
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic enantioselective total synthesis of antitubercular agents (–)-bedaquiline and (–)-sudapyridine enabled by dynamic kinetic resolution-asymmetric transfer hydrogenation†
Jiyao
Han
b,
Dongliang
Zhang
b,
Yuan
Tao
c,
Pei
Tang
*d and
Fen-er
Chen
 *abc
*abc
aCollege of Chemistry and Chemical Engineering, Jiangxi Normal University, Nanchang 330022, China. E-mail: rfchen@fudan.edu.cn
bSchool of science, Harbin Institute of Technology (Shenzhen), Shenzhen 518055, China
cEngineering Center of Catalysis and Synthesis for Chiral Molecules, Department of Chemistry, Fudan University, Shanghai 200433, China
dSichuan Research Center for Drug Precision Industrial Technology, West China School of Pharmacy, Sichuan University, Chengdu 610041, China. E-mail: peitang@scu.edu.cn
First published on 25th June 2025
Abstract
(–)-Bedaquiline [(–)-BDQ] is considered to be one of the most promising new therapeutic agents for tuberculosis for over 50 years. However, there are limited general and highly stereocontrolled asymmetric synthetic methods available for (–)-BDQ and its analogues due to the challenge of forging their vicinal stereocenters. Herein, we report a concise and stereocontrolled synthetic route to (–)-BDQ in six steps with an overall yield of 34%, integrating a Rh-catalyzed dynamic kinetic resolution-asymmetric transfer hydrogenation (DKR-ATH) with TADDOL-mediated diastereoselective allylation. The reactivity and stereoselectivity of DKR-ATH were systematically investigated using a range of sterically hindered N-hetero-1,2,2-triarylethanones. This approach offers a robust and reliable method for synthesizing N-hetero-1,2,2-triarylethanols featuring two continuous stereocenters, which serve as crucial chiral building blocks for pharmaceutical applications. Furthermore, the aforementioned two-stage protocol has been successfully applied to the synthesis of (–)-sudapyridine, a tuberculosis drug candidate currently in phase III clinical trials. This study presents a versatile and generalizable strategy for the synthesis of BDQ-type architectures, which hold significant interest for both medicinal and process chemists.
Introduction
Tuberculosis (TB) is an infectious disease primarily caused by Mycobacterium tuberculosis, with approximately one-third of the global population having been infected with latent TB.1 Despite being curable, TB remains a significant public health threat and causes 1.3 million deaths annually, making it one of the most lethal infectious diseases worldwide.1b The primary factors impeding access to a cure encompass (i) high cost of a complete TB drug regimen; (ii) poor treatment adherence; and (iii) the emergence of multidrug-resistant tuberculosis (MDR-TB).2 To make matters worse, the proportion of new TB cases due to MDR-TB has been progressively increasing over time, further aggravating this global health crisis.3(–)-Bedaquiline [1, (–)-BDQ, trade name Sirturo, shown in Fig. 1], was approved by the Food and Drug Administration in 2012 as the first new antitubercular agent for over 50 years, making it a practical treatment for MDR-TB.4 By leveraging an innovative mechanism of action, (–)-BDQ offers a more efficacious and time-efficient treatment regimen for TB, particularly for MDR-TB.5 Furthermore, the high efficiency of (–)-BDQ has stimulated the development of additional analogues, such as (–)-sudapyridine (2), TBAJ-587 (3), TBAJ-876 (4) etc.4b,6 Structurally, the (–)-BDQ family is characterized by a 1-ethanamine-1,2-diaryl-2-quinolylethanol framework that incorporates two adjacent stereocenters with a (1S,2R)-configuration, one of which is a sterically hindered oxa-quaternary center.7 Given its high efficacy against MDR-TB, developing efficient synthetic strategies for (–)-BDQ remains a highly worthwhile pursuit for organic and medicinal chemists.
 | ||
| Fig. 1 Antitubercular agent (–)-BDQ and its analogues. | ||
Over the past few decades, several synthetic methods have been reported for producing (–)-BDQ.8–11 The first enantioselective synthesis of (–)-BDQ was investigated by Shibasaki, employing enantioselective proton migration as the pivotal catalytic transformation (Scheme 1a).8 Subsequently, Chandrasekhar9 and Aggarwal10 independently developed synthetic routes for (–)-BDQ utilizing Sharpless asymmetric epoxidation and sulfur ylide-mediated asymmetric epoxidation, respectively (Scheme 1b and c). In their approaches, stereoselective allylation of the common ketone intermediate was not achieved, leading to a lack of predominant formation of the desired diastereoisomer. Additionally, Naicker reported a diastereoselective modification of the industrial route7 using a chiral base as a ligand (Scheme 1d).11a Zhang also investigated the industrial route based on strategies involving promotion of non-covalent interactions and bimetallic activation (Scheme 1d).11b However, due to the challenges of forging vicinal stereocenters and low reactivity of the substrates, the reported syntheses of (–)-BDQ still suffered from low yield, excessive use of additives, and the lack of general methods toward structurally related molecules. Consequently, there remains an urgent requirement for catalytic and unified strategies that can effectively prepare (–)-BDQ and its structurally related analogues with precise stereochemical control.
 | ||
| Scheme 1 Previous syntheses of (–)-BDQ. | ||
Herein, we report a concise six-step catalytic asymmetric synthesis of (–)-BDQ. In this process, dynamic kinetic resolution-asymmetric transfer hydrogenation (DKR-ATH) serves as the key transformation, followed by TADDOL-mediated diastereoselective allylation of the resultant α-chiral ketone to construct the (1S,2R) adjacent stereocenters with high stereochemical control. Notably, the reactivity and stereoselectivity of DKR-ATH were systematically evaluated using a series of sterically hindered N-hetero-1,2,2-triarylethanones, validating this approach as one of the most efficient strategies for constructing chiral N-hetero-1,2,2-triarylethanols. Moreover, our streamlined two-stage protocol enables the successful synthesis of (–)-sudapyridine, a compound currently in phase III clinical trials as a promising candidate for tuberculosis treatment.11b
Results and discussion
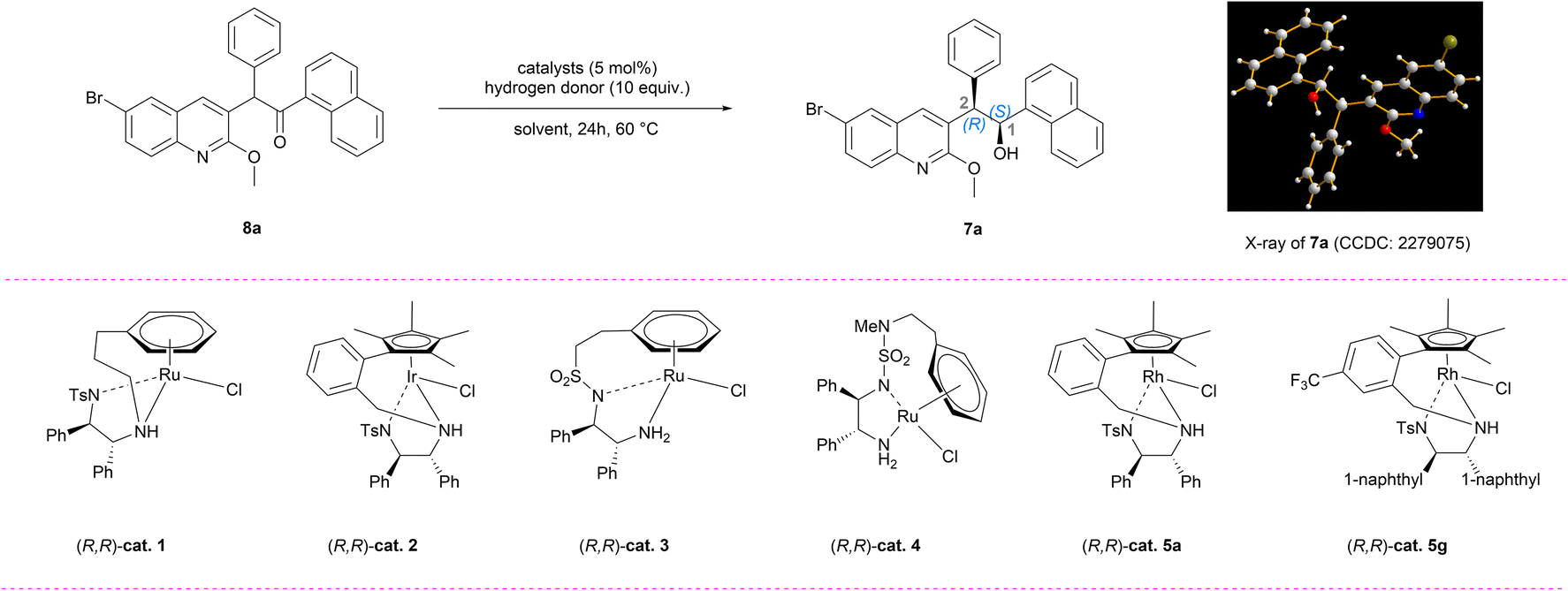
From a retrosynthetic perspective (Fig. 2), (–)-BDQ (1) can be synthesized through oxidative cleavage and reductive amination of homoallyl alcohol 5a. The crucial chiral scaffold 5a can be established via diastereoselective allylation of α-chiral ketone 6a. To achieve efficient access to this key structure, we devised a DKR-ATH followed by oxidation of the resultant chiral alcohol 7a starting from the racemic ketone 8a. This intermediate can be traced back to the quinoline 9a and naphthoyl building block through an aroylation reaction. A critical aspect for the successful implementation of this synthetic route lies in achieving high stereocontrol during the generation of secondary alcohol 7a. | ||
| Fig. 2 Retrosynthetic analysis of (–)-BDQ. | ||
Based on this synthetic design, we initially investigated the aroylation reaction between quinoline fragment 9a (ref. 12) and various naphthoyl electrophiles using LiHMDS as the base.13 Upon evaluating multiple electrophiles, naphthoyl pyrrole 10d exhibited relatively superior performance, affording 8a with 45% yield (Tables 1, entries 1–5, S1 and S2†). Additionally, the LC-MS analysis did not detect any debrominated byproduct, thereby demonstrating the practicality of utilizing silyl amide bases in this transformation.11 This outcome has prompted us to further investigate various silyl amide bases (Table 1, entries 6–7). A significant byproduct 8a′ was obtained when NaHMDS or KHMDS was used as the base. According to Baell's work,14 the higher temperature and increased basicity may promote the cyclization of 8a, leading to the formation of furan 8a′ (see the ESI†). Through extensive optimization involving temperature, additives, mole ratio, etc., the yield of 8a can be improved to 91% by employing LiHMDS as the base with 1.2 equivalents of 10d at a temperature of 50 °C (Table 1, entry 8).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Sub | Base | Temp. (°C) | Conv. (%) | Yield (8a) (%) |
| a Conversion and yield were determined by LC-MS analysis. b Toluene as the solvent and Cs2CO3 as the additive. c Base (4 equiv), 10d (1.2 equiv). d Isolated yield. | |||||
| 1 | 10a | LiHMDS | 25 | 23 | 11 |
| 2b | 10b | LiHMDS | 110 | 31 | 13 |
| 3b | 10c | LiHMDS | 110 | 23 | Trace |
| 4 | 10d | LiHMDS | 80 | 79 | 45 |
| 5 | 10e | LiHMDS | 80 | 37 | 18 |
| 6 | 10d | NaHMDS | 80 | >99 | <10 |
| 7 | 10d | KHMDS | 80 | >99 | <10 |
| 8c | 10d | LiHMDS | 50 | >99 | 96 (91)d |

With 8a in hand, we embarked on a systematic investigation into the DKR-ATH methodology. The DKR-ATH approach has been recognized as an exceptionally robust strategy for the synthesis of enantiopure alcohols, achieving remarkable success in both industrial applications and academic research.15 Notably, previously reported DKR-ATH mainly focused on various racemic ketones including acyclic ketones with α-electron-withdrawing groups,16 α-ketoesters with β-labile stereocenters,17 and α-substituted cyclic ketones,18 among others. However, the DKR-ATH transformation of sterically hindered N-hetero-1,2,2-triarylethanones for general synthesis of chiral N-hetero-1,2,2-triarylethanols remains unreported. Several intrinsic challenges have hindered progress in this area.19 First, the strong coordination ability of N-heteroarene scaffolds can lead to the deactivation of chiral catalysts. Second, the increased steric bulk of the substrates significantly reduces their reactivity. Third, achieving precise control over both enantioselectivity and diastereoselectivity by differentiating among three structurally similar aryl groups represents a formidable challenge.
We initially conducted an investigation into the racemization process of (R)-8a (93% ee), which resulted in complete racemization occurring within 1 hour in the presence of triethylamine (Scheme S1†). This result encouraged us to further explore the DKR-ATH reaction conditions. After an extensive screening of chiral catalysts and ligands (Tables S3–S7†), five Noyori–Ikariya type catalysts were identified as capable of this challenging transformation, while maintaining the integrity of the bromo group in 8a (Table 2, entries 1–5). Among them, the tethered rhodium catalyst (R,R)-cat. 5a (ref. 20) exhibited the highest level of stereoselectivity (90% ee and 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 dr) in ethyl acetate (EA) utilizing an azeotropic mixture (HCOOH:Et3N = 3:2) as the hydrogen donor at a temperature of 60 °C, albeit with limited conversion (Table 2, entry 5). The single-crystal X-ray diffraction (SCXRD) analysis of 7a confirmed the absolute configuration as (1S,2R), which could be directly used in the asymmetric synthesis of (–)-BDQ. To further enhance the conversion efficiency, we conducted an investigation into the impact of hydrogen donors and solvents on the reaction.21 Four azeotropic mixtures with varying ratios were screened using EA as the solvent, resulting in lower observed conversion rates (Table 2, entries 6–10). The reactivity using HCOONa or HCOONH4 as the hydrogen donor was also found to be very poor (Table 2, entries 11–12, <5% conv.). To our great delight, the utilization of HCOOK as a hydrogen donor facilitated a smooth reaction yielding 7a with over 98% conversion, 91% ee and 82:18 dr with EA as the solvent (Table 2, entry 13). We speculated that a 16-electron amide active species was more easily formed for this catalytic transformation in the presence of HCOOK.21c Different solvents were also investigated (Table S4†). For instance, when employing THF and toluene as solvents, the conversions could be maintained at 95% and 88%, respectively, with comparable stereoselectivity (Table 2, entries 14–15). In contrast, the utilization of the polar aprotic solvent DMF or nonpolar aprotic solvent hexane resulted in poor conversion (Table 2, entries 16–17). Various derivatives of (R,R)-cat. 5a were also tested to improve the stereoselectivity, and it was found that (R,R)-cat. 5g exhibited the highest performance with a remarkable 96% ee and 88:12 dr (Table 2, entry 18 and Table S5†). Finally, the effects of temperature, catalyst loading, and equivalents of HCOOK were evaluated (Table 2, entries 19–21), and the optimal conditions were determined to be using (R,R)-cat. 5g as a catalyst in EA solvent with HCOOK as a hydrogen donor at 60 °C for 24 hours.
:20 dr) in ethyl acetate (EA) utilizing an azeotropic mixture (HCOOH:Et3N = 3:2) as the hydrogen donor at a temperature of 60 °C, albeit with limited conversion (Table 2, entry 5). The single-crystal X-ray diffraction (SCXRD) analysis of 7a confirmed the absolute configuration as (1S,2R), which could be directly used in the asymmetric synthesis of (–)-BDQ. To further enhance the conversion efficiency, we conducted an investigation into the impact of hydrogen donors and solvents on the reaction.21 Four azeotropic mixtures with varying ratios were screened using EA as the solvent, resulting in lower observed conversion rates (Table 2, entries 6–10). The reactivity using HCOONa or HCOONH4 as the hydrogen donor was also found to be very poor (Table 2, entries 11–12, <5% conv.). To our great delight, the utilization of HCOOK as a hydrogen donor facilitated a smooth reaction yielding 7a with over 98% conversion, 91% ee and 82:18 dr with EA as the solvent (Table 2, entry 13). We speculated that a 16-electron amide active species was more easily formed for this catalytic transformation in the presence of HCOOK.21c Different solvents were also investigated (Table S4†). For instance, when employing THF and toluene as solvents, the conversions could be maintained at 95% and 88%, respectively, with comparable stereoselectivity (Table 2, entries 14–15). In contrast, the utilization of the polar aprotic solvent DMF or nonpolar aprotic solvent hexane resulted in poor conversion (Table 2, entries 16–17). Various derivatives of (R,R)-cat. 5a were also tested to improve the stereoselectivity, and it was found that (R,R)-cat. 5g exhibited the highest performance with a remarkable 96% ee and 88:12 dr (Table 2, entry 18 and Table S5†). Finally, the effects of temperature, catalyst loading, and equivalents of HCOOK were evaluated (Table 2, entries 19–21), and the optimal conditions were determined to be using (R,R)-cat. 5g as a catalyst in EA solvent with HCOOK as a hydrogen donor at 60 °C for 24 hours.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Hydrogen donor | Solvent | Conversion (%) | ee (%) | dr |
| a Reaction conditions: the reactions were carried out with a substrate (0.05 mmol) in 0.5 mL of solvent, conversions and dr were determined by 1H NMR analysis, and ee was determined by chiral HPLC analysis. b 50 °C. c cat. 5g (2 mol%). d HCOOK (5 equiv.). | ||||||
| 1 | cat. 1 | HCOOH/Et3N (3:2) |
EA | 57 | 88 | 52:48 |
| 2 | cat. 2 | HCOOH/Et3N (3:2) |
EA | 11 | 89 | 78:22 |
| 3 | cat. 3 | HCOOH/Et3N (3:2) |
EA | 51 | 87 | 59:41 |
| 4 | cat. 4 | HCOOH/Et3N (3:2) |
EA | 32 | 28 | 57:43 |
| 5 | cat. 5a | HCOOH/Et3N (3:2) |
EA | 17 | 90 | 80:20 |
| 6 | cat. 5a | HCOOH/Et3N (5:2) |
EA | <5 | — | — |
| 7 | cat. 5a | HCOOH/Et3N (1:1) |
EA | 20 | 91 | 80:20 |
| 8 | cat. 5a | HCOOH/DBU (1:1) |
EA | <5 | — | — |
| 9 | cat. 5a | HCOOH/DIPEA (1:1) |
EA | <5 | — | — |
| 10 | cat. 5a | HCOOH/DABCO (1:1) |
EA | <5 | — | — |
| 11 | cat. 5a | HCOONH4 | EA | <5 | — | — |
| 12 | cat. 5a | HCOONa | EA | <5 | — | — |
| 13 | cat. 5a | HCOOK | EA | >98 | 91 | 82:18 |
| 14 | cat. 5a | HCOOK | THF | 95 | 90 | 80:20 |
| 15 | cat. 5a | HCOOK | PhMe | 88 | 91 | 78:22 |
| 16 | cat. 5a | HCOOK | DMF | <5 | — | — |
| 17 | cat. 5a | HCOOK | n Hex | <5 | — | — |
| 18 | cat. 5g | HCOOK | EA | >98 | 96 | 88:12 |
| 19b | cat. 5g | HCOOK | EA | 80 | 96 | 88:12 |
| 20c | cat. 5g | HCOOK | EA | 67 | 96 | 88:12 |
| 21d | cat. 5g | HCOOK | EA | 86 | 96 | 88:12 |
With the optimal conditions established, we proceeded to investigate the generality of this DKR-ATH reaction by synthesizing a diverse range of racemic N-hetero-1,2,2-triarylethanones and subjecting them to comprehensive evaluation. The results obtained are presented in Scheme 2. First, substrates bearing diverse substituents on the quinoline framework were evaluated. Electron-withdrawing groups such as Cl, Br, and F, or electron-donating groups like MeO and Me at various positions of the quinoline moiety exhibited good compatibility, leading to the formation of the corresponding syn-products in high yields with excellent enantioselectivity and satisfactory diastereoselectivity (Scheme 2, 7a–7g: 75–84% yields, 92–98% ee, 83: 17–90: 10 dr). Notably, the integrity of the quinoline core remained unaffected throughout all transformations.15 The diastereoselectivity significantly decreases (67:33 dr) upon substitution of quinoline with pyridine (Scheme 2, 7h), while the enantioselectivity remains consistently high (95% ee). In contrast, favorable stereoselectivity is achieved for the substrate bearing a 4-chlorophenyl group at the 5-position of pyridine (Scheme 2, 7i, 97% ee, 85:15 dr), and the resulting syn-product can serve as a crucial intermediate in (–)-sudapyridine synthesis.16 These results (7hvs.7i) highlight the significant impact of steric hindrance differences between benzene and quinoline on diastereoselective control.
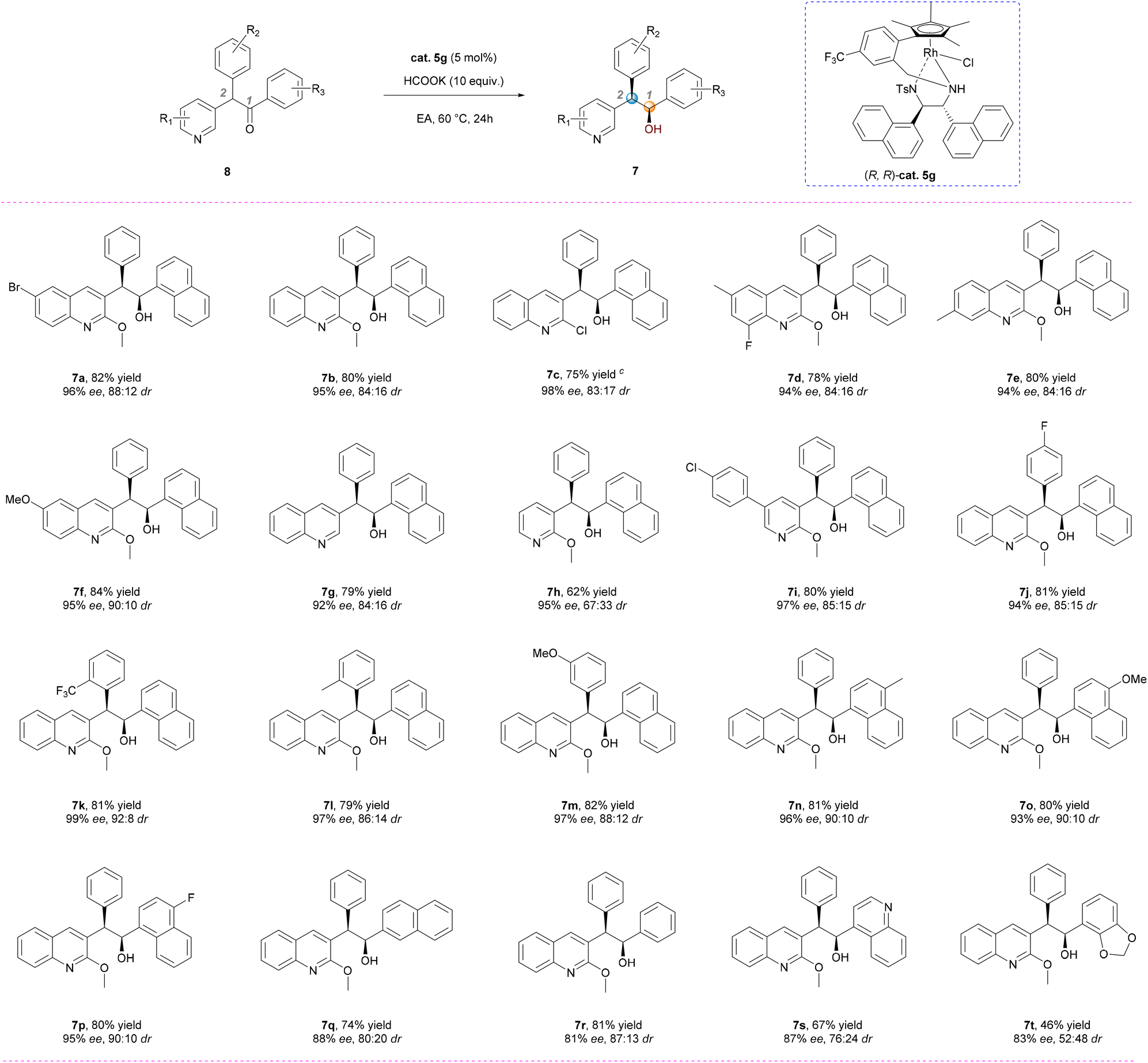 | ||
| Scheme 2 Substrate scope of the DKR-ATH.a,b aReaction conditions: the reactions were carried out with a catalyst/substrate (0.1 mmol) ratio of 1:20 in 1.0 mL of EA, HCOOK (10 equiv.) at 60 °C for 24 h; ball yields were of isolated products; dr values were determined via1H NMR analysis of the reaction mixtures; ee values were determined via HPLC analysis on a chiral stationary phase; c50 °C, 48 h. | ||
Subsequently, we investigated the impact of substituents on benzene and naphthalene. Various functional groups, including F, CF3, MeO, and Me at different positions, were found to be compatible with this transformation (Scheme 2, 7j–7p), affording yields ranging from 79% to 82%, enantiomeric excesses between 93% and 99%, and diastereomeric ratios from 85:15 to 92:8. Moreover, substrates bearing a phenyl or a 2-naphthyl group at the C1 position could also be smoothly transformed; however, slightly reduced control over enantioselectivity was observed (Scheme 2, 7q–7r). Nevertheless, the reaction of 8s or 8t with quinoline or benzo[d][1,3]dioxole at the C1 position of the substrates resulted in the formation of alcohols 7s or 7t with limited diastereoselectivities and moderate enantioselectivities. Introducing additional Lewis basic heterocycles into the substrate may lead to competitive coordination with the rhodium catalyst, thereby interfering with the chiral ligand's control over the metal center. This interference can reduce both catalytic efficiency and the level of chiral induction.
With the abundant key intermediate 7a in hand, chiral ketone 6a can be efficiently obtained using Dess–Martin periodinane (DMP) as the oxidant, without compromising enantioselectivity (93% yield, 96% ee). The SCXRD analysis of compound 6a further confirmed its (R)-configuration (Fig. S1†). After establishing the tertiary carbon stereocenter, our focus shifted towards constructing the quaternary stereocenter through a diastereoselective allylation of 6a. In accordance with Chandrasekhar's work,9 the allylation product can be delivered with the desired diastereomer as the minor isomer by employing allylzinc bromide as a nucleophile (Table 3, entry 1). To enhance the yield and selectivity towards the desired isomer, we conducted an extensive screening of diverse ligands and commercially available allylation reagents commonly employed in asymmetric nucleophilic addition of carbonyl compounds (Fig. S4†).22 Regrettably, thorough evaluation of diastereoselective allylation using 6a as a substrate clearly demonstrated that the majority of these ligands exhibited no reactivity, with only two reaction systems being confirmed as effective for this transformation (Table 3, entries 2–3). The allylzinc reagent with bisoxazoline ligand L8 exhibited a relatively low yield (38%) and suboptimal diastereoselectivity (52:48 dr). Conversely, when employing TADDOL-type ligand L9a as the ligand and allylmagnesium chloride as the nucleophile, the allylation product was obtained in a 65:35 dr (desired diastereomer 5a as the major isomer) despite still yielding at a lower level (Table 3, entry 3).22a It is noteworthy that this transformation did not affect the configuration of tertiary carbon. Using ent-L9a as the ligand, the allylation product 5a’ as the major isomer was obtained with 78:22 dr (Table 3, entry 4). Subsequently, we explored other dihydroxy ligands such as SPINEL, H8BINOL, and BINOL ligands (L10–L12) in conjunction with allylmagnesium chloride as the nucleophile; however, none of them yielded the desired products (Table 3, entries 5 and 6). These findings underscore the key role of L9a in controlling reactivity and diastereoselectivity.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | X | Ligand | Sol. | Conv.b (%) | Yieldb (%) | drc |
| a Reaction conditions: the reactions were carried out with 6a (0.1 mmol), nucleophile (3 equiv.), ligand (1 equiv.). b Conversions and yields (four isomers) were determined by 1H NMR analysis. c dr was determined by chiral HPLC analysis. d r. t. and CuBr·Me2S as the additive. e Allylmagnesium bromide (1.0 M in Et2O). f 0 °C. g Allylmagnesium chloride (2.0 M in THF). h L9a (2 equiv.), allylmagnesium chloride (6 equiv.). i L9a (3 equiv.), allylmagnesium chloride (9 equiv.). | ||||||
| 1d | ZnBr | — | THF | 57 | 32 | 47:53 |
| 2 | ZnBr | L8 | THF | 61 | 38 | 52:48 |
| 3 | MgCl | L9a | THF | 46 | 34 | 65:35 |
| 4 | MgCl | ent-L9a | THF | 43 | 30 | 22:78 |
| 5 | MgCl | L10 | THF | 0 | — | — |
| 6 | MgCl | L11 | THF | 0 | — | — |
| 7 | MgCl | L12 | THF | 0 | — | — |
| 8e | MgBr | L9a | THF | 41 | 33 | 52:48 |
| 9 | MgCl | L9a | DCM | 45 | 33 | 82:18 |
| 10 | MgCl | L9a | Et2O | 0 | — | — |
| 11f | MgCl | L9a | DCM | 0 | — | — |
| 12g | MgCl | L9a | DCM | 42 | 31 | 89:11 |
| 13g | MgCl | L9b | DCM | 0 | — | — |
| 14g | MgCl | L9c | DCM | 0 | — | — |
| 15g,h | MgCl | L9a | DCM | 84 | 72 | 91:9 |
| 16g,i | MgCl | L9a | DCM | 99 | 86 | 91:9 |
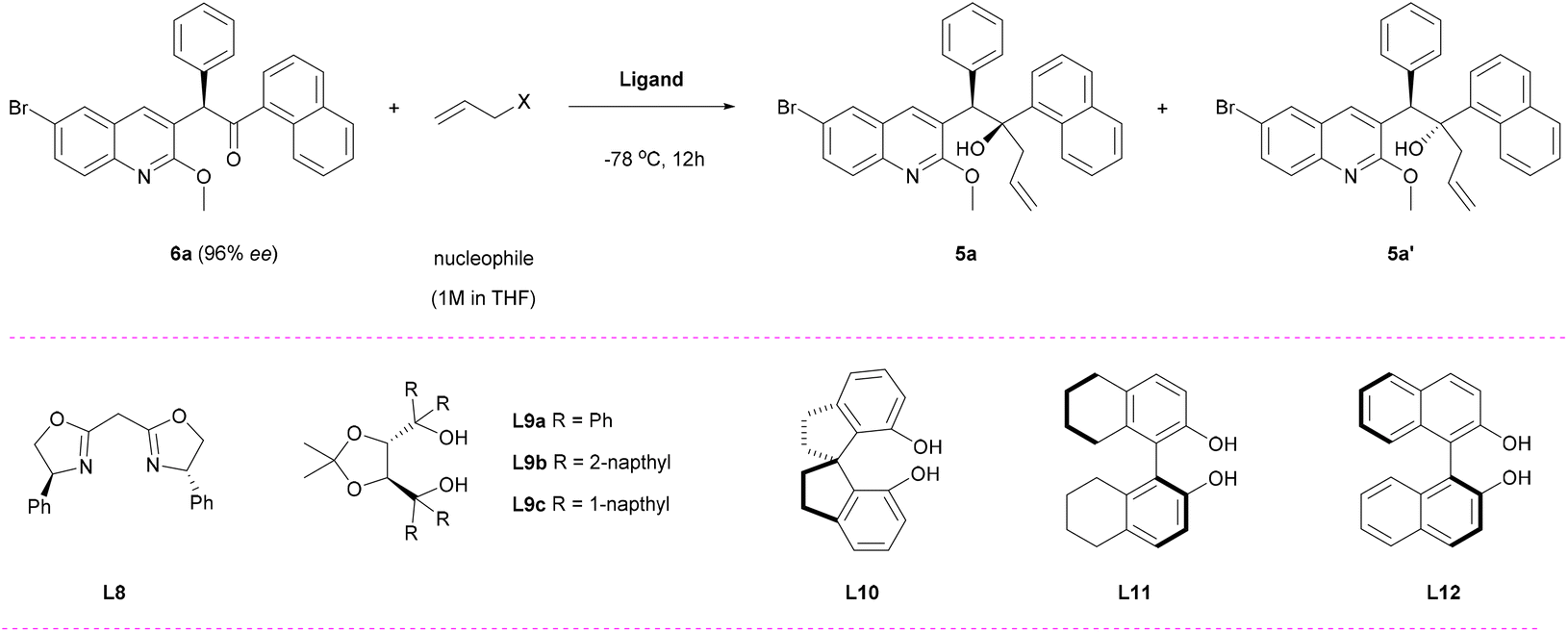
According to previous reports, the stereochemical outcome can be dramatically influenced by various factors such as counterions, solvents, temperature, etc. when employing allylmagnesium reagents as nucleophiles.22c Thus, we conducted a study on different reaction parameters (Table 3, entries 7–12, Table S8†). To our great satisfaction, the allylation product was obtained with 89:11 dr using dichloromethane (DCM) as the solvent and allylmagnesium chloride (2.0 M in THF) as the nucleophile (Table 3, entry 12). Unexpectedly, the reaction failed to occur when L9b and L9c were used as ligands, which could be attributed to steric hindrance causing inhibition of reactivity by the ligands (Table 3, entries 13 and 14). We further investigated the impact of nucleophile and ligand dosage (Table 3, entries 15 and 16). Finally, the optimal reaction conditions were determined to be using L9a as the ligand (3 equiv.), DCM as the solvent, and allylmagnesium chloride (9 equiv.) as the nucleophile, resulting in a desired isomer yield of 86% with 91:9 dr.
After successfully constructing the key functionalized homoallyl alcohol 5a, which possesses two desired adjacent stereocenters, our focus shifted toward completing the total synthesis of (–)-BDQ (1) (Scheme 3a). The oxidative cleavage of 5a using K2OsO4·H2O/NaIO4 resulted in the formation of an intermediate aldehyde. Without undergoing purification, the crude product was directly subjected to treatment with dimethylamine and Na(OAc)3BH to obtain the final product in 58% yield over two steps.
 | ||
| Scheme 3 Completion of the asymmetric synthesis of (a) (–)-BDQ and (b) (–)-sudapyridine. | ||
The generality of this tactic could be further demonstrated in the synthesis of (–)-sudapyridine (Scheme. 3b). Initially, the palladium-catalyzed Heck reaction between 10i and 9i under reaction conditions developed by Baell and co-workers in toluene proceeded smoothly, affording 8i in 74% yield.14 Following a similar pathway to that of (–)-BDQ (1), (–)-sudapyridine (2) was obtained with 18% overall yield in 6 steps. We have successfully accomplished the highly stereoselective synthesis of (–)-BDQ and its analogues (with 96% ee and 94% ee, respectively) using this strategy for the first time. More importantly, this synthetic route achieves the highest overall yields (34% and 18%, respectively) compared to all previously reported methods.8–11
Conclusions
In summary, we successfully accomplished a 6-step catalytic asymmetric synthesis of (–)-bedaquiline (1) and (–)-sudapyridine (2), employing Rh-catalyzed DKR-ATH and TADDOL ligand-mediated diastereoselective allylation as two practical transformations, with 34% and 18% overall yields, respectively. Additionally, the reactivity and stereoselectivity of DKR-ATH for sterically hindered N-hetero-1,2,2-triarylethanones were thoroughly evaluated. In most cases, excellent enantioselectivities ranging from 92% to 99% ee and diastereomeric ratios between 84:16 to 92:8 were achieved, providing a robust and straightforward approach for the production of enantiomerically pure N-hetero-1,2,2-triarylethanols. It is worth noting that all reaction conditions employed in this strategy are convenient, thus highlighting its utility in the synthesis of bedaquiline and related analogues for drug discovery.
Data availability
All experimental and characterization data, and NMR spectra are available in the ESI.† Crystallographic data for compound 7a and 6a have been deposited at the Cambridge Crystallographic Data Centre under accession number CCDC 2279075 and 2278094.Author contributions
Jiyao Han carried out the experimental work and wrote the first draft. Dongliang Zhang and Yuan Tao performed experimental testing. Pei Tang revised the paper. Fener Chen designed the project and supervised the work. All the authors discussed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2021YFF0600704).Notes and references
- (a) S. Keshavjee and P. E. Farmer, N. Engl. J. Med., 2012, 367, 931 CrossRef CAS PubMed; (b) T. Smith and B. Aldridge, Science, 2019, 364, 1234 CrossRef CAS PubMed; (c) K. Q. Abdool and K. S. S. Abdool, Science, 2020, 369, 366 CrossRef PubMed; (d) World Health Organization, 2023, https://www.who.int/news-room/fact-sheets/detail/tuberculosis, accessed 2023-11-07 Search PubMed.
- J. M. S. Robey, S. Maity, S. L. Aleshire, A. Ghosh, A. K. Yadaw, S. Roy, S. J. Mear, T. F. Jamison, G. Sirasani, C. H. Senanayake, R. W. Stringham, B. F. Gupton, K. O. Donsbach, R. C. Nelson and C. S. Shanahan, Org. Process Res. Dev., 2023, 27, 2146 CrossRef CAS PubMed.
- G. F. S. Fernandes, A. M. Thompson, D. Castagnolo, W. A. Denny and J. L. Dos Santos, J. Med. Chem., 2022, 65, 7489 CrossRef CAS PubMed.
- (a) K. Andries, P. Verhasselt, J. Guillemont, H. W. H. Gohlmann, J.-M. Neefs, H. Winkler, J. Van Gestel, P. Timmerman, M. Zhu, E. Lee, P. Williams, D. de Chaffoy, E. Huitric, S. Hoffner, E. Cambau, C. Truffot-Pernot, N. Lounis and V. Jarlier, Science, 2005, 307, 223 CrossRef CAS PubMed; (b) A. S. T. Tong, P. J. Choi, A. Blaser, H. S. Sutherland, S. K. Y. Tsang, J. Guillemont, M. Motte, C. B. Cooper, K. Andries, W. Van Den Broeck, S. G. Franzblau, A. M. Upton, W. A. Denny, B. D. Palmer and D. Conole, ACS Med. Chem. Lett., 2017, 8, 1019 CrossRef CAS PubMed.
- (a) P. J. Choi, D. Conole, H. S. Sutherland, A. Blaser, A. S. T. Tong, C. B. Cooper, A. M. Upton, B. D. Palmer and W. A. Denny, Molecules, 2020, 25, 1423 CrossRef CAS PubMed; (b) M. B. Calvert, D. P. Furkert, C. B. Cooper and M. A. Brimble, Bioorg. Med. Chem. Lett., 2020, 30, 127 CrossRef PubMed.
- (a) T. Ahmad, F. Gao, J. Li, Z. Zhang, T. Song, Q. Yuan and W. Zhang, J. Org. Chem., 2023, 88, 7601 CrossRef CAS PubMed; (b) J. Li, F. Gao, T. Ahmad, Y. Luo, Z. Zhang, Q. Yuan and W. Zhang, Chin. J.Chem., 2023, 41, 1319 CrossRef CAS.
- J. Guillemont, C. Meyer, A. Poncelet, X. Bourdrez and K. Andries, Future Med. Chem., 2011, 3, 1345 CrossRef CAS PubMed.
- Y. Saga, R. Motoki, S. Makino, Y. Shimizu, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2010, 132, 7905 CrossRef CAS PubMed.
- S. Chandrasekhar, G. S. K. Babu and D. K. Mohapatra, Eur. J. Org Chem., 2011, 2011, 2057 CrossRef.
- M. Bashir, M. Arshad, R. Begum and V. K. Aggarwal, Org. Lett., 2023, 25, 4281 CrossRef CAS PubMed.
- (a) H. Lubanyana, P. I. Arvidsson, T. Govender, H. G. Kruger and T. Naicker, ACS Omega, 2020, 5, 3607 CrossRef CAS PubMed; (b) F. Gao, J. Li, T. Ahmad, Y. Luo, Z. Zhang, Q. Yuan, X. Huo, T. Song and W. Zhang, Sci. China: Chem., 2022, 65, 1968 CrossRef CAS.
- S. J. Mear, T. Lucas, G. P. Ahlqvist, J. M. S. Robey, J.-P. Dietz, P. V. Khairnar, S. Maity, C. Williams, D. R. Snead, R. C. Nelson, T. Opatz and T. F. Jamison, Chem.–Eur. J., 2022, 28, e202201311 CrossRef CAS PubMed.
- (a) D. A. Evans, G. Borg and K. A. Scheidt, Angew. Chem., Int. Ed., 2002, 41, 3188 CrossRef CAS; (b) F. Yang, D. Zou, S. Chen, H. Wang, Y. Zhao, L. Zhao, L. Li, J. Li and P. J. Walsh, Adv. Synth. Catal., 2020, 362, 3423 CrossRef CAS; (c) C.-C. Bao, Y.-L. Luo, H.-Z. Du and B.-T. Guan, Sci. China: Chem., 2021, 64, 1349 CrossRef CAS; (d) Y. Gu, Z. Zhang, Y.-E. Wang, Z. Dai, Y. Yuan, D. Xiong, J. Li, P. J. Walsh and J. J. Mao, Org. Chem., 2022, 87, 406 CrossRef CAS PubMed.
- D. L. Priebbenow, L. Barbaro and J. B. Baell, Org. Biomol. Chem., 2016, 14, 9622 RSC.
- (a) R. Noyori and T. Ohkuma, Angew. Chem., Int. Ed., 2001, 40, 40 CrossRef CAS; (b) J.-P. Genet, Acc. Chem. Res., 2003, 36, 908 CrossRef CAS PubMed; (c) T. Ikariya and A. J. Blacker, Acc. Chem. Res., 2007, 40, 1300 CrossRef CAS PubMed; (d) J. Lu, J. Dimroth and M. Weck, J. Am. Chem. Soc., 2015, 137, 12984 CrossRef CAS PubMed; (e) Y. Yuki, T. Touge, H. Nara, K. Matsumura, M. Fujiwhara, Y. Kayaki and T. Ikariya, Adv. Synth. Catal., 2018, 360, 568 CrossRef CAS; (f) V. K. Vyas, G. J. Clarkson and M. Wills, Angew. Chem., Int. Ed., 2020, 59, 14265 CrossRef CAS PubMed; (g) H. Wang, J. Wen and X. Zhang, Chem. Rev., 2021, 121, 7530 CrossRef CAS PubMed.
- (a) G. Sun, Z. Zhou, Z. Luo, H. Wang, L. Chen, Y. Xu, S. Li, W. Jian, J. Zeng, B. Hu, X. Han, Y. Lin and Z. Wang, Org. Lett., 2017, 19, 4339 CrossRef CAS PubMed; (b) L. Zhang, Z. Wang, Z. Han and K. Ding, Angew. Chem., Int. Ed., 2020, 59, 15565 CrossRef CAS PubMed; (c) M. Sterle, M. Hus, M. Lozinsek, A. Zega and A. E. Cotman, ACS Catal., 2023, 13, 6242 CrossRef CAS PubMed.
- (a) K. M. Steward, E. C. Gentry and J. S. Johnson, J. Am. Chem. Soc., 2012, 134, 7329 CrossRef CAS PubMed; (b) K. M. Steward, M. T. Corbett, C. G. Goodman and J. S. Johnson, J. Am. Chem. Soc., 2012, 134, 20197 CrossRef CAS PubMed; (c) M. T. Corbett and J. S. Johnson, J. Am. Chem. Soc., 2013, 135, 594 CrossRef CAS PubMed; (d) B. He, L.-S. Zheng, P. Phansavath and V. Ratovelomanana-Vidal, ChemSusChem, 2019, 12, 3032–3036 CrossRef CAS PubMed.
- (a) V. K. Vyas and B. M. Bhanage, Org. Lett., 2016, 18, 6436 CrossRef CAS PubMed; (b) A. E. Cotman, B. Modec and B. Mohar, Org. Lett., 2018, 20, 2921 CrossRef CAS PubMed; (c) D. Yang, A.-J. Yang, Y. Chen, J.-H. Xie and Q.-L. Zhou, Org. Lett., 2021, 23, 1616 CrossRef CAS PubMed; (d) Y. Xu, Y. Luo, J. Ye, D. Liu and W. Zhang, J. Am. Chem. Soc., 2023, 145, 21176 CrossRef CAS PubMed.
- L.-X. Ruan, B. Sun, J.-M. Liu and S.-L. Shi, Science, 2023, 379, 662 CrossRef CAS PubMed.
- (a) D. S. Matharu, D. J. Morris, A. M. Kawamoto, G. J. Clarkson and M. Wills, Org. Lett., 2005, 7, 5489 CrossRef CAS PubMed; (b) P.-G. Cheverria, C. Férard, P. Phansavath and V. Ratovelomanana-Vidal, Catal. Commun., 2015, 62, 95 CrossRef; (c) L.-S. Zheng, Q. Llopis, P.-G. Echeverria, C. Ferard, G. Guillamot, P. Phansavath and V. Ratovelomanana-Vidal, J. Org. Chem., 2017, 82, 5607 CrossRef CAS PubMed.
- (a) T. Wang, L.-G. Zhuo, Z. Li, F. Chen, Z.-Y. Ding, Y. He, Q.-H. Fan, J. Xiang, Z.-X. Yu and A. S. C. Chan, J. Am. Chem. Soc., 2011, 133, 9878 CrossRef CAS PubMed; (b) W. Ma, J. Zhang, C. Xu, F. Chen, Y.-M. He and Q.-H. Fan, Angew. Chem., Int. Ed., 2016, 55, 12891 CrossRef CAS PubMed; (c) F. Wang, Z. Zhang, Y. Chen, R.-V. Virginie, P. Yu, G.-Q. Chen and X. Zhang, Nat. Commun., 2022, 13, 7794 CrossRef CAS PubMed.
- (a) B. Weber and D. Seebach, Tetrahedron, 1994, 50, 6117 CrossRef CAS; (b) R. Wada, K. Oisaki, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2004, 126, 8910 CrossRef CAS PubMed; (c) N. D. Bartolo, J. A. Read, E. M. Valentín and K. A. Woerpel, Chem. Rev., 2020, 120, 1513 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: All experimental and characterization data, and NMR spectra. See DOI: https://doi.org/10.1039/d5sc03865k |
| This journal is © The Royal Society of Chemistry 2025 |