
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDifluoroenol phosphinates as difluoroenolate surrogates: synthesis and applications in defluorination and deoxygenative coupling†
Miki B.
Kurosawa‡
,
Shuhei
Shimoyama‡
,
Hiroki
Tanaka
and
Junichiro
Yamaguchi
 *
*
Department of Applied Chemistry, Waseda University, 513 Wasedatsurumakicho, Shinjuku, Tokyo 162-0041, Japan. E-mail: junyamaguchi@waseda.jp
First published on 20th June 2025
Abstract
We report defluorinative and deoxygenative functionalization reactions of trifluoromethyl ketones mediated by the phospha-Brook rearrangement, offering a streamlined approach to selectively modifying fluorinated compounds. Trifluoromethyl ketones react with phosphine oxides to undergo a phospha-Brook rearrangement followed by β-fluoride elimination, providing difluoromethyl ketones in good yields. By tuning the reaction conditions, we achieved the selective one-pot synthesis of monofluoromethyl ketones and methyl ketones, demonstrating the method's versatility across a range of fluorine-containing derivatives. Furthermore, we successfully demonstrated a range of deoxygenative transformations of key intermediates, such as difluoroenol phosphinates, showcasing their potential as building blocks for diverse functionalizations. These findings not only expand the synthetic toolbox for fluorine-containing molecules but also highlight the utility of phosphinate intermediates in developing novel reaction pathways.
Introduction
The carbon–fluorine bond is a cornerstone of modern medicinal chemistry and organic materials science, critically influencing the properties and functions of fluorinated molecules.1 Its high bond dissociation energy and unique electronic characteristics render fluorinated compounds distinct in terms of physicochemical and biological properties. Among fluorinated substituents, the trifluoromethyl group (CF3) is the most widely employed, but difluoromethyl (CF2H), monofluoromethyl (CFH2), and their methylene derivatives (–CF2–, –CHF–) are increasingly found in functional molecules due to their unique hydrogen bonding and steric properties.2 This shift has spurred substantial interest in developing efficient synthetic methods to access these groups.3 Strategies for synthesizing fluorinated compounds generally fall into three categories: (1) conversion of existing functional groups into fluorinated analogs, (2) direct fluorination, and (3) introduction of carbon chains into pre-existing fluorinated scaffolds. Among these, defluorinative functionalization reactions, where fluorine atoms are sequentially removed from trifluoromethyl groups, have gained attention as a precise and controlled approach for diversifying fluorinated motifs. Despite the robustness of carbon–fluorine bonds, recent advances have enabled their selective cleavage under carefully designed conditions.4Trifluoromethyl carbonyl compounds have emerged as versatile substrates for such transformations. Notably, single-electron transfer (SET)-based methodologies employing photoredox catalysis or mild reductants have been applied to achieve defluorinative functionalization of esters and amides (Fig. 1A).5 However, these methods are unsuitable for trifluoromethyl ketones due to competing reduction of the carbonyl group, leading to undesired byproducts. As a result, only a few examples of radical-based defluorinative functionalization of trifluoromethyl ketones have been reported to date.6
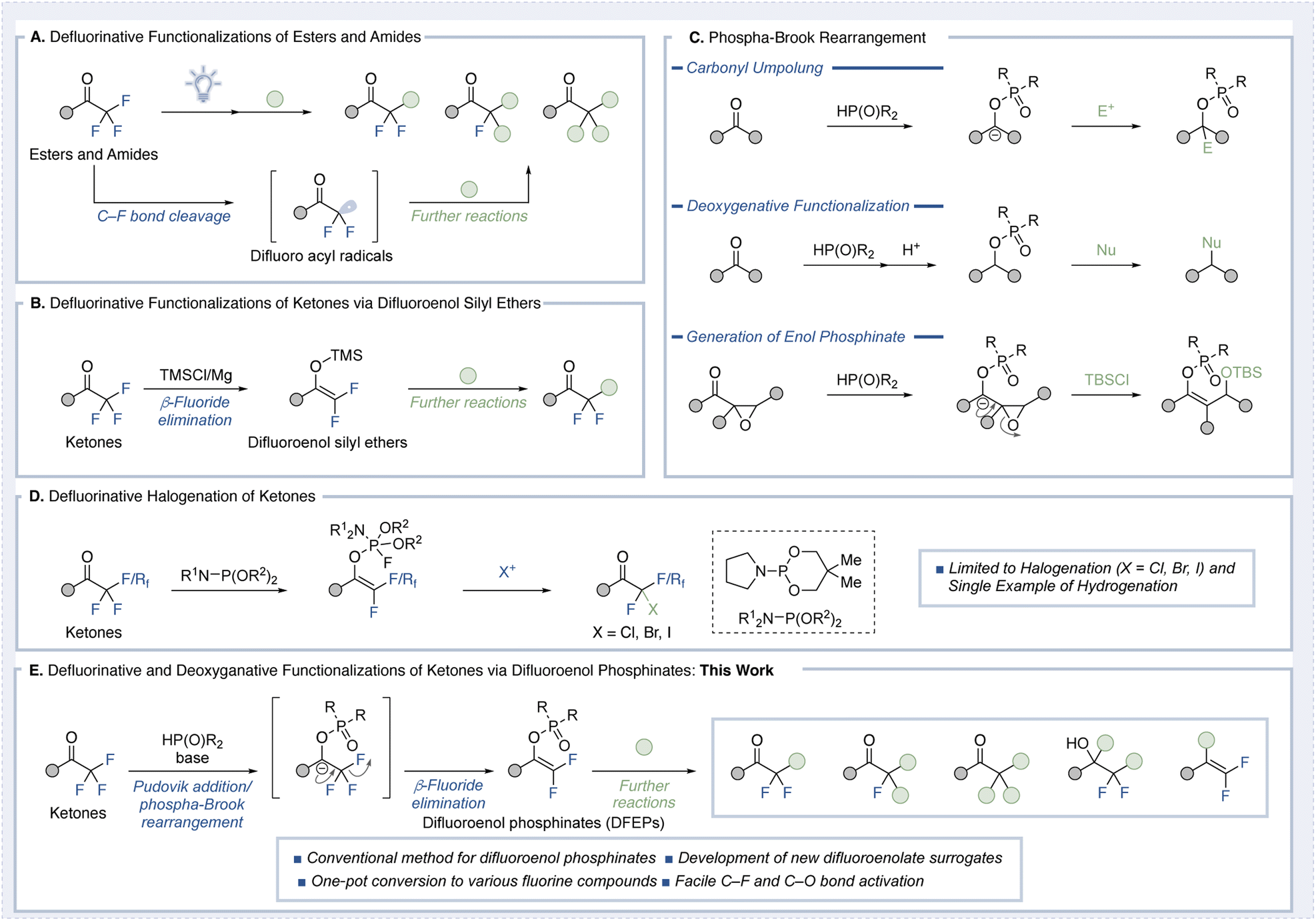 | ||
| Fig. 1 Strategies for the defluorinative functionalization of trifluoromethyl carbonyl compounds. (A) Recent advances in single-electron transfer (SET)-based defluorinative transformations of trifluoromethyl esters and amides. (B) Established method for generating difluoroenol silyl ethers from trifluoromethyl ketones, enabling α-functionalization with various electrophiles. (C) Phospha-Brook rearrangement: umpoluning, deoxyganative and formation of enolates (D) defluorinative halogenation of ketones (E) this work: development of a facile synthesis of difluoroenol phosphinates as difluoroenolate surrogates. | ||
For trifluoromethyl ketones, the most established approach involves the generation of difluoroenol silyl ethers, as demonstrated by Uneyama and Amii (Fig. 1B).7 In this method, trifluoromethyl ketones are treated with magnesium and TMSCl to form difluoroenol silyl ethers, which can react at the α-position with various electrophiles to produce difluoromethyl compounds.8 While effective, this strategy requires the difluoroenol silyl ether to be prepared in a separate step, limiting its practical application.9 While other difluoroenolate surrogates have been reported,10,11 they are typically unstable and cannot be isolated. Furthermore, they lack the versatile reactivity exhibited by difluoroenol silyl ethers. Moreover, methods for converting these intermediates into monofluoromethyl ketones or methyl ketones remain unexplored.
