
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric intramolecular reductive coupling of bisimines templated by chiral diborons†
Tian
Chen‡
a,
Hao-Yang
Wang‡
c,
Ronghua
Xu
b,
Guangqing
Xu
b,
He
Yang
*b,
Jiangtao
Sun
 *a,
Lung Wa
Chung
*c and
Wenjun
Tang
*ad
*a,
Lung Wa
Chung
*c and
Wenjun
Tang
*ad
aJiangsu Key Laboratory of Advanced Catalytic Materials & Technology, School of Petrochemical Engineering, Changzhou University, 1 Gehu Road, Changzhou 213164, China. E-mail: jtsun@cczu.edu.cn
bState Key Laboratory of Chemical Biology, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, 345 Ling Ling Road, Shanghai 200032, China. E-mail: yanghe@sioc.ac.cn; tangwenjun@sioc.ac.cn
cShenzhen Grubbs Institute, Department of Chemistry and Guangdong Provincial Key Laboratory of Catalysis, Southern University of Science and Technology, Shenzhen 518055, China. E-mail: oscarchung@sustech.edu.cn
dSchool of Chemistry and Materials Science, Hangzhou Institute for Advanced Study, University of Chinese Academy of Sciences, 1 Sub-lane Xiangshan, Hangzhou 310024, China
First published on 17th June 2025
Abstract
An asymmetric intramolecular reductive coupling of bisimines has been accomplished for the first time under mild conditions with bis((+)-pinanediolato)diboron as the template, providing the unprecedented chiral dihydrophenanthrene-9,10-cis-diamines in high yields and excellent enantioselectivities. The chiral exocyclic cis-diamine products have served as effective chiral ligands for asymmetric catalysis. A DFT study highlights the crucial roles of the uncommon twisted-boat pathway (instead of the common chair type) and the steric effect in exclusively forming the cis-diamines and achieving high enantioselectivity. This reductive coupling protocol represents a significant expansion of the diboron-promoted [3,3]-sigmatropic rearrangement.
Introduction
Chiral exocyclic vicinal cis-diamines1 belong to a unique class of chiral vicinal diamines that are found in the structures of a number of biologically important natural products and therapeutic agents (Fig. 1a). For example, biotin,2 containing a chiral cis-diamine substructure on a tetrahydrothiofuran ring, is involved in a wide range of metabolic processes as one of the B vitamins; edoxaban,3 a factor Xa inhibitor used as an anti-coagulant medication, possesses a chiral cyclohexane cis-diamine scaffold; saxitoxin,4 bearing a cis-diamine structure on a cyclic guanidine, is a neurotoxin that acts as a sodium channel blocker. In addition, enantiomerically pure exocyclic vicinal cis-diamines serve as conformationally rigid and sterically bulky backbones for transition-metal catalysts in asymmetric catalysis.5 However, despite the significant progress in the synthesis of chiral acyclic vicinal diamines6 and exocyclic trans-diamines,7 the construction of chiral exocyclic vicinal cis-diamines, mainly based on a cyclohexane skeleton, remains a significant challenge.8 The current reported methods suffered from either lengthy synthetic sequences8d or low diastereo- and/or enantioselectivities. | ||
| Fig. 1 Chiral cis-diamines and their enantioselective syntheses by intramolecular asymmetric reductive coupling of bisimine. (a) The prevalence of chiral cis-diamines. (b) Intermolecular asymmetric reductive coupling of imines templated by chiral diborons. (c) This study on intramolecular asymmetric reductive coupling of imines. | ||
We have recently developed an efficient method for the synthesis of chiral vicinal diamines through asymmetric intermolecular reductive homocoupling of imines templated by chiral diborons (Fig. 1b).9 The featured diboron-promoted [3,3]-sigmatropic rearrangement has enabled the single-step synthesis of chiral tetrahydro-bisisoquinolines, bis(cyclic amine)s, acyclic vicinal disubstituted diamines, and acyclic vicinal tetrasubstituted diamines in excellent yields, diastereoselectivities, and enantioselectivities. By using bisaldimines as starting materials, polymeric chiral diamines were synthesized in a highly stereoselective fashion with Mn ranging from 5000 to 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000.9d In contrast, efficient asymmetric intramolecular reductive coupling of bisimines has not been realized. Such transformation would lead to chiral exocyclic vicinal diamines in a step-economic fashion.
000.9d In contrast, efficient asymmetric intramolecular reductive coupling of bisimines has not been realized. Such transformation would lead to chiral exocyclic vicinal diamines in a step-economic fashion.
There are several challenges for such an unprecedented transformation: (1) chemoselectivity: how to inhibit the intermolecular polymerization and enforce the intramolecular cyclization? (2) Diastereoselectivity: would the intramolecular diboron-promoted [3,3]-sigmatropic rearrangement proceed through a chair-like six-membered transition state to form a trans-diamine or a cis-diamine? (3) Enantioselectivity: can a chiral exocyclic diamine be constructed in a single step from a readily available starting material? In this study, we have successfully addressed the above challenges by judicious selection of bisimine substrates and chiral diborons, and accomplished the first and asymmetric intramolecular reductive coupling of bisimines (Fig. 1c). The coupling protocol has enabled one-step synthesis of unprecedented dihydrophenanthrene-9,10-cis-diamines and provided excellent yields, exclusive cis selectivities, and almost perfect enantioselectivities, by employing chiral bis(pinanediolato)diboron as the template.
Results and discussion
At the outset of our study, the feasibility of intramolecular reductive coupling of bisimines was investigated with the hexane-1,6-diimine substrate (1a), which was in situ prepared from adipaldehyde and ammonia (Fig. 2a). Treatment of 1a with (Bpin)2 at rt provided a mixture of polyamines without detection of intramolecular cyclization product 2a. We assumed that the intermolecular coupling was more favorable than the intramolecular coupling in the case of 1a. A structurally more rigid diimine 1b could be more suitable for intramolecular coupling. Thus, compound 1b was prepared in situ from [1,1′-biphenyl]-2,2′-dicarbaldehyde and ammonia and treated with (Bpin)2 at rt. Indeed, a trace amount of the intramolecular coupling product 2b was formed according to LC-MS, and the major isolated product was 5-hydroxy-5H-dibenzo[c,e]azepin (2b′).10 These results showed that the intramolecular coupling of bisimine could be accomplished by inhibiting the polymerization process and other side reactions, including hydrolysis and formation of a stable dibenzo[c,e]azepin derivative, by employing substrates with increased rigidity and steric hindrance. Pleasingly, the efficiency of the intramolecular coupling was significantly enhanced when N-substituents were introduced into the substrate. Thus, 1c was treated with (Bpin)2 under similar conditions, and the resulting cyclization cis-diamine product 2c was formed in 80% yield, whose relative configuration was confirmed by the X-ray crystal structure of its urea derivative 3c (Fig. 2b).11 Notably, the unexpected formation of cis-diamine 2c was intriguing since no meso products were formed in our previous reports on various intermolecular reductive homocouplings of imines.9 Despite the symmetry of cis-diamine 2c, we proposed that a chiral cis-diamine would be constructed for the first time if an unsymmetrical bisimine based on the [1,1′-biphenyl]-2,2′-dicarbaldehyde skeleton was employed, and an effective chiral diboron template could be discovered. Hence, 3-methoxy-[1,1′-biphenyl]-2,2′-dicarbaldehyde (4a) was treated with MeNH2·HCl and triethylamine in situ to form bisimine 5a, which was then treated with chiral diborons in THF at rt for 48 h (Fig. 2c). Encouragingly, the chiral diboron with four phenyl substituents DB1 provided 6a in 85% yield and 22% ee. This proof-of-concept result demonstrated the feasibility of forming 6a in high enantioselectivity by screening various chiral diborons. While DB2 with bromo substituents showed no reactivity, DB3 with six phenyl substituents led to 6a in 84% yield and 55% ee. Further modifications of substituents on the aryl groups in DB4-10 all provided good yields, but with moderate enantioselectivities. Finally, employment of chiral bis(pinanediolato)diboron DB11 led to the formation of 6a in 90% yield and 99% ee. Further solvent screening showed that the cyclization proceeded with similarly high enantioselectivities in various solvents, indicating the robustness of the transformation (see the ESI† for more details). | ||
| Fig. 2 Asymmetric intramolecular reductive coupling of bisimine 5a templated by various chiral diborons. (a) Attempts on diboron templated intramolecular reductive coupling of bisimines. (b) Formation of cis-diamine by diboron templated intramolecular reductive coupling. (c) The development of asymmetric intramolecular reductive coupling protocol. | ||
The substrate scope of intramolecular reductive coupling was studied. As can be seen from Table 1, unsymmetrical [1,1′-biphenyl]-2,2′-bisimine substrates with various 3-substituents were employed, providing chiral dihydrophenanthrene-9,10-cis-diamines 6a–h in moderate to high yields. While most products 6d–g were formed in almost perfect enantioselectivities regardless of their electronic properties, the small-size fluoro-substituted product 6c was obtained in 56% ee, indicating that the size of the substituent might be influential to the enantioselectivity. In addition, product 6h with a dioxolane ring was less selective (75% ee). The presence of 3-substitution (ortho-substitution) appeared to be crucial for the enantioselectivity, as substrates with 4- or 5-methoxy substituents provided very low enantioselectivities (see the ESI† for more details).
| a Unless otherwise specified, the reactions were carried out at rt with 5a–aj (0.2 mmol, 1.0 equiv.), DB11 (0.2 mmol, 1.0 equiv.) in THF (10 mL) for 48 h. Isolated yields. The ee values were determined by chiral HPLC analysis. |
|---|

|
By fixing the upper aryl ring with a methoxy substituent at the 3′ position, the substitution effect on the lower aryl ring was studied. The excellent enantioselectivities (91–99% ee) obtained for 6i–u demonstrated that the reaction was compatible with various substituents at 3′, 4′, and 5′ positions regardless of their electronic properties. It was interesting that products 6o and 6p with ortho-substituents on both upper and lower rings were obtained in excellent ee's. The synthesis of product 6v with a thiophene ring was extremely enantioselective (99% ee). A substrate with a naphthalene upper ring was also compatible, forming 6w in 88% yield and 99% ee. Surprisingly, product 6x with a dioxolane ring at both 4′ and 5′ positions was afforded in a low ee (30%). Substrates with multiple substituents on the upper aryl ring were also suitable. While the optical purity of product 6y with ortho-fluoro substituent was inferior (47% ee), product 6z with ortho-bromo substituent was obtained in 99% ee. The intramolecular reductive coupling was not limited to N-methyl substituents. A substrate with N-ethyl substituents was also compatible, leading to 6ab in 36% yield and 71% ee. Finally, substrates with 3-aryl substituents were employed, forming a series of ortho-aryl dihydrophenanthrene-9,10-cis-diamines 6ac–aj in good yields (53–98%) and excellent enantioselectivities (95–99% ee).
The exclusive formation of uncommon cis-diamine products and excellent enantioselectivities from the intramolecular reductive coupling called for a plausible mechanistic rationale. To gain insights into the reaction mechanism and the origin of the enantioselectivity, a systematic DFT study (SMD M06-2X-D3/def2-TZVP//SMD M06-2X-D3/BS1 method mainly) was first conducted by using the chiral diboron agent DB11 and the bisimine substrate 5e (R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Br; Fig. 3).11 Compared with our previous studies on the intermolecular coupling systems,9a,c,11e our current computational results reveal a distinct feature for this intramolecular coupling of bisimines. This intramolecular coupling adopts uncommon twisted boat transition states during the [3,3]-sigmatropic rearrangement to afford the synthetically challenging chiral vicinal cis-diamines, due to the ring strain of the bisimine tether (Fig. 3b, S2 and S3†). In contrast, the previous intermolecular coupling systems follow the common chair-type pathway.9a,c,11e A total of 16 conformers for the coupling transition states (TSs) (Tables S8 and S10, Fig. S2 and S3†) were located for 5e through our comprehensive conformational search, with eight of them yielding the major product (P) and the others yielding the enantiomer product (Pent). As shown in Fig. 3a, the most favorable TS1F leading to the desired coupling product (P) was computed to be lower in free energy than that giving the minor product PentviaTS1Aent by 1.8 kcal mol−1. This result qualitatively explains the observed stereochemistry. The similar free energy difference (ΔΔG≠) is also supported by a few different common computational methods (Tables S9 and S11†).
Br; Fig. 3).11 Compared with our previous studies on the intermolecular coupling systems,9a,c,11e our current computational results reveal a distinct feature for this intramolecular coupling of bisimines. This intramolecular coupling adopts uncommon twisted boat transition states during the [3,3]-sigmatropic rearrangement to afford the synthetically challenging chiral vicinal cis-diamines, due to the ring strain of the bisimine tether (Fig. 3b, S2 and S3†). In contrast, the previous intermolecular coupling systems follow the common chair-type pathway.9a,c,11e A total of 16 conformers for the coupling transition states (TSs) (Tables S8 and S10, Fig. S2 and S3†) were located for 5e through our comprehensive conformational search, with eight of them yielding the major product (P) and the others yielding the enantiomer product (Pent). As shown in Fig. 3a, the most favorable TS1F leading to the desired coupling product (P) was computed to be lower in free energy than that giving the minor product PentviaTS1Aent by 1.8 kcal mol−1. This result qualitatively explains the observed stereochemistry. The similar free energy difference (ΔΔG≠) is also supported by a few different common computational methods (Tables S9 and S11†).
 | ||
| Fig. 3 (a) Calculated Gibbs free energy profile of the reaction of DB11 with 5e (RBr) at the SMD M06-2X-D3/BS1 (L1) and SMD M06-2X-D3/def2-TZVP//SMD M06-2X-D3/BS1 (L2) levels (see ref. 11a). (b) Computed structures and key geometric parameters (distances in Å and dihedrals in italics and degrees) of the three key coupling transition states with their relative Gibbs free energies. Unimportant H atoms were omitted for clarity. (c) The effect of R substituents on the relative Gibbs free energy (in kcal mol−1) of the two key transition states to form the minor product with respect to their corresponding lowest-energy TS1F forms leading to the major product at the SMD M06-2X-D3/def2-TZVP//SMD M06-2X-D3/BS1 level. #TS1Gent-H has the same geometry as TS1F-H. | ||
Additional calculations were conducted to examine the size effect of R (F, Cl, Br or H) on the stereoselectivity (Fig. 3c, S4 and Tables S12–S17†). Likewise, the same favorable coupling TSs (TS1F-Cl and TS1Aent-Cl) and free energy difference (ΔΔG≠ ∼1.8 kcal mol−1) for 5d (RCl) were also found. In contrast, 5c (RF) adopts another and lower-energy conformer for the minor-coupling TS (TS1Gent-F), which reduces the free energy difference between the two stereo-determining coupling TSs (ΔΔG≠) to ∼0.9 kcal mol−1 only. To examine the steric effect of R on the energetics of the three key coupling TSs, tBu was employed and found to further increase the free energy difference (ΔΔG≠ ∼2.4–6.1 kcal mol−1vs. 1.8–4.4 kcal mol−1 for RBr; Table S18†). Overall, these computational results are in qualitative agreement with the experimental observation, highlighting the essential role of the steric effect of the R substituent in controlling stereoselectivity (particularly for the TS1Gent type TSs).
As shown in Fig. 3b and S4,† the major coupling TSs (TS1F, TS1F-Cl and TS1F-F) can avoid steric congestion between the halogen substituent and diboron as well as minimize H–H repulsion inside the diboron part (especially on the methylene bridge; see Fig. S6†). In contrast, the minor coupling TSs (e.g.TS1Aent and TS1Gent) suffer from more steric hindrance between R and the diboron as well as more severe H–H repulsion in the diboron part. Generally, such steric crowdedness slightly increases the dihedral angle along the biphenyl C–C single bond (diminishing delocalization) and decreases the dihedral angles of the boat-shaped [3,3]-sigmatropic rearrangement TS structure (Table S19†). The TS1Gent and TS1F form TSs adopt the essentially same conformation except for the substituent positioned in different arene rings. For a very small substituent (RF or H), the TS1Gent form TSs are more energetically favorable to form the minor product than the TS1Aent form TSs and lead to a smaller free energy difference with the TS1F form TSs. However, the TS1Gent form TSs become unfavorable than the TS1Aent and TS1F form TSs with a larger R (RCl or Br), since the R substituent has the closest contact with the diboron in the TS1Gent form TSs and experiences more steric repulsion. In addition, the most favorable TS1F type TSs have a lesser B–B bond elongation than the other two minor-type TSs. Distortion/interaction analysis12 further revealed that the TS1F type TSs have smaller distortion energy than the two types of minor TSs (ΔΔEdist = 2.7–4.0 kcal mol−1 for RF and 3.3–9.1 kcal mol−1 for RBr; Table S20†), which plays a vital role in the stereoselectivity. Local distortion analysis13 on the 10-atom diboron core part also gave comparable relative distortion energies for these key TSs (ΔΔEdist = 1.5–5.3 kcal mol−1 for RBr; Fig. S5†).
To demonstrate the practicality of this transformation, the reductive cyclization of 5e was performed in THF at rt for 48 h in the presence of DB11 at a 2.5 gram scale and product 6e was obtained in 92% yield and 99% ee as a single diastereomer (Scheme 1a). Treatment of 6e with triphosgene and TEA provided 7e, whose absolute stereochemistry was confirmed by its X-ray crystal structure. The yields from 5e to 6e were monitored by NMR studies, and the intramolecular reductive coupling was found to be a much slower process, in contrast to the previously reported intermolecular homocoupling of isoquinolines.9a The chiral bromo-substituted product 7e was a versatile intermediate for further derivatization (Scheme 1b). The Suzuki–Miyaura coupling of 7e with phenylboronic acid or prop-1-en-2-ylboronic acid catalyzed by Pd/SPhos afforded products 7ac and 8a in 50% yield and 99% yield, respectively, without erosion of enantiomeric purities (99% ee). Moreover, the Suzuki–Miyaura coupling of 7e with 9-anthrylboronic acid proceeded with Pd(OAc)2 and BIDIME as the catalyst system to give 7aj in 69% yields. A Buchwald–Hartwig coupling of 7e with aniline catalyzed by Pd/BIDIME delivered 8b in 93% yield. In addition, Sonogashira coupling of 7e with phenylacetylene led to 8c in 62% yield and 99% ee.
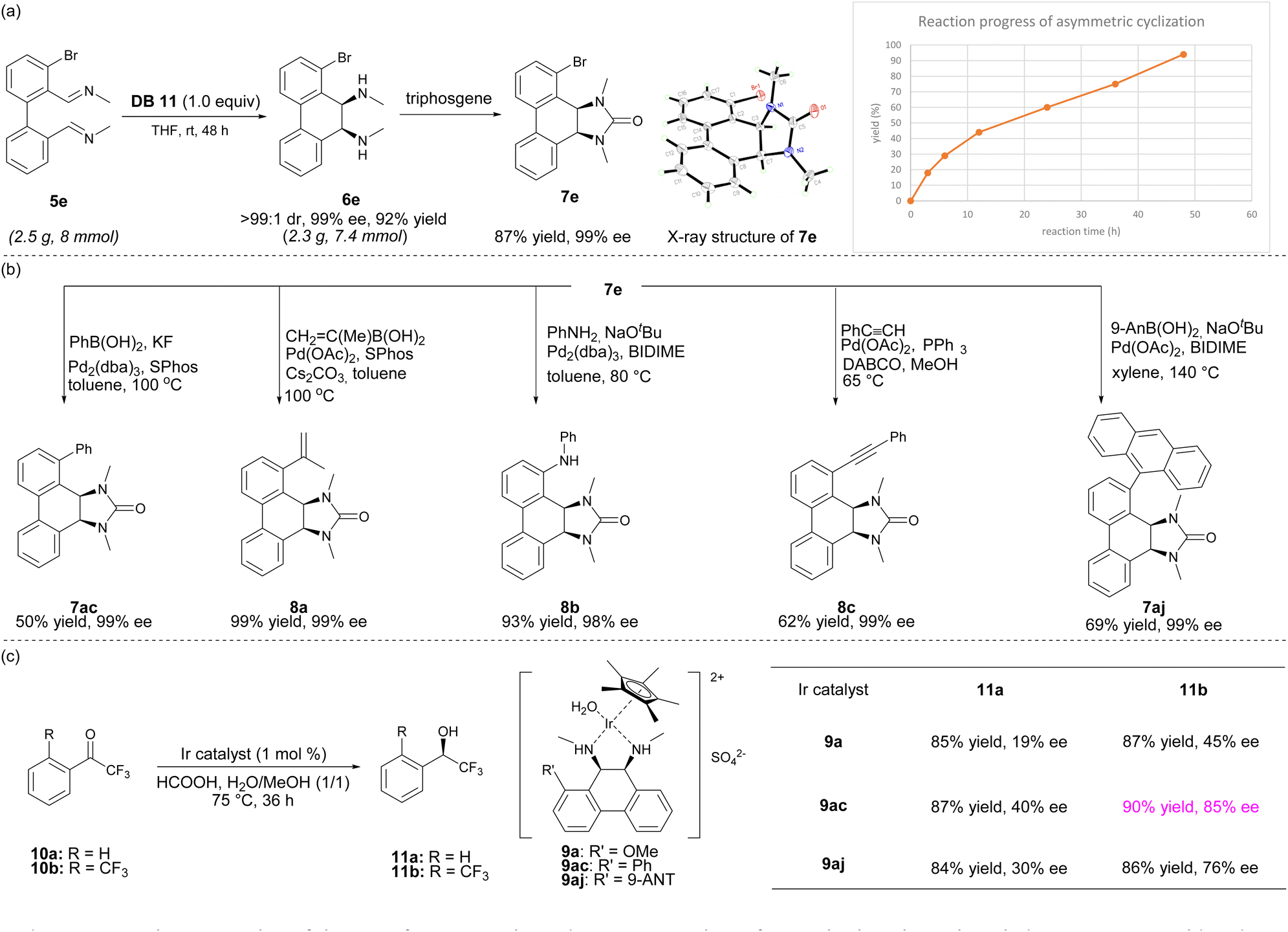 | ||
| Scheme 1 Derivatization and catalytic applications of chiral cis-diamines. (a) Gram scale reaction and kinetic profile. (b) Transformation of coupling product. (c) Utility of chiral cis-diamine product in asymmetric transfer hydrogenation. | ||
The conformationally rigid chiral cis-diamine products were suitable ligands for asymmetric catalysis. We envisioned that its η5 iridium complex could be suitable for asymmetric transfer hydrogenation.9d,14 Therefore, iridium complexes 9a, 9ac, and 9aj were prepared and applied to the asymmetric transfer hydrogenation of trifluoroacetophenones 10a and 10b (Scheme 1c). All reductions were found to be enantioselective. Notably, the asymmetric transfer hydrogenation of 10b with catalyst 9ac provided 11b in 90% yield and 85% ee, demonstrating the effectiveness of these chiral cis-diamines as chiral ligands in asymmetric catalysis.
Conclusions
In summary, we have developed an asymmetric intramolecular reductive coupling of bisimines under mild conditions using chiral bis(pinanediolato)diboron as the template, providing the unprecedented chiral dihydrophenanthrene-9,10-cis-diamines in high yields and excellent enantioselectivities. Our systematic computational investigation has revealed the vital roles of a less common twisted-boat pathway (rather than the common chair pathway) and steric effect in exclusively forming the cis-diamines and achieving high enantioselectivity for this first intramolecular reductive coupling. The chiral exocyclic cis-diamine products are effective ligands for asymmetric catalytic reactions. This method signifies the broad scope of the diboron-promoted [3,3]-sigmatropic rearrangement and enriches the chemistry of chiral vicinal cis-diamines.Data availability
The ESI† is available and includes full experimental details, synthesis protocols, characterization data, and spectroscopic details. CCDC 2428416 (compound 3c), 2428418 (compound 7a), and 2428420 (compound 7e) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge viahttps://www.ccdc.ca-m.ac.uk/data_request/cif, or by emailing E-mail: data_request@ccdc.cam.ac.uk, or by contacting the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.Author contributions
W. T., G. X. and H. Y. conceptualised the work. T. C., R. X. and H. Y. conducted experiments. H.-Y. W. and L. W. C. performed DFT calculations. The manuscript was written through contributions of all authors. W. T., L. W. C., J. S. and H. Y. supervised the work, secured funding, edited and finalised the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The work was supported by the National Key R&D Program of China (2021YFF0701600), NSFC (82188101, 22122104, and 22193023), the Youth Innovation Promotion Association, CAS, the Shenzhen Nobel Prize Scientists Laboratory Project (C17783101), the Guangdong Provincial Key Laboratory of Catalysis (2020B121201002) and the Science and Technology Commission of Shanghai Municipality (23ZR1476500). The authors thank the Center for Computational Science and Engineering of Southern University of Science and Technology and CHEM HPC at SUSTech for partly supporting this work.Notes and references
- (a) D. Lucet, T. Le Gall and C. Mioskowski, Angew. Chem., Int. Ed., 1998, 37, 2580–2627 CrossRef CAS; (b) F. Foubelo, C. Nájera, M. G. Retamosa, J. M. Sansano and M. Yus, Chem. Soc. Rev., 2024, 53, 7983–8085 RSC; (c) S. R. S. S. Kotti, C. Timmons and G. Li, Chem. Boil. Drug. Des., 2006, 67, 101–114 CrossRef CAS; (d) Z. L. Tao and S. E. Denmark, Synthesis, 2021, 53, 3951–3962 CrossRef CAS; (e) A. K. Weber, J. Schachtner, R. Fichtler, T. M. Leermann, J. M. Neudörfl and A. J. von. Wangelin, Org. Biomol. Chem., 2014, 12, 5267–5277 RSC; (f) B. Yang, A. Yu and Y. Wang, ChemCatChem, 2023, 15, e202300141 CrossRef CAS.
- (a) A. Marquet, Pure Appl. Chem., 1993, 65, 1249–1252 CrossRef CAS; (b) C. A. Perry and T. A. Butterick, Adv. Nutr., 2024, 15, 100251 CrossRef CAS PubMed.
- (a) M. Poulakos, J. N. Walker, U. Baig, T. David and Am. J. Health, Syst Pharm., 2017, 74, 117–129 CAS; (b) G. E. Raskob, N. van. Es, P. Verhamme, M. Carrier, M. D. Nisio, D. Garcia, M. A. Grosso, A. K. Kakkar, M. J. Kovacs, M. F. Mercuri, G. Meyer, A. Segers, M. Shi, T.-F. Wang, E. Yeo, G. Zhang, J. I. Zwicker, J. I. Weitz and H. R. Büller, N. Engl. J. Med., 2018, 378, 615–624 CrossRef CAS.
- (a) V. Pratheepa and V. Vasconcelos, Mini. Rev. Med. Chem., 2017, 17, 320–327 CrossRef CAS; (b) A. P. Thottumkara, W. H. Parsons and J. D. Bois, Angew. Chem., Int. Ed., 2014, 53, 5760–5784 CrossRef CAS; (c) L. E. Llewellyn, Nat. Prod. Rep., 2006, 23, 200–222 RSC; (d) K. D. Cusick and G. S. Sayler, Mar. Drugs, 2013, 11, 991–1018 CrossRef CAS.
- (a) H. Zhou, Y. Z. Geng and S. W. Yan, Acta Phys. Sin., 2023, 72, 068702 CrossRef; (b) T. A. Davis and J. N. Johnston, Chem. Sci., 2011, 2, 1076–1079 RSC.
- (a) Y.-W. Zhong, K. Izumi, M.-H. Xu and G.-Q. Lin, Org. Lett., 2004, 6, 4747–4750 CrossRef CAS PubMed; (b) E. P. Vanable, J. L. Kennemur, L. A. Joyce, R. T. Ruck, D. M. Schultz and K. L. Hull, J. Am. Chem. Soc., 2019, 141, 739–742 CrossRef CAS PubMed; (c) J. Chen, X. Gong, J. Li, Y. Li, J. Ma, C. Hou, G. Zhao, W. Yuan and B. Zhao, Science, 2018, 360, 1438–1442 CrossRef CAS PubMed; (d) P. Zhou, X. Shao and S. J. Malcolmson, J. Am. Chem. Soc., 2021, 143, 13999–14008 CrossRef CAS PubMed; (e) Y. Chen, Y. Pan, Y. M. He and Q. H. Fan, Angew. Chem., Int. Ed., 2019, 58, 16831–16834 CrossRef CAS; (f) L. Yu and P. Somfai, Angew. Chem., Int. Ed., 2019, 58, 8551–8555 CrossRef CAS; (g) Z. Tao, B. B. Gilbert and S. E. Denmark, J. Am. Chem. Soc., 2019, 141, 19161–19170 CrossRef CAS PubMed; (h) S. Roland, P. Mangeney and A. Alexakis, Synthesis, 1999, 2, 228–230 CrossRef; (i) S. Y. M. Chooi, P. Leung, S. Ng, G. H. Quek and K. Y. Sim, Tetrahedron: Asymmetry, 1991, 2, 981–982 CrossRef CAS.
- (a) F. Liu, G. Zhao, W. Cai, D. Xu and B. Zhao, Org. Biomol. Chem., 2018, 16, 7498–7502 RSC; (b) M. Chandrasekhar, G. Sekar and V. K. Singh, Tetrahedron Lett., 2000, 41, 10079–10083 CrossRef CAS; (c) H. S. Chong, X. Sun, Y. Chen and M. Wang, Tetrahedron Lett., 2015, 56, 946–948 CrossRef CAS; (d) M. R. Monaco, B. Poladura, M. D. L. Bernardos, M. Leutzsch, R. Goddard and B. List, Angew. Chem., Int. Ed., 2014, 53, 7063–7067 CrossRef CAS PubMed; (e) S.-Z. Lin and T.-P. You, Synth. Commun., 2009, 39, 4133–4138 CrossRef CAS; (f) Z. Chai, P. J. Yang, H. Zhang, S. Wang and G. Yang, Angew. Chem., Int. Ed., 2017, 56, 650–654 CrossRef CAS; (g) Y. H. Cho, V. Zunic, H. Senboku, M. Olsen and M. Lautens, J. Am. Chem. Soc., 2006, 128, 6837–6846 CrossRef CAS; (h) N. Taniguchi, T. Hata and M. Uemura, Angew. Chem., Int. Ed., 1999, 38, 1232–1235 CrossRef CAS.
- (a) F. Orsini, G. Sello and G. Bestetti, Tetrahedron: Asymmetry, 2001, 12, 2961–2969 CrossRef CAS; (b) S. H. Jung and H. Kohn, J. Am. Chem. Soc., 1985, 107, 2931–2943 CrossRef CAS; (c) R. Annunziata, M. Benaglia, M. Caporale and L. Raimondi, Tetrahedron: Asymmetry, 2002, 13, 2727–2734 CrossRef CAS; (d) D. Yuan, Q. Peng, W. Xia, W. Yu, L. Ruan, Y. Wang, Y. Chen and X. Pan, Org. Process Res. Dev., 2024, 28, 2623–2634 CrossRef CAS.
- (a) D. Chen, G. Xu, Q. Zhou, L. W. Chung and W. Tang, J. Am. Chem. Soc., 2017, 139, 9767–9770 CrossRef CAS; (b) M. Zhou, K. Li, D. Chen, R. Xu, G. Xu and W. Tang, J. Am. Chem. Soc., 2020, 142, 10337–10342 CrossRef CAS; (c) M. Zhou, Y. Lin, X.-X. Chen, G. Xu, L. W. Chung and W. Tang, Angew. Chem., Int. Ed., 2023, 62, e202300334 CrossRef CAS; (d) Y. Lin, G. Xu and W. Tang, J. Am. Chem. Soc., 2024, 146, 27736–27744 CrossRef CAS PubMed.
- J. O. Hawthorne, E. L. Mhielic, M. S. Morgan and M. H. Wilt, J. Org. Chem., 1963, 28, 2831–2835 CrossRef CAS.
- (a) See ESI† for the computational details.; (b) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed; (c) A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS; (d) G. Luchini, J. V. Alegre-Requena, I. Funes-Ardoiz and R. S. Paton, F1000 Research, 2020, 9, 291–305 Search PubMed; (e) Q. Zhou, W. Tang and L. W. Chung, J. Organomet. Chem., 2018, 864, 97–104 CrossRef CAS; (f) J. Lan, X. Li, Y. Yang, X. Zhang and L. W. Chung, Acc. Chem. Res., 2022, 55, 1109–1123 CrossRef CAS PubMed; (g) Z. Ding, Z. Liu, Z. Wang, T. Yu, M. Xu, J. Wen, K. Yang, H. Zhang, L. Xu and P. Li, J. Am. Chem. Soc., 2022, 144, 8870–8882 CrossRef CAS PubMed; (h) J. Cao, G. Wang, L. Gao, X. Cheng and S. Li, Chem. Sci., 2018, 9, 3664–3671 RSC; (i) L. Zhang and L. Jiao, Chem. Sci., 2018, 9, 2711–2722 RSC; (j) J. Liu, M. Nie, Q. Zhou, L. W. Chung, W. Tang and K. Ding, Chem. Sci., 2017, 8, 5161–5165 RSC; (k) J. L. Koniarczyk, J. W. Greenwood, J. V. Alegre-Requena, R. S. Paton and A. McNally, Angew. Chem., Int. Ed., 2019, 58, 14882–14886 CrossRef CAS PubMed.
- (a) S. Nagase and K. Morokuma, J. Am. Chem. Soc., 1978, 100, 1666–1672 CrossRef CAS; (b) D. H. Ess and K. N. Houk, J. Am. Chem. Soc., 2007, 129, 10646–10647 CrossRef CAS PubMed; (c) F. M. Bickelhaupt and K. N. Houk, Angew. Chem., Int. Ed., 2017, 56, 10070–10086 CrossRef CAS PubMed; (d) I. Fernández and F. M. Bickelhaupt, Chem. Soc. Rev., 2014, 43, 4953–4967 RSC.
- Z. Yan, Y. S. Liao, X. Li and L. W. Chung, Chem. Sci., 2025, 16, 2351–2362 RSC.
- H. Vμzquez-Villa, S. Reber, M. A. Ariger and E. M. Carreira, Angew. Chem., Int. Ed., 2011, 50, 8979–8981 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2428416, 2428418, 2428420. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc03633j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |