni and Zn also exhibit lower migration barriers compared to Pt and Co, with mean values of 0.97 eV for Ni and 0.68 eV for Zn, respectively. Importantly, the presence of these dopants similarly reduces the barriers for Pt and Co diffusion (Pt2CoNi:Pt/Co = 1.06/1.06 eV; Pt2CoZn:Pt/Co = 0.95/1.01 eV), indicating that the barrier-lowering effect is not unique to Cu but may be generalized to other dopants (Table S7†).
 | ||
| Fig. 4 Kinetic insights into the disorder-to-order transition of PtCoCu. (a) Comparison between the vacancy migration barrier of PtCo calculated using DFT and the machine learning potential. Distribution of atomic migration energy barriers for Pt, Co, and Cu. (b) Schematic representation of two extreme atomic migration modes. In one case, one of the twelve possible paths dominates with directional migration, R = 0; in the other case, all twelve paths have equal probability in a random migration scenario, R = 1. (c) Statistical distribution of machine learning predicted atomic migration energy barriers for Pt, Co, and M in PtCo, Pt2CoCu, Pt2CoNi and Pt2CoZn; the horizontal dashed line in the figure represents the average migration energy. (d) The distribution of R of PtCo and Pt2CoCu at different temperatures: 400 K, 800 K and 1800 K. (e) Migration multiplicity Var(R) as a function of temperature for PtCo and Pt2CoCu. | ||
The probability of vacancy migration along each path is given by:
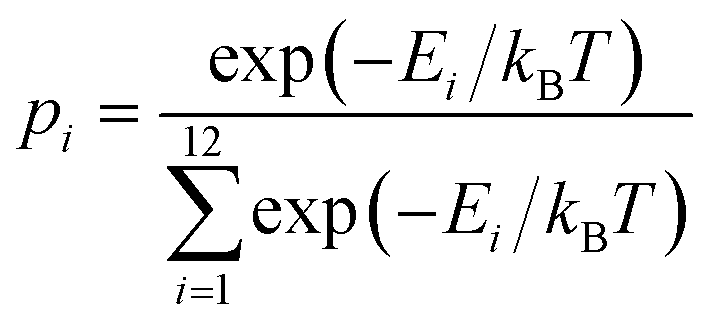 | (2) |
| R = 1 − σ(p)/max(σ(p)) | (3) |
Conclusions
In conclusion, by fine-tuning a pretrained large atomic model, we developed a machine learning potential model that accurately predicts the energy of PtCoM ternary alloys with various chemical orderings. This model serves as a powerful tool for uncovering thermodynamic and kinetic insights into the disorder-to-order transition in PtCoM alloys. Rather than building separate machine learning models for each PtCoM composition, as commonly done in earlier work, we employ a transferable, fine-tuned machine learning potential capable of handling a broad range of elements across the periodic table. This enhanced generality enables efficient screening not only of the 16 elements considered in previous studies but also of most transition metals in the 3rd, 4th, and 5th periods, thereby substantially expanding the discovery space for new alloy candidates.Thermodynamically, we find that introducing third elements such as Ni, Cu, and Zn increases both the thermodynamic driving force and the critical transition temperature for the disorder-to-order transformation compared to binary PtCo. Our screening results identify several promising PtCoM alloy candidates—PtCoTi, PtCoV, PtCoNi, PtCoCu, PtCoZn, PtCoNb, PtCoAg, PtCoCd, PtCoTa, PtCoAu, and PtCoHg—that exhibit a stronger thermodynamic driving force for ordering than PtCo. These findings highlight the important role of chemical ordering between the third element M and Co in enhancing the overall ordering tendency of PtCoM alloys.
Kinetically, migration barrier calculations reveal that Cu not only possesses a lower migration energy barrier compared to Pt and Co but also facilitates the migration of neighboring atoms. This enhanced atomic mobility promotes structural rearrangement and accelerates the ordering process during annealing. These insights not only deepen our understanding of the underlying atomic mechanisms but also provide valuable guidance for the design of high-performance, cost-effective multicomponent catalysts for energy conversion and storage applications.
Data availability
Data for this article, including the training dataset, the trained machine learning model, the code for Monte Carlo simulations, and the raw data for the figures are available in the GitHub repository at https://github.com/ZhangLabTHU/disorder-to-order.Author contributions
X. N.: data curation, investigation, methodology, visualization, writing – original draft. S. Z., R. Z., and J. L.: project discussion. L. Z.: conceptualization, supervision, methodology, writing – review & editing, project administration and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Program of Beijing Huairou Laboratory (ZD2022006A), the National Natural Science Foundation of China (22373055) the Tsinghua University Initiative Scientific Research Program and the Center of High Performance Computing.References
- M. K. Debe, Electrocatalyst approaches and challenges for automotive fuel cells, Nature, 2012, 486, 43–51 CrossRef CAS PubMed.
- K. Kodama, T. Nagai, A. Kuwaki, R. Jinnouchi and Y. Morimoto, Challenges in applying highly active Pt-based nanostructured catalysts for oxygen reduction reactions to fuel cell vehicles, Nat. Nanotechnol., 2021, 16, 140–147 CrossRef CAS PubMed.
- K. Jiao, J. Xuan, Q. Du, Z. Bao, B. Xie, B. Wang, Y. Zhao, L. Fan, H. Wang and Z. Hou, et al., Designing the next generation of proton-exchange membrane fuel cells, Nature, 2021, 595, 361–369 CrossRef CAS PubMed.
- Y. Wang, Y. Pang, H. Xu, A. Martinez and K. S. Chen, PEM fuel cell and electrolysis cell technologies and hydrogen infrastructure development–a review, Energy Environ. Sci., 2022, 15, 2288–2328 RSC.
- S. Ye, Y. Hou, X. Li, K. Jiao and Q. Du, Pore-scale investigation of coupled two-phase and reactive transport in the cathode electrode of proton exchange membrane fuel cells, Trans. Tianjin Univ., 2023, 29, 1–13 CrossRef CAS.
- X. Tian, X. F. Lu, B. Y. Xia and X. W. D. Lou, Advanced electrocatalysts for the oxygen reduction reaction in energy conversion technologies, Joule, 2020, 4, 45–68 CrossRef CAS.
- L. Huang, S. Zaman, X. Tian, Z. Wang, W. Fang and B. Y. Xia, Advanced platinum-based oxygen reduction electrocatalysts for fuel cells, Acc. Chem. Res., 2021, 54, 311–322 CrossRef CAS.
- S. Zaman, L. Huang, A. I. Douka, H. Yang, B. You and B. Y. Xia, Oxygen reduction electrocatalysts toward practical fuel cells: progress and perspectives, Angew. Chem., 2021, 133, 17976–17996 CrossRef.
- V. R. Stamenkovic, B. Fowler, B. S. Mun, G. Wang, P. N. Ross, C. A. Lucas and N. M. Markovic, Improved oxygen reduction activity on Pt3Ni (111) via increased surface site availability, Science, 2007, 315, 493–497 CrossRef CAS PubMed.
- Y. Bing, H. Liu, L. Zhang, D. Ghosh and J. Zhang, Nanostructured Pt-alloy electrocatalysts for PEM fuel cell oxygen reduction reaction, Chem. Soc. Rev., 2010, 39, 2184–2202 RSC.
- X. Huang, Z. Zhao, L. Cao, Y. Chen, E. Zhu, Z. Lin, M. Li, A. Yan, A. Zettl and Y. M. Wang, et al., High-performance transition metal-doped Pt3Ni octahedra for oxygen reduction reaction, Science, 2015, 348, 1230–1234 CrossRef CAS PubMed.
- M. Liu, Z. Zhao, X. Duan and Y. Huang, Nanoscale structure design for high-performance Pt-based ORR catalysts, Adv. Mater., 2019, 31, 1802234 CrossRef.
- X. Tian, X. Zhao, Y.-Q. Su, L. Wang, H. Wang, D. Dang, B. Chi, H. Liu, E. J. Hensen and X. W. Lou, et al., Engineering bunched Pt-Ni alloy nanocages for efficient oxygen reduction in practical fuel cells, Science, 2019, 366, 850–856 CrossRef CAS PubMed.
- M. Zhou, C. Li and J. Fang, Noble-metal based random alloy and intermetallic nanocrystals: syntheses and applications, Chem. Rev., 2020, 121, 736–795 CrossRef PubMed.
- C.-L. Yang, L.-N. Wang, P. Yin, J. Liu, M.-X. Chen, Q.-Q. Yan, Z.-S. Wang, S.-L. Xu, S.-Q. Chu and C. Cui, et al., Sulfur-anchoring synthesis of platinum intermetallic nanoparticle catalysts for fuel cells, Science, 2021, 374, 459–464 CrossRef CAS PubMed.
- T.-W. Song, M.-X. Chen, P. Yin, L. Tong, M. Zuo, S.-Q. Chu, P. Chen and H.-W. Liang, Intermetallic PtFe electrocatalysts for the oxygen reduction reaction: ordering degree-dependent performance, Small, 2022, 18, 2202916 CrossRef CAS.
- Y. Ma, J. Peng, J. Tian, W. Gao, J. Xu, F. Li, P. Tieu, H. Hu, Y. Wu and W. Chen, et al., Highly stable and active catalyst in fuel cells through surface atomic ordering, Sci. Adv., 2024, 10, eado4935 CrossRef CAS PubMed.
- Y. Ma, et al., Highly stable and active catalyst in fuel cells through surface atomic ordering, Sci. Adv., 2024, 10, eado4935 CrossRef CAS PubMed.
- J. Liang, F. Ma, S. Hwang, X. Wang, J. Sokolowski, Q. Li, G. Wu and D. Su, Atomic arrangement engineering of metallic nanocrystals for energy-conversion electrocatalysis, Joule, 2019, 3, 956–991 CrossRef CAS.
- F. Lin, M. Li, L. Zeng, M. Luo and S. Guo, Intermetallic nanocrystals for fuel-cells-based electrocatalysis, Chem. Rev., 2023, 123, 12507–12593 CrossRef CAS PubMed.
- J. Li and S. Sun, Intermetallic nanoparticles: synthetic control and their enhanced electrocatalysis, Acc. Chem. Res., 2019, 52, 2015–2025 CrossRef CAS PubMed.
- Z. Yang, H. Yang, L. Shang and T. Zhang, Ordered PtFeIr intermetallic nanowires prepared through a silica-protection strategy for the oxygen reduction reaction, Angew. Chem., 2022, 134, e202113278 CrossRef.
- T.-W. Song, C. Xu, Z.-T. Sheng, H.-K. Yan, L. Tong, J. Liu, W.-J. Zeng, L.-J. Zuo, P. Yin and M. Zuo, et al., Small molecule-assisted synthesis of carbon supported platinum intermetallic fuel cell catalysts, Nat. Commun., 2022, 13, 6521 CrossRef CAS PubMed.
- M. Cui, C. Yang, S. Hwang, M. Yang, S. Overa, Q. Dong, Y. Yao, A. H. Brozena, D. A. Cullen and M. Chi, et al., Multi-principal elemental intermetallic nanoparticles synthesized via a disorder-to-order transition, Sci. Adv., 2022, 8, eabm4322 CrossRef CAS PubMed.
- W. Zhang, F. Li, Y. Li, A. Song, K. Yang, D. Wu, W. Shang, Z. Yao, W. Gao and T. Deng, et al., The role of surface substitution in the atomic disorder-to-order phase transition in multi-component core–shell structures, Nat. Commun., 2024, 15, 9762 CrossRef CAS PubMed.
- R.-Y. Shao, X.-C. Xu, Z.-H. Zhou, W.-J. Zeng, T.-W. Song, P. Yin, A. Li, C.-S. Ma, L. Tong and Y. Kong, et al., Promoting ordering degree of intermetallic fuel cell catalysts by low-melting-point metal doping, Nat. Commun., 2023, 14, 5896 CrossRef CAS.
- W. Zhao, Z. Hu, M. Li and S. Hu, Asymmetrical Promotional Effect in the Disorder-to-Order Transition of Au-Pt-Cu Ternary Alloy Electrocatalysts, Adv. Funct. Mater., 2024, 2409481 CrossRef CAS.
- J. Liang, Y. Wan, H. Lv, X. Liu, F. Lv, S. Li, J. Xu, Z. Deng, J. Liu and S. Zhang, et al., Metal bond strength regulation enables large-scale synthesis of intermetallic nanocrystals for practical fuel cells, Nat. Mater., 2024, 1–9 Search PubMed.
- M. Chi, C. Wang, Y. Lei, G. Wang, D. Li, K. L. More, A. Lupini, L. F. Allard, N. M. Markovic and V. R. Stamenkovic, Surface faceting and elemental diffusion behaviour at atomic scale for alloy nanoparticles during in situ annealing, Nat. Commun., 2015, 6, 8925 CrossRef CAS PubMed.
- G. Rossi, R. Ferrando and C. Mottet, Structure and chemical ordering in CoPt nanoalloys, Faraday Discuss., 2008, 138, 193–210 RSC.
- D. Alloyeau, C. Ricolleau, C. Mottet, T. Oikawa, C. Langlois, Y. Le Bouar, N. Braidy and A. Loiseau, Size and shape effects on the order–disorder phase transition in CoPt nanoparticles, Nat. Mater., 2009, 8, 940–946 CrossRef CAS PubMed.
- J. M. Rahm and P. Erhart, Understanding chemical ordering in bimetallic nanoparticles from atomic-scale simulations: the competition between bulk, surface, and strain, J. Phys. Chem. C, 2018, 122, 28439–28445 CrossRef CAS.
- L. Li, X. Li, Z. Duan, R. J. Meyer, R. Carr, S. Raman, L. Koziol and G. Henkelman, Adaptive kinetic Monte Carlo simulations of surface segregation in PdAu nanoparticles, Nanoscale, 2019, 11, 10524–10535 RSC.
- H. Wang, L. Zhang, J. Han and E. Weinan, DeePMD-kit: a deep learning package for many-body potential energy representation and molecular dynamics, Comput. Phys. Commun., 2018, 228, 178–184 CrossRef CAS.
- Y.-L. Liao, B. Wood, A. Das and T. Smidt, Equiformerv2: improved equivariant transformer for scaling to higher-degree representations, in International Conference on Learning Representations (ICLR), 2024, https://openreview.net/forum?id=mCOBKZmrzD Search PubMed.
- D. Zhang, H. Bi, F.-Z. Dai, W. Jiang, X. Liu, L. Zhang and H. Wang, Pretraining of attention-based deep learning potential model for molecular simulation, npj Comput. Mater., 2024, 10, 94 CrossRef.
- P. Yin, X. Niu, S.-B. Li, K. Chen, X. Zhang, M. Zuo, L. Zhang and H.-W. Liang, Machine-learning-accelerated design of high-performance platinum intermetallic nanoparticle fuel cell catalysts, Nat. Commun., 2024, 15, 415 CrossRef CAS PubMed.
- Y. Yang, Z. Guo, A. J. Gellman and J. R. Kitchin, Simulating segregation in a ternary Cu–Pd–Au alloy with density functional theory, machine learning, and Monte Carlo simulations, J. Phys. Chem. C, 2022, 126, 1800–1808 CrossRef CAS.
- L. Chen, Y. Tian, X. Hu, S. Yao, Z. Lu, S. Chen, X. Zhang and Z. Zhou, A universal machine learning framework for electrocatalyst innovation: a case study of discovering alloys for hydrogen evolution reaction, Adv. Funct. Mater., 2022, 32, 2208418 CrossRef CAS.
- P. A. Santos-Florez, S.-C. Dai, Y. Yao, H. Yanxon, L. Li, Y.-J. Wang, Q. Zhu and X.-X. Yu, Short-range order and its impacts on the BCC MoNbTaW multi-principal element alloy by the machine-learning potential, Acta Mater., 2023, 255, 119041 CrossRef CAS.
- W. Qi, Y. Li, S. Xiong and S.-T. Lee, Modeling size and shape effects on the order–disorder phase-transition temperature of CoPt nanoparticles, Small, 2010, 6, 1996–1999 CrossRef CAS PubMed.
- Q. He, P. Tang, H. Chen, S. Lan, J. Wang, J. Luan, M. Du, Y. Liu, C. Liu and C. Pao, et al., Understanding chemical short-range ordering/demixing coupled with lattice distortion in solid solution high entropy alloys, Acta Mater., 2021, 216, 117140 CrossRef CAS.
- S. Moniri, Y. Yang, J. Ding, Y. Yuan, J. Zhou, L. Yang, F. Zhu, Y. Liao, Y. Yao and L. Hu, et al., Three-dimensional atomic structure and local chemical order of medium-and high-entropy nanoalloys, Nature, 2023, 624, 564–569 CrossRef CAS PubMed.
- L.-Y. Tian, H. Levämäki, O. Eriksson, K. Kokko, Á. Nagy, E. K. Délczeg-Czirják and L. Vitos, Density functional theory description of the order-disorder transformation in Fe-Ni, Sci. Rep., 2019, 9, 8172 CrossRef PubMed.
- L. Zhang, A. S. Raman and A. Vojvodic, Reviving inert oxides for electrochemical water splitting by subsurface engineering, Chem. Mater., 2020, 32, 5569–5578 CrossRef CAS.
- W. Zhao, B. Chi, L. Liang, P. Yang, W. Zhang, X. Ge, L. Wang, Z. Cui and S. Liao, Optimizing the electronic structure of ordered Pt–Co–Ti ternary intermetallic catalyst to boost acidic oxygen reduction, ACS Catal., 2022, 12, 7571–7578 CrossRef CAS.
- B. N. Wanjala, B. Fang, S. Shan, V. Petkov, P. Zhu, R. Loukrakpam, Y. Chen, J. Luo, J. Yin and L. Yang, et al., Design of ternary nanoalloy catalysts: effect of nanoscale alloying and structural perfection on electrocatalytic enhancement, Chem. Mater., 2012, 24, 4283–4293 CrossRef CAS.
- Z. Chen, J. Liu, B. Yang, M. Lin, C. Molochas, P. Tsiakaras and P. Shen, Two-stage confinement derived small-sized highly ordered L10-PtCoZn for effective oxygen reduction catalysis in PEM fuel cells, J. Colloid Interface Sci., 2023, 652, 388–404 CrossRef CAS PubMed.
- M. Cui, C. Yang, S. Hwang, B. Li, Q. Dong, M. Wu, H. Xie, X. Wang, G. Wang and L. Hu, Rapid atomic ordering transformation toward intermetallic nanoparticles, Nano Lett., 2021, 22, 255–262 CrossRef PubMed.
- B. Xing, T. J. Rupert, X. Pan and P. Cao, Neural network kinetics for exploring diffusion multiplicity and chemical ordering in compositionally complex materials, Nat. Commun., 2024, 15, 3879 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc04043d |
| This journal is © The Royal Society of Chemistry 2025 |