Meanwhile, the Pudovik addition followed by a phospha-Brook rearrangement is a well-established and increasingly explored strategy for the umpolung of carbonyl compounds, enabling access to carbanion reactivity from electrophilic ketones and aldehydes (Fig. 1C).12 Upon rearrangement, the resulting carbanion can be trapped by electrophiles, enabling C–C bond formation. Alternatively, protonation yields a phosphinate, which, owing to its inherent electrophilicity, can act as a leaving group in deoxygenative transformations involving nucleophiles.13 If the carbanion bears a β-leaving group (e.g., in the form of an epoxide) or unsaturated moiety, it can undergo β-elimination to generate enol phosphinates, thereby accessing enolate-type reactivity.14 A very recent study by Choi and Chung employed a trivalent phosphine-mediated Perkow-type transformation to selectively dehalogenate trifluoromethyl ketones (Fig. 1D).15 However, their approach remains limited to halogen incorporation and a single example of hydrogenation, thus lacking generality.
Inspired by the enolate-forming strategy outlined in Fig. 1C, we hypothesized that the nucleophilic addition of phosphine oxides or phosphites to trifluoromethyl ketones could initiate a phospha-Brook rearrangement, followed by β-fluoride elimination prior to protonation (Fig. 1E). This would generate difluoroenol phosphinates (DFEPs)—a new class of isolable, functional difluoroenolate surrogates.16,17 These intermediates are structurally analogous to difluoroenol silyl ethers, yet possess a C–O bond that is significantly more labile than the Si–O bond, suggesting potential for downstream transformations, particularly C–O bond activation. We further envisioned that these DFEPs could serve as branching intermediates in sequential defluorination, enabling stepwise access to monofluoromethyl and methyl ketones from trifluoromethyl ketones. In addition, the electrophilic nature of the phosphinate moiety was expected to facilitate cross-coupling reactions, providing access to gem-difluoroalkenes and other fluorinated architectures that are challenging to construct via conventional enolate chemistry.18
In this work, we report the synthesis of DFEPs from trifluoromethyl ketones via phospha-Brook rearrangement, their structural characterization including single-crystal X-ray analysis, and their application in diverse α-functionalization and C–O bond activations. Together, these findings establish DFEPs as a robust and flexible platform for selective defluorination and molecular diversification within the realm of organofluorine chemistry.
Results and discussion
We began our investigation by synthesizing difluoroenol phosphinate (Fig. 2A). Treatment of commercially available 4-methoxytrifluoroacetyl ketone (1A) with diphenylphosphine oxide (1.2 equiv.) and potassium carbonate (2.0 equiv.) afforded difluoromethyl ketone 3A in 80% yield (Condition A: entry 1). Minor side products included the over-defluorinated compound 4A and the addition product 5A, resulting from the phospha-Brook rearrangement. These findings are consistent with the proposed mechanism in Fig. 1C, where fluoride elimination occurs after the phospha-Brook rearrangement, with the resulting HF promoting protonation. We hypothesized that efficient HF trapping could enable the formation of difluoroenol phosphinate 2Aa. However, neither increasing the base equivalents nor screening various additives successfully afforded 2Aa. Remarkably, the addition of molecular sieves (MS4Å) proved critical, enabling the formation of 2Aa in 80% yield (entry 2). HF trapping by 4 Å molecular sieves was confirmed by 19F NMR analysis, which showed clear evidence of HF capture. Although we also considered the possibility that MS4Å might be trapping byproducts such as KF or KHCO3 formed from the reaction with base, little to no effect was observed in those cases (see ESI† for details). Therefore, we propose that MS4Å effectively scavenges HF, thereby facilitating the reaction. While the synthesis of difluoroenol phosphates has been previously reported, the methods typically involve multiple synthetic steps or rely on chlorodifluoroketones as starting materials.16 Notably, no direct transformation from trifluoromethyl ketones to difluoroenol phosphinates has been reported.17 Furthermore, this work represents the first successful isolation of the adduct 2Aa between a trifluoromethyl ketone and diphenylphosphine oxide. | ||
| Fig. 2 (A) Synthesis of difluoroenol phosphinate. (B) Scope of the method and crystallographic analysis. (C) Sequential defluorination to monofluoromethyl ketones and methyl ketones. | ||
To extend the scope of the methodology, we investigated the use of alternative phosphorus reagents (Fig. 2B). Substitution of diphenylphosphine oxide with diethyl phosphite afforded 2Ab in high yield, whereas the use of diphenyl phosphite resulted in a significantly lower yield (2Ac: 47%). To enable X-ray crystallographic analysis, tert-butyl groups were introduced at the para position to enhance the crystallization properties of the resulting enol phosphinates (2Bb and 2Ba). Recrystallization attempts with these derivatives were successful, and single crystals of 2Ba were obtained, allowing its structure to be determined via X-ray diffraction. Furthermore, although in moderate yield, trifluoromethyl ketones bearing alkyl substituents could also be converted into the corresponding enol phosphinate 2Cb using the same methodology.
