
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synergistic C–H bond activation across molybdenum–iridium multiply bonded complexes: a cascade of transformations†
Zachary
Dubrawski
 a,
Iker
Del Rosal
b,
Erwann
Jeanneau
c,
Laurent
Maron
b,
Chloé
Thieuleux
a and
Clément
Camp
*a
a,
Iker
Del Rosal
b,
Erwann
Jeanneau
c,
Laurent
Maron
b,
Chloé
Thieuleux
a and
Clément
Camp
*a
aLaboratory of Catalysis, Polymerization, Processes and Materials (CP2M UMR 5128), CNRS, Universite Claude Bernard Lyon 1, CPE-Lyon, Institut de Chimie de Lyon, 43 Bd du 11 Novembre 1918, F-69616 Villeurbanne, France. E-mail: clement.camp@univ-lyon1.fr
bLPCNO, Université de Toulouse, INSA Toulouse, 135 Avenue de Rangueil, Toulouse 31077, France
cCentre de Diffractométrie Henri Longchambon, Universite Claude Bernard Lyon 1, 5 Rue de la Doua, 69100 Villeurbanne, France
First published on 7th July 2025
Abstract
Heterobimetallic compounds offer unique opportunities for activating substrates through cooperative interactions between two distinct metal centers, potentially leading to catalytic reactivity beyond the reach of monometallic systems. The pairing of molybdenum with iridium has recently shown remarkable performance in heterogeneous catalytic and electrocatalytic processes, yet remains surprisingly unexplored in the context of homogeneous catalysis and well-defined molecular complexes. In this work, we address this gap by reporting the synthesis and characterization of complexes featuring rare molybdenum–iridium multiple bonds: Cp*Ir(H)Mo(NMe2)3, 1 and (Cp*IrH2)2Mo(NMe2)2, 2. Both complexes undergo electrophilic insertions of heteroallenes (CO2, tBuNCO) into the ancillary amido ligands. In the case of compound 1, the insertion of tert-butyl isocyanate initiates a unique cascade of bond breaking/forming events – including triple C–H activations and Csp3–Csp3 coupling – ultimately yielding a Mo(VI) metallacyclopropane complex, 5. Addition of multiple equivalents of isocyanate instead interrupts this mechanism, leading to C–N and C–O bonds cleavage and the formation of aminocarbyne and imido ligands bridging the Ir and Mo centers, along with a molybdenum(VI)-oxo moiety. This unprecedented reactivity, mediated by a key reaction intermediate featuring an unsupported Mo–Ir quadruple bond, illustrates the interest of heterobimetallic compounds for complex bond activation and reorganization.
Introduction
The first discovery of dimolybdenum paddlewheel complexes by Mason in 1965 launched a near-constant excitement for interesting multiple bonding in metal complexes.1–5 Since then, many supported multiply bonded bimetallic complexes have been reported (i.e. with a ligand bridging both transition metals in the complex), however unsupported systems present a particular challenge.6–8 Ligands must be small enough to allow for efficient orbital overlap between transition metal centers and avoid bridging across the metals, yet large enough to stabilize the often reactive transition metal complexes.9,10 Perhaps due to added complexity, species featuring unsupported multiple bonding between two different transition metals have rarely been investigated, with only a few reports in the literature.11–13This is surprising, given the growing interest in heterobimetallic transition metal complexes for promoting challenging bond activations.14–16 Indeed, by utilizing the electrons localized within metal–metal bonds and the inherent polarity of these architectures, such systems can access unique reactivity which is often beyond that of monometallic or homobimetallic congeners.17 A prototypical example is the phosphinoamide supported Zr–Co complexes from Thomas and coworkers, which can oxidatively add electrophiles such as CO2 or methyl iodide across the two metal centers.18 Building on this strategy, this group19 and others have since employed with success heterobimetallic complexes in a variety of catalytic reactions.20–23 However, many heterobimetallic systems featuring multiple metal–metal bonds rely on polydentate ligands to drive their assembly. While effective for structural organization, these bulky ligands can significantly hinder reactivity, particularly when concerted substrate activation across both metal centers is required, as ligand steric encumbrance can block access to the reactive site.
Unsupported metal–metal bonded systems, or those bridged by small hydride ligands, offer a promising strategy to overcome this limitation and access more reactive species. For example, in our prior work, we demonstrated that Ta–Ir species are highly active hydrogen isotope exchange catalysts as a result of a cooperative C–H bond activation across the two metals.28 In pursuit of a more covalent metal–metal bonding arrangement, we identified molybdenum as a promising candidate for the development of heterobimetallic complexes. While the Mo–Ir pairing has recently shown exceptional reactivity in heterogeneous catalysis and electrocatalysis,29–33 it remains surprisingly underexplored in the context of homogeneous catalysis and molecular complexes, with very few well-defined Mo–Ir compounds reported to date.34,35 To address this gap in literature, we report here the synthesis and characterization of rare molybdenum–iridium complexes featuring short, multiple metal–metal bonds. We further explore their reactivity towards electrophilic heteroallenes (CO2, tert-butyl isocyanate), which triggers unexpected cascade transformations involving multiple C–H, C–C, C–N, and C–O bond-breaking or -forming events. The underlying reaction mechanisms were rationalized through a combination of experimental studies and computational analysis.
Results and discussion
Addition of one equivalent of tetrakis(dimethylamido)molybdenum(IV), Mo(NMe2)4, to tetrahydrido(pentamethylcyclopentadienyl)iridium(V), Cp*IrH4, generates complex Cp*Ir(H)Mo(NMe2)3, 1, in 75% yield (Scheme 2). Complex 1 is diamagnetic with NMR data consistent with the structural assignment. The hydride signal in the 1H NMR spectrum (−6.5 ppm in C6D6) integrates for only one proton, suggesting the loss of three hydrides during the reaction (Fig. S2†). One hydride is lost in the protonolysis of one dimethylamido ligand, releasing one equiv. of free dimethylamine, as can be observed by monitoring the reaction by 1H NMR (Fig. S6†). This reaction is typical of the Brønsted acidic Cp*IrH4 starting material and has been used in our lab with great success to prepare Ir-based heterobimetallic compounds from metal-alkyl reagents.11,22,36,37 Here, we show that this approach can be extended to amido metallic species, expanding the scope of this synthetic strategy. Performing the reaction in a J-Young NMR tube fully filled with C6D6 allows for the detection and quantitation of H2 generated, and confirming the release of a stoichiometric amount of dihydrogen (see Fig. S6†).Two resonance structures, both obeying the 18-electron rule for each metal center, can be proposed for 1, as shown on Scheme 1. According to Green's formalism, where the hydride is treated as X-type, the amidos as XL-type and the Cp* ligand as an XL2-type ligands, the total oxidation state across both metals in compound 1 is +5 (total d electron count of 10). Starting from monometallic precursors with a combined formal oxidation state of +9, this decrease is consistent with the net double reductive elimination of H2 and HNMe2. How this total oxidation state is partitioned between the two metals, however, remains open to interpretation. One approach is to consider the metal–metal bonds as nonpolar, leading to formal oxidation states of +2 for Ir (d7) and +3 for Mo (d3) in the neutral resonance form 1a, or +1 for Ir (d8) and +4 for Mo (d2) considering the zwitterion form 1b. Alternatively, given the higher electronegativity of Ir compared to Mo and the polar nature of the metal–metal bonding, an uneven electron distribution across the metal–metal bond can be proposed. In the extreme case, this would correspond to Mo in the +6 state (d0) and Ir in the −1 state (d10), a description that is chemically questionable but could offer a rationale for the diamagnetism observed in compound 1. While oxidation state formalism plays a valuable role in our understanding of chemical bonding and reactivity, it remains an inherently arbitrary method for partitioning electrons between atomic centers. For this reason, we refrain assigning specific oxidation states to the metals in such compounds throughout the remainder of this article.
 | ||
| Scheme 1 Representative examples of molybdenum-based homo- and heterobimetallic complexes reported in the literature. Bond lengths and structural representations are taken from the respective article as described in the original text. A table with further examples is located in the ESI (Table S1).†24–27 | ||
Another point of discussion is whether the hydride is terminal (resonance form 1a in Scheme 2), or engages in a three-center two-electron (3c–2e) interaction, in which the iridium hydride bond donates electron density to the molybdenym center, in a Ir–H− → Mo+ agostic-type of bonding. The latter scenario is illustrated in form 1b in Scheme 2, using a half-arrow notation originally introduced by Parkin and Green,38 and subsequently applied to related systems.39–41 Depending on the limiting bonding scenario considered, the metal–metal bond order is adjusted (triple or double) to satisfy the 18-electron rule. Further characterization of complex 1 was thus carried out to gain a deeper understanding of its structure.
 | ||
| Scheme 2 Synthesis of the multiply bonded iridium–molybdenum complexes. | ||
Single crystals of 1 can be obtained by cooling a saturated pentane solution of complex 1 to −40 °C (Fig. 1). The most pertinent feature of the solid-state structure is the very short metal–metal bond distance of 2.2889(7) Å. Formal Shortness Ratio (FSR, = MM′ distance/sum of MM′ covalent radii) introduced by Cotton et al. can be a good indicator for the qualitative identification of multiply bonded species from structural data.42 Structural analysis of 1 gives an FSR of only 0.89, suggestive of a Mo–Ir bond of 2 or above. While other Mo–Ir heterobimetallic complexes have been reported in the literature, to our knowledge, none of the other crystallographically characterized examples available in the CCDC database exhibit an FSR below 1.34,35,43–47 The sum of the Mo–N–C and C–N–C angles at each amido ligand is close to 360° (359.6(1)°, 358.1(1)° and 359.7(1)°), consistent with sp2 hybridization of the nitrogen atoms, which enables them to act as 3e− donors (LX-type ligands) to maintain a 18-valence electron configuration.
 | ||
| Fig. 1 X-ray diffraction crystal structures of complexes 1 (top) and 2 (bottom) with thermal ellipsoids drawn at 50% probability and hydrogens removed for clarity. Relevant structural parameters for complex 1: Mo–Ir = 2.2889(7) Å, FSRMoIr = 0.89, Mo–N = 1.935(5)–1.955(5) Å. ∠Cp*centroid–Ir–Mo = 156.0(1)°, τ4 = 0.93. Relevant structural parameters for complex 2: Mo–Ir = 2.3946(5)–2.4075(5) Å, FSRMoIr = 0.93–0.94, Mo–N = 1.945(4)–1.948(5) Å. ∠Cp*centroid–Ir1–Mo = 154.0(1)°, ∠Cp*centroid–Ir2–Mo = 151.5(1)°, τ4 = 0.93. | ||
Although assigning hydrides adjacent to the heavy nuclei in X-ray crystallography was not possible, the significant tilting of the Cp* ligand with respect of the Mo–Ir multiple bond (Cp*centroid–Ir–Mo angle of 156.08°) is indicative of the presence of a hydride on the Ir center.48 The presence of the hydride is further supported by the Diffuse Reflectance Infrared Fourier Transform (DRIFT) spectrum for 1, which exhibits one signal typical of a iridium hydride stretch (1990 cm−1, Fig. S33†).49,50
The geometry of complex 1 was optimized at the DFT level (B3PW91 functional, Fig. S44†). The optimized structure compares well with the experimental one and among others the Mo–Ir distance is perfectly reproduced (2.30 Å vs. 2.29 Å experimentally). Bonding analysis confirms the presence of metal–metal bonding interactions at the Natural Bonding Orbital (NBO level). Indeed, two almost purely covalent Mo–Ir bonds (50–50 and 55–45) are found at the NBO level. One Mo–Ir bond involves overlap between sd hybrid orbitals while the second one implies pure d atomic orbitals (see Fig. S44†). The associated Wiberg Bond Indexes (WBI) are 1.67 indicating the presence of a highly covalent Mo–Ir double bond. A substantial donation from an iridium d lone pair into an empty d orbital at Mo is observed at the second order. The latter may explain the experimental observation of a bond order above two, but a backdonation from an occupied lone pair on Ir to an antibonding Mo–Ir bond is also observed so that complex 1 is better described with a Mo–Ir double bond.
The computed Mo–Ir–H angle in 1 is estimated to be 65°, which is compatible with the presence of a 3c–2e interaction. The Mo–H distance in 1 (2.19 Å) is significantly longer than typical values observed in classical μ-hydride molybdenum complexes (1.83–1.87 Å)51,52 while the computed Ir–H bond is notably shorter at 1.65 Å, despite Ir having a larger atomic radius than Mo. This suggests an asymmetric hydride interaction, with stronger bonding to Ir. The latter is corroborated by an analysis of the Wiberg bond indexes (WBIs, Fig. S44†). The Ir–H WBI is 0.57, in line with a mainly covalent interaction, whereas the Mo–H WBI is 0.30, in line with some electron delocalization from the Ir–H bond onto an acceptor orbital on Mo. These data support the description of 1 as stabilized by partial Ir–H bond donation to the [Mo(NMe2)3]+ fragment. Assuming an η2 interaction between the Ir–H bond and Mo, and that each terminal amido group donates three electrons, this necessitates a Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Ir double bond to satisfy the 18-electron rule – an assignment consistent with natural bond orbital (NBO) analysis. Additionally, the Ir–H− → Mo+ donation shortens the Ir–Mo distance further, in agreement with the XRD data. Naturally, the bonding lies along a continuum between the two limiting forms 1a and 1b, as illustrated in Scheme 2.
Ir double bond to satisfy the 18-electron rule – an assignment consistent with natural bond orbital (NBO) analysis. Additionally, the Ir–H− → Mo+ donation shortens the Ir–Mo distance further, in agreement with the XRD data. Naturally, the bonding lies along a continuum between the two limiting forms 1a and 1b, as illustrated in Scheme 2.
The trinuclear complex (Cp*IrH2)2Mo(NMe2)2, 2, is similarly synthesized in 94% yield through the addition of two equivalents of Cp*IrH4 to Mo(NMe2)4 and the release of one equivalent of dihydrogen and two equivalents of dimethylamine (Scheme 2). The X-ray crystal structure of complex 2, shown on Fig. 1-bottom, reveals a trinuclear MoIr2 core with a tetrahedral molybdenum center (τ4 = 0.93). The Mo–Ir bond distances range from 2.3946(5) to 2.4075(5) Å (FSR = 0.93–0.94) indicating some multiple bonding character but less than that of 1. Again, the hydrides could not be located from the Fourier difference map of the X-ray crystallography experiment, however the hydride signals in the 1H NMR spectrum for 2 (−10.23 ppm, 4H, Fig. S7†) and the clear hydride stretching frequencies in the DRIFT spectrum (2022 and 1929 cm−1, Fig. S34†) confirm their presence. Similarly, the Cp*centroid–Ir–Mo angles (151.5(1)°–154.0(1)°) give a qualitative understanding of the hydride positions in the solid state structure.
Complex 2 was also investigated computationally (Fig. S45†). The optimized structure compares well with the experimental one. As for complex 1, the Mo–Ir distances are correctly reproduced (2.44 Å vs. 2.40–2.41 Å experimentally). The bonding analysis at the NBO level indicates the presence of two single covalent Mo–Ir bonds (55–45) with an associated WBI of 1.0, as a result of overlap between d orbitals on Mo and Ir. At the second-order level, there is evidence of donation from an occupied Mo orbital to vacant orbitals on Ir, which could suggest a bond order greater than one. However, simultaneous backdonation into the Mo–Ir bonding orbitals is also observed. As a result, the bonding is more accurately described as comprising two polarized Mo–Ir single bonds. The bonding situation for the hydrides in 2 is similar to that in 1 (see Fig. S45†).
Carbon dioxide rapidly reacts with complex 1 to give intractable mixtures in which the major component is paramagnetic (Fig. S10†). This complex mixture of products persists across different reaction conditions, including changes in solvents, in the absence of light, varying the concentration or stoichiometric amounts of CO2 and reduction of the temperature. In contrast, complex 2 cleanly reacts with CO2via insertion of CO2 into the dimethylamido ligands, affording the carbamato complex, 3, in 95% yield (Scheme 3). 1H NMR spectroscopic analysis shows a shift of the dimethylamido signals from 3.49 ppm to 2.37 ppm, with the appearance of a resonance in the 13C NMR spectrum at +169 ppm, corresponding to the carbamato carbonyl (Fig. S12†). The DRIFT spectrum for 3 exhibits a characteristic υCO signal at 1570 cm−1, further confirming the formation of carbamato moieties (Fig. S35†). The hydride 1H NMR signal shifts from −10.23 ppm in 2 to −6.54 ppm in 3, however the integral of this signal falls to two protons (Fig. S11†) indicating that the CO2 insertion is coupled with an irreversible loss of one equivalent of dihydrogen (as confirmed by 1H NMR reaction monitoring, see Fig. S15†). Interestingly, we see no indication of CO2 insertion into the M–H bonds of 2 despite the literature precedent for Mo and Ir hydride insertion.53,54
 | ||
| Scheme 3 Reaction of complexes 1 and 2 with CO2. | ||
This CO2 insertion also leads to a surprising structural rearrangement as revealed by the single crystal X-ray structure of complex 3 (Fig. 2). Here, the iridium centers bend down toward one another, reducing the Ir–Mo–Ir′ angle from 108.8(1)° in 2 to 71.4(1)° in 3. The long interatomic distance between the Ir centers (2.7722(3) Å, FSR = 1.10) suggests an absence of formal Ir–Ir′ bonding interaction, a conclusion further supported by computational analysis (see below). We instead propose that the close proximity of the Ir centers is maintained by two bridging hydrides. The Mo–Ir bond distance (2.3759(6) Å, FSR = 0.93) still gives a FSR well below 1, suggesting that the multiple bonding character of the Mo–Ir bonding is maintained through this structural change. Note that the carbamato oxygens are solely bound to molybdenum.
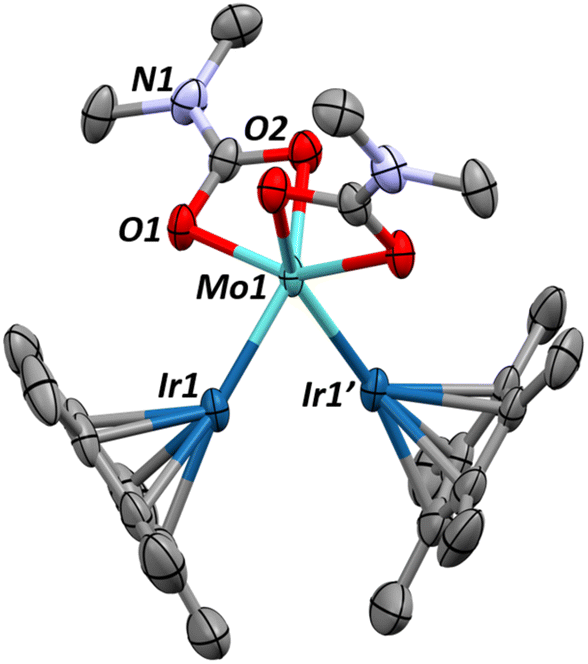 | ||
| Fig. 2 X-ray crystal structure of complex 3, grown from a saturated pentane solution, reveals a major structural change upon CO2 insertion into the dimethylamido ligands. Thermal ellipsoids shown at 50% and hydrogens removed or clarity. Relevant structural parameters are as follows: Mo–Ir = 2.37659(6) Å, FSRMoIr = 0.93, Ir–Ir = 2.7722(3) Å, FSR = 1.10, Mo–O = 2.199–2.149 Å, ∠Ir–Mo–Ir = 71.3(1)°, ∠Cp*centroid–Ir–Mo = 155.7(1)°, ∠Cp*centroid–Ir1–Ir1′ = 139.5(1)°. | ||
As already carried out for complexes 1 and 2, DFT optimizations were performed for complex 3. The resulting optimized geometry compares satisfactorily with the experimental one (Fig. S46†). Among other, the Mo–Ir distances are nicely reproduced (2.32 and 2.36 Å vs. 2.37 Å experimentally). The Ir–Ir distance is a bit longer than the experimental one (2.85 Å vs. 2.78 Å experimentally), but still in an acceptable range. NBO-level analysis of bonding indicates the presence of two Mo–Ir double bonds, which are polarized toward the Ir center (60%) and primarily imply d–d orbital overlap. These polarized double bonds explain the relatively low Mo–Ir WBI of 1.3–1.5. In contrast, the Ir–Ir interaction is only detected at the second order donor–acceptor level, which is in line with the low WBI of 0.35.
We hypothesize that 1 undergoes a similar CO2 insertion as 2 but does not stop at the carbamato ligated product. Accordingly, we sought alternatives to further explore this chemistry. Isocyanates are isoelectronic with CO2 and often used to investigate mechanisms in CO2 activation.55–57 Addition of one equivalent of tert-butylisocyanate (tBuNCO) to 1 quickly generates a new product, 4, in 95% yield as quantified by 1H NMR spectroscopy. Complex 4 was isolated (65% recovered yield after recrystallization) by quickly (within 10 min) removing the volatiles from a room temperature reaction mixture of 1 and one equivalent of tBuNCO. X-ray quality single crystals of complex 4, obtained from a saturated pentane solution of this reaction mixture cooled to −40 °C, confirms the insertion of tBuNCO into a dimethylamido ligand generating the κ2-ureate complex 4 (Fig. 3 – top), reminiscent of isocyanate insertions into alkoxide ligands of structurally similar molybdenum complexes.58,59 Computational analysis of this insertion confirms the low enthalpic barrier (19.9 kcal mol−1, Scheme 5) through a classical C–N double insertion transition state (TS), previously explored in the context of polymerization catalysis.60 The very short Mo–Ir bond length (2.2646(7) Å, FSR = 0.89) has not significantly changed from 1, indicating the electronic nature of the complex has not been significantly altered. The Cp* ligand tilting (Cp*–Ir–Mo angle of 167.1(2)°) indicates that the terminal hydride is still present. The presence of the hydride is further confirmed by DRIFT spectroscopy, revealing an intense signal at 2153 cm−1, assigned to ν(Ir–H) (Fig. S36†).49
 | ||
| Fig. 3 X-ray diffraction crystal structures of complexes 4 (top) and 5 (bottom). Thermal ellipsoids are shown at 50% probability and most hydrogens are removed for clarity. Relevant structural parameters for complex 4: Mo–Ir = 2.2646(7) Å, FSRMoIr = 0.89, Mo–N1 = 1.957(4) Å, Mo–N2 = 2.286(4) Å, Mo–N3 = 2.139(4) Å, ∠Cp*centroid–Ir–Mo = 167.1(2)°. Relevant structural parameters for complex 5: Mo–Ir = 2.6229(4) Å, FSRMoIr = 1.04, Mo–N1 = 1.724(3) Å, Mo–N2 = 2.080(4) Å, Mo–C1 = 2.175(5) Å, Mo–C2 = 2.169(4) Å, C1–C2 1.419(7) Å, ∠Cp*centroid–Ir–Mo = 175.3(1)°. | ||
A clean 1H NMR spectrum could be obtained before significant conversion to complex 5 (see below). This 1H NMR spectrum reveals a hydride resonance shift from −6.40 ppm to −9.85 ppm and significant broadening of the N-methyl signal of both the dimethylamido and ureate ligands (3.49 ppm and 2.89 ppm respectively, Fig. S16†). The formation of 4 is computed to be only lightly exothermic (−5.6 kcal mol−1, Scheme 5) and given the low barrier we anticipate a chemical equilibrium between 1 and 4. Exchange spectroscopy (EXSY) 2D NMR experiments show a broad cross peak between the two signals and those of remaining 1 in solution (Fig. S19†), hinting at chemical exchange on the NMR timescale. Unfortunately, due to the low barrier to further transformation, Van't Hoff analysis on the equilibrium could not be performed (see below). VT-NMR spectra collected in toluene-d8 allow some deconvolution of these signals into four broad singlets, each integrating for 3H, but assignment remains unclear (Fig. S17†). A 13C NMR spectrum could also be obtained at −25 °C and features a characteristic ureato signal at +164 ppm (Fig. S18†), allowing for confirmation of the isocyanate insertion.61
Complex 4 is metastable and slowly converts at room temperature into complex 5 (Scheme 4). The 1H NMR spectrum of 5 shows a shift of the hydride signal from −9.85 to −12.55 ppm with an increase in the integral from one to three protons. Three characteristic signals, corresponding to the diastereotopic protons of a metallacyclopropyl fragment, are observed in the alkyl region at +4.20 ppm (doublet of doublets), +2.69 ppm (pseudotriplet) and +2.17 ppm (doublet of doublets). Each of these metallacyclopropyl signals integrates for one H showing typical 2J and 3J coupling constants (6.5 and 9.7 Hz, respectively, see Fig. S20†).
 | ||
| Scheme 4 Complex 1 reacts with one equivalent of tBuNCO in a clean dimethylamine insertion to yield complex 4, which then undergoes a triple C–H activation process to yield the new product 5. Addition of 5 equivalents of tBuNCO to 1 instead leads to multiple CN and CO bond activations to yield complex 6. | ||
A single crystal of 5, suitable for X-ray diffraction, was grown from a saturated pentane solution cooled to −40 °C. The resulting structure (Fig. 3-bottom) unequivocally confirms the product, formed in an apparent triple C–H activation process combined with Csp3–Csp3 bond formation to give a tert-butyl-imido metallacyclopropane complex. The Ir–Mo distance in 5 is substantially lengthened from 2.2889(7) Å in 1 to 2.6224(4) Å (FSR = 1.04), in 5, precluding multiple bonding between Mo and Ir. The Cp* ligand is nearly perpendicular to the Mo–Ir axis, with a Cp*centroid–Ir–Mo angle of 175.3°, as a result of three hydrides bridging the two metals, consistent with architectures previously reported in our group.37,50,62,63
The presence of three hydrides is confirmed by 1H NMR (Fig. S20†) and the DRIFT spectrum of 5, displaying several metal-hydride stretching signals between 1971 and 1780 cm−1 (Fig. S37†). The newly formed carbon–carbon bond between C1 and C2 (Fig. 3) is 1.419(7) Å long, which lies between typical C–C single and double bond lengths.64 The considerable C–C bond elongation compared to free ethylene (1.34 Å) suggests that the structure is better described as a metallacyclopropane complex rather than olefin-type bonding arrangement.65,66 The Mo–C bond lengths (Mo–C = 2.175(5) Å, 2.169(4) Å) agree with this assignment as well with typical metallacyclopropane Mo–C lengths of 2.209 Å.67 Previous examples of α-N-metallacyclopropanes are extremely limited, however a previously reported tantalum complex does show diasterotopic 1H NMR resonances for the M–CH2 at about 4 ppm and a C1–C2, bond distance of 1.468(8) Å, agreeing well with our results.68 The molybdenum imido bond length (Mo–N1 = 1.724(3) Å) is consistent with other examples of Mo(VI) alkylimido complexes.69,70
DFT calculations were carried out on complex 5 (Fig. S48†). The optimized geometry compares well with the experimental one (Mo–Ir distance of 2.66 Å vs. 2.62 Å experimentally and two Mo–C bonds of 2.16 Å vs. 2.17 Å experimentally). The NBO analysis indicates the presence of one polarized Mo–Ir single bond and two Mo–C single bonds (WBIs of 0.6–0.7). This further support the metallacyclopropane description of this complex proposed experimentally. Two hydrides appear to engage in 3c–2e bridging interactions (Ir–H distances of 1.65–1.66 Å and Mo–H distances of 2.02–2.03 Å) while the third hydride is best described as terminal (longer Mo–H distance of 2.24 Å). This description is in agreement with an 18 electron count for Ir (assuming a XL2-type Cp* ligand, 3 X-type hydrides and the metal–metal bond) and an 18 electron count for Mo (assuming 2 X-type alkyls, a X-type amido, a X2L-type imido, 2 agostic Ir–H interations and the Mo–Ir bond).
Computational analysis to examine the mechanism of 4 going to 5 was found to be quite complex and necessitated a deeper investigation into the initial stability of complex 4 (Scheme 5). Given the loss of both a dimethylamido and a hydrido ligand in 5, the initial loss of dimethylamine from complex 4 was hypothesized but found to be exergonically inaccessible at room temperature (ΔGr = 41 kcal mol−1, Scheme 5-red). However, since 1 and 4 are in equilibrium, there exists a concentration of free tBuNCO available in solution for a second NMe2 insertion. The associated barrier is similar to the one found for the formation of complex 4 (23.8 kcal mol−1) and yields a rather unstable intermediate (Int 3, 16.4 kcal mol−1) which further reacts through hydride abstraction from the Ir center with a kinetically accessible barrier (6.4 kcal mol−1 from Int 3, 28.4 kcal mol−1 from complex 4). This gives a Mo urea adduct which is not very stable (Int 4, 13.5 kcal mol−1 with respect to the entrance channel, see Scheme 5), with loss of urea from the coordination sphere inducing an extra energetic destabilization of 7.6 kcal mol−1. This results in the formation of the key reaction intermediate, Int 5, whose NBO and AIM analysis reveal an unsupported metal–metal quadruple bond (Table S3†), in line with the 18-electron rule. Note that the computed Mo–Ir bond length in Int 5 (2.210 Å, FSR = 0.86) is considerably shorter than that in 1 (2.2889(7) Å).
 | ||
| Scheme 5 Computed mechanism for the formation of complex 4 and the key intermediate, Int 5. | ||
Int 5 can then undergo two subsequent intramolecular hydrogen abstractions from a methyl group of the bound ureate ligand with relatively low barriers (7.3 and 18.7 kcal mol−1, Scheme 6) to yield a bridging dihydride iridium–molybdenum methylidene complex, which is the first intermediate more stable than complex 4 (Int 7, −9.4 kcal mol−1, Scheme 6). This finding accounts for the slow conversion of complex 4 to complex 5 found experimentally. Although stable, this amido–methylidene complex (Int 7) undergoes another hydrogen abstraction from the methyl group of the amido ligand this time with a low barrier (16.3 kcal mol−1) to yield a surprisingly stable trishydride–alkyl–methylidene complex (Int 8, −10.3 kcal mol−1). The changes in the metal–metal bond orders during the transformation from Int 5 to Int 8, in which the bridging hydrides are consecutively formed, is shown by the gradual decrease in the Ir–Mo WBI (2.41 in Int 5, 1.63 in Int 6, 1.14 in Int 7, 0.62 in Int 8; see Table S4† for more details). This trend highlights the pivotal role of the Ir–Mo bond in mediating this reactivity. The heterobimetallic core functions as a dynamic platform for electron storage and release via the Mo–Ir multiple bond, enabling multiple C–H activations and hydride transfers.
 | ||
| Scheme 6 Calculated mechanism for the formation of 5. | ||
A C–C coupling reaction can then be done between the alkyl and the methylidene ligand with almost no barrier (4.9 kcal mol−1) followed by a C–N bond breaking (barrier of 6.1 kcal mol−1) to yield the very stable complex 5 (−30.3 kcal mol−1). Gratifyingly, NMR reaction monitoring shows, over time, the increase in concentration of 1 (as the pre-equilibrium shifts to the left), the formation of one equivalent of N,N-dimethyl-N′-tert-butyl-urea (as complex 4 converts to Int 5) and the formation of 5 (see Fig. S26†).
Alternative mechanisms were also explored, including the β-methyl migration (Scheme 6, red) to first form a Mo imide (Int 7′) before C–C coupling, but these were found to have prohibitively high activation barriers and so were excluded. Despite literature precedent showing isocyanate insertion into molybdenum imides, complex 5 does not undergo further reaction with tBuNCO, even under heating nor extended reaction times.71
Interestingly, the room temperature addition of five equivalents of tBuNCO to complex 1 generates a different complex, 6, in 82% isolated yield (Scheme 4). X-ray quality crystals of this product, grown from an approximately 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 toluene:pentane saturated solution, allow for an unambiguous assignment of ligand identity following this reaction (Fig. 4). The iridium–molybdenum distance is 2.6339(5) Å, corresponding to an FSR of 1.03. The elongated metal–metal distance is most likely due to the presence of two bridging ligands. Indeed, the two metals are bridged by an N,N-dimethylaminocarbyne ligand and an N-tBu imido ligand, as a result of the insertion of the reactive tBuNCO into the last remaining dimethylamido ligand in Int 5 (see below). The C1–N1 bond length (1.342(8) Å) is typical of bridging aminocarbyne ligands (1.284–1.442 Å) with C1–M distances (1.962(6)–1.965(6) Å) also in line with previous reports on dinuclear systems (2.012 Å for a Mo2 μ-aminocarbyne).72–75
:1 toluene:pentane saturated solution, allow for an unambiguous assignment of ligand identity following this reaction (Fig. 4). The iridium–molybdenum distance is 2.6339(5) Å, corresponding to an FSR of 1.03. The elongated metal–metal distance is most likely due to the presence of two bridging ligands. Indeed, the two metals are bridged by an N,N-dimethylaminocarbyne ligand and an N-tBu imido ligand, as a result of the insertion of the reactive tBuNCO into the last remaining dimethylamido ligand in Int 5 (see below). The C1–N1 bond length (1.342(8) Å) is typical of bridging aminocarbyne ligands (1.284–1.442 Å) with C1–M distances (1.962(6)–1.965(6) Å) also in line with previous reports on dinuclear systems (2.012 Å for a Mo2 μ-aminocarbyne).72–75
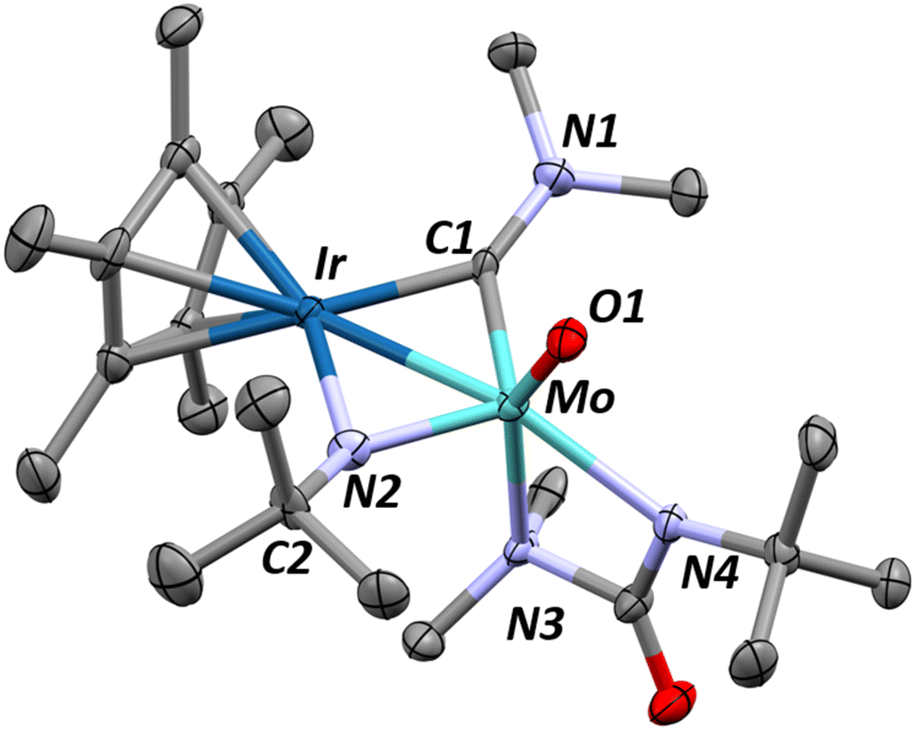 | ||
| Fig. 4 X-ray diffraction crystal structure of complex 6 with thermal ellipsoids represented at 50% probability and hydrogens removed for clarity. Relevant structural parameters are as follows: Mo–Ir = 2.6339(5) Å, FSRMoIr = 1.03, Ir–C1 = 1.962(6), Mo–C1 = 1.965(6) Å, Ir–N2 = 2.040(6) Å, Mo–N2 = 1.860(6) Å, C1–N1 = 1.342(8) Å, N2–C2 = 1.464(9) Å, Mo–O1 = 1.738(5) Å, ∠Cp*centroid–Ir–Mo = 173.3(1)°. | ||
The methyl groups of the aminocarbyne moiety of complex 6 are equivalent in the 1H NMR spectrum, recorded in C6D6 solution at room temperature, which is suggestive of free rotation around the C1–N1 bond (Fig. S28†). The presence of the aminocarbyne moiety in 6 is confirmed by the characteristic 13C resonance found at +295 ppm (Fig. S29†). Note that whereas bridging aminocarbynes on homobimetallic molybdenum complexes typically appear further downfield (+305–390 ppm), the shift is highly metal dependant and homobimetallic iridium complexes show this resonance at +255 ppm.74–77 The carbyne 13C NMR chemical shift in 6 is thus an intermediate case between the Mo and Ir extremes. Neither the 1H NMR nor the DRIFT spectra (Fig. S28 and S38†) show signals typical of hydrides for this complex, in agreement with the proposed structure.
Complex 6 has a vibrant blue colour due to an intense absorption at 585 nm (ε = 2760 M−1 cm−1, Fig. S42†), consistent with previous reports of aminocarbyne LMCT bands.78 Altogether, the spectroscopic and the structural data point more to a μ-carbyne description (in contrast to a μ-iminium), where the bonding is more akin to a Schrock-type carbyne.75,79
Computational analysis shows that the HOMO is localized on the π bonding between the Mo–Ir core and empty orbitals from the aminocarbyne (Fig. S49†). The Mo–O bond length in the XRD structure, of 1.738(5) Å, is typical of Mo(VI) oxo complexes and precludes the possibility of a hydroxo ligand which tend to have much longer Mo–O bond distances (1.9–2.2 Å).80–82 Similarly, the bridging imido ligand does not have a proton on N2 as the geometry around nitrogen is more trigonal planar than tetrahedral (C2–N2–MoIrplane = 153° vs. 109° for a tetrahedral geometry), agreeing with literature on bridging imido vs. amido ligands in binuclear systems.83–86 The Ir–N2 bond length of 2.040(6) Å also agrees well with the assignment of a bridging imido, with typical values of 1.88–2.10 Å, compared to bridging amido ligands which show longer bond distances (2.15–2.30 Å).85,87–90
As discussed above, the formation of Int 5 is accompanied by the release of one equivalent of N,N-dimethyl-N′-tert-butyl-urea, isolated from the reaction mixture by trituration with pentane (92% isolated yield from moles of 1). This co-product was unequivocally identified through both 1H NMR and X-ray crystallography (Fig. S30 and S43,† respectively), which shows a proton attached to the urea nitrogen, confirming the computed mechanism for the formation of Int 5 in Scheme 5.
Due to the surprising ligand rearrangement occurring in this reaction, we turned to computational analysis to understand the mechanism at play (Scheme 7). Addition of another equivalent of tBuNCO to Int 5 results in a third CN insertion into the Mo–Namido bond. This insertion intermediate (Int 12) undergoes a C–N bond breaking reaction (barrier of 21.1 kcal mol−1) to yield a very stable bridging imido-carbamoyl complex (Int 13, −12.3 kcal mol−1). In this intermediate, the carbamoyl ligand is bound to the Ir center with an extra stabilization by the interaction between the η2 CO double bond and the Mo center. This π-type interaction allows a C–O bond breaking reaction to occur at the Mo center with an accessible barrier of 25.2 kcal mol−1, yielding complex 6 which is thermodynamically stable (−33.0 kcal mol−1). This side-on carbamoyl ligand has been previously characterized in the context of decarbonylation chemistry but never seen to instead undergo oxygen atom transfer to make a Mo-oxo.91–93 This C–O bond cleavage is reminiscent of Chisholm's work, in which the C–O triple bond of carbon monoxide is cleaved across a ditungsten complex, leading to the formation of a tungsten-oxo moiety and insertion of the carbon atom into an alkylidyne ligand to generate a bridging alkynyl species.94 While aminocarbyne ligands have been well explored, this is the first reported synthesis from the oxygen atom transfer of a carbamoyl ligand. The calculated mechanism only requires one equivalent of tBuNCO from Int 5 (3 equiv. in total from moles of 1). However, a slightly larger excess (5 equiv. with respect to 1) was found to be required experimentally to avoid the competitive formation of complex 5.
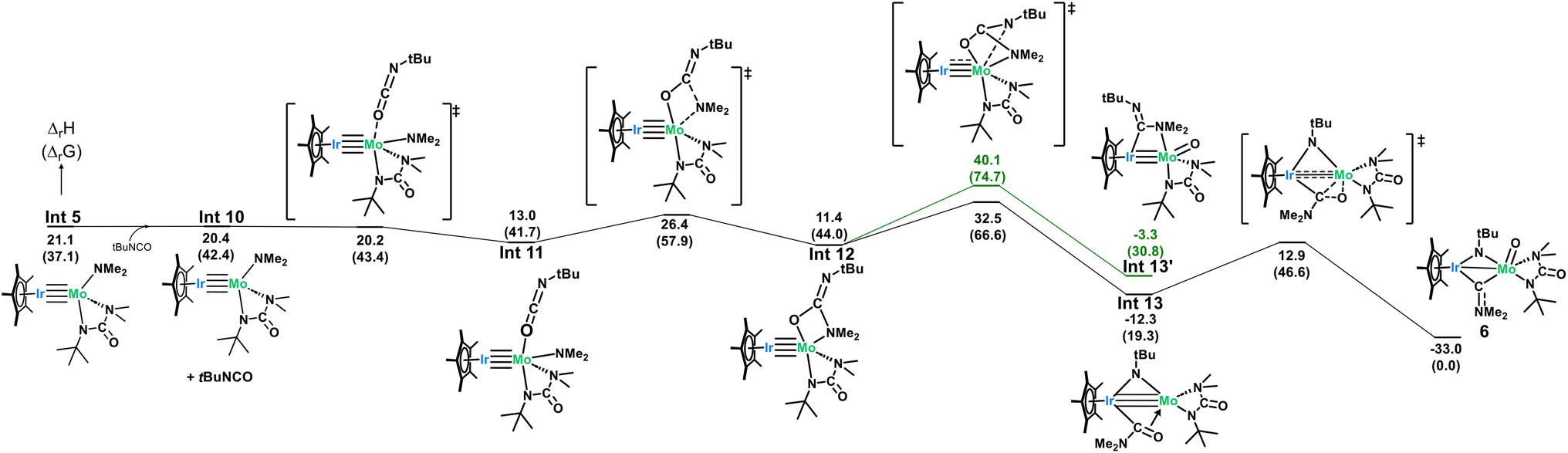 | ||
| Scheme 7 Computed mechanism for the formation of 6. | ||
Conclusions
We report the synthesis and characterization of two novel iridium–molybdenum heterobimetallic complexes, Cp*Ir(H)Mo(NMe2)3 (1) and (Cp*IrH2)2Mo(NMe2)2 (2), prepared via protonolysis reactions between Cp*IrH4 and Mo(IV) amido complexes. To the best of our knowledge, these compounds represent the first crystallographically characterized examples of Mo–Ir multiple bonds. Both compounds react with CO2; however, only the trinuclear complex 2 yields a well-defined product. This transformation proceeds via a combination of H2 reductive elimination and CO2 insertion into the dimethylamido ligands, resulting in the formation of the carbamato complex 3. Complex 1 was found to react cleanly with tert-butylisocyanate (tBuNCO). In this transformation, insertion of the isocyanate into the amido ligand generates the intermediate 4, from which a urea ligand can be decoordinated, yielding a highly reactive, low-coordinate Mo–Ir quadruply bonded species, intermediate Int 5. This key species initiates a cascade of intriguing reactivity. In the absence of additional equivalents of isocyanate, this activation pathway leads to an unexpected triple C–H bond activation, through subsequent α-hydride abstractions promoted by the metal–metal bond, and a C–C coupling event. This ultimately yields a Mo(VI) metallacyclopropane complex, 5. However, this mechanism is interrupted if the key intermediate, Int 5, reacts with another equivalent of isocyanate. This results instead in C–N and C–O bonds cleavage and the formation of aminocarbyne and imido ligands bridging the Ir and Mo centers, along with a molybdenum(VI)-oxo moiety, yielding complex 6. The mechanisms of these complex transformations have been elucidated in details through DFT studies, which agree with the experimental observations and reveal the role of the bimetallic assembly in mediating hydride transfer and serving as a reservoir for electron storage and release via the Mo–Ir multiple bond. This unprecedented reactivity illustrates the promise of heterobimetallic compounds for enabling complex bond activations and reorganizations.This work naturally leads to a discussion of alternative ligands as a strategy to tune the reactivity of the Mo–Ir multiple bond. Previous studies have demonstrated that modifying the ligand environment around Mo centers in multiply bonded dimolybdenum systems can profoundly influence their reactivity. Investigations are currently ongoing in our group to further research this topic.
Data availability
Crystallographic data for complexes 1–6 and N,N-dimethyl-N′-tert-butyl-urea has been deposited at the Cambridge Crystallographic Date Centre under accession codes 2410538–2410544 and can be obtained at https://www.ccdc.cam.ac.uk/data_request/cif.Author contributions
The manuscript was written through contributions of all authors. ZD performed the experimental work. LM and IDR directed the computational work. EJ contributed the X-ray structure determinations. CC and ZD conceptualized the research. CC found the funds and administrated the project. CC and CT supervised the work. ZD and CC wrote the initial version of the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the European Union (ERC StG grant DUO, Project Number 01041762). Views and opinions expressed are however those of the authors only and do not necessarily reflect those of the European Union or the European Research Council Executive Agency. Neither the European Union nor the granting authority can be held responsible for them. Sylvie Guibert, Anne Baudouin and Emmanuel Chefdeville are acknowledged for their help in acquiring the NMR data. LM is a senior member of the Institut Universitaire de France. CalMip is acknowledged for a generous grant of computing time. The referees are acknowledged for helpful discussions regarding the bonding in these compounds.Notes and references
- D. Lawton and R. Mason, The Molecular Structure of Molybdenum(II) Acetate, J. Am. Chem. Soc., 1965, 87(4), 921–922, DOI:10.1021/ja01082a046.
- Multiple Bonds Between Metal Atoms, ed. F. A. Cotton, C. A. Murillo and R. A. Walton, Springer US, Boston, MA, 2005, DOI:10.1007/b136230.
- Molecular Metal-Metal Bonds: Compounds, Synthesis, Properties, ed. S. T. Liddle, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2015 Search PubMed.
- M. Pérez-Jiménez, J. Campos, J. López-Serrano and E. Carmona, Reactivity of a Trans-[H–MoMo–H] Unit towards Alkenes and Alkynes: Bimetallic Migratory Insertion, H-Elimination and Other Reactions, Chem. Commun., 2018, 54(66), 9186–9189, 10.1039/C8CC04945A.
- D. Rütter, N. Nöthling, M. Leutzsch, A. A. Auer and A. Fürstner, Tripodal Silanolate Ligands Expand [MoX3] Chemistry Beyond Its Traditional Borders, J. Am. Chem. Soc., 2025, 147(16), 13871–13884, DOI:10.1021/jacs.5c02178.
- N. H. Hunter, J. E. Stevens, C. E. Moore and C. M. Thomas, One Bridge, Three Bonds: A Frontier in Multiple Bonding in Heterobimetallic Complexes, Inorg. Chem., 2023, 62(2), 659–663, DOI:10.1021/acs.inorgchem.2c03716.
- L. P. H. Lopez, R. R. