
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of monodisperse inorganic polyphosphate polyP10via a photocaging strategy†
Sandra
Moser
a,
Gloria
Hans
a,
Jiahui
Ma
a,
Thomas
Haas
a,
Nikolaus
Jork
a,
Felix
Bauer
a,
Bernhard
Breit
 a and
Henning J.
Jessen
*ab
a and
Henning J.
Jessen
*ab
aInstitute of Organic Chemistry, Albert-Ludwigs-Universität Freiburg, Albertstraße 21, 79104 Freiburg im Breisgau, Germany. E-mail: henning.jessen@oc.uni-freiburg.de
bCIBSS – Centre for Integrative Biological Signalling Studies, Albert-Ludwigs-Universität Freiburg, Schänzlestraße 18, 79104 Freiburg im Breisgau, Germany
First published on 10th July 2025
Abstract
Inorganic polyphosphate (polyP), a linear biopolymer composed only of orthophosphate units, has emerged as a molecule of critical biological importance across species. While commercially available polyPs are polydisperse mixtures – irrespective of their origin (chemical, biochemical) – recent strategies have focused on the bottom-up synthesis of monodisperse polyPs that have distinct advantages in mechanistic studies. However, until now, syntheses have been limited to defined chains of up to eight phosphate units due to challenges in deprotection-associated degradation and purification. Here, we disclose a new strategy based on two terminal coumarin photocages to synthesize the longest monodisperse polyP chain available to date: polyP10. The photoremovable protecting groups facilitate purification and enable efficient deprotection with light. By tuning the photocage, we achieve control over uncaging wavelengths, integrate targeting modifications and incorporate 18O-labels. This is the first example of a photouncaging strategy in which an 18O-labeled photocage is specifically designed to release an 18O-labeled metabolite for downstream applications. During the uncaging, we observe an unprecedented aromatic substitution reaction from a cleaved coumarin photocage cation onto the second photocage that is still attached to the polyP chain. This suggests a π-stacking facilitated loop-like arrangement of caged polyP in water that is supported by DFT calculations.
Introduction
Inorganic polyphosphate (polyP), a linear polymer composed of three up to thousands of orthophosphates, has evolved from its former status as a “forgotten polymer” 30 years ago1 to a molecule of critical biological and technological importance. Today it is known that this conserved biopolymer is involved in fundamental cellular processes2 such as energy metabolism,3 stress response4 and DNA damage repair.5 Additionally, it holds biomedical relevance6 as it plays a role in blood clotting,7 inflammation,8 bone regeneration9 and bacterial virulence.10 PolyP has been shown to covalently and non-covalently bind to certain protein domains.11 Commercially available polyP is manufactured through two main methods: chemical synthesis and enzymatic synthesis.In the chemical process, sodium monophosphate is heated to 700–1000 °C with subsequent rapid cooling.12 This technique yields a glass-like mixture of polyphosphates with different chain lengths,13 known as Graham's salt or – misguiding because of its linear structure – as sodium hexametaphosphate.14 By varying the temperature and vapor pressure, the average chain length can be adjusted.12 Moreover, different modifications have been obtained in the solid state, usually showing a helical arrangement of the polymer; based on these reports, such arrangements have also been suggested in solution.15
PolyP can also be synthesized enzymatically. Certain organisms, including yeast, bacteria and algae store high quantities of polyphosphates, which can be isolated through methods like phenol/chloroform extraction.12 Recently, an optimized extraction protocol has also become available for mammalian cells.16 While not yet commercially available, innovative biotechnological methods have demonstrated the potential to use Saccharomyces cerevisiae to convert phosphate-rich wastewater17 or de-oiled seeds and bran18 into sodium polyphosphate. These production methods have all in common, that they provide polyP samples with mixed chain lengths, making it hard to apply precise analytical techniques like mass spectrometry.
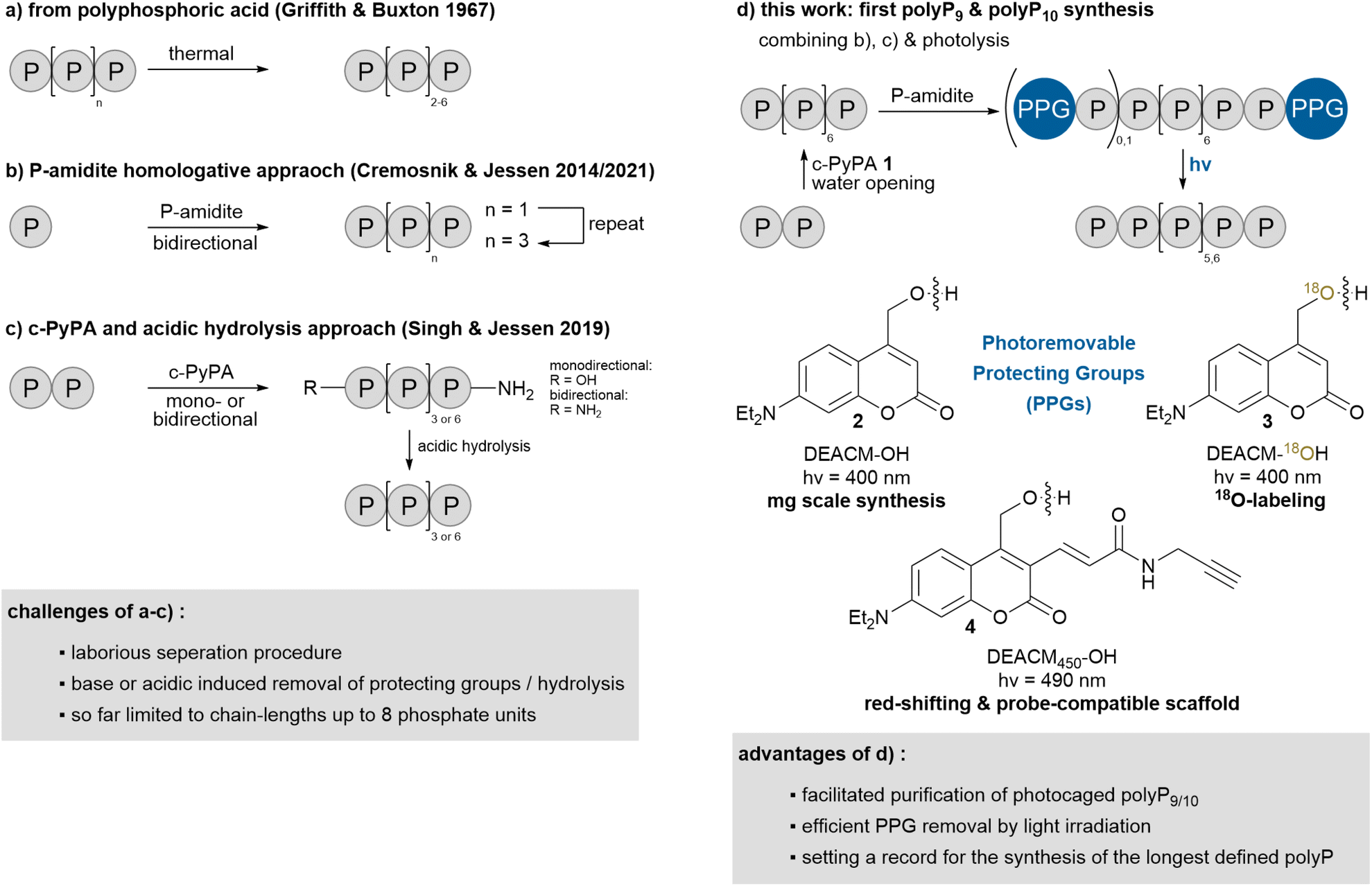
The isolation of polyPs with a defined chain-length is currently not possible with the methods described above, but recent advances have enabled the synthesis of short-chain polyPs up to eight phosphate units on mg to g scale (Scheme 1). Pure tetra- to octapolyphosphates are accessible by heating polyphosphoric acid, resulting in a polyP mixture with an average chain length of around five (Scheme 1a).19 Separation is achieved through multiple extraction steps combined with cation- and anion-exchange chromatography making this process highly labor-intensive.19 The bottom-up synthesis of defined polyPs is possible using a P-amidite homologative approach (Scheme 1b).20 This process builds on three steps: activation/coupling, oxidation and base-induced deprotection, which can be performed in a single flask, and can be repeated iteratively.20a,21 To enhance efficiency, an improved approach was developed using the triphosphorylation reagent cyclic pyrophosphoryl P-amidite 1 (c-PyPA, Scheme 1c and 2).20b,22
 | ||
| Scheme 1 Overview of chemical synthesis strategies for defined unmodified short-chain polyPs. | ||
 | ||
| Scheme 2 Syntheses of polyP86, polyP910 and polyP1011. (a) One-step synthesis of polyP86via the bidirectional c-PyPA approach with water-induced ring opening. (b) Attempted synthesis of polyP9 and polyP10 using the bidirectional P-amidite method with a standard P-amidite 7a followed by piperidine deprotection,20b yielding a non-separable polyP8, polyP9 and polyP10 mixture using SAX. (c) Successful synthesis of polyP910 and polyP1011via photolysis of their photocaged derivatives 8 and 9, which are separable by SAX, employing a photocaged P-amidite 7b. Abbreviations: TBA: tetrabutylammonium, ETT: 5-(ethylthio)-1H-tetrazole, mCPBA: meta-chloroperbenzoic acid, Fm: fluorenylmethyl, PPG: photoremovable protecting group, DBU: 1,8-diazabicyclo[5.4.0]undec-7-ene. | ||
This reagent enables the simultaneous incorporation of three phosphate units in a monodirectional approach or six in a bidirectional approach. The three-steps – activation/coupling, oxidation and linearization by nucleophiles – can be carried out in one pot as well. Amines are particularly effective for linearization. Starting from pyrophosphate, one then has direct access to symmetrical polyP8-diamidates, which can be hydrolyzed in acidic conditions to yield unmodified monodisperse polyP8.22a,23 While additional polyphosphorylation reagents exist for the synthesis of terminally modified oligophosphates,24 including nucleoside tetra- to heptaphosphates,25 dinucleoside tetra- and penta-phosphates24c,25a,26 as well as oligophosphorylated peptides,27 none of them have yet been used to synthesize unmodified polyP. To date, no monodisperse polyPs with chain lengths longer than eight units have been successfully synthesized. Achieving this would be an important step towards covering additional biological polyP structures, thus enabling more precise analysis28 and enhancing our understanding of their metabolic functions and topology. Defined longer chains will serve for precise analytical assignments and can help to understand and quantify polyP binding to proteins.
The synthetic limitation arises from the complexity of acquiring suitably long phosphate precursors and the increasing difficulty in purifying and isolating well-defined, elongated, non-UV active polyphosphates.29
Herein, we address these challenges by developing a novel strategy, which extends one-step-synthesized polyP8 to monodisperse and unmodified polyP9 and polyP10via photocaged polyP9 and polyP10 (Scheme 1d). The photoremovable protecting groups are crucial for their separation and enable efficient deprotection by light irradiation, avoiding decomposition observed with chemically triggered deprotection. Additionally, by tuning the photoremovable protecting groups, our approach allows for adjustment of the uncaging wavelength, the addition of clickable residues for probe development and the incorporation of 18O-labels to create heavy derivatives of polyP9 and polyP10 underlining the versatility of the approach. During the photouncaging experiments, we observed an unprecedented aromatic substitution on one photocage by the primary coumarin cation of another, indicating the formation of a loop-like structure in the polyP chain, likely stabilized by π-stacking interactions between the coumarin cages, aligning them in close proximity.
Results and discussion
Chemical synthesis of polyP9 and polyP10
PolyP86 was synthesized in a single step starting from pyrophosphate 5, using the bidirectional approach with c-PyPA 1 (Scheme 2a). Direct water-mediated ring opening to polyP86 has been low-yielding previously, while amine-induced linearization followed by acidic hydrolysis was more effective.22a The modified approach described herein provides direct access to unmodified polyP8 by quenching the reaction mixture into excess of water, resulting in clean linearization to polyP86.To extend this readily available polyP8 (235 mg synthesized in a single step) to polyP10, the bidirectional P-amidite approach can be used.20b However, this strategy presents two main challenges: first, the basic conditions required to remove the fluorenylmethyl (Fm) protecting groups from the newly introduced terminal phosphates can lead to partial degradation of the polyP chain. Second, the resulting mixture of polyP8, polyP9 and polyP10 is difficult to separate effectively by strong anion exchange chromatography (SAX) and hard to assign analytically (Scheme 2b). To overcome these limitations, a P-amidite bearing only one Fm group and a photoremovable protecting group (photocage) was employed enabling milder and orthogonal cleavage (Scheme 2c). The coumarin derivative DEACM-OH 2 was chosen due to its well-established photocleavage mechanism and its straightforward three-step synthesis.30 Reaction of the P-amidite 7b (2.5 eq.) with polyP86 followed by oxidation and careful Fm removal gave a mixture of mono-photocaged polyP98 (26% yield) and bis-photocaged polyP109 (19% yield) in a ratio of approx. 1.4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Increasing the amount of P-amidite 7b to 3 eq. shifted the product distribution strongly in favour of bis-photocaged polyP109 with a product ratio of 8 to 9 of approx. 0.1:1. The two products were readily separated by SAX. UV-activity of these compounds greatly facilitated purification. While their proton NMR chemical shifts are very similar, 31P-NMR provided a clear distinction: unsymmetrically mono-caged polyP98 shows an integration ratio of 1:1:7, whereas 9 shows a 2:8 pattern. This confirms the symmetrical dual-caged structure of the latter (ESI,† for a 31P-NMR chemical shift table for condensed phosphates see Accounts Chem. Res.20b). Irradiation of 8 and 9 at λ = 400 nm released defined polyP910 or polyP1011, respectively, without requiring acid or base treatment. The reaction proceeded cleanly; however, precipitation followed by extensive washing failed to completely remove the DEACM-OH 2 cleavage product, resulting in the isolation of a yellow-brown precipitate. Therefore, purification via SAX was required, which led to significant losses and ultimately reduced the yields to 38% for 10 and 26% for 11, respectively. Importantly, this new synthesis approach conceptually allows for further extension of the polyP chain by using polyP9 or polyP10 as starting materials.
:1. Increasing the amount of P-amidite 7b to 3 eq. shifted the product distribution strongly in favour of bis-photocaged polyP109 with a product ratio of 8 to 9 of approx. 0.1:1. The two products were readily separated by SAX. UV-activity of these compounds greatly facilitated purification. While their proton NMR chemical shifts are very similar, 31P-NMR provided a clear distinction: unsymmetrically mono-caged polyP98 shows an integration ratio of 1:1:7, whereas 9 shows a 2:8 pattern. This confirms the symmetrical dual-caged structure of the latter (ESI,† for a 31P-NMR chemical shift table for condensed phosphates see Accounts Chem. Res.20b). Irradiation of 8 and 9 at λ = 400 nm released defined polyP910 or polyP1011, respectively, without requiring acid or base treatment. The reaction proceeded cleanly; however, precipitation followed by extensive washing failed to completely remove the DEACM-OH 2 cleavage product, resulting in the isolation of a yellow-brown precipitate. Therefore, purification via SAX was required, which led to significant losses and ultimately reduced the yields to 38% for 10 and 26% for 11, respectively. Importantly, this new synthesis approach conceptually allows for further extension of the polyP chain by using polyP9 or polyP10 as starting materials.
An unexpected quasi-intramolecular reaction during the uncaging process
It is possible to track the uncaging process (Fig. 1a) in water by 31P-NMR. This requires high concentrations (approx. 10 mM) to detect the phosphate resonances of the termini. As expected, during polyP9 release from mono-caged polyP98, the ratio of the free phosphate signal to the caged phosphate signal gradually shifted from 1:1 to 2:0 over time (Fig. 1b and ESI-2a†). However, this process was slow, taking approximately 7 h to completion, as the cleaved DEACM-OH 2 chromophore competes for light absorption and is poorly water soluble, leading to precipitation and an opaque reaction mixture. A more practical approach was to conduct the reaction at lower concentration, such as 2 mM or less, and track its progress by capillary electrophoresis coupled to mass spectrometry31 (CE-MS, Fig. 1c and ESI-2b†). Dilution significantly shortens the reaction time to 90 min.
 | ||
| Fig. 1 (a) Photorelease of polyP910 from 8 at 400 nm. (b) At 10 mM in H2O, 31P-NMR monitoring indicated complete photorelease after approx. 7 h. (c) At a lower concentration of 2 mM in H2O, CE-MS monitoring (with sample dilution to 500 μM prior to analysis), showed completion of cleavage after approx. 90 min. Detailed time courses are available in Fig. ESI-2.† | ||
A similar behaviour as discussed above was expected for the bis-photocaged polyP109 (Scheme 3a). The reaction should proceed through the mono-photocaged polyP10 intermediate 12. However, interestingly, a new main distinct peak appeared after a very short irradiation time in the CE-MS profile in addition to the peaks for the starting material 9, mono-caged polyP1012 and the free polyP1011 both at 2 mM of 9 (Scheme 3b and Fig. ESI-3a†), as well as at 100 μM of 9 (Fig. ESI-3b†). This new peak represents a constitutional isomer with the exact same mass as the starting material 9. The new isomer could still be further cleaved under irradiation to yield polyP1011 albeit at a reduced rate. The reaction (2 mM) was complete after approximately 5 h of irradiation. Literature reports that coumarins can undergo reversible [2 + 2] cycloadditions30c,32 or decarboxylation30c,33 under UV light, the latter ruled out by the requirement of identical mass. Thus, the [2 + 2] cycloaddition would be a viable explanation. However, the expected cycloadduct would no longer function as a photocage, which contradicts our observations. Yet, regeneration of the cage and cleavage might be a result of a [2 + 2] cycloreversion. To identify the intermediate, it was generated by stopping the reaction after 1 h irradiation time, when it had accumulated next to polyP1011, followed by isolation via SAX. Full NMR characterization suggested the formation of the substitution product 13 (Scheme 3). The proposed mechanism, illustrated in Scheme 3c, is also supported by DFT calculations (Fig. 2, [BP86/def2SVP-D3BJ-SMD(water)]). It starts with the photolysis of bis-DEACM-caged polyP109. Coumarin photocages are believed to operate through the heterolysis of the DEACM–OP bond in the excited state. This generates a contact ion pair34 consisting of the primary DEACM cation 15 and its conjugated base, the anion of the leaving group 16.35 Unlike typical pathways where the cation would either quickly recombine with the anion or be intercepted by the solvent water after escape from the contact ion pair,3515 and 16 instead undergo an electrophilic aromatic substitution (ArSE). The reaction proceeds through Wheland complex 17, in which a phosphate group oxygen abstracts the aromatic proton (Fig. 2). For this quasi-intramolecular reaction to occur, the two DEACM residues must be in close proximity, as otherwise the primary cation generated during photoheterolysis rapidly reacts with water.34 This suggests that in the starting material 9, the polyP10 chain adopts a loop-like conformation, stabilized by π- stacking interactions between the DEACM-modifications in the polar solvent water. Since the newly formed intermediate 13 retains the coumarin photocage structure, prolonged irradiation leads to the release of unmodified polyP1011, albeit more slowly, potentially as a result of more efficient relaxation pathways. Indeed, HRMS analysis of the fully deprotected polyP10 reaction mixture – lyophilized after light irradiation and redissolved in DCM – revealed the presence of the final cleavage product 14 alongside DEACM–OH 2 (Fig. ESI-4†). Examples in which the contact ion pair of a coumarin-caged compound undergoes reaction pathways beyond simple recombination to the starting material or solvent trapping have been reported. These include an intramolecular cyclization rearrangement in styryl-substituted coumarins36 and deprotonation of a tertiary coumarin cation yielding an alkene rather than the expected alcohol.34 Photocleavage of a trimethylsilyl-substituted coumarin-based photocage also affords an alkene by either intramolecular silylcarbonylation, hydrolysis or Peterson-type desilylation after photoexcitation.37 Photo-Claisen rearrange-ments that impair release efficiency have also been observed in coumarin-caged tyrosine38 and 4-hydroxytamoxifen analogues.39 ArSE reactions have not been previously described.
 | ||
| Scheme 3 (a) Photorelease of polyP1011 from 9 (2 mM in H2O) at 400 nm. (b) CE-MS reaction monitoring (sample dilution to 500 μM prior to analysis) revealed complete photorelease within 5 h, proceeding through two intermediates, 12 and 13. (c) The formation of 13 is proposed to be a quasi-intramolecular ArSE reaction, occurring after the cleavage of one of the two photoremovable protecting groups from a loop-like pre-oriented structure (see Fig. 2). Evidence for the release of the byproducts 2 and 14 is provided in Fig. ESI-4.† | ||
 | ||
| Fig. 2 Calculated transition state of the proton abstraction (red bonds) in Wheland complex 17 supporting a loop-like structure of the polyP10 chain (P: orange, O: red). The counterions are Na+ (purple). [BP86/def2SVP-D3BJ-SMD(water)]. | ||
To further demonstrate that the light-induced heterolysis of the DEACM–OP bond is essential for side-product formation in our system, bis-DEACM–CH2–protected polyP1023 was synthesized (Scheme 4a) and irradiated at λ = 400 nm (Scheme 4b and Fig. ESI-5†). In this derivative, the DEACM carbon chain at position 4 is extended by one CH2 group, eliminating its uncaging pathways, while still potentially enabling the known [2 + 2] cycloaddition of coumarins. Electropherograms showed no additional peak with the same mass (Scheme 4b and Fig. ESI-5†), confirming that ArSE product formation did not occur under these conditions and requires primary cation generation in a contact ion pair. Extended irradiation (beyond 2 h) led to decomposition, resulting in a mixture of undefined products. The synthesis of DEACM–CH2–OH 21 was accomplished in three steps, starting from commercially available coumarin 18via enamine formation, hydrolysis and reduction (Scheme 4a). The protected polyP1023 exhibits an absorption maximum at 381 nm and two fluorescence maxima of 437 nm and 474 nm. The presence of multiple emission maxima, not only in 23, but also in the mono-DEACM-caged polyP98 and bis-DEACM-caged polyP109 (Table 1), may be explained by the existence of chromophore π-stacking interactions. This supports not only a loop-like arrangement of the polyP chain in bis-caged molecules, but highlights the potential for intermolecular interactions leading to supramolecular aggregation.40 Bis-DEACM–CH2 protected polyP1023 is the first example of a fluorophore end-labeled monodisperse double-digit polyP and may have versatile applications, such as fluorescence-based tracking of cellular uptake and direct fluorescent detection of polyphosphorylated proteins on gels without the need for staining methods.
 | ||
| Scheme 4 (a) Synthesis of bis-DEACM–CH2–protected polyP1023 as non-cleavable control. (b) CE-MS reaction monitoring shows no formation of an isobaric product. Abbreviations: DMF-DMA: N,N-dimethylformamide dimethyl acetal, DMF: dimethylformamide, pTsOH: p-toluenesulfonic acid, THF: tetrahydrofuran, Fm: fluorenylmethyl, ETT: 5-(ethylthio)-1H-tetrazole, DCM: dichloromethane, mCPBA: meta-chloroperbenzoic acid, DBU: 1,8-diazabicyclo[5.4.0]undec-7-ene. | ||
| polyP | Absorption λmaxa/nm | ε max /(M−1 cm−1) | Emission λmaxc/nm |
|---|---|---|---|
| a Wavelength of the absorption maximum, 50 μM. b Molar extinction coefficient at the absorption maximum λmax. c Wavelength of the emission maxima upon excitation at the absorption maximum, 100 nM. | |||
| DEACM-polyP98 | 386 | 16340 |
445, 491, 527 |
| 18O-DEACM-polyP934 | 386 | 16220 |
445, 491, 527 |
| Bis-DEACM-polyP109 | 382 | 20280 |
440, 490, 527 |
| Bis-18 O-DEACM-polyP1035 | 382 | 26440 |
440, 490, 527 |
| Bis-DEACM–CH2–polyP1023 | 381 | 21060 |
437, 474 |
| Bis-DEAC450-polyP1024 | 438 | 28474 |
523, 543 |
| Bis-TPP-DEAC450-polyP1027 | 441 | 16975 |
520, 542 |
Synthesis of clickable, red-shifted photocaged polyP10
Bis-DEACM-photocaged polyP109 has an absorption maximum at 382 nm (Table 1) and can be cleaved with a 400 nm LED. However, for specific applications, such as cellular studies, red-shifted activation is preferred to high-energy UV light. Visible light has better tissue penetration and has reduced photo-toxicity to cells.41 By using different photocages, our synthetic approach allows for easy tuning of these parameters. Moreover, targeting moieties and modifications that enhance cellular uptake could be installed.42 Intracellular delivery of polyP into cells has previously been achieved using polycationic molecular transporters by noncovalent polyplex formation.43 Choosing clickable42 DEAC450–OH444 instead of DEACM-OH 2 as photocage on the P-amidite, a photocaged polyP1024 with an absorption maximum around 438 nm was obtained (Fig. 3a and Table 1). Additionally, it features a clickable handle for further probe development, such as organelle-specific targeting.45 In-terestingly, during the uncaging process with 490 nm light, the direct photorelease again competed with an ArSE reaction as identified by CE-MS, LC-MS and NMR (Fig. 3b and ESI-7†). | ||
| Fig. 3 Synthesis and uncaging of red-shifted and modified caged polyP10's. (a) Synthesis of bis-DEAC450 caged polyP1024. A: 1. (iPr)2N–P(OFm)(ODEAC450) (3.0 eq.), ETT (20 eq.), MeCN/DCM, r.t., 30 min. 2. mCPBA (3.0 eq.), 0 °C, 20 min. 3. DBU (5 vol%), 0 °C → r.t., 1 h. (b) Photorelease of polyP1011 from 24 (100 μM in H2O) at 490 nm. CE-MS reaction monitoring demonstrated that the photorelease proceeds through two intermediates, 25 and 26. The formation of 25 follows the mechanism suggested in Scheme 3. The photorelease process was slowed down through 25 and incomplete even after 8 h of irradiation. (c) Synthesis of bis-TPP-DEAC450 caged polyP1027. B: (4-azidobutyl) triphenyl phosphonium bromide (2.0 eq.), CuSO4·5H2O (1.0 eq.), THPTA (5.0 eq.), sodium ascorbate (10 eq.), 100 mM TEAA/DMSO, r.t., 3 h. (d) Photorelease of polyP1011 from 27 (100 μM in H2O) at 490 nm. CE-MS reaction monitoring revealed complete photorelease within 20 min via mono-caged polyP1029. The isobaric intermediate 28, formed from 27, was not isolated and identified. Abbreviations: Fm: fluorenylmethyl, ETT: 5-(ethylthio)-1H-tetrazole, DCM: dichloromethane, DMSO: dimethyl sulfoxide, mCPBA: meta-chloroperbenzoic acid, DBU: 1,8-diazabicyclo[5.4.0]undec-7-ene, TPP: triphenyl-phosphonium, THPTA: tris[(1-hydroxy-propyl-1H-1,2,3-triazol-4-yl)methyl]amine, TEAA: triethylammonium acetate. | ||
It likely follows the mechanism and required loop pre-arrangement proposed in Scheme 3c. While the bis-DEACM-polyP10 ArSE intermediate 13 fully released free polyP1011 (see Fig. ESI-3b†) within 60 min at a concentration of 100 μM, the formation of the ArSE intermediate of bis-DEAC450-polyP1025 significantly slowed down the polyP1011 release, preventing complete polyP10 liberation even after 8 h irradiation at 100 μM (Fig. 3b and ESI-6†). This may in part be attributed to the lower output power of the 490 nm LED (140 (mW)3) compared to the 400 nm LED (265 (mW)3), yet we surmise that additional rotational degrees of freedom for excited state inactivation and potentially energy transfer between the two chromophores followed by dissipation are also playing a role here. Notwithstanding, the reaction proceeded cleanly (see Fig. 3b), but at a much slower rate.
As an example for an organelle-targeting modification, we clicked46 the mitochondria-targeting group triphenyl-phosphonium (TPP+) to the bis-DEAC450-polyP1024 to obtain 27 (Fig. 3c). PolyP has been proposed to be produced by the mitochondrial F0F1-ATP synthase in mammalian cells,47 and consequently its subcellular targeting for biological studies would be beneficial. However, the slow-release kinetics described above would be a major obstacle for further tool development. Even so, steric hindrance in the TPP+ modified caged polyP in combination with coulombic repulsion of positive charges might reduce or obliterate the ArSE side reaction. Indeed, a 100 μM solution of 27 exposed to 490 nm light fully released unmodified polyP1011 within only 20 min (Fig. 3d and ESI-8†). The isobaric intermediate 28, formed from 27, did not significantly affect the photorelease kinetics and was not further characterized. These results indicate that 27 is, in principle, suitable for uncaging in living cells.
Synthesis of 18O-labeled polyP9 and polyP10
We have recently demonstrated the use of 18O-labeled phosphorylated metabolites as internal standards for quantitative CE-MS analysis.48 Among available isotope labeling strategies, 18O-labeling represents the only suitable approach for polyP, as oxygen is the only element with stable isotopes present in polyP. While Haas et al. achieved 18O-labeling of polyP4 using a base-labile P-amidite,48 our synthesis method now enables the straightforward incorporation of 18O-labels into the terminal phosphates of polyP9/10 (and by extension beyond) by using 18O-labeled DEACM-OH 3 as protecting group (Scheme 5). This heavy photocage was synthesized via Mitsunobo esterification of DEACM-OH 2 with 18O-labeled 4-nitrobenzoic acid 31, followed by hydrolysis based on a 18O- labeling strategy for alcohols from Beddoe et al.49 The corresponding photocaged P-amidite 33 was synthesized according to standard procedures.50 Coupling 2.5 eq. of P-amidite 33 to polyP86 yielded a mixture of mono-18 O-DEACM-polyP934 (20% yield) and bis-18 O-DEACM-polyP1035 (39% yield) which were well separable by SAX. Deprotection of 34 and 35 with 400 nm at 2 mM for 2 or 8 h, respectively, yielded 18O-labeled polyP936 with 97% 18O-isotope enrichment (5.8% natural abundance) or 18O-labeled polyP1037 with 95:5 (18O2:18O) isotope ratio (6.4% natural abundance). Importantly, these are the first examples of an 18O-labeled photocage designed specifically to release 18O-labeled metabolites for downstream use, unlike previous strategies, where the label is incorporated during uncaging via18O-enriched water,35,51 pre-installed in the biomolecule52 or where the focus lies solely on the uncaging mechanism itself.53 This 18O-labeling approach can be adapted for the synthesis of diverse 18O-labeled phosphorylated metabolites for use in biology, medicine and environmental science.
 | ||
| Scheme 5 Synthesis of 18O-labeled polyP936 and 18O-labeled polyP1037. Abbreviations: DIAD: diisopropyl azodicarboxylate, THF: tetrahydrofuran, Fm: fluorenylmethyl, ETT: 5-(ethylthio)-1H-tetrazole, DCM: dichloromethane, mCPBA: meta-chloroperbenzoic acid, DBU: 1,8-diazabicyclo[5.4.0]undec-7-ene. | ||
Conclusions
This study discloses the synthesis of two monodisperse polyphosphates, polyP9 and polyP10. It follows a new strategy to access for the first time polyP in the two-digit range. By generating photocaged versions, an effective separation of the different chain lengths in solution becomes possible. The uncaging process selectively releases the pure polyPs with defined chain length. It can be tracked conveniently with CE-MS in an aqueous environment.During our study, we identified a novel photolysis side reaction involving quasi-intramolecular electrophilic aromatic substitution in bis-DEACM and bis-DEAC450 caged polyP10, slowing down photorelease. This discovery suggests a loop-like structure of the caged polyP10 starting materials, possibly stabilized through π-stacking in water. The unique deactivation mechanism via ArSE for coumarin type photocages from the contact ion pair state34 has not been described previously and is supported by DFT calculations. Importantly, the side-reaction can be reduced by introducing larger substituents on the photocage, such as TPP+.
Additionally, varying the photocage enables tailoring the uncaging wavelength and incorporating handles for further functional modifications. Both are paving the way for organelle-specific delivery of polyP10 that can be released by light irradiation within cells, a focus for our future studies. Furthermore, utilizing an 18O-labeled photocage allows for the introduction of 18O into polyP9 and polyP10, which can be applied as internal references in mass spectrometry. This strategy is readily extendable to the synthesis of other 18O-labeled phosphorylated metabolites to allow their identification and quantification in complex biological samples via quantitative CE-MS. Their light-controlled release in living cells offers a tool to study intracellular dynamic phosphate turnovers and perturb cellular polyP metabolism.54
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
S. Moser and H. Jessen designed the molecules. S. Moser and G. Hans synthesized most of the compounds. In addition, J. Ma, T. Haas, and N. Jork provided precursors. S. Moser characterized the compounds. F. Bauer performed DFT calculations. S. Moser drafted the initial manuscript and prepared the schemes and figures. B. Breit and H. Jessen reviewed the manuscript draft. H. Jessen conceived the project and provided feedback.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank G. Liu, M. Lu and I. Prucker, A. Shukla from the Jessen group for help with CE and HRMS measurements. We also would like to thank Dr S. Braukmüller, Dr M. Keller and C. Warth from the Analytical Service Team of the University of Freiburg for NMR and HRMS measurements, respectively. This project was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, project number 445698446) in collaboration with the Indian Department of Biotechnology (DBT). Moreover, this work was supported by the Deutsche Forschungsgemeinschaft (DFG) under Germany's Excellence Strategy (CIBSS-EXC-2189-Project ID 390939984, to HJJ).Notes and references
- A. Kornberg, J. Bacteriol., 1995, 177, 491–496 CrossRef CAS.
- L. Xie and U. Jakob, J. Biol. Chem., 2019, 294, 2180–2190 CrossRef CAS.
- W. E. G. Müller, S. Wang, M. Neufurth, M. Kokkinopoulou, Q. Feng, H. C. Schröder and X. Wang, J. Cell Sci., 2017, 130, 2747–2756 CrossRef.
- M. J. Gray and U. Jakob, Curr. Opin. Microbiol., 2015, 24, 1–6 CrossRef CAS.
- S. Bru, B. Samper-Martín, E. Quandt, S. Hernández-Ortega, J. M. Martínez-Laínez, E. Garí, M. Rafel, J. Torres-Torronteras, R. Martí, M. P. C. Ribeiro, J. Jiménez and J. Clotet, DNA Repair, 2017, 57, 171–178 CrossRef CAS PubMed.
- X. Wang, H. C. Schröder and W. E. G. Müller, J. Mater. Chem. B, 2018, 6, 2385–2412 RSC.
- (a) S. A. Smith, N. J. Mutch, D. Baskar, P. Rohloff, R. Docampo and J. H. Morrissey, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 903–908 CrossRef CAS PubMed; (b) D. Kudela, S. A. Smith, A. May-Masnou, G. B. Braun, A. Pallaoro, C. K. Nguyen, T. T. Chuong, S. Nownes, R. Allen, N. R. Parker, H. H. Rashidi, J. H. Morrissey and G. D. Stucky, Angew. Chem., Int. Ed., 2015, 54, 4018–4022 CrossRef CAS.
- S. A. Smith, S. H. Choi, J. N. R. Collins, R. J. Travers, B. C. Cooley and J. H. Morrissey, Blood, 2012, 120, 5103–5110 CrossRef CAS PubMed.
- S. Omelon, J. Georgiou, Z. J. Henneman, L. M. Wise, B. Sukhu, T. Hunt, C. Wynnyckyj, D. Holmyard, R. Bielecki and M. D. Grynpas, PLoS One, 2009, 4, e5634 CrossRef.
- M. H. Rashid, K. Rumbaugh, L. Passador, D. G. Davies, A. N. Hamood, B. H. Iglewski and A. Kornberg, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 9636–9641 CrossRef CAS.
- (a) C. Azevedo, J. Singh, N. Steck, A. Hofer, F. A. Ruiz, T. Singh, H. J. Jessen and A. Saiardi, ACS Chem. Biol., 2018, 13, 1958–1963 CrossRef CAS PubMed; (b) C. Azevedo, F. Borghi, X. B. Su and A. Saiardi, Mol. Cell, 2024, 84, 1811–1815 CrossRef CAS PubMed; (c) N. Neville, K. Lehotsky, K. A. Klupt, M. Downey and Z. Jia, Mol. Cell, 2024, 84, 1802–1810 CrossRef CAS PubMed; (d) A. Celik, F. Schöpf, C. E. Stieger, J. A. M. Morgan, S. Lampe, M. Ruwolt, F. Liu, C. P. R. Hackenberger, D. Roderer and D. Fiedler, bioRxiv, 2024 DOI:10.1101/2024.07.29.605581.
- B. Lorenzi and H. C. Schroder, in Inorganic Polyphosphates Biochemistry, Biology, Biotechnology, ed. H. C. Schröder and W. E. G. Müller, Mainz, Germany, 1st edn, 1999 Search PubMed.
- C. F. Callis, J. R. Van Wazer and P. G. Arvan, Chem. Rev., 1954, 54, 777–796 CrossRef CAS.
- (a) T. Graham, Philos. Trans. R. Soc. London, 1833, 123, 253–284 CrossRef; (b) J. F. McCullough, J. R. V. Wazer and E. J. Griffith, J. Am. Chem. Soc., 1956, 78, 4528–4533 CrossRef CAS.
- (a) L. E. Jackson, B. M. Kariuki, M. E. Smith, J. E. Barralet and A. J. Wright, Chem. Mater., 2005, 17, 4642–4646 CrossRef CAS; (b) K. Förg and H. A. Höppe, Dalton Trans., 2015, 44, 19163–19174 RSC; (c) R. Schoeppe, M. Waldmann, H. J. Jessen and T. Renné, Biomolecules, 2024, 14, 937 CrossRef CAS PubMed; (d) T. Glonek, Origins Life Evol. Biospheres, 2021, 51, 1–60 CrossRef.
- B. Lázaro, A. Sarrias, F. J. Tadeo, J. Marc Martínez-Láinez, A. Fernández, E. Quandt, B. Depares, T. Dürr-Mayer, H. Jessen, J. Jiménez, J. Clotet and S. Bru, Methods, 2025, 234, 211–222 CrossRef PubMed.
- J. Fees, J. J. Christ, S. Willbold and L. M. Blank, Biotechnol. Bioeng., 2023, 120, 456–464 CrossRef CAS PubMed.
- K. R. Herrmann, J. Fees, J. J. Christ, I. Hofmann, C. Block, D. Herzberg, S. Bröring, B. Reckels, C. Visscher, L. M. Blank, U. Schwaneberg and A. J. Ruff, EFB Bioeconomy J., 2023, 3, 100048 CrossRef CAS.
- E. J. Griffith and R. L. Buxton, J. Chem. Soc., 1967, 89, 2884–2890 CrossRef CAS.
- (a) G. S. Cremosnik, A. Hofer and H. J. Jessen, Angew. Chem., Int. Ed., 2014, 53, 286–289 CrossRef CAS; (b) H. J. Jessen, T. Dürr-Mayer, T. M. Haas, A. Ripp and C. C. Cummins, Acc. Chem. Res., 2021, 54, 4036–4050 CrossRef CAS PubMed.
- H. J. Jessen, N. Ahmed and A. Hofer, Org. Biomol. Chem., 2014, 12, 3526–3530 RSC.
- (a) J. Singh, N. Steck, D. De, A. Hofer, A. Ripp, I. Captain, M. Keller, P. A. Wender, R. Bhandari and H. J. Jessen, Angew. Chem., Int. Ed., 2019, 58, 3928–3933 CrossRef CAS; (b) J. Singh, A. Ripp, T. M. Haas, D. Qiu, M. Keller, P. A. Wender, J. S. Siegel, K. K. Baldridge and H. J. Jessen, J. Am. Chem. Soc., 2019, 141, 15013–15017 CrossRef CAS PubMed.
- S. M. Hacker, M. Mex and A. Marx, J. Org. Chem., 2012, 77, 10450–10454 CrossRef CAS.
- (a) J. Y. Liao, S. Bala, A. K. Ngor, E. J. Yik and J. C. Chaput, J. Am. Chem. Soc., 2019, 141, 13286–13289 CrossRef CAS PubMed; (b) S. M. Shepard and C. C. Cummins, J. Am. Chem. Soc., 2019, 141, 1852–1856 CrossRef CAS PubMed; (c) S. M. Shepard, H. Kim, Q. X. Bang, N. Alhokbany and C. C. Cummins, J. Am. Chem. Soc., 2021, 143, 463–470 CrossRef CAS PubMed.
- (a) S. Mohamady and S. D. Taylor, Org. Lett., 2013, 15, 2612–2615 CrossRef CAS; (b) G. Park, E. C. Wralstad, N. Faginas-Lago, K. Qian, R. T. Raines, G. Bistoni and C. C. Cummins, ACS Cent. Sci., 2024, 10, 1415–1422 CrossRef CAS PubMed.
- (a) Y. Ahmadibeni and K. Parang, Org. Lett., 2007, 9, 4483–4486 CrossRef CAS PubMed; (b) Q. Han, B. L. Gaffney and R. A. Jones, Org. Lett., 2006, 8, 2075–2077 CrossRef CAS.
- K. Qian, B. Hanf, C. Cummins and D. Fiedler, Angew. Chem., Int. Ed., 2025, e202419147 CAS.
- S. K. Roy, S. Moser, T. Dürr-Mayer, R. Hinkelmann and H. J. Jessen, Org. Biomol. Chem., 2025, 23, 1373–1379 RSC.
- S. M. Shepard, H. J. Jessen and C. C. Cummins, J. Am. Chem. Soc., 2022, 144, 7517–7530 CrossRef CAS PubMed.
- (a) V. Hagen, J. Bendig, S. Frings, T. Eckardt, S. Helm, D. Reuter and U. B. Kaupp, Angew. Chem., Int. Ed., 2001, 40, 1045–1048 CrossRef; (b) T. Weinrich, M. Gränz, C. Grünewald, T. F. Prisner and M. W. Göbel, Eur. J. Org. Chem., 2017, 2017, 491–496 CrossRef CAS; (c) L. J. G. W. Van Wilderen, C. Neumann, A. Rodrigues-Correia, D. Kern-Michler, N. Mielke, M. Reinfelds, A. Heckel and J. Bredenbeck, Phys. Chem. Chem. Phys., 2017, 19, 6487–6496 RSC; (d) C. Hamerla, C. Neumann, K. Falahati, J. Von Cosel, L. J. G. W. Van Wilderen, M. S. Niraghatam, D. Kern-Michler, N. Mielke, M. Reinfelds, A. Rodrigues-Correia, A. Heckel, J. Bredenbeck and I. Burghardt, Phys. Chem. Chem. Phys., 2020, 22, 13418–13430 RSC.
- D. Qiu, M. S. Wilson, V. B. Eisenbeis, R. K. Harmel, E. Riemer, T. M. Haas, C. Wittwer, N. Jork, C. Gu, S. B. Shears, G. Schaaf, B. Kammerer, D. Fiedler, A. Saiardi and H. J. Jessen, Nat. Commun., 2020, 11, 6035 CrossRef CAS PubMed.
- (a) G. Ciamician and P. Silber, Ber. Dtsch. Chem. Ges., 1902, 35, 3849–4571 CrossRef; (b) T. Wolff and H. Görner, Phys. Chem. Chem. Phys., 2004, 6, 368–376 RSC; (c) S. Inacker, J. Fanelli, S. I. Ivlev and N. A. Hampp, Macromolecules, 2022, 55, 8461–8471 CrossRef CAS.
- N. Kus, S. Breda, I. Reva, E. Tasal, C. Ogretir and R. Fausto, Photochem. Photobiol., 2007, 83, 1237–1253 CrossRef CAS PubMed.
- A. M. Schulte, G. Alachouzos, W. Szymanski and B. L. Feringa, Chem. Sci., 2024, 15, 2062–2073 RSC.
- B. Schade, V. Hagen, R. Schmidt, R. Herbrich, E. Krause, T. Eckardt and J. Bendig, J. Org. Chem., 1999, 64, 9109–9117 CrossRef CAS.
- Q. Lin, L. Yang, Z. Wang, Y. Hua, D. Zhang, B. Bao, C. Bao, X. Gong and L. Zhu, Angew. Chem., Int. Ed., 2018, 57, 3722–3726 CrossRef CAS PubMed.
- M. Yoshimura, R. Sasayama, T. Kajiwara, C. Mori, Y. Nakasone and T. Inose, Angew. Chem., Int. Ed., 2025, e202502376 CAS.
- C. Maller, E. Marouda and M. Köhn, ChemBioChem, 2024, 25, e202400561 CrossRef CAS PubMed.
- P. T. Wong, E. W. Roberts, S. Tang, J. Mukherjee, J. Cannon, A. J. Nip, K. Corbin, M. F. Krummel and S. K. Choi, ACS Chem. Biol., 2017, 12, 1001–1010 CrossRef CAS PubMed.
- C. Ji, L. Lai, P. Li, Z. Wu, W. Cheng and M. Yin, Aggregate, 2021, 2, e39 CrossRef CAS.
- M. Bojtár, A. Kormos, K. Kis-Petik, M. Kellermayer and P. Kele, Org. Lett., 2019, 21, 9410–9414 CrossRef.
- J. Ma, J. Wehrle, D. Frank, L. Lorenzen, C. Popp, W. Driever, R. Grosse and H. J. Jessen, Chem. Sci., 2024, 15, 6478–6487 RSC.
- G. M. Fernandes-Cunha, C. J. McKinlay, J. R. Vargas, H. J. Jessen, R. M. Waymouth and P. A. Wender, ACS Cent. Sci., 2018, 4, 1394–1402 CrossRef CAS.
- J. P. Olson, H.-B. Kwon, K. T. Takasaki, C. Q. Chiu, M. J. Higley, B. L. Sabatini and G. C. R. Ellis-Davies, J. Am. Chem. Soc., 2013, 135, 5954–5957 CrossRef CAS PubMed.
- Q. Ding, Z. Zhang, M. Li, J.-H. Zhu, W. Fu, M. He, Y. Bai, Z. Zhang, S. Li, L. Wang, C. Deng, X. Hong, Y. Xiao and J. S. Kim, Cell Biomater, 2025, 1, 100001 CrossRef.
- J. M. Baskin and C. R. Bertozzi, QSAR Comb. Sci., 2007, 26, 1211–1219 CrossRef CAS.
- A. Y. Baev, P. R. Angelova and A. Y. Abramov, Biochem. J., 2020, 477, 1515–1524 CrossRef CAS PubMed.
- T. M. Haas, S. Mundinger, D. Qiu, N. Jork, K. Ritter, T. Dürr-Mayer, A. Ripp, A. Saiardi, G. Schaaf and H. J. Jessen, Angew. Chem., Int. Ed., 2022, 61, e202112457 CrossRef CAS.
- R. H. Beddoe, D. C. Edwards, L. Goodman, H. F. Sneddon and R. M. Denton, Chem. Commun., 2020, 56, 6480–6483 RSC.
- A. Hofer, G. S. Cremosnik, A. C. Müller, R. Giambruno, C. Trefzer, G. Superti-Furga, K. L. Bennett and H. J. Jessen, Chem.–Eur. J., 2015, 21, 10116–10122 CrossRef CAS PubMed.
- B. Amit, D. A. Ben-Efraim and A. Patchornik, J. Chem. Soc., Perkin Trans. 1, 1976, 1, 57–63 RSC.
- X. Du, H. Frei and S. H. Kim, J. Biol. Chem., 2000, 275, 8492–8500 CrossRef CAS PubMed.
- J. E. T. Corrie and G. P. Reid, J. Labelled Compd. Radiopharm., 1995, 36, 205–306 CrossRef.
- G.-D. Kim, G. Liu, D. Qiu, M. G. De Leo, N. Gopaldass, J. Hermes, J. Timmer, A. Saiardi, A. Mayer and H. J. Jessen, J. Am. Chem. Soc., 2025, 147, 17626–17641 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc04037j |
| This journal is © The Royal Society of Chemistry 2025 |