Building on the observation that monofluoroketones could be obtained under the conditions shown in Fig. 2A, we hypothesized that a one-pot method could be developed to selectively convert trifluoromethyl ketones into difluoromethyl ketones, monofluoromethyl ketones, or methyl ketones (Fig. 2C). By optimizing the equivalents of diphenylphosphine oxide and the reaction temperature (details provided in the ESI†), we successfully devised conditions for each transformation. Specifically, using 2.5 equiv. of diphenylphosphine oxide at 130 °C provided monofluoromethyl ketone 4A in 67% yield (Condition B). Further increasing the amount of diphenylphosphine oxide to 4.0 equiv. and raising the temperature to 160 °C resulted in the formation of methyl ketone 6A in moderate yield (47%) (Condition C).
These results highlight that, starting from a trifluoromethyl ketone, simply varying the reaction conditions allows for the selective, one-pot synthesis of three distinct compounds—difluoromethyl ketones, monofluoromethyl ketones, and methyl ketones—offering a streamlined and versatile approach to functional group manipulation. It should be noted that potassium carbonate is insoluble in 1,4-dioxane, making efficient stirring crucial for consistent reaction performance. Insufficient stirring significantly reduced the yields, emphasizing the importance of proper agitation during the reaction.
Using the optimized conditions (Condition A) for the selective synthesis of difluoromethyl ketones, monofluoromethyl ketones, and methyl ketones, we investigated the substrate scope of various trifluoromethyl ketones (Fig. 3). Substrates containing a benzene ring (1B), alkyl groups at the C4 position (1C and 1D), or a phenyl group (1E) successfully produced the corresponding difluoromethyl ketones 3B–3E in moderate yields. Functional groups such as chloro (3F), thiomethyl (3G), benzyloxy (3H), amine (3I and 3J), and dioxo (3K) groups were well-tolerated, with the reactions proceeding smoothly. Remarkably, substrates known to quench photoredox catalysis, such as those containing a naphthyl group (1L and 1M) or anthracenyl group (1N), also afforded the corresponding difluoromethyl ketones 3L–3N. Furthermore, heterocyclic ketones, including pyrrole (1O and 1P), quinoline (1Q), indole (1R), benzofuran (1S), and benzothiophene (1T), underwent the transformation to produce the corresponding difluoromethyl ketones 3O–3T. For most compounds, the reaction conditions were generally based on Condition A, with slight modifications such as reducing the equivalents of diphenylphosphine oxide, slightly increasing the reaction temperature, or extending the reaction time. However, in the case of quinoline (1Q), the yield of product 3Q was limited to 20%, with the corresponding monofluorinated compound 4Q obtained in 35% yield. It is known that difluoroenol phosphinates derived from phosphine oxides exhibit weaker O–P bonds compared to those derived from dialkyl phosphites. To address this issue, we employed dibutyl phosphite to generate a more stable difluoroenol phosphinate. The increased stability of the difluoroenol phosphinate derived from dibutyl phosphite prevented the formation of 4Q, leading to a significant improvement in the yield of 3Q to 53%. Notably, the reaction was not limited to aromatic ketones; aliphatic trifluoromethyl ketone 1U also participated, albeit with lower yields, affording difluoromethyl ketone 3U. Additionally, an aliphatic trifluoromethyl ketone derived from the pharmaceutical compound naproxen 1V afforded the corresponding difluoromethyl ketone 3V in 57% yield.
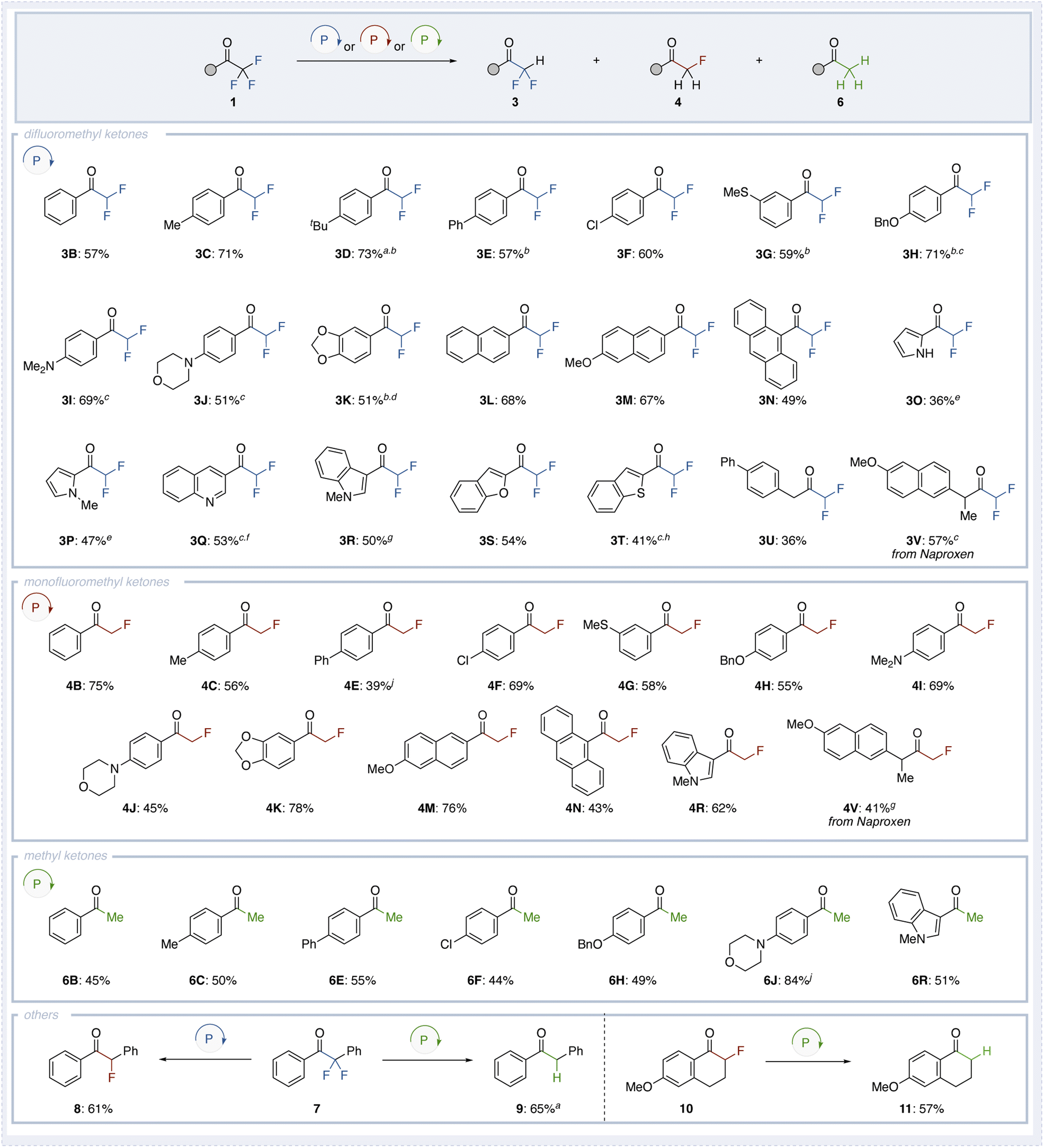 | ||
| Fig. 3 Substrate scope for defluorinations. Condition A: 1 (0.20 mmol), HP(O)Ph2 (0.24 mmol), K2CO3 (0.40 mmol), 80 °C, 6 h. Condition B: 1 (0.20 mmol), HP(O)Ph2 (0.50 mmol), K2CO3 (0.40 mmol), 130 °C, 6 h. Condition C: 1 (0.20 mmol), HP(O)Ph2 (0.80 mmol), K2CO3 (0.40 mmol), 160 °C, 6 h. aReaction time extended to 24 h. bHP(O)Ph2 (0.20 mmol) was used. cReaction conducted at 90 °C. dReaction time extended to 12 h. eReaction conducted at 150 °C. fHP(O)(OnBu)2 (0.20 mmol) was used. gReaction conducted at 140 °C. hHP(O)(OnBu)2 (0.24 mmol) was used. iToluene (1.0 mL) was used as the solvent. jHP(O)(OnBu)2 (0.80 mmol) was used. | ||
Although the methodology showed broad substrate generality, in the case of alkyl trifluoromethyl ketones and electron-deficient heteroaromatics such as pyridines, the anionic intermediate generated after the phospha-Brook rearrangement appears to be unstable. As a result, a significant amount of protonated byproduct (e.g., compound 5a) is formed, leading to reduced yields. Additionally, substrates bearing strongly electron-withdrawing groups failed to undergo the initial Pudovik addition and were recovered unchanged. This is likely due to the strong electron-withdrawing effect promoting hydration of the carbonyl group to form a hemiacetal, which interferes with the nucleophilic substitution step.
Next, we investigated the substrate scope of trifluoromethyl ketones under the optimal reaction conditions for monofluoromethyl ketone synthesis (Condition B). When substrates such as unsubstituted (1B), methyl-substituted (1C), phenyl-substituted (1E), were tested, the corresponding monofluoromethyl ketones 4B, 4C and 4E were obtained in moderate to high yields. Similar to the synthesis of difluoromethyl ketones, compounds bearing chloro (4F), thiomethyl (4G), benzyloxy (4H), amino (4I and 4J), or dioxo (4K) groups were successfully converted, affording the products in moderate yields and demonstrating excellent functional group tolerance. Moreover, trifluoromethyl ketones containing naphthalene (1M) or anthracene (1N) also underwent the reaction to give 4M and 4N. Ketones bearing indole (1R) moieties gave the corresponding products 4R in moderate to high yields. Additionally, the pharmaceutical compound naproxen (1V) was successfully converted, producing the corresponding monofluoromethyl ketone 4V.
Finally, using Condition C, various trifluoromethyl ketones 1 were converted into methyl ketones through defluorination. Substrates such as unsubstituted 1B, methyl-substituted 1C, phenyl-substituted 1D, and chloro-substituted 1E afforded the corresponding methyl ketones 6B–6E in moderate yields. When trifluoromethyl ketones bearing electron-donating groups at the para position (1H and 1J) were employed, the desired products 6H and 6J were obtained in moderate to high yields. Additionally, ketone 1S containing an indole moiety was successfully converted to 6S, with a yield of 51%.
In addition, although only one example was tested, the reaction was found to proceed not only with trifluoromethyl ketones but also with ketones 7 bearing a difluoromethylene group at the α-position. When 7 was subjected to Condition A, an α-monofluoroketone 8 was obtained in 61% yield. However, when 7 was treated under Condition C, further defluorination occurred, resulting in a mixture of monofluoroketone and methyl ketone 9. In contrast, applying Condition C to 7 yielded methyl ketone 9 in good yield (65%). Moreover, this reaction was also applicable to cyclic ketones. When cyclic ketone 10 was treated under Condition C, the corresponding tetralone derivative 11 was obtained in 57% yield.
To elucidate the reaction mechanism underlying this defluorination process, mechanistic experiments were conducted (Fig. 4). First, separately prepared 5A was treated with potassium carbonate at 80 °C (Fig. 4A). If 5A were an intermediate, 3A would be expected as the product; however, no 3A was observed. This suggests that 5A is a side product formed via protonation before β-fluoride elimination. These findings imply that β-fluoride elimination proceeds directly from one of the anionic intermediates proposed in the mechanism.
 | ||
| Fig. 4 Mechanistic investigations of the defluorinations. (A) Testing 5A as a potential intermediate (B) conversion of 3A to 4A and enol phosphinate formation. (D) Competition experiment, and time-course study: monitoring by 19F NMR. (E) Proposed reaction pathway. | ||
Next, 3A was subjected to the reaction conditions of Condition B, yielding monofluoromethyl ketone 4A quantitatively (Fig. 4B). This result confirms that monofluoromethyl ketones are generated via the intermediate difluoromethyl ketones. To further investigate, we examined the formation of enol phosphinates as intermediates. When MS4Å was added under Condition B, the desired monofluoromethyl ketones were replaced by enol phosphinates, which were obtained as E/Z mixtures. These E and Z isomers were easily separable by silica gel column chromatography, and their structures were determined by NOE analysis. Using diethyl phosphite as the phosphorus reagent afforded 12Bb in high yield, while diarylphosphine oxide (Ar = p-MeOC6H4-) also yielded the corresponding enol phosphinate 12Bd, albeit in moderate yield. However, when diphenylphosphine oxide was used, the enol phosphinate could not be isolated, likely due to the weaker O–P bond. Instead, complete conversion to monofluoromethyl ketone 4A was observed, preventing the isolation of the desired enol phosphinate. Although enol phosphinates have been previously reported,19 their synthesis typically involves lengthy and multi-step procedures. In contrast, our approach enables the direct, one-pot synthesis of these compounds starting from difluoromethyl ketones, significantly simplifying the process.
We also performed a competition experiment (Fig. 4C). Structurally similar trifluoromethyl ketone 1W and difluoromethyl ketone 3A were reacted under Condition A, which is optimized for the synthesis of difluoromethyl ketones. The results revealed that only 1W was converted into difluoromethyl ketone 3W, while 3A was recovered in 84% yield, and no monofluoromethyl ketone 4A was formed. This indicates that under these reaction conditions, difluoromethyl ketones do not undergo further transformations.
Furthermore, a competition experiment between difluoromethyl ketone 3W and monofluoromethyl ketone 4A was conducted. When subjected to Condition B at a slightly lower temperature (110 °C instead of 130 °C), monofluoromethyl ketone 4W was selectively obtained, while methyl ketone 6A was not formed, and 4A was recovered in 68% yield. These results demonstrate that even in the presence of both substrates, selective transformation into the desired fluorinated ketone can be achieved simply by adjusting the reaction conditions.
To monitor the reaction progress, time-course experiments were conducted using 19F NMR spectroscopy (Fig. 4D). Shortly after the reaction began, an addition product 13, which we attribute to 13 was observed, and it was nearly consumed within 90 min. By 30 min, difluoroenol phosphinate 2Aa, the desired difluoromethyl ketone 3A, and phospha-Brook rearrangement product 5A were already detected. Over time, 2Aa was gradually consumed as the yield of 3A increased, while the amount of 5A remained relatively constant. Additionally, small amounts of fluorinated byproducts 14 containing phosphorus, along with aldol adducts 15 and 16 were observed but were eventually consumed. Based on these results, the proposed reaction pathway is depicted in Fig. 4E. Trifluoromethyl ketones undergo Pudovik addition followed by a phospha-Brook rearrangement, leading to β-fluoride elimination and the formation of enol phosphinate and HF. In the presence of MS4Å, HF is efficiently trapped, favoring the formation of difluoroenol phosphinate 2Aa. In the absence of a trapping agent, protonation occurs, yielding difluoromethyl ketone 3A. The significant reactivity difference between 1 and 3 ensures that under mild conditions (Condition A), the reaction halts at this stage. However, increasing the equivalents of phosphine oxide and raising the temperature drives a second β-fluoride elimination, forming monofluorophosphinate 12, which leads to monofluoromethyl ketone 4. Under harsher conditions, further defluorination occurs, resulting in the formation of methyl ketone 6.
Next, we explored the defluorinative functionalization of trifluoromethyl ketones as an alternative to hydrodefluorination. A one-pot approach for α-functionalization and ketone transformation of trifluoromethyl ketones was investigated, enabling transformations that are difficult to achieve via difluoroenol silyl ethers (Fig. 5A). Initially, treatment under Condition A converted the trifluoromethyl ketone into a difluoromethyl ketone, which was subsequently reacted with chloroarenes in the presence of a palladium catalyst, affording α-arylated compounds 17 in moderate yields.20 Furthermore, the use of a rhodium catalyst with arylboronic acids enabled 1,2-addition to the ketone, affording alcohol 18.21 Next, we examined the defluorinative allylation of trifluoromethyl ketones (Fig. 5B). Direct transformation of trifluoromethyl ketones with allylating agents was challenging, prompting the use of difluoroenol phosphinate 2Aa as the substrate. Treatment of 2Aa with an allylating agent in the presence of 1.0 mol% Pd(allyl)Cl dimer, BINAP, and TBAT in toluene at 40 °C for 12 h afforded an unexpected O-allylated product 19 in 62% yield.22 Interestingly, purification of 19 by PTLC partially converted it into the α-allylated compound 20, suggesting that 19 underwent a [3,3]-sigmatropic rearrangement on silica gel. This transformation is likely facilitated by the gem-difluoro substitution at the vinyl position, which is known to accelerate Claisen-type rearrangements at relatively mild temperatures, in contrast to non-fluorinated analogues.23
 | ||
| Fig. 5 α-Functionalization and transformations of trifluoromethyl ketones via difluoromethyl intermediates. (A) One-pot conversion of trifluoromethyl ketones to difluoromethyl ketones followed by α-arylation and arylation. (B) Defluorinative allylation of difluoroenol phosphinate. (C) Aldol-type reactions of difluoroenol phospate with benzaldehyde. (D) Radical addition of difluoroenol phosphinate 2Ab. | ||
We further explored aldol-type reactions using 2Ab with aldehydes and nucleophiles (Fig. 5C).24 When 2Ab was treated with HBF4 and benzaldehyde, aldol adduct 21A was obtained in 59% yield. Replacing HBF4 with TMSOTf produced the ethyl ether 21B in 49% yield. Notably, the ethoxy group in 21B originates from diethyl phosphite. During the course of our investigation, we discovered that the reaction proceeds via intermediate 21′, in which the aldol reaction occurs first, followed by migration of the phosphite moiety. Based on this finding, we hypothesized that the resulting ethoxide could be trapped while introducing an alternative nucleophile.
To test this, trifluoroacetic anhydride (TFAA) was employed as a scavenger for ethoxide, and methyl tosylamide or benzoic acid was added as nucleophiles. As a result, the tosylamide 21C was obtained in 83% yield, while the carboxylate 21D was obtained in 32% yield. Theoretically, any nucleophile that does not react with TFAA could be introduced using this approach. This type of three-component coupling is a unique feature enabled by the use of difluoroenol phosphinate.
Furthermore, we explored radical-mediated alkylation and heteroatom incorporation reactions of enol phosphinate 2Ab (Fig. 5D). Inspired by previous reports on the photoredox-catalyzed functionalization of difluoroenol silyl ethers,25 we subjected 2Ab to visible-light irradiation with blue LEDs in the presence of catalytic amounts of PC1 and redox-active ester. This reaction proceeded smoothly, affording alkylated product 22 in good yield. Additionally, in an attempt to introduce heteroatoms, we investigated a thiolation reaction under metallaphotoredox conditions.26 Unexpectedly, we discovered that the reaction proceeded efficiently even in the absence of a transition metal catalyst. Specifically, treatment of 2Ab with anisyl thiol under blue-light irradiation in the presence of PC2 led to the formation of the desired coupling product 23 in high yield.
Thus, we have discovered new reactions of trifluoromethyl ketones and difluoroenol phosphinates, demonstrating that these compounds enable defluorinative functionalization at a level comparable to or even superior to that of difluoroenol silyl ethers.
Although the functionalization reactions described above could also be achieved using conventional difluoroenol silyl ethers, C–O bond activation reactions remain particularly challenging for these compounds. Exploiting the pronounced electrophilicity of the phosphinate group, we investigated C–O bond activation reactions unique to difluoroenol phosphinates (Fig. 6). As previously mentioned, 2Aa is less stable than 2Ab, with C–P bond cleavage being more favorable. Thus, all C–O bond transformation reactions were performed using 2Ab. For example, treating 2Ab with an arylboronic acid in the presence of a nickel catalyst enabled a Suzuki–Miyaura coupling reaction, producing the arylated product 24.27 Furthermore, palladium-catalyzed reaction of 2Ab with trimethylaluminum afforded the methylated compound 25. Notably, replacing trimethylaluminum (AlMe3) with triethylaluminum (AlEt3) led to the hydrogenated product 26.28 Additionally, treatment with separately prepared aluminum acetylides enabled the synthesis of the alkyne derivative 27.29 Reaction of 2Ab with a chloro(vinyl)silane and manganese as a reductant afforded vinylsilane 28, albeit in a low yield.30 Finally, the cross-electrophile coupling of enol phosphinate 2Ab with benzyl chloride under nickel catalysis enabled C–O bond benzylation, affording compound 29 in 43% yield.27
 | ||
| Fig. 6 C–O bond activation of difluoroenol phosphinates. | ||
The C–O bond activation reactions described here represent unprecedented transformations that are inaccessible with conventional difluoroenol silyl ethers. These findings highlight the unique reactivity of difluoroenol phosphinates, providing a platform for the synthesis of structurally diverse fluorinated compounds.
Conclusions
In this study, we developed a series of defluorinative and deoxygenative functionalization reactions of trifluoromethyl ketones, utilizing difluoroenol phosphinates as functional intermediates.30 Central to these transformations is the phospha-Brook rearrangement, which enabled the selective synthesis of difluoromethyl ketones, monofluoromethyl ketones, and methyl ketones under tunable conditions. Notably, difluoroenol phosphinates demonstrated unique reactivity in C–O bond activation reactions compared to conventional difluoroenol silyl ethers. The newly discovered C–O bond activation processes—including Suzuki–Miyaura coupling, methylation, hydrogenation, alkynylation, reductive vinylation, and alkylation—demonstrate the potential of difluoroenol phosphinates as a synthetic platform for constructing structurally diverse fluorinated compounds. These transformations are unprecedented and leverage the electrophilic nature of the phosphinate group, enabling reactions that are inaccessible with traditional silyl ether analogs.31This work significantly expands the synthetic toolbox for fluorinated molecules, offering new avenues for exploring the unique reactivity of difluoroenol phosphinates. The findings pave the way for further investigations into novel reaction pathways and applications in pharmaceuticals, materials science, and beyond.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
J. Y. directed the projects and designed the experiments. M. K. and S. S. mainly performed experiments and H. T. synthesized some starting materials. All authors contributed to data analysis. J. Y. wrote the manuscript with feedback from the other authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI Grant Numbers JP21H05213 (Digi-TOS) (to J. Y.). This work was partly supported by JST CREST Grant Number JPMJCR24T3 (to J. Y.). We thank Dr Ryohei Doi (Tokyo Metropolitan University) for valuable discussions and insights regarding defluorination. We also thank Dr Kenta Kato (FUJIFILM) for helping X-ray crystallographic analysis. Materials Characterization Central Laboratory in Waseda University is acknowledged for the support of NMR and HRMS measurement.Notes and references
- (a) D. O'Hagan, Chem. Soc. Rev., 2007, 37, 308–319 RSC; (b) R. Berger, G. Resnati, P. Metrangolo, E. Weber and J. Hulliger, Chem. Soc. Rev., 2011, 40, 3496–3508 RSC; (c) Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422–518 CrossRef CAS PubMed; (d) J. Han, L. Kiss, H. Mei, A. M. Remete, M. Ponikvar-Svet, D. M. Sedgwick, R. Roman, S. Fustero, H. Moriwaki and V. A. Soloshonok, Chem. Rev., 2021, 121, 4678–4742 CrossRef CAS PubMed; (e) D. O'Hagan and R. J. Young, Med. Chem. Res., 2023, 32, 1231–1234 CrossRef.
- (a) O. O. Grygorenko, K. P. Melnykov, S. Holovach and O. Demchuk, ChemMedChem, 2022, 17, e202200365 CrossRef CAS PubMed; (b) D. O'Hagan, Y. Wang, M. Skibinski and A. M. Z. Slawin, Pure Appl. Chem., 2012, 84, 1587–1595 CrossRef; (c) R. Hevey, Chem.–Eur. J., 2021, 27, 2240–2253 CrossRef CAS PubMed; (d) Q. Wang, H. Song and Q. Wang, Chin. Chem. Lett., 2022, 33, 626–642 CrossRef CAS; (e) W. Zhang, P. Guo, Y. Zhang, Q. Zhou, Y. Sun and H. Xu, J. Agric. Food Chem., 2024, 72, 21344–21363 CrossRef CAS PubMed.
- (a) X. Ma and Q. Song, Chem. Soc. Rev., 2020, 49, 9197–9219 RSC; (b) J. B. I. Sap, C. F. Meyer, N. J. W. Straathof, N. Iwumene, C. W. am Ende, A. A. Trabanco and V. Gouverneur, Chem. Soc. Rev., 2021, 50, 8214–8247 RSC; (c) D. R. Carvalho and A. H. Christian, Org. Biomol. Chem., 2021, 19, 947–964 RSC; (d) K. Sathish, S. Jain, N. Sihag and M. R. Yadav, Org. Biomol. Chem., 2024, 22, 8078–8096 RSC; (e) M. Reichel and K. Karaghiosoff, Angew. Chem., Int. Ed., 2020, 59, 12268–12281 CrossRef CAS PubMed; (f) R. Britton, V. Gouverneur, J.-H. Lin, M. Meanwell, C. Ni, G. Pupo, J.-C. Xiao and J. Hu, Nat. Rev. Methods Primers, 2021, 1, 47 CrossRef CAS; (g) Z. Han and C. Zhang, Adv. Synth. Catal., 2020, 362, 4256–4292 CrossRef CAS; (h) Z. Feng, Y.-L. Xiao and X. Zhang, Acc. Chem. Res., 2018, 51, 2264–2278 CrossRef CAS PubMed.
- (a) L. V. Hooker and J. S. Bandar, Angew. Chem., Int. Ed., 2023, 62, e202308880 CrossRef CAS PubMed; (b) F. Zhao, W. Zhou and Z. Zuo, Adv. Synth. Catal., 2022, 364, 234–267 CrossRef CAS; (c) Z. Wang, Y. Sun, L.-Y. Shen, W.-C. Yang, F. Meng and P. Li, Org. Chem. Front., 2021, 9, 853–873 RSC; (d) S. Li and W. Shu, Chem. Commun., 2021, 58, 1066–1077 RSC.
- (a) T. T. Simur, T. Ye, Y.-J. Yu, F.-L. Zhang and Y.-F. Wang, Chin. Chem. Lett., 2022, 33, 1193–1198 CrossRef CAS; (b) S.-S. Yan, S.-H. Liu, L. Chen, Z.-Y. Bo, K. Jing, T.-Y. Gao, B. Yu, Y. Lan, S.-P. Luo and D.-G. Yu, Chem, 2021, 7, 3099–3113 CrossRef CAS; (c) M. W. Campbell, V. C. Polites, S. Patel, J. E. Lipson, J. Majhi and G. A. Molander, J. Am. Chem. Soc., 2021, 143, 19648–19654 CrossRef CAS PubMed; (d) J. Ye, P. Bellotti, C. Heusel and F. Glorius, Angew. Chem., Int. Ed., 2022, 61, e202115456 CrossRef CAS PubMed; (e) C. Liu, N. Shen and R. Shang, Nat. Commun., 2022, 13, 354 CrossRef CAS PubMed; (f) Y.-J. Yu, F.-L. Zhang, T.-Y. Peng, C.-L. Wang, J. Cheng, C. Chen, K. N. Houk and Y.-F. Wang, Science, 2021, 371, 1232–1240 CrossRef CAS; (g) H. Zhang, J.-X. Chen, J.-P. Qu and Y.-B. Kang, Nature, 2024, 635, 610–617 CrossRef CAS PubMed; (h) J. Jia, K. Murugesan, C. Zhu, H. Yue, S.-C. Lee and M. Rueping, Chin. Chem. Lett., 2025, 36, 109866 CrossRef CAS.
- (a) S. Ghosh, Z. Qu, S. Pradhan, A. Ghosh, S. Grimme and I. Chatterjee, Angew. Chem., Int. Ed., 2022, 61, e202115272 CrossRef CAS PubMed; (b) S. Ghosh, Z. Qu, S. Roy, S. Grimme and I. Chatterjee, Chem.–Eur. J., 2023, 29, e202203428 CrossRef CAS PubMed.
- H. Amii, T. Kobayashi, Y. Hatamoto and K. Uneyama, Chem. Commun., 1999, 1323–1324 RSC.
- Selective examples, see: (a) G. K. S. Prakash, J. Hu and G. A. Olah, J. Fluorine Chem., 2001, 112, 355–360 CrossRef; (b) O. Lefebvre, T. Brigaud and C. Portella, J. Org. Chem., 2001, 66, 1941–1946 CrossRef CAS PubMed; (c) K. Uneyama, H. Tanaka, S. Kobayashi, M. Shioyama and H. Amii, Org. Lett., 2004, 6, 2733–2736 CrossRef CAS PubMed; (d) F. Huguenot, A. Billac, T. Brigaud and C. Portella, J. Org. Chem., 2008, 73, 2564–2569 CrossRef CAS; (e) Y. Guo and J. M. Shreeve, Chem. Commun., 2007, 3583–3585 RSC; (f) Y.-L. Liu and J. Zhou, Chem. Commun., 2012, 48, 1919–1921 RSC; (g) J. Yu, Y. Liu, J. Tang, X. Wang and J. Zhou, Angew. Chem., Int. Ed., 2014, 53, 9512–9516 CrossRef CAS PubMed; (h) X. Jiang, D. Meyer, D. Baran, M. A. C. González and K. J. Szabó, J. Org. Chem., 2020, 85, 8311–8319 CrossRef CAS PubMed.
- The direct hydrodefluorination of trifluoromethyl ketones via electrochemical methods has been previously reported. See: J. R. Box, A. P. Atkins and A. J. J. Lennox, Chem. Sci., 2021, 12, 10252–10258 RSC.
- M. Decostanzi, J.-M. Campagne and E. Leclerc, Org. Biomol. Chem., 2015, 13, 7351–7380 RSC.
- (a) R. Doi, K. Kikushima, M. Ohashi and S. Ogoshi, J. Am. Chem. Soc., 2015, 137, 3276–3282 CrossRef CAS PubMed; (b) R. Doi, M. Ohashi and S. Ogoshi, Angew. Chem., Int. Ed., 2016, 55, 341–344 CrossRef CAS; (c) T. Brigaud, P. Doussot and C. Portella, J. Chem. Soc., Chem. Commun., 1994, 2117–2118 RSC.
- (a) R. Kaur and R. P. Singh, J. Org. Chem., 2023, 88, 10325–10338 CrossRef CAS PubMed; (b) A. Kondoh and M. Terada, Bull. Chem. Soc. Jpn., 2021, 94, 339–356 CrossRef CAS.
- (a) M. B. Kurosawa, R. Isshiki, K. Muto and J. Yamaguchi, J. Am. Chem. Soc., 2020, 142, 7386–7392 CrossRef CAS PubMed; (b) M. B. Kurosawa, K. Kato, K. Muto and J. Yamaguchi, Chem. Sci., 2022, 13, 10743–10751 RSC; (c) M. Sakihara, M. B. Kurosawa, M. Watanabe, S. Shimoyama and J. Yamaguchi, J. Org. Chem., 2024, 89, 8157–8167 CrossRef CAS PubMed; (d) M. Sakihara, S. Shimoyama, M. B. Kurosawa and J. Yamaguchi, Bull. Chem. Soc. Jpn., 2024, 97, uoae078 CrossRef CAS; (e) F. Liu, J. Dong, R. Cheng, S.-F. Yin, L. Chen, L. Su, R. Qiu, Y. Zhou, L.-B. Han and C.-J. Li, Sci. Adv., 2025, 11, eads4626 CrossRef CAS PubMed.
- (a) Q. Lin, S. Zheng, L. Chen, J. Wu, J. Li, P. Liu, S. Dong, X. Liu, Q. Peng and X. Feng, Angew. Chem., Int. Ed., 2022, 61, e202203650 CrossRef CAS; (b) A. Kondoh, T. Aoki and M. Terada, Chem.–Eur. J., 2017, 23, 2769–2773 CrossRef CAS PubMed; (c) A. Ali, H. K. Harit, M. Devi, D. Ghosh and R. P. Singh, J. Org. Chem., 2022, 87, 16313–16327 CrossRef CAS PubMed; (d) A. Kondoh, N. Tasato, T. Aoki and M. Terada, Org. Lett., 2020, 22, 5170–5175 CrossRef CAS PubMed.
- After our submission to ChemRxiv, a related study was published. The authors reported the synthesis of enol phosphinates from trifluoromethyl ketones, followed by hydrogenation and halogenation. See: H. E. Kim, J.-H. Choi and W. Chung, JACS Au, 2025, 5, 1007–1015 CrossRef CAS PubMed.
- X.-J. Zhang, Y.-M. Cheng, X.-W. Zhao, Z.-Y. Cao, X. Xiao and Y. Xu, Org. Chem. Front., 2021, 8, 2315–2327 RSC.
- (a) T. Ishihara, M. Yamana and T. Ando, Tetrahedron Lett., 1983, 24, 5657–5660 CrossRef CAS; (b) T. Ishihara, M. Yamana, T. Maekawa, M. Kuroboshi and T. Ando, J. Fluorine Chem., 1988, 38, 263–277 CrossRef CAS; (c) A. S. Demir and S. Eymur, J. Org. Chem., 2007, 72, 8527–8530 CrossRef CAS PubMed.
- The synthesis of enol phosphinates from trifluoromethylated enones has been recently reported: L.-W. Sun, Y.-F. Hu, W.-J. Ji, P.-Y. Zhang, M. Ma, Z.-L. Shen and X.-Q. Chu, Org. Lett., 2023, 25, 3745–3749 CrossRef CAS PubMed.
- P. Beier, R. Pohl and A. Alexandrova, Synthesis, 2009, 957–962 CAS.
- S. Ge, W. Chaładaj and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 4149–4152 CrossRef CAS PubMed.
- L. S. Dobson and G. Pattison, Chem. Commun., 2016, 52, 11116–11119 RSC.
- É. Bélanger, K. Cantin, O. Messe, M. Tremblay and J.-F. Paquin, J. Am. Chem. Soc., 2007, 129, 1034–1035 CrossRef PubMed.
- M. R. Garayt and J. M. Percy, Tetrahedron Lett., 2001, 42, 6377–6380 CrossRef CAS.
- S. Sasaki, T. Suzuki, T. Uchiya, S. Toyota, A. Hirano, M. Tanemura, H. Teramoto, T. Yamauchi and K. Higashiyama, J. Fluorine Chem., 2016, 192, 78–85 CrossRef CAS.
- H. Song, R. Cheng, Q.-Q. Min and X. Zhang, Org. Lett., 2020, 22, 7747–7751 CrossRef CAS PubMed.
- (a) M. S. Oderinde, M. Frenette, D. W. Robbins, B. Aquila and J. W. Johannes, J. Am. Chem. Soc., 2016, 138, 1760–1763 CrossRef CAS PubMed; (b) P. Sorrentino, R. M. Herrick, M. K. Abd El-Gaber, A. Z. Abdelazem, A. Kumar and R. A. Altman, J. Org. Chem., 2022, 87, 16676–16690 CrossRef PubMed.
- B. Du, C.-M. Chan, P.-Y. Lee, L.-H. Cheung, X. Xu, Z. Lin and W.-Y. Yu, Nat. Commun., 2021, 12, 412 CrossRef CAS PubMed.
- K. Takai, K. Oshima and H. Nozaki, Tetrahedron Lett., 1980, 21, 2531–2534 CrossRef CAS.
- M. Sato, K. Takai, K. Oshima and H. Nozaki, Tetrahedron Lett., 1981, 22, 1609–1612 CrossRef CAS.
- J. Duan, K. Wang, G. Xu, S. Kang, L. Qi, X. Liu and X. Shu, Angew. Chem., Int. Ed., 2020, 59, 23083–23088 CrossRef CAS PubMed.
- An initial version of this work was deposited in ChemRxiv on January 22, 2025, DOI:10.26434/chemrxiv-2025-1rklr.
Footnotes |
| † Electronic supplementary information (ESI) available: For experimental procedures, spectroscopic data for compounds including 1H and 13C NMR spectra. CCDC 2404597. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc03915k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |