
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAluminacyclopropene–phosphine complex: a carbene-exchange reagent†
Huihui Xu‡
a,
José Miguel León Baeza‡ab,
Antoine Baceiredoa,
René Segundo Rojas Guerrero *b,
Nathalie Saffon-Merceronc,
Vicenç Branchadelld and
Tsuyoshi Kato*a
*b,
Nathalie Saffon-Merceronc,
Vicenç Branchadelld and
Tsuyoshi Kato*a
aLaboratoire Hétérochimie Fondamentale et Appliquée (UMR 5069), Université de Toulouse, CNRS, 118 Route de Narbonne, F-31062 Toulouse, France. E-mail: tsuyoshi.kato@utoulouse.fr
bDepartamento de Química Inorganica, Facultad de Química y de Farmacia, Pontificia Universidad Catolica de Chile, Casilla 306, Santiago 22, Chile. E-mail: rrojasg@uc.cl
cInstitut de Chimie de Toulouse (FR 2599), Université de Toulouse, CNRS, 118 Route de Narbonne, F-31062 Toulouse, France
dDepartament de Química, Universitat Autònoma de Barcelona, 08193 Bellaterra, Spain
First published on 30th June 2025
Abstract
N-heterocyclic carbenes (NHCs) are extensively used as auxiliary ligands or organocatalysts thanks to their remarkable stability. However, due to their high structural stability, applications involving skeletal modifications without losing the low-valent nature (carbene exchange reactions) remain extremely rare. We report here that aluminacyclopropene–phosphine complex 1 promotes original “carbene-to-carbene” transformations of stable carbenes. Indeed, the Al(III)–phosphine Lewis pair complex 1, due to its high ring strain, is able to react with NHC 2 to produce a cyclopropenylidene (4), via atomic carbon transfer from the NHC to the η2-coordinated alkyne fragment, within the coordination sphere of the Al(III) center. Moreover, the complex 1 transforms the stable (diamino)cyclopropenylidene 7 into a more reactive acyclic (amino)carbene 8, which has been isolated in crystalline form.
Introduction
Stable carbenes, featuring a divalent carbon center well stabilized by substitution and structural effects, have become indispensable tools in chemistry due to their ease of handling and synthetic usefulness as reagents with the properties of great modularity through substitution patterns. A large variety of stable carbenes, such as N-heterocyclic carbenes (NHCs), are now available and found in applications, mainly as chemical reagents, ligands1 and organocatalysts.2 Usually, their reactions take place at the divalent carbene center, accompanied by its oxidation [C(II) → C(IV) transformation], without skeletal modifications. Although still rare, reactions involving skeletal modifications, which enable them to be used as unique low-valent carbon sources, are also emerging. Indeed, Tobisu et al. have recently demonstrated that the C(II) atom of NHCs can be used as a formal atomic carbon [“C(0)”] source, showing the reaction of NHCs with acrylamides via single carbon atom doping annulation to form a γ-lactam and release of diimine (Fig. 1(a)).3 More recently, Radius' group also described the reaction of NHCs as an atomic carbon source with Me3N·AlH3 to produce ethylene and a diaminoalane dimer (Fig. 1(b)).4 Such behavior of NHCs is rather surprising, given their high framework stability and limited examples of ring structure modifications,5–8 in agreement with their current extensive applications as persistent ligands and organocatalysts.1,2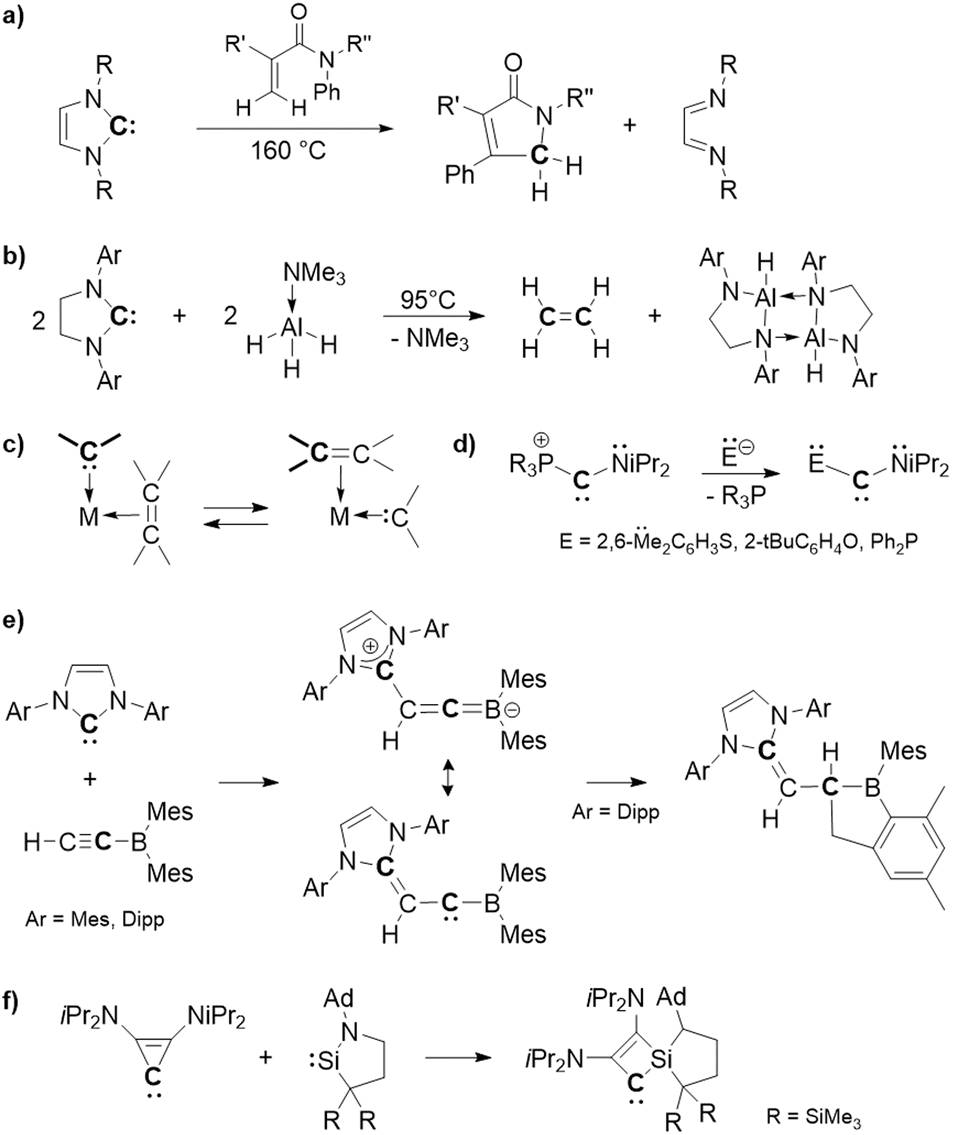 | ||
| Fig. 1 Applications of stable cyclic carbenes as atomic carbon sources (a and b) and carbene-to-carbene transformations (c–f). | ||
In addition to these [C(II)/“C(0)” → C(IV)] transformations, reactions without losing their divalent nature could be expected, allowing the synthesis of new carbenes (carbene-to-carbene transformations). However, although the olefin metathesis is a well-known catalytic process that promotes the interchange of carbenes in the coordination sphere of a transition metal (Fig. 1(c)), such a transformation with stable carbenes is rather uncommon. A rare example is the substitution reaction at the carbene center of (phosphonio)(amino)carbene, reported by Bertrand et al., allowing the synthesis of various stable push–push carbenes from a single carbene precursor (Fig. 1(d)).9 Braunschweig described a reaction of an NHC with a borylacetylene to generate a zwitterionic allene, whose central carbon exhibits carbene-type reactivity, despite no skeletal modification involved in this process (Fig. 1(e)).10 Koike and Iwamoto also successfully synthesized a stable cyclobutenylidene via carbene-to-carbene ring expansion of stable diaminocyclopropenylidene through its reaction with a cyclic (amino)(alkyl)silylene (Fig. 1(f)).11 We report here a unique behavior of strained Al(III)–phosphine Lewis pair complex 1 being able to promote “carbene-to-carbene” transformations of stable carbenes (NHC 2 and cyclopropenylidenDe 7) via skeletal recasting.
Results and discussion
As demonstrated by the work of Radius' group, the use of Al(III) compounds is a viable strategy to achieve skeletal modifications of NHCs by overcoming their excessive framework stability.4–8,12 Hence, we considered the use of phosphine-aluminocyclopropene Lewis pair complex 1, expecting greater reactivity thanks to the high ring strain of the three-membered cyclic structure.13 Indeed, aluminocyclopropene 1, a formal η2-alkyne–alumylene complex,14,15 reacts at room temperature (RT) with NHC 2 to give the Al(III) complex 3 featuring a phosphine-supported cyclopropenylidene moiety (Scheme 1). Formally, to obtain 3, both C–N bonds of the NHC were cleaved and the divalent carbon atom was transferred to the η2-coordinated alkyne fragment at the aluminum center to generate the transient cyclopropenylidene–Al(III) complex 4, which is stabilized by the coordination of the phosphine ligand. The resulting complex 3 is stable at RT and was isolated as brown crystals in good yield (58%) from a pentane solution at −30 °C. | ||
| Scheme 1 The synthesis of phosphine-stabilized cyclopropenylidene Al(III) complex 3 and its isomerization into polycyclic Al(III) complex 5. | ||
The 31P-NMR spectrum of 3 displays only a sharp signal at 68.0 ppm, indicating that the reaction proceeds in a diastereoselective manner. In the 13C-NMR spectrum, the signal of the sp2-C atom attached to the Al center could not be observed at either 25 °C or −70 °C, probably due to a broadening of the signal induced by the C–Al connection. However, a HMBC experiment yielded a correlation between a carbon signal at 119.2 ppm and the proton signal of CH2–ethyl groups. The other three-membered ring sp2- and sp3-carbon atoms were observed at 130.1 ppm (singlet) and at 26.3 ppm (doublet, 1JCP = 108.0 Hz), respectively. The X-ray structure of 3 confirms the presence of an Al center substituted by diamino and cyclopropenyl moieties and the migration of the phosphine ligand from Al to the carbon center of the cyclopropenyl group to form a seven-membered ring (Fig. 2-left). The Al–C1 bond distance [1.959(1) Å] corresponds to covalent Al–C(sp2) single bonds. In contrast, the elongated Al–N1 bond [1.936(1) Å] relative to other Al–N bonds [Al–N2 1.841(1) Å and Al–N3 1.842(1) Å] is probably due to its dative bond character enhanced by the π-attracting effect of the phosphonio group in addition to the steric repulsion between the two bulky groups, the Dipp substituent and the diamino moiety. As expected, the computationally optimized structure of cyclopropenylidene complex 4 exhibits an elongated Al–C1 bond [2.031 Å] and a shorter Al–N1 bond [1.889 Å] (Fig. 2, right) compared to those observed for 3. Certainly, due to the aromatic character of the cyclopropenylidene fragment in 4,16 the stabilization energy gained by the phosphine coordination to give 3 is relatively small (ΔG4→3 = −16.2 kcal mol−1, Fig. 3), suggesting a possible equilibrium between complexes 3 and 4. However, all efforts to find a suitable reactivity of 3 to experimentally prove the generation of intermediate 4 in order to use it as a source of cyclopropenylidene have failed.
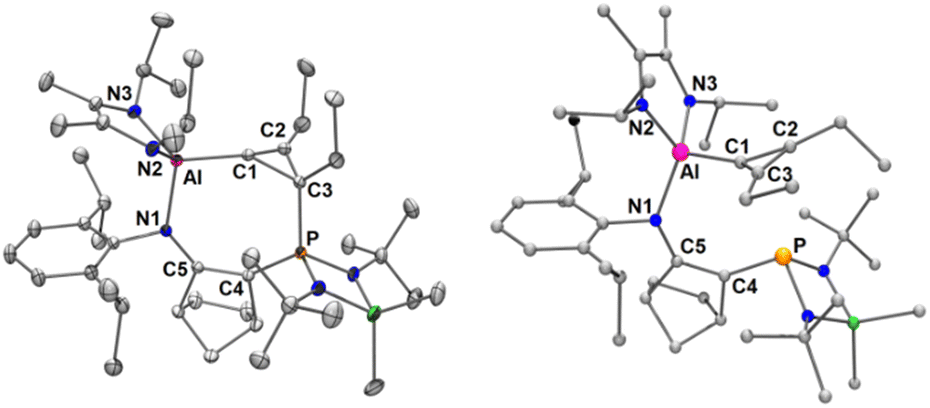 | ||
| Fig. 2 Molecular structure of 3 obtained by X-ray diffraction analysis (left). Thermal ellipsoids represent 30% probability. Structure of 4 calculated at the M06-2X/6-31G(d) level of theory (right). H and disordered atoms are omitted from both structures for clarity. Selected bond lengths [Å] and angles [°]: 3: Al–N2 1.841(1), Al–N3 1.842(1), N1–Al 1.936(1), Al–C1 1.959(1), C1–C2 1.298(2), C2–C3 1.501(2), C3–C1 1.583(2), C3–P 1.789(1), C3–C1–C2 61.89(8), C1–C2–C3 68.44(9), C2–C3–C1 49.67(7), N1–Al–C1 99.10(4), C1–C3–P 114.98(8), C2–C3–P 118.76(9), C3–P–C4 113.47(5), P–C4–C5 138.62(9), C4–C5–N1 135.32(10), C5–N1–Al 131.69(7). Σ°N2 = 340.90°. Σ°N3 = 352.62°. Σ°C1 = 355.39°. 4: Al–N2 1.831, Al–N3 1.840, N1–Al 1.889, Al–C1 2.031, C1–C2 1.392, C2–C3 1.346, C3–C1 1.397. Σ°C1 = 355.39°. | ||
 | ||
| Fig. 3 Calculated pathway for the reaction of 1 with NHC 2 (M06-2X/Def2TZVP//M06-2X/6-31G(d) level). Gibbs energies of intermediates (and transition states) are in kcal mol−1. Compounds 3, 5 and 6, outlined by a dotted line, are isolable. | ||
The transformation of 1/NHC into 4 can be regarded as a single atomic carbon transfer from the NHC to the η2-coordinated alkyne ligand of the Al(III) complex 1, resulting in a formal carbene exchange reaction from NHC to cyclopropenylidene (Fig. 3). DFT calculations predict that the reaction starts with the coordination of NHC to the Al center of 1 (INT1), which triggers two ring expansion reactions consecutively, first in the three-membered aluminocyclopropene ring and then in the NHC-five-membered ring, affording bicyclic intermediate INT3. Further isomerization of INT3 via two successive ring contractions, involving two 1,2-migrations of ethylene carbon (Al → C) (INT4) and the amino group (C → Al) in opposite directions from the previous ones, promoted by the phosphine ligand, to generate cyclopropenylidene INT5 formally stabilized by an Al(III)–phosphine Lewis pair. Then, the phosphine ligand migration, via the intermediacy of cyclopropenylidene–Al(III) complex 4, gives the more stable isomer 3. All steps, except for the transformation of INT3 to INT4 via a ring-contraction to form a three-membered ring (ΔGINT3→INT4 = +15.0 kcal mol−1), are exergonic processes, and this carbene exchange reaction is thermodynamically favored (ΔG1+2→3 = −20.3 kcal mol−1). A related reaction of NHC-supported aluminocyclopropene, proceeding in a similar way, has been recently reported by Liu et al.17 As expected by the following endergonic and rate-determining steps (INT3 → INT4 and INT5 → 4, respectively) for further evolution, INT3 should be a relatively long-lived intermediate. Indeed, monitoring the reaction by 31P-NMR spectroscopy allowed the detection of a signal at 61.0 ppm corresponding to the fused-tricyclic compound 6, which is the phosphine-stabilized form of INT3. Complex 6 has been successfully isolated by crystallization from the reaction mixture in pentane solution at −30 °C, and its structure was confirmed by X-ray diffraction analysis (Fig. 4, right). Given the poorly exergonic nature of INT3 → 6 transformation (ΔGINT3→6 = −7.3 kcal mol−1), this stabilization process is expected to be reversible. Indeed, although 6 is stable at −40 °C in THF and can be fully characterized by NMR, it isomerizes into Al(III) complex 3 at RT in C6D6. This experimental result is in good agreement with the reaction pathway predicted by calculations.
 | ||
| Fig. 4 Molecular structures of 5 (left) and 6 (right). Thermal ellipsoids represent 30% probability. H and disordered atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: 5: Al–N1 1.886(2), Al–N2 1.850(2), Al–N3 2.026(2), Al–C1 1.895(2), C1–C3 1.463(2), C3–C2 1.361(3), C2–N3 1.507(2), C1–P 1.670(2), N3–Al–C1 90.96(7), N3–Al–N2 86.66(6), N1–Al–C1 108.23(7), N1–Al–N2 120.12(7), Al–C1–C3 104.68(12), Al–C1–P 122.76(10), P–C1–C3 131.03(13). 6: C1–N3 1.398(9), C1–Al 1.849(8), Al–N2 1.821(7), Al–C3 2.145(8), C3–C2 1.534(10), C2–C1 1.400(10), C3–P 1.824(8), N1–Al 1.865(6), Al–C3–C2 82.6(4), C3–C2–C1 106.5(7), C2–C1–N3 135.2(8), C2–C1–Al 98.0(5), C1–Al–C3 71.7(3). | ||
Complex 3 is stable at room temperature, but it evolves above 60 °C (4 h at 80 °C) via a fusion of three- and five-membered rings, affording the tricyclic Al(III)-substituted phosphonium ylide 5 (Scheme 1). The 31P-NMR chemical shift of 5 (10.7 ppm) is high-field shifted compared to those of isomers 3 and 6 (68.0 and 61.0 ppm, respectively). The 13C-NMR spectrum exhibits a large doublet signal at 56.1 ppm, corresponding to the Al-ylidic carbon atom, with a large phosphorus–carbon coupling constant (1JCP = 115.1 Hz). The X-ray structure of 5 reveals a fused tricyclic structure with a tetracoordinated Al center (Fig. 4, left). Calculations on the reaction pathway predict that the reaction starts with the reformation of less stable isomer INT5 from 3 with the migration of the phosphine ligand through 4 and then INT5 undergoes a ring fusion via a formal substitution reaction at the vinyl carbon of the cyclopropylidene moiety by the adjacent amino group of the diamino ligand to directly produce the final tricyclic Al(III) complex 5 (Fig. 3). Careful analysis of the transition state (TSINT5→5) indicates that the ring fusion is triggered by ring opening of cyclopropylidene, formally activated by an Al/P Lewis pair, via heterolytic C–C bond cleavage (Fig. 3) and therefore, the high ring strain of the cyclopropylidene moiety is an important driving force for such a peculiar rearrangement.
In marked contrast to the reaction of aluminocyclopropene–phosphine complex 1 with NHC 2 generating a carbene complex, its reaction with stable (diamino)cyclopropenylidene 7 affords an acyclic (amino)carbene 8, as a mixture of two diastereomers (80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20). Carbene 8 is stable at RT and was isolated as red crystals from a concentrated pentane solution at −25 °C (yield: 85%). The 31P-NMR spectrum exhibits two singlet signals corresponding to the two diastereomers at 20.1 ppm and 22.5 ppm, respectively, with an 80:20 ratio. In the 13C-NMR spectrum, two doublet signals corresponding to the carbene carbon atom appear at 296 ppm (JCP = 17.6 Hz) and 301 ppm (JCP = 17.6 Hz), respectively, chemical shifts within the range of (amino)carbenes.18–21 The X-ray structure of 8 (Fig. 5) exhibits a short C3–N2 bond length [1.285(3) Å], indicating double bond character, and a planar geometry around the N2 atom (ΣN2°: 360°). These are consistent with the stabilization of carbene by π-electron donation from the amino group. The (amino)carbene 8 also shows a widened carbene angle [125.7(2)°] and a shorter C2–C3 bond [1.415(3) Å] compared to those of previously reported (amino)(aryl)carbenes [Caryl–Ccarbene–N: 117.9–121.0° and Caryl–Ccarbene: 1.453–1.469 Å].18 This suggests an enhanced π-interaction of the carbene lone pair with the adjacent phosphonio-substituted Al-heterocycle with a strong π-attracting character. Indeed, the 13C-NMR signal for the C1 atom adjacent to the P/Al atoms (102.0 ppm) is upfield shifted compared to those observed for its derivatives 9–11 (137.8–140.1 ppm). Hence, 8 is regarded as a push–pull carbene rather than a push–spectator carbene. As expected, with a singlet-triplet energy gap (ΔES–T = 31.0 kcal mol−1) of 8 much smaller than that of the starting cyclopropenylidene 7 (ΔES–T = 70.6 kcal mol−1), the (amino)carbene 8 exhibits enhanced ambiphilic reactivity.
:20). Carbene 8 is stable at RT and was isolated as red crystals from a concentrated pentane solution at −25 °C (yield: 85%). The 31P-NMR spectrum exhibits two singlet signals corresponding to the two diastereomers at 20.1 ppm and 22.5 ppm, respectively, with an 80:20 ratio. In the 13C-NMR spectrum, two doublet signals corresponding to the carbene carbon atom appear at 296 ppm (JCP = 17.6 Hz) and 301 ppm (JCP = 17.6 Hz), respectively, chemical shifts within the range of (amino)carbenes.18–21 The X-ray structure of 8 (Fig. 5) exhibits a short C3–N2 bond length [1.285(3) Å], indicating double bond character, and a planar geometry around the N2 atom (ΣN2°: 360°). These are consistent with the stabilization of carbene by π-electron donation from the amino group. The (amino)carbene 8 also shows a widened carbene angle [125.7(2)°] and a shorter C2–C3 bond [1.415(3) Å] compared to those of previously reported (amino)(aryl)carbenes [Caryl–Ccarbene–N: 117.9–121.0° and Caryl–Ccarbene: 1.453–1.469 Å].18 This suggests an enhanced π-interaction of the carbene lone pair with the adjacent phosphonio-substituted Al-heterocycle with a strong π-attracting character. Indeed, the 13C-NMR signal for the C1 atom adjacent to the P/Al atoms (102.0 ppm) is upfield shifted compared to those observed for its derivatives 9–11 (137.8–140.1 ppm). Hence, 8 is regarded as a push–pull carbene rather than a push–spectator carbene. As expected, with a singlet-triplet energy gap (ΔES–T = 31.0 kcal mol−1) of 8 much smaller than that of the starting cyclopropenylidene 7 (ΔES–T = 70.6 kcal mol−1), the (amino)carbene 8 exhibits enhanced ambiphilic reactivity.
 | ||
| Fig. 5 Molecular structure of 8. Thermal ellipsoids represent 30% probability. H and disordered atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: Al–C1 1.992(2), C1–C2 1.401(3), C1–P 1.708(2), C2–C3 1.415(3), C3–N2 1.285(3), C2–C4 1.518(3), C4–C5 1.355(3), C5–Al 1.990(2), N1–Al 1.917(2), Al–N3 1.845(2), C2–C3–N2 125.7(2). Torsion angles [°]: C1–C2–C3–N3 104.5(3). | ||
Indeed, 8 reacts with phenylacetylene and catecholborane, through C–H and B–H bond activation, respectively, to give the corresponding 1,1-adducts 9 and 10 (Scheme 2), in a manner similar to that of previously reported (amino)carbenes.22,23 Their formation was easily monitored using the characteristic 1H-NMR signals of the newly formed C–H group at the carbene center [9:5.20 ppm and 10:4.53 ppm]. The structure of 9 was confirmed by X-ray diffraction analysis (see ESI, Fig. S6†). The reaction of 8 with CuCl at RT affords the corresponding Cu(I)-complex 11, which has been isolated as orange crystals from a toluene solution at −30 °C (yield: 82%). The 13C-NMR spectrum exhibits a characteristic signal for the Cu(I)-complexed (amino)carbene at 244.4 ppm (JCP = 14.4 Hz), upfield shifted relative to that of the free-carbene 8 (296 and 301 ppm).24 The molecular structure of complex 11 was confirmed by X-ray diffraction analysis (see ESI, Fig. S7†).
 | ||
| Scheme 2 The reaction of 1 with stable cyclopropenylidene 7 and reactions of (amino)carbene 8. | ||
According to DFT calculations on the reaction pathway, the reaction starts with a formal insertion of cyclopropenylidene 7 into the Al–C bond of aluminocyclopropene 1, which proceeds in two steps, through the carbene → Al coordination (INT6) followed by the expansion of the aluminacyclopropene ring via the 1,2-migration of the vinyl carbon from Al to the Ccarbene atoms to form the corresponding spirocyclic intermediate INT7 (Fig. 6). Of particular interest, although slightly endergonic (ΔGINT7→INT8 = 2.5 kcal mol−1), the amino-substituted cyclopropene ring in INT7 readily opens to generate the corresponding (vinyl)(amino)carbene INT8, which then intramolecularly reacts with the adjacent aluminocyclobutene moiety via formal C–C bond activation to produce a bicyclic cyclopropene INT9. The resulting INT9 then undergoes the migration of the amino group towards the Al center to generate the zwitterionic cyclopropenium ion INT10. Subsequent stabilization of INT10 by the phosphine coordination again generates an amino-substituted cyclopropene intermediate INT11, analogous to INT7, which also opens to give the experimentally obtained stable (amino)carbene 8. Certainly, due to the ring strain release of two three-membered rings, the reaction is strongly exergonic (ΔG1+7→8 = −47.2 kcal mol−1) and all steps, except for the carbene formation step (INT7 → INT8), are exergonic processes with a highest reaction barrier of 24.4 kcal mol−1 (INT9 → INT10), which is in good agreement with the experimentally observed reaction at RT to form 8.
 | ||
| Fig. 6 Calculated pathway for the reaction of 1 with bis(diisopropylamino)cycopropenylidene 7 (M06-2X/Def2TZVP//M06-2X/6-31G(d) level). Gibbs energies of intermediates (and transition states) are in kcal mol−1. | ||
Although several transition metal-involved skeletal modifications of stable (amino)cyclopropenylidene 7 are known, none of them affords a carbene derivative.25 To date, only one example of “carbene-to-carbene” transformation of 7 has been reported by Koike and Iwamoto (Fig. 1(f)).11 Without any doubt, the most interesting transformation found in this pathway is the easy generation of (amino)carbenes (INT8 and 8) from amino-substituted cyclopropene precursors (INT7 and INT11), also regarded as donor–acceptor complexes of cyclopropenylidenes. This implies the possibility to use such a transformation to develop an original way to activate stable (amino)cyclopropenylidenes using Lewis pairs.
Conclusions
In conclusion, we have demonstrated that the Lewis pair complex of aluminacyclopropene with phosphine 1 can be used as a carbene-exchange reagent, which promotes “carbene-to-carbene” transformations of stable carbenes via skeletal recasting. Indeed, 1 reacts with a structurally stable NHC to produce a “more strained” cyclopropenylidene (4) via atomic carbon transfer from the NHC to the η2-coordinated alkyne fragment, within the coordination sphere of the Al(III) center. DFT calculations indicate that the transformation proceeds step-by-step ring expansions and contractions around the transferring carbon atom, and, indeed, one of the intermediates, 6 (“frozen” form of INT6 by phosphine coordination), was successfully isolated and fully characterized. Moreover, the Al(III)–phosphine complex 1 is also able to transform the stable (diamino)cyclopropenylidene 7 into an isolable acyclic (amino)carbene 8, which exhibits enhanced ambiphilic reactivity. As demonstrated by the numerous studies on stable mono-amino carbenes, featuring a broad range of electronic properties, the new stable amino-carbene 8, readily available from (diamino)cyclopropenylidene 7, has an enhanced ambiphilic character and holds promising applications in synthesis and catalysis. Further studies on the applications of this new method to activate stable carbenes, through “carbene-to-carbene” transformations promoted by Al/P Lewis pair complexes, are under active investigation. In addition, the expansion of reactivity of new carbenes (4) (the equilibrium species of 3)/8 is currently being studied.Data availability
All experimental and computational data associated with this work are available in the ESI.†Author contributions
Huihui Xu and José Miguel León Baeza carried out the synthetic experiments and analyzed the experimental data. Nathalie Saffon-Merceron assisted in the XRD refinement of the compounds. Vicenç Branchadell performed the DFT calculations. Tsuyoshi Kato, Antoine Baceiredo and René Segundo Rojas Guerrero supervised the work and edited the manuscript. The manuscript was written through the contribution of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to CNRS, the ANR (ChaMo and Ide-Al), ECOS-Sud Chili (No. C19E04), ANID-FONDECYT (No. 1200748 and 1230537), ANID (Scholarship to J. M. León Baeza, No. 21220559) and grant PID2023-150881NB-I00 funded by AEI/10.13039/501100011033 for financial support of this work. Access to the computational facilities of CSUC is acknowledged.Notes and references
- Selected reviews on the applications of NHC as organocatalysts: D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Chem. Rev., 2015, 115, 9307–9387 CrossRef CAS.
- Selected reviews on the applications of NHC as ligands: (a) S. P. Nolan, N-Heterocyclic Carbenes: Effective Tools for Organometallic Synthesis, John Wiley & Sons, 2014 CrossRef; (b) S. Diez-Gonzalez, N-Heterocyclic Carbenes: From Laboratory to Curiosities to Efficient Synthetic Tools, Royal Society of Chemistry, 2016 RSC; (c) P. L. Arnold and I. J. Casely, Chem. Rev., 2009, 109, 3599–3611 CrossRef CAS PubMed; (d) V. Nesterov, D. Reiter, P. Bag, P. Frisch, R. Holzner, A. Porzelt and S. Inoue, Chem. Rev., 2018, 118, 9678–9842 CrossRef CAS; (e) A. Doddi, M. Peters and M. Tamm, Chem. Rev., 2019, 119, 6994–7112 CrossRef CAS PubMed.
- (a) M. Kamitani, B. Nakayasu, H. Fujimoto, K. Yasui, T. Kodama and M. Tobisu, Science, 2023, 379, 484–488 CrossRef CAS PubMed; (b) H. Fujimoto, B. Nakayasu and M. Tobisu, J. Am. Chem. Soc., 2023, 145, 19518–19522 CrossRef CAS PubMed.
- L. Werner and U. Radius, Angew. Chem., Int. Ed., 2024, e202403639 CAS.
- (a) A. W. Waltman, T. Ritter and R. H. Grubbs, Organometallics, 2006, 25, 4238–4239 CrossRef CAS; (b) S. K. Bose, K. Fucke, L. Liu, P. G. Steel and T. B. Marder, Angew. Chem., Int. Ed., 2014, 53, 1799–1803 CrossRef CAS; (c) S. Pietsch, U. Paul, I. A. Cade, M. J. Ingleson, U. Radius and T. B. Marder, Chem.–Eur. J., 2015, 21, 9018–9021 CrossRef CAS PubMed.
- (a) D. Schmidt, J. H. J. Berthel, S. Pietsch and U. Radius, Angew. Chem., Int. Ed., 2012, 51, 8881–8885 CrossRef CAS PubMed; (b) M. Arrowsmith, M. S. Hill, G. Kociok-Kohn, D. J. MacDougall and M. F. Mahon, Angew. Chem., Int. Ed., 2012, 51, 2098–2100 CrossRef CAS; (c) S. M. I. Al-Rafia, R. McDonald, M. J. Ferguson and E. Rivard, Chem.–Eur. J., 2012, 18, 13810–13820 CrossRef CAS PubMed; (d) D. Franz and S. Inoue, Chem.–Asian J., 2014, 9, 2083–2087 CrossRef CAS; (e) T. Wang and D. W. Stephan, Chem.–Eur. J., 2014, 20, 3036–3039 CrossRef CAS; (f) M. D. Anker, A. L. Colebatch, K. J. Iversen, D. J. D. Wilson, J. L. Dutton, L. García, M. S. Hill, D. J. Liptrot and M. F. Mahon, Organometallics, 2017, 36, 1173–1178 CrossRef CAS; (g) H. Schneider, A. Hock, R. Bertermann and U. Radius, Chem.–Eur. J., 2017, 23, 12387–12398 CrossRef CAS.
- (a) K. J. Iversen, D. J. D. Wilson and J. L. Dutton, Organometallics, 2013, 32, 6209–6217 CrossRef CAS; (b) K. J. Iversen, D. J. D. Wilson and J. L. Dutton, Dalton Trans., 2015, 44, 3318–3325 RSC; (c) K. J. Iversen, J. L. Dutton and D. J. D. Wilson, Chem.–Asian J., 2017, 12, 1499–1508 CrossRef CAS PubMed.
- (a) K. J. Iversen, D. J. D. Wilson and J. L. Dutton, Dalton Trans., 2014, 43, 12820–12823 RSC; (b) S. Würtemberger-Pietsch, U. Radius and T. B. Marder, Dalton Trans., 2016, 45, 5880–5895 RSC.
- N. Merceron-Saffon, A. Baceiredo, H. Gornitzka and G. Bertrand, Science, 2003, 301, 1223–1225 CrossRef CAS.
- R. Bertermann, H. Braunschweig, C. K. L. Brown, A. Damme, R. D. Dewhurst, C. Hörl, T. Kramer, I. Krummenacher, B. Pfaffinger and K. Radacki, Chem. Commun., 2014, 50, 97–99 RSC.
- T. Koike and T. Iwamoto, J. Am. Chem. Soc., 2023, 145, 9264–9272 CrossRef CAS.
- L. Werner and U. Radius, Dalton Trans., 2024, 53, 16436–16454 RSC.
- Al-containing three-membered cyclic compounds are known to be highly reactive: C. Yan and R. Kinjo, J. Am. Chem. Soc., 2023, 145, 12967–12986 CrossRef CAS.
- J. M. L. Baeza, H. Xu, S. Takahashi, A. Baceiredo, R. S. R. Guerrero, D. Hashizume, N. Saffon-Merceron, V. Branchadell and T. Kato, Angew. Chem., Int. Ed., 2025, e202505181 CAS.
- R. L. Falconer, K. M. Byrne, G. S. Nichol, T. Krämer and M. J. Cowley, Angew. Chem., Int. Ed., 2021, 60, 24702–24708 CrossRef CAS PubMed.
- The geometry of the cyclopropylidene moiety in 4 is similar to that observed for the chromium complex reported previously: T. Kurogi, K. Irifune and K. Takai, Chem. Sci., 2021, 12, 14281–14287 RSC.
- X. Zhang, H. Wang, L. Kong and L. L. Liu, Organometallics, 2024, 43, 2392–2396 CrossRef CAS.
- Acyclic (amino)(aryl) carbenes: (a) S. Solé, H. Gornitzka, W. W. Schoeller, D. Bourissou and G. Bertrand, Science, 2001, 292, 1901–1903 CrossRef; (b) X. Cattoën, H. Gornitzka, D. Bourissou and G. Bertrand, J. Am. Chem. Soc., 2004, 126, 1342–1343 CrossRef PubMed; (c) D. Magis, J. J. Cabrera-Trujillo, J. Vignolle, J.-M. Sotiropoulos, D. Taton, K. Miqueu and Y. Landais, J. Am. Chem. Soc., 2024, 146, 16802–16813 CrossRef CAS.
- Cyclic (amino)(aryl) carbene: J. Lorkowski, P. Yorkgitis, M. R. Serrato, M. Gembicky, C. Pietraszuk, G. Bertrand and R. Jazzar, Angew. Chem., Int. Ed., 2024, e202401020 CAS.
- Acyclic (Amino)(Alkyl)carbenes: V. Lavallo, J. Mafhouz, Y. Canac, B. Donnadieu, W. W. Schoeller and G. Bertrand, J. Am. Chem. Soc., 2004, 126, 8670–8671 CrossRef CAS.
- Cyclic (amino)(alkyl)carbenes: (a) V. Lavallo, Y. Canac, C. Präsang, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2005, 44, 5705–5709 CrossRef CAS; (b) R. Jazzar, R. D. Dewhurst, J.-B. Bourg, B. Donnadieu, Y. Canac and G. Bertrand, Angew. Chem., Int. Ed., 2007, 46, 2899–2902 CrossRef CAS; (c) R. Jazzar, J.-B. Bourg, R. D. Dewhurst, B. Donnadieu and G. Bertrand, J. Org. Chem., 2007, 72, 3492–3499 CrossRef CAS; (d) X. Zeng, G. D. Frey, R. Kinjo, B. Donnadieu and G. Bertrand, J. Am. Chem. Soc., 2009, 131, 8690–8696 CrossRef CAS; (e) E. Tomás-Mendivil, M. M. Hansmann, C. M. Weinstein, R. Jazzar, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2017, 139, 7753–7756 CrossRef; (f) C. M. Weinstein, G. P. Junor, D. R. Tolentino, R. Jazzar, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2018, 140, 9255–9260 CrossRef CAS PubMed.
- Z. R. Turner, Chem.–Eur. J., 2016, 22, 11461–11468 CrossRef CAS.
- (a) S. Würtemberger-Pietsch, H. Schneider, T. B. Marder and U. Radius, Chem.–Eur. J., 2016, 22, 13032–13036 CrossRef PubMed; (b) K. K. Manar, V. K. Porwal, R. S. Kamte, M. Adhikari, S. K. Thakur, D. Bawari, A. R. Choudhury and S. Singh, Dalton Trans., 2019, 48, 17472–17478 RSC.
- (a) X. Hu, M. Soleilhavoup, M. Melaimi, J. Chu and G. Bertrand, Angew. Chem., Int. Ed., 2015, 54, 6008–6011 CrossRef CAS; (b) Y. D. Bidal, M. Lesieur, M. Melaimi, F. Nahra, D. B. Cordes, K. S. Athukorala Arachchige, A. M. Z. Slawin, G. Bertrand and C. S. J. Cazin, Adv. Synth. Catal., 2015, 357, 3155–3161 CrossRef CAS; (c) J. Chu, D. Munz, R. Jazzar, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2016, 138, 7884–7887 CrossRef CAS PubMed; (d) E. A. Romero, T. Zhao, R. Nakano, X. Hu, Y. Wu, R. Jazzar and G. Bertrand, Nat. Catal., 2018, 1, 743–747 CrossRef CAS; (e) H. Kim, M. Kim, H. Song and E. Lee, Chem.–Eur. J., 2021, 27, 3849–3854 CrossRef CAS; (f) A. S. Romanov, D. Di, L. Yang, J. Fernandez-Cestau, C. R. Becker, C. E. James, B. Zhu, M. Linnolahti, D. Credgington and M. Bochmann, Chem. Commun., 2016, 52, 6379–6382 RSC; (g) J. Lorkowski, M. Krahfuß, M. Kubicki, U. Radius and C. Pietraszuk, Chem.–Eur. J., 2019, 25, 11365–11374 CrossRef CAS; (h) D. Pichon, M. Soleilhavoup, J. Morvan, G. P. Junor, T. Vives, C. Crévisy, V. Lavallo, J.-M. Campagne, M. Mauduit, R. Jazzar and G. Bertrand, Chem. Sci., 2019, 10, 7807–7811 RSC; (i) F. Chotard, V. Sivchik, M. Linnolahti, M. Bochmann and A. S. Romanov, Chem. Mater., 2020, 32, 6114–6122 CrossRef CAS; (j) K. K. Manar, S. Chakrabortty, V. K. Porwal, D. Prakash, S. K. Thakur, A. R. Choudhury and S. Singh, ChemistrySelect, 2020, 5, 9900–9907 CrossRef CAS; (k) M. Deng, N. F. M. Mukthar, N. D. Schley and G. Ung, Angew. Chem., Int. Ed., 2020, 59, 1228–1231 CrossRef CAS; (l) M. R. Serrato, M. Melaimi and G. Bertrand, Chem. Commun., 2022, 58, 7519–7521 RSC; (m) A. Madron du Vigné and N. Cramer, Organometallics, 2022, 41, 2731–2741 CrossRef; (n) W. Chu, T. Zhou, E. Bisz, B. Dziuk, R. Lalancette, R. Szostak and M. Szostak, Chem. Commun., 2022, 58, 13467–13470 RSC; (o) J. Volk, M. Heinz, M. Leibold, C. Bruhn, T. Bens, B. Sarkar, M. C. Holthausen and U. Siemeling, Chem. Commun., 2022, 58, 10396–10399 RSC; (p) S. Baguli, A. Kundu, S. Nath, D. Adhikari and D. Mukherjee, Org. Lett., 2023, 25, 3141–3145 CrossRef CAS PubMed.
- (a) G. T. Kent, S. L. Staun, G. Wu and T. W. Hayton, Organometallics, 2020, 39, 2375–2382 CrossRef CAS; (b) T. H. Nguyen, X. Yu, G. T. Kent, J. Autschbach and T. W. Hayton, Organometallics, 2023, 42, 1005–1012 CrossRef CAS; (c) L. N. V. Le, G. A. Bailey, A. G. Scott and T. Agapie, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2109241118 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2441055–2441061. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc03846d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |