
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper-catalysed oxy-alkylation of styrenes enabled by halogen-atom transfer
Qiujian
Tan†
ab,
Xiang
Lyu†
 c,
Yu
Zhao
a,
Huaquan
Fang
*b,
Xianxiu
Xu
*a and
Zhongyan
Hu
*a
c,
Yu
Zhao
a,
Huaquan
Fang
*b,
Xianxiu
Xu
*a and
Zhongyan
Hu
*a
aCollege of Chemistry, Chemical Engineering and Materials Science, Shandong Normal University, Jinan 250014, China. E-mail: xuxx677@sdnu.edu.cn; huzy@sdnu.edu.cn
bSchool of Pharmaceutical Sciences, Chongqing University, Chongqing 401331, China. E-mail: fanghq@cqu.edu.cn
cSchool of Pharmaceutical Sciences, Institute of Materia Medica, Shandong First Medical University, Jinan 250117, China
First published on 26th August 2025
Abstract
Herein, we present a mechanistically distinct approach to the multicomponent difunctionalisation of styrenes with alkyl halides and oxygen by integrating α-aminoalkyl radical-mediated halogen-atom transfer (XAT) with copper catalysis under air-equilibrated conditions. This strategy eliminates the need for external peroxides or photoredox conditions, offering a streamlined and efficient alternative. Mechanism studies uncover a unique role for the copper catalyst: instead of directly activating alkyl halides, inexpensive CuCl2 oxidizes a tertiary amine to generate an α-aminoalkyl radical, which then drives XAT to release alkyl radicals. These radicals subsequently add to alkenes, facilitating efficient difunctionalisation. This method accommodates a broad range of alkyl radical precursors, including fluoroalkyl- and alkyl halides, and demonstrates compatibility with diverse styrenes, enabling the modular synthesis of β-(fluoro)alkylated ketones with excellent functional group tolerance under mild conditions. Notably, the strategy's practical utility is exemplified through the late-stage functionalisation of biologically active molecules and pharmaceuticals, showcasing its potential for rapid, efficient access to structurally complex molecules.
Introduction
The accessibility and versatile reactivity of carbon–carbon double bonds make alkenes a sought-after class of feedstocks in chemical synthesis.1 Over the past few decades, numerous efficient strategies have been developed for their functionalisation.2 Among these, the catalytic difunctionalisation of styrenes has emerged as a particularly powerful approach for enhancing molecular complexity by simultaneously installing two distinct functional groups across the double bond in a single transformation.3 Alkyl halides, due to their structural diversity and synthetic accessibility, have become some of the most widely employed reagents in organic chemistry, serving as excellent precursors for generating alkyl radicals.4 In this context, the catalytic alkylation of styrenes with alkyl halides has become a vibrant area of research in both transition metal and photoredox catalysis (Scheme 1a). These approaches typically rely on direct single-electron transfer (SET) between metal (e.g., Ni,5,6 Co,7,8 Pd,9,10 Cu,11–13 and Fe14) or photoredox catalyst15,16 and the alkyl halides to generate the corresponding alkyl radicals.5–16 However, their general applicability is often constrained by substrate-specific reactivity, with optimal performance typically observed for activated halides such as ethyl bromodifluoroacetate. In contrast, unactivated alkyl halides present a substantial challenge due to their intrinsically more negative reduction potentials (<−2 V vs. SCE),17–20 necessitating strongly reducing conditions that are often incompatible with mild radical initiation. Seminal work by the groups of Huang,5,8 Lei,7 Zhang,6 and others5–16 has demonstrated that cobalt-, nickel-, or palladium-catalysed processes, employing electron-shuttle catalysis,5,8 halogen-atom transfer (XAT),7 photoinduced systems,10 or organolithium reagents as coupling partners,6 can promote electron transfer from low-valent metal species to alkyl halides, thereby facilitating alkene functionalisation. While these advances represent significant progress, the pursuit of a low-cost and green alternative strategy for styrene alkylation, capable of enabling simpler, milder, and more efficient activation of alkyl halides with broad functional group compatibility, remains a crucial and highly sought-after objective.21–23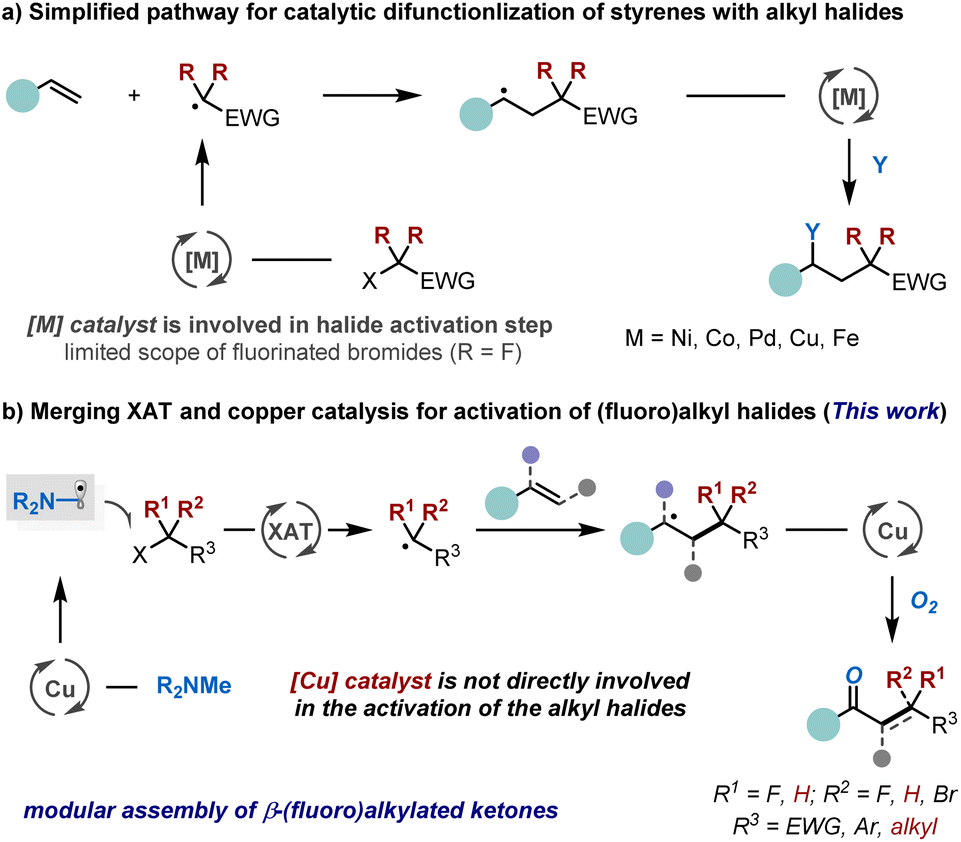 | ||
| Scheme 1 Radical-mediated difunctionalisation of styrenes with alkyl halides. | ||
Recent studies have revealed that alkyl radicals can be efficiently generated from their corresponding halides by leveraging α-aminoalkyl radicals to trigger XAT reactions.18–20,24–26 This strategic approach leverages strong polar effects in the transition state during halide abstraction, thereby facilitating alkyl radical formation.18–20,24–30 As a result, the alkyl halide is not required to engage in direct SET with the metal,19 nor coordination with it for subsequent abstraction or oxidative addition,13,31,32 thereby enabling broader synthetic applications. Building on these insights, we hypothesized a conceptually distinct strategy where the copper catalyst facilitates the formation of α-aminoalkyl radical without directly activating the alkyl halide.33–37 This approach could offer significant synthetic advantages in assembling challenging small-molecule building blocks. The proposed catalytic mechanism, illustrated in Scheme 1b, involves the copper catalyst preferentially oxidizing tertiary amines to generate α-aminoalkyl radicals, which then mediate XAT from alkyl halides, forming the corresponding alkyl radicals. These radicals subsequently add to alkenes, generating a new C-centered radical. The resulting intermediate undergoes recombination with oxygen (O2) in the presence of the copper catalyst,38 ultimately yielding the desired difunctionalisation product. The copper catalyst is continuously regenerated through electron exchange with the tertiary amine in the presence of O2 from air. A key mechanistic distinction of this approach, compared to traditional transition-metal and photocatalysis systems, is that carbon–halogen bond activation occurs outside the copper catalytic cycle, independent of the nature of the copper catalyst and alkyl halide. This unique feature also eliminates the need for expensive catalysts and additional photocatalysis,39,40 which we anticipate will broaden the substrate scope and enhance the versatility of this transformation.
Results and discussion
Reaction development
To optimize the reaction conditions, a model reaction between styrene (1) and ethyl bromodifluoroacetate (2) was performed under air-equilibrated conditions (Table 1). After extensive screening (see the SI for details), we were pleased to find that ethyl (Z)-2-fluoro-4-oxo-4-phenylbut-2-enoate (3) was obtained in 73% yield with excellent regioselectivity (Z/E > 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) when CuCl2 was used as the catalyst and N-cyclohexyl-N-methylcyclohexanamine (Cy2NMe) was employed as the XAT reagent in MeCN at 25 °C (entry 1). Control experiments revealed that both CuCl2 and Cy2NMe were essential for the fluoroalkylation reaction (entries 2 and 3), and the absence of PPh3 led to slightly decrease in yield (entry 4). Substituting CuCl2 with other copper salts, such as CuCl or CuBr2, resulted in lower yields, while other transition metals (e.g., CoCl2, PdCl2) proved ineffective (entries 5–7). Notably, functionalized tertiary amines with less steric hindrance produced inferior results or failed entirely, underscoring the critical role of the sterically bulky Cy2NMe (entries 8–11). Notably, air-equilibrated conditions are essential, no reaction occurs under a N2 atmosphere (entry 12), while an O2 atmosphere results in a decreased yield (entry 13). Finally, other difluoroalkyl radical precursors were examined. Chlorides proved unreactive, likely due to the stronger C–Cl bond.17,41 Although more reactive, iodides afforded product 3 with reduced efficiency, likely due to the formation of Heck-type by-products, as evidenced by 19F NMR analysis (Fig. S1).42 We propose that, in the case of iodides, the newly radical formed via the initial addition step, undergoes rapid iodine atom transfer to generate an alkyl iodide, which subsequently undergoes dehydroiodination under the reaction conditions (Table S9).43 These findings confirm that difluoroalkyl bromides deliver the best overall performance.
:1) when CuCl2 was used as the catalyst and N-cyclohexyl-N-methylcyclohexanamine (Cy2NMe) was employed as the XAT reagent in MeCN at 25 °C (entry 1). Control experiments revealed that both CuCl2 and Cy2NMe were essential for the fluoroalkylation reaction (entries 2 and 3), and the absence of PPh3 led to slightly decrease in yield (entry 4). Substituting CuCl2 with other copper salts, such as CuCl or CuBr2, resulted in lower yields, while other transition metals (e.g., CoCl2, PdCl2) proved ineffective (entries 5–7). Notably, functionalized tertiary amines with less steric hindrance produced inferior results or failed entirely, underscoring the critical role of the sterically bulky Cy2NMe (entries 8–11). Notably, air-equilibrated conditions are essential, no reaction occurs under a N2 atmosphere (entry 12), while an O2 atmosphere results in a decreased yield (entry 13). Finally, other difluoroalkyl radical precursors were examined. Chlorides proved unreactive, likely due to the stronger C–Cl bond.17,41 Although more reactive, iodides afforded product 3 with reduced efficiency, likely due to the formation of Heck-type by-products, as evidenced by 19F NMR analysis (Fig. S1).42 We propose that, in the case of iodides, the newly radical formed via the initial addition step, undergoes rapid iodine atom transfer to generate an alkyl iodide, which subsequently undergoes dehydroiodination under the reaction conditions (Table S9).43 These findings confirm that difluoroalkyl bromides deliver the best overall performance.
|
|
|||
|---|---|---|---|
| Entry | Variation from standard conditions | Yield of 3b (%) | Z/Eb |
| a Reaction conditions: 1 (0.3 mmol), 2 (1.2 mmol), CuCl2 (0.06 mmol), PPh3 (0.06 mmol), and Cy2NMe (1.2 mmol) in CH3CN (2 mL) at 25 °C under air atmosphere for 24 h. b Yields and Z/E ratios were determined by 19F NMR analysis of the crude reaction mixtures using CF3Ph as the internal standard. c Yield of isolated product. | |||
| 1 | None | 73 (71)c | >22:1 |
| 2 | No CuCl2 | 0 | — |
| 3 | No Cy2NMe | 0 | — |
| 4 | No PPh3 | 62 | >22:1 |
| 5 | CuCl instead of CuCl2 | 37 | >22:1 |
| 6 | CuBr2 instead of CuCl2 | 54 | >22:1 |
| 7 | CoCl2/PdCl2 instead of CuCl2 | 0 | — |
| 8 | Cy2NEt instead of Cy2NMe | 17 | >22:1 |
| 9 | Pempidine instead of Cy2NMe | 47 | >22:1 |
| 10 | iPr2NMe instead of Cy2NMe | 34 | >22:1 |
| 11 | Et2NMe instead of Cy2NMe | 0 | — |
| 12 | N2 atmosphere | 0 | — |
| 13 | O2 atmosphere | 31 | >22:1 |

|
|||
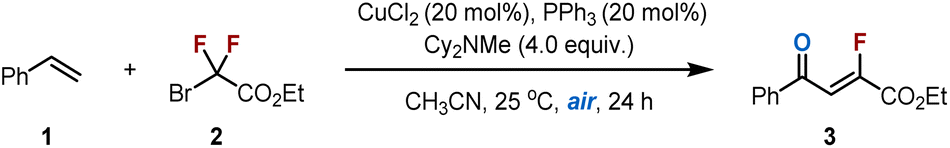
Oxo-fluoroalkylation of styrenes
With the optimized reaction conditions established, the substrate scope of the fluoroalkylation was initially explored using fluorinated alkyl bromides (Scheme 2). This transformation demonstrated compatibility with a broad range of styrene-type olefins, including those with varying electronic properties at the para, ortho, or meta positions, affording the corresponding β-fluoroenone products 3–18 in moderate to good yields. Styrenes containing labile and synthetically versatile functional groups, including amines (6), halides (8–10, 18), alcohols (11), silanes (12), boronic esters (13), alkenes (14), and alkynes (15),44 were well-tolerated, offering opportunities for further derivatisation. Additionally, polysubstituted styrenes were also compatible, yielding the desired products (19–20) smoothly. Styrenes featuring fused arenes and heterocycles, such as naphthalene (21), thiophene (22), thiazole (23), benzofuran (24), benzothiophene (25–26), and carbazole (27) also proved compatible with this protocol, delivering the target products in moderate yields. Remarkably, radical precursors containing amides afforded the desired product 28, while phosphonate groups resulted in the formation of β-fluoroenone 29 and an unexpected α-trifluoromethyl ketone 30 in moderate yields (Fig. S39).45 Remarkably, 1,3-dienes, cyclic internal styrenes, and 1,2-diphenylethenes were all compatible with the reaction conditions, delivering the corresponding β-fluoroenone products (31–34) in acceptable yields. The observed stereoselectivity of 34 can be rationalized by comparing the transition states energies leading to each isomer during the anti-fluoride elimination, as illustrated in Fig. S40. The transition state leading to (E)-34 is energetically favored, as it avoids steric repulsion between the phenyl (Ph–) and alkoxycarbonyl (EtO2C–) groups.46 Interestingly, when (E)-β-methylstyrenes were empolyed under the standard condition, the non-defluorinated products (35–36) were obtained, likely due to steric hindrance that impedes the E2 elimination pathway.47 Additionally, 1,1-disubstituted and 1,1,2-trisubstituted styrenes furnished the corresponding Heck-type products (37–38). This outcome is consistent with previous observations involving other catalytic systems utilizing α-aminoalkyl radical-mediated XAT strategies.48 This method was also effective for monofluorinated substrates, as exemplified by the reaction of styrene with ethyl 2-bromo-2-fluoroacetate, which afforded the desired (E)-α,β-unsaturated ester 39. Whereas ethyl dibromofluoroacetate led to the formation of product 3 through debromination, rather than defluorination. However, unactivated olefins, such as 4-phenyl-1-butene and 1-octene, proved to be inefficient in this transformation, typically leading to the recovery of over 90% of the starting olefins (Fig. S17). Pyridine-containing olefins (e.g., 4-vinylpyridine), which can act as ligands and interfere with the metal-catalytic system, also failed to undergo efficient transformation. Additionally, olefins with unique structural features, such as 1,3-enynes (e.g., but-3-en-1-yn-1-ylbenzene) and allenes (e.g., 1-phenylallene), led to complex reaction mixtures, complicating purification efforts. Biologically relevant olefins, derived from compounds such as indomethacin, ibuprofen, fenbufen, loxoprofen, and 2-propylpentanoic acid, were smoothly converted into β-fluoroenones 40–44 in moderate yields. Similarly, α-trifluoromethyl ketone derivatives 45–49 performed well under standard conditions, highlighting the potential utility of this method for late-stage diversification of pharmaceutical agents. | ||
| Scheme 2 Oxo-fluoroalkylation of styrenes with fluoroalkyl halides. Reaction conditions: styrenes (0.3 mmol), fluoroalkyl bromides (1.2 mmol), CuCl2 (0.06 mmol), PPh3 (0.06 mmol), and Cy2NMe (1.2 mmol) in CH3CN (2 mL) at 25 °C under air atmosphere for 24 h. aStyrenes (0.3 mmol), fluoroalkyl bromides (0.6 mmol), CuCl2 (0.06 mmol), PPh3 (0.06 mmol), and Cy2NMe (0.6 mmol) in CH3CN (2 mL) at 25 °C under air atmosphere for 24 h. Yield of isolated product, Z/E ratios were determined by 19F NMR analysis of the crude reaction mixtures using CF3Ph as the internal standard. bYield based on recovered starting material. cE/Z > 22:1, the E-isomer is the major product. dNo reaction, rsm = recovered of starting material. | ||
Synthetic transformations
To demonstrate the scalability of this fluoroalkylation, the reaction between 1 and 2 was scaled up to 1 mmol, yielding the desired product 3 in moderate yield (Scheme 3). While β-fluoroenones are intrinsically valuable,49 their potential as synthetic intermediates were further showcased by converting 3 into versatile enaminones (50–56)50 in high yields with excellent E-selectivity via a transition metal-free defluorination/amination or ammonolysis sequence.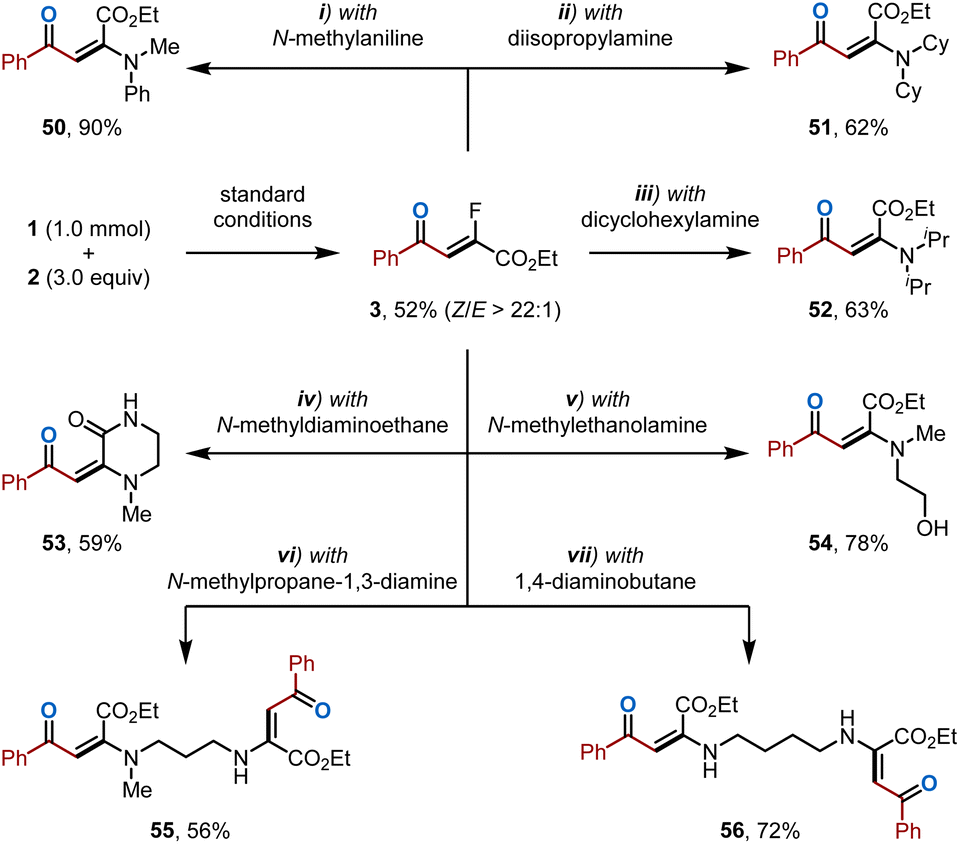 | ||
| Scheme 3 Scale-up reaction and synthetic transformations. | ||
Oxo-alkylation of styrenes
Building on the initial success, we next investigated the scope of the reaction with respect to unfluorinated alkyl halides (Scheme 4). We were pleased to observe that a broad range of alkyl bromides successfully participate in the reaction with various styrenes, leading to the formation of a diverse array of γ-ketoesters (57–68) in moderate to good yields. Notably, γ-ketoester 57 could be obtained on a large-scale with a 60% isolated yield. Furthermore, α-bromo amides were also compatible with the reaction, providing the desired products 69–70, which feature both oxo and amino groups, making them valuable for subsequent transformation. Additionally, a variety of α-bromoketones was incorporated into the method, yielding structurally diverse 1,4-dicarbonyl compounds (71–74). These compounds present a synthetic challenge due to the intrinsic polarity mismatch of the substrates,51 demonstrating the broad applicability of the protocol. α-Bromonitrile was also a suitable partner, delivering the synthetically valuable β-nitrile ketone 75 in moderate yield. Ethyl dibromoacetate also participated in this transformation, resulting in the formation of the debromination product 39. Notably, unactivated alkyl halides, such as benzyl bromides (e.g., (bromomethyl)benzene) and alkyl iodides (e.g., 1-fluoro-2-iodoethane, 1-iodo-3,3,3-trifluoropropane, and 1-iodo-4,4,4-trifluorobutane), were successfully incorporated into this platform, affording the desired products 76–79. However, most of other alkyl bromides proved inefficient in this transformation, with the aminomethylation28 or E2 elimination30 by-products detected by high-resolution mass spectrometry (HRMS) analysis (Fig. S18–S29). Attempts to improve reactivity by using more electrophilic Michael acceptors (e.g., ethyl acrylate, acrylonitrile, 1-phenylprop-2-en-1-one, and cinnamonitrile) were also unsuccessful under the current conditions (Fig. S30–S33). We speculate that in these cases, α-aminoalkyl radical-mediated XAT is slowed due to the stronger nature of the C–Br bond,41 making the direct Giese addition of alkyl radicals to the acceptor more competitive.18,43 This reactivity gap is especially pronounced under aerobic conditions.52 Finally, this protocol was shown to be effective in late-stage modification of complex molecules, as exemplified by compounds 80–86, where biorelevant groups were incorporated to afford the desired 1,4-dicarbonyl products in moderate to high yields. | ||
| Scheme 4 Oxo-alkylation of styrenes with alkyl halides. Reaction conditions: styrenes (0.3 mmol), alkyl halides (1.2 mmol), CuCl2 (0.06 mmol), PPh3 (0.06 mmol), and Cy2NMe (1.2 mmol) in CH3CN (2 mL) at 25 °C under air atmosphere for 24 h. a1.0 mmol scale experiment, alkenes (1.0 mmol), alkyl halides (2.0 mmol), CuCl2 (0.06 mmol), PPh3 (0.06 mmol), and Cy2NMe (2.0 mmol) in CH3CN (4 mL) at 25 °C under air atmosphere for 24 h. Yield of isolated product. bYield based on recovered starting material. c20% benzaldehyde isolated as a by-product. dAlkyl iodides were used. | ||
Mechanistic investigations
To gain insight into the reaction mechanism, a series of control experiments were conducted. First, radical trapping experiments were performed using 2,2,6,6-tetramethylpiperidinyl-1-oxide (TEMPO) and 2,6-di-tert-butyl-4-methylphenol (BHT) as radical scavengers each separately (Scheme 5a). The reaction was completely inhibited in the presence of TEMPO, while the formation of product 3 was significantly reduced in the presence of BHT. In addition, several radical-adducts were observed and characterized, including difluoroalkyl-TEMPO (S1), α-aminoalkyl-TEMPO (S2), difluoroalkyl-BHT (S4), and α-aminoalkyl-BHT (S5), through 19F NMR and HRMS analysis (Fig. S2 and S3).6,53 These findings suggest that the reaction likely proceeds via a radical pathway. Further radical trapping studies using 1,1-diphenylethylene revealed the presence of a C-centered radical species (S6) and oxo-difluoroalkylation products (3′), with the difluoroacetate adduct 37 isolated in 34% yield (Scheme 5b and Fig. S4).54 Additionally, the detection of iminium ion S3, ammonium salts S7 and S8 suggests that Cy2NMe is involved in both the XAT and defluorination steps. To further confirm the involvement of a difluoroalkyl radical in the reaction, a radical-clock experiment was conducted. As shown in Scheme 5c, treatment of compound 87, known to undergo radical rearrangement, with 1 and 2 under standard conditions afforded the ring-expanded product 88 in 40% yield (determined by 19F NMR spectroscopy), along with 33% yield of 3 (Fig. S5).55–57 These results support the involvement of an ethyl difluoroacetate radical and confirm that the reaction proceeds via a radical-mediated Giese-type mechanism.58 Consistent with this mechanistic hypothesis, intermediate 3′ was detected by 19F NMR and HRMS upon lowering Cy2NMe and shortening the reaction time (Fig. S6), supporting its conversion to β-fluoroenones 3via dehydrofluorination.59 | ||
| Scheme 5 Mechanistic studies. | ||
To further investigate the conversion of fluoroalkyl halides and amines, electron paramagnetic resonance (EPR) experiments were conducted using phenyl N-tert-butylnitrone (PBN). In the reaction of 1 with 2 under standard conditions, the formation of the PBN-spin adduct R1 indicates the selective generation of the ethyl difluoroacetate radical (Scheme 5d).6,54,60 When 2 was reacted alone, a new PBN-spin adduct (R2) was observed, which was assigned as the α-aminoalkyl radical based on its spectral characteristics,61,62 confirmed through HRMS analysis (Fig. S7 and S8). Furthermore, in the presence of both CuCl2 and Cy2NMe, the PBN-spin adduct R3, generated from the reaction of amine radical cation with PBN, was entrapped (Scheme 5e and Fig. S9).63,64 In contrast, no radical species were observed when CuCl2 was replaced by CuCl under the same conditions (Fig. S10). Notably, when a mixture of CuCl2, 2, and PBN was used, no difluoroacetate radicals were observed in the EPR spectrum, and a band with g-values around 2.134 was observed, corresponding to Cu(II) and (Fig. S11).65 Although PPh3 is not essential for product formation, its presence enhance the reaction yield. To elucidate its role, we conducted cyclic voltammetry studies (Fig. S35). The oxidation peak of Cy2NMe was observed at +0.71 V (vs. Ag/AgCl) in CH3CN,28,66 while CuCl2 exhibited an oxidation peak at +0.61 V (vs. Ag/AgCl),67 indicating that the SET oxidation of Cy2NMe by CuCl2 is thermodynamically unfavorable. Notably, mixing CuCl2 with PPh3, resulted in a new peak at + 0.99 V (vs. Ag/AgCl), suggesting the formation of a Cu(II)Ln complex as the active oxidant at the onset of the reaction.68 We further performed 31P NMR analysis of the reaction mixture to probe the fate of PPh3 at the end of the reaction. As shown in Fig. S36, the spectrum displayed a single resonance at δ ≈ 27 ppm, corresponding to triphenylphosphine oxide (OPPh3), with no detectable signal for unreacted PPh3.69,70 This indicates complete oxidation of PPh3 to OPPh3 under the reaction conditions, likely mediated by the copper catalyst and molecular oxygen, consistent with previous reports on copper-catalysed aerobic phosphine oxidation.71 Accordingly, the complete conversion of PPh3 to OPPh3 supports a copper-mediated oxidation mechanism. We cannot exclude the possibility that copper–dioxygen species function as one-electron oxidizing agents for the tertiary amine during the process.34,35,72 Taken together, these results suggest that the amine radical cation is formed via single-electron oxidation of Cy2NMe by Cu(II) species, and the difluoroacetate radical is generated via an XAT process from the α-aminoalkyl radical, rather than from a Cu(II) complex. Additionally, the reaction of the ATRA product 89 did not yield the expected difunctionalisation products 3 or 3′ under standard conditions (Scheme 5f), ruling out an ATRA mechanism.5,21,73 Finally, the reaction of 1 and 2 with a CuCl catalyst, conducted in the absence of Cy2NMe under standard conditions, was found to be inefficient (Scheme 5g and Fig. S15). Moreover, no EPR signal was observed in the mixture of CuCl, 2, and PBN, either in the presence or absence of PPh3 under these conditions (Fig. S12 and S13). In addition, stirring a mixture of CuCl, 2, and PPh3 in CH3CN at 25 °C for 24 h under air-equilibrated conditions resulted in quantitative recovery of 2, indicating no detectable consumption (Fig. S14). Taken together, these findings suggest that Cu(I) is unlikely to engage in direct oxidative addition74,75 or SET5,21,73 with 2 to generate the ethyl difluoroacetate radical under the tested conditions.
Based on our experimental observations and previous insights, a catalytic cycle that accounts for this transformation is outlined in Scheme 5h. The cycle initiates with a Cu(II)-mediated SET oxidation of Cy2NMe,33,36,72,76 leading to the formation of a Cu(I) complex and the amine radical cation I. Cu(I) complexes are reoxidized by atmospheric O2, regenerating Cu(II) complexes.77 Meanwhile, the amine radical cation rapidly undergoes deprotonation in the presence of base to form the α-aminoalkyl radical II.66,78 A subsequent XAT between the α-aminoalkyl radical II and ethyl bromodifluoroacetate (2) produces the ethyl difluoroacetate radical III and the iminium ion S3.18,26,29,79 The ethyl difluoroacetate radical III then adds to styrene (1), forming a new C-centered radical IV,79,80 which recombines with Cu(I) and O2 to yield the Cu(III)-peroxyl radical V.81 The Cu(III) species V undergoes rapid rearrangement to produce 3′ and regenerate the Cu(II) species.81,82 Finally, (Z)-β-fluoroenone 3 is formed via a dehydrofluorination process in the presence of Cy2NMe.83
Additionally, the formation of Cu(I) species was confirmed through UV-vis spectral analysis (Scheme 5i).19,82,84 The UV-vis spectrum of CuCl2 shows a band at 460 nm, which disappears upon the addition of Cy2NMe, accompanied by the appearance of a new band at 230 nm. Similarly, CuCl in the presence of Cy2NMe exhibits a similar spectrum with a λmax at 230 nm. This spectral change supports the idea that Cu(II) preferentially oxidizes Cy2NMe to form Cu(I) and the amine radical cation I, which participates in the reaction, consistent with the EPR spin-trapping results. To further investigate the oxo-alkylation process, a Hammett analysis was performed (Scheme 5j). Competition experiments between phenylacetylene and para-substituted styrene derivatives revealed a linear relationship between log(kX/kH) and σ, with a negative slope (ρ = −0.23), suggesting that the transition state is stabilized by electron-donating substituents. This finding supports a mechanism involving radical addition to the double bond, followed by the formation of a Cu(III) intermediate.85,86 Moreover, the reaction of Cy2NMe with CuCl2 and 2 at 25 °C in CD3CN was monitored by NMR spectroscopy (Scheme 5k and Fig. S37). As expected, 1H NMR revealed the liberation of a proton (H/D exchange forming CH3CN) during the deprotonation of the amine radical cation I, along with the generation of iminium ion S3 during the XAT process. These results further support the involvement of the α-aminoalkyl radical in the XAT process. Moreover, density functional theory (DFT) calculations suggest that the XAT activation of fluoroalkyl reagents by an α-aminoalkyl radical is favorable both kinetically and thermodynamically (Fig. S41). In addition, the iminium ion S3 could be isolated by filtration and was well-characterized by NMR and HRMS analysis (Fig. S37 and S38), providing additional evidence that the ethyl difluoroacetate radical is generated via XAT mediated by the α-aminoalkyl radical, rather than by Cu(II) species.
Conclusions
In summary, we have developed a copper-catalysed, α-aminoalkyl radical-mediated XAT protocol for the difunctionalisation of diverse styrenes with alkyl halides under air-equilibrated conditions. This method enables divergent access to β-(fluoro)alkylated ketones with broad substrate scope, excellent functional group tolerance, and suitability for late-stage modification of complex bioactive molecules. The reaction is scalable and operationally simple, demonstrating its practical utility. Compared to recently developed catalytic methods that utilize ligated boryl radicals under photoredox conditions,17,87,88 this straightforward approach oxidizing readily available amines with inexpensive Cu(II) facilitates the formation of α-aminoalkyl radicals, offering an alternative strategy for directly activating fluoroalkyl- and alkyl halides without requiring peroxides19,25,89 or photoredox conditions.21 We anticipate this modular approach will be valuable to both academic and industrial chemists. Ongoing efforts in our laboratory are focused on further investigating and expanding the scope of these transformations.Author contributions
H. F., X. X. and Z. H. conceived and supervised the project. Q. T. and Y. Z. performed the synthetic experiments and analyzed the data. X. L. participated in theoretical calculations. X. L., H. F. and Z. H. participated in the discussions. H. F. and Z. H. co-wrote the manuscript. H. F., X. X. and Z. H. revised and polished the manuscript. All author discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
Supplementary information: The data supporting this article have been included as part of the SI. See DOI: https://doi.org/10.1039/d5sc03774c.Acknowledgements
The authors are grateful for the financial support from the National Natural Science Foundation of China (22101159, 22171168), National Science Foundation of Shandong Province (ZR2022YQ11), and Hongshen Young Scholars Program from Chongqing University (0247005203005). Special thanks to Prof. Hanmin Huang (USTC), Prof. Zhi-Xiang Yu (PKU) and Prof. Deshuang Tu (NJU) for their helpful discussions and suggestions. The authors are grateful to the supports from Shandong Key Laboratory of next-generation high-performance OLED display material.Notes and references
- X. Wu and C. Zhu, Acc. Chem. Res., 2020, 53, 1620–1636 CrossRef CAS PubMed.
- M. Beller, J. Seayad, A. Tillack and H. Jiao, Angew. Chem., Int. Ed., 2004, 43, 3368–3398 CrossRef CAS PubMed.
- Z.-L. Li, G.-C. Fang, Q.-S. Gu and X.-Y. Liu, Chem. Soc. Rev., 2020, 49, 32–48 RSC.
- S. Crespi and M. Fagnoni, Chem. Rev., 2020, 120, 9790–9833 CrossRef CAS PubMed.
- C. Rao, T. Zhang, H. Liu and H. Huang, Nat. Catal., 2023, 6, 847–857 CrossRef CAS.
- F.-F. Tong, Y.-C. Luo, H.-Y. Zhao, X.-P. Fu and X. Zhang, Angew. Chem., Int. Ed., 2025, 64, e202417858 CrossRef CAS PubMed.
- S. Wang, D. Ren, Z. Liu, D. Yang, P. Wang, Y. Gao, X. Qi and A. Lei, Nat. Synth., 2023, 2, 1202–1210 CrossRef CAS.
- C. Rao, T. Zhang and H. Huang, Nat. Commun., 2024, 15, 7924 CrossRef CAS PubMed.
- Z.-P. Bao, Y. Zhang and X.-F. Wu, Chem. Sci., 2022, 13, 9387–9391 RSC.
- J. Wang, Q. Zhou, L. Zhou and Z. Zhang, ACS Catal., 2024, 14, 18499–18506 CrossRef CAS.
- D. Lu, Y. Li, P. Wang, Z. Wang, D. Yang and Y. Gong, ACS Catal., 2022, 12, 3269–3278 CrossRef CAS.
- J. Yu, N.-Y. Yang, J.-T. Cheng, T.-Y. Zhan, C. Luan, L. Ye, Q.-S. Gu, Z.-L. Li, G.-Q. Chen and X.-Y. Liu, Org. Lett., 2021, 23, 1945–1949 CrossRef CAS PubMed.
- A. Reichle, M. Koch, H. Sterzel, L.-J. Großkopf, J. Floss, J. Rehbein and O. Reiser, Angew. Chem., Int. Ed., 2023, 62, e202219086 CrossRef CAS PubMed.
- X. Hou, H. Liu and H. Huang, Nat. Commun., 2024, 15, 1480 CrossRef CAS PubMed.
- R. K. Dhungana, A. Granados, V. Ciccone, R. T. Martin, J. Majhi, M. Sharique, O. Gutierrez and G. A. Molander, ACS Catal., 2022, 12, 15750–15757 CrossRef CAS.
- R. Giri, E. Zhilin and D. Katayev, Chem. Sci., 2024, 15, 10659–10667 RSC.
- C.-Z. Fang, B.-B. Zhang, Y.-L. Tu, Q. Liu, Z.-X. Wang and X.-Y. Chen, J. Am. Chem. Soc., 2024, 146, 26574–26584 CrossRef CAS PubMed.
- T. Constantin, M. Zanini, A. Regni, N. S. Sheikh, F. Juliá and D. Leonori, Science, 2020, 367, 1021–1026 CrossRef CAS PubMed.
- Z. Zhang, B. Górski and D. Leonori, J. Am. Chem. Soc., 2022, 144, 1986–1992 CrossRef CAS PubMed.
- X. Sun and K. Zheng, Nat. Commun., 2023, 14, 6825 CrossRef CAS PubMed.
- T. D. Ho, B. J. Lee, C. Tan, J. A. Utley, N. Q. Ngo and K. L. Hull, J. Am. Chem. Soc., 2023, 145, 27230–27235 CrossRef CAS PubMed.
- G. Liu, K. Gao, T. Yao, H. Hu and Z. Wang, Angew. Chem., Int. Ed., 2025, 64, e202500781 CrossRef CAS PubMed.
- D. Wu, S. Wang, H. Zhang, H. Ke, Z. Sun, S. Xie, Y. Gao, J. Yang, B. Wang and X. Lei, J. Am. Chem. Soc., 2025, 147, 25508–25516 CrossRef PubMed.
- R. K. Neff, Y.-L. Su, S. Liu, M. Rosado, X. Zhang and M. P. Doyle, J. Am. Chem. Soc., 2019, 141, 16643–16650 CrossRef CAS PubMed.
- B. Górski, A.-L. Barthelemy, J. J. Douglas, F. Juliá and D. Leonori, Nat. Catal., 2021, 4, 623–630 CrossRef.
- L. Caiger, H. Zhao, T. Constantin, J. J. Douglas and D. Leonori, ACS Catal., 2023, 13, 4985–4991 CrossRef CAS.
- Y.-L. Su, L. Tram, D. Wherritt, H. Arman, W. P. Griffith and M. P. Doyle, ACS Catal., 2020, 10, 13682–13687 CrossRef CAS.
- T. Zhang and H. Huang, Angew. Chem., Int. Ed., 2023, 62, e202310114 CrossRef CAS PubMed.
- W. Suo, J.-Q. Qi, J. Liu, S. Sun, L. Jiao and X. Guo, J. Am. Chem. Soc., 2024, 146, 25860–25869 CrossRef CAS PubMed.
- H. Zhao, A. J. McMillan, T. Constantin, R. C. Mykura, F. Juliá and D. Leonori, J. Am. Chem. Soc., 2021, 143, 14806–14813 CrossRef CAS PubMed.
- Z.-Y. Xu, Y.-Y. Jiang, W. Su, H.-Z. Yu and Y. Fu, Chem.–Eur. J., 2016, 22, 14611–14617 CrossRef CAS PubMed.
- D. Kurandina, M. Parasram and V. Gevorgyan, Angew. Chem., Int. Ed., 2017, 56, 14212–14216 CrossRef CAS PubMed.
- M. L. Deb, S. S. Dey, I. Bento, M. T. Barros and C. D. Maycock, Angew. Chem., Int. Ed., 2013, 52, 9791–9795 CrossRef CAS PubMed.
- Y. Li, R. Wang, T. Wang, X.-F. Cheng, X. Zhou, F. Fei and X.-S. Wang, Angew. Chem., Int. Ed., 2017, 56, 15436–15440 CrossRef CAS PubMed.
- S. Nakai, T. Yatabe, K. Suzuki, Y. Sasano, Y. Iwabuchi, J.-Y. Hasegawa, N. Mizuno and K. Yamaguchi, Angew. Chem., Int. Ed., 2019, 58, 16651–16659 CrossRef CAS PubMed.
- W. Li, W. Liu, D. K. Leonard, J. Rabeah, K. Junge, A. Brückner and M. Beller, Angew. Chem., Int. Ed., 2019, 58, 10693–10697 CrossRef CAS PubMed.
- V. O. Nyagilo, S. C. Mallojjala and J. S. Hirschi, ACS Catal., 2024, 14, 4683–4689 CrossRef CAS PubMed.
- F. Pünner, Y. Sohtome, Y. Lyu, D. Hashizume, M. Akakabe, M. Yoshimura, Y. Yashiroda, M. Yoshida and M. Sodeoka, Angew. Chem., Int. Ed., 2024, 63, e202405876 CrossRef PubMed.
- J. A. Leitch, T. Rossolini, T. Rogova, J. A. P. Maitland and D. J. Dixon, ACS Catal., 2020, 10, 2009–2025 CrossRef CAS.
- K. Sachidanandan, B. Niu and S. Laulhé, ChemCatChem, 2023, 15, e202300860 CrossRef CAS.
- N. Sanosa, B. Peñín, D. Sampedro and I. Funes-Ardoiz, Eur. J. Org Chem., 2022, 34, e202200420 CrossRef.
- L. Zhao, Y. Huang, Z. Wang, E. Zhu, T. Mao, J. Jia, J. Gu, X.-F. Li and C.-Y. He, Org. Lett., 2019, 21, 6705–6709 CrossRef CAS PubMed.
- V. S. Kostromitin, A. O. Sorokin, V. V. Levin and A. D. Dilman, Chem. Sci., 2023, 14, 3229–3234 RSC.
- X. Xu and X. Li, Org. Lett., 2009, 11, 1027–1029 CrossRef CAS PubMed.
- M. Briand, E. Magnier, E. Anselmi and G. Dagousset, Adv. Synth. Catal., 2024, 366, 3494–3499 CrossRef CAS.
- P. Cherkupally, A. Slazhnev and P. Beier, Synlett, 2011, 3, 331–334 Search PubMed.
- Z. Zhang, X. Li and D. Shi, Adv. Synth. Catal., 2021, 363, 3348–3353 CrossRef CAS.
- S. Arora, P. Katiyar, T. Singh and A. Singh, Org. Lett., 2025, 27, 3617–3621 CrossRef CAS PubMed.
- S. Morand, P. Jubault, J.-P. Bouillon and S. Couve-Bonnaire, Chem.–Eur. J., 2021, 27, 17273–17292 CrossRef CAS PubMed.
- Y. Han, L. Zhou, C. Wang, S. Feng, R. Ma and J.-P. Wan, Chin. Chem. Lett., 2024, 35, 108977 CrossRef CAS.
- M. Lemmerer, M. Schupp, D. Kaiser and N. Maulide, Nat. Synth., 2022, 1, 923–935 CrossRef CAS.
- V. Soulard, G. Villa, D. P. Vollmar and P. Renaud, J. Am. Chem. Soc., 2018, 140, 155–158 CrossRef CAS PubMed.
- G. Tu, C. Yuan, Y. Li, J. Zhang and Y. Zhao, Angew. Chem., Int. Ed., 2018, 57, 15597–15601 CrossRef CAS PubMed.
- W.-T. Fan, Y. Li, D. Wang, S.-J. Ji and Y. Zhao, J. Am. Chem. Soc., 2020, 142, 20524–20530 CrossRef CAS PubMed.
- E. Shirakawa, X. Zhang and T. Hayashi, Angew. Chem., Int. Ed., 2011, 50, 4671–4674 CrossRef CAS PubMed.
- Z. Feng, Q.-Q. Min, H.-Y. Zhao, J.-W. Gu and X. Zhang, Angew. Chem., Int. Ed., 2015, 54, 1270–1274 CrossRef CAS PubMed.
- N. Rao, Y.-Z. Li, Y.-C. Luo, Y. Zhang and X. Zhang, ACS Catal., 2023, 13, 4111–4119 CrossRef CAS.
- A. L. G. Kanegusuku and J. L. Roizen, Angew. Chem., Int. Ed., 2021, 60, 21116–21149 CrossRef PubMed.
- W. Li, R. Liu, R. Li, S. Wang, D. Li and J. Yang, Adv. Synth. Catal., 2021, 363, 5266–5271 CrossRef CAS.
- H.-Y. Zhao, Z. Feng, Z. Luo and X. Zhang, Angew. Chem., Int. Ed., 2016, 55, 10401–10405 CrossRef CAS PubMed.
- A. Alberti, M. Benaglia and D. Macciantelli, Org. Lett., 2000, 2, 1553–1555 CrossRef CAS PubMed.
- G. R. Buettner, Free Radical Biol. Med., 1987, 3, 259–303 CrossRef CAS PubMed.
- P. Biswas, A. Maity, M. T. Figgins and D. C. Powers, J. Am. Chem. Soc., 2024, 146, 30796–30801 CrossRef CAS PubMed.
- X. Guo, H. Wang, Z. Hu, Y. Zhao, J. Dong, K. Zhong, Y. Lan and X. Xu, ACS Catal., 2025, 15, 5307–5317 Search PubMed.
- Y. Abderrazak and O. Reiser, ACS Catal., 2024, 14, 4847–4855 Search PubMed.
- P.-F. Yuan, Z. Yang, S.-S. Zhang, C.-M. Zhu, X.-L. Yang and Q.-Y. Meng, Angew. Chem., Int. Ed., 2024, 63, e202313030 CrossRef CAS PubMed.
- L. Xu, J. Xu, J. Zhu, Z. Yao, N. Yu, W. Deng, Y. Wang and B.-L. Lin, Angew. Chem., Int. Ed., 2019, 58, 6070–6073 CrossRef CAS PubMed.
- F. M. Dennis, A. R. Arenas, M. Shanmugam and B. M. Partridge, Chem. Eur. J., 2024, 30, e202303636 CrossRef CAS PubMed.
- X. Zhang, H. Liu, X. Hu, G. Tang, J. Zhu and Y. Zhao, Org. Lett., 2011, 13, 3478–3481 CrossRef CAS PubMed.
- J. Xiao, J. Wang, H. Zhang, J. Zhang and L.-B. Han, J. Org. Chem., 2023, 88, 3909–3915 CrossRef CAS PubMed.
- H.-J. Zhang, A. W. Schuppe, S.-T. Pan, J.-X. Chen, B.-R. Wang, T. R. Newhouse and L. Yin, J. Am. Chem. Soc., 2018, 140, 5300–5310 CrossRef CAS PubMed.
- J.-S. Tian and T.-P. Loh, Angew. Chem., Int. Ed., 2010, 49, 8417–8420 CrossRef CAS PubMed.
- S. Engl and O. Reiser, Chem. Soc. Rev., 2022, 51, 5287–5299 RSC.
- M.-C. Belhomme, D. Dru, H.-Y. Xiong, D. Cahard, T. Besset, T. Poisson and X. Pannecoucke, Synthesis, 2014, 46, 1859–1870 CrossRef.
- D. Li, T. Mao, J. Huang and Q. Zhu, J. Org. Chem., 2018, 83, 10445–10452 CrossRef CAS PubMed.
- T. P. Nicholls, G. E. Constable, J. C. Robertson, M. G. Gardiner and A. C. Bissember, ACS Catal., 2016, 6, 451–457 CrossRef CAS.
- B. Zhu, T. Shen, X. Huang, Y. Zhu, S. Song and N. Jiao, Angew. Chem., Int. Ed., 2019, 58, 11028–11032 CrossRef CAS PubMed.
- Y. Shen and T. Rovis, J. Am. Chem. Soc., 2021, 143, 16364–16369 CrossRef CAS PubMed.
- W.-J. Yue, C. S. Day, A. J. B. Rucinski and R. Martin, Org. Lett., 2022, 24, 5109–5114 CrossRef CAS PubMed.
- S. Jana and N. Cramer, J. Am. Chem. Soc., 2024, 146, 35199–35207 CrossRef CAS PubMed.
- H. Y. Kim and K. Oh, Org. Biomol. Chem., 2021, 19, 3569–3583 RSC.
- N. Katta, Q.-Q. Zhao, T. Mandal and O. Reiser, ACS Catal., 2022, 12, 14398–14407 CrossRef CAS PubMed.
- K. Fuchibe, H. Hatta, K. Oh, R. Oki and J. Ichikawa, Angew. Chem., Int. Ed., 2017, 56, 5890–5893 CrossRef CAS PubMed.
- T. D. Ho, B. J. Lee, T. L. Buchanan, M. E. Heikes, R. M. Steinert, E. G. Milem, S. T. Goralski, Y.-N. Wang, S. Lee, V. M. Lynch, M. J. Rose, K. R. Mitchell-Koch and K. L. Hull, J. Am. Chem. Soc., 2024, 146, 25176–25189 CrossRef CAS PubMed.
- W. S. Yoon, W. J. Jang, W. Yoon, H. Yun and J. Yun, Nat. Commun., 2022, 13, 2570 CrossRef CAS PubMed.
- J. Ren, K. Liu, N. Wang, X. Kong, J. Li and K. Li, ACS Catal., 2023, 13, 11001–11011 CrossRef CAS.
- Z. Zhang, M. J. Tilby and D. Leonori, Nat. Synth., 2024, 3, 1221–1230 CrossRef CAS.
- T. Wan, L. Capaldo, D. Ravelli, W. Vitullo, F. J. de Zwart, B. de Bruin and T. Noël, J. Am. Chem. Soc., 2023, 145, 991–999 CrossRef CAS PubMed.
- Z. Zhang, L. Poletti and D. Leonori, J. Am. Chem. Soc., 2024, 146, 22424–22430 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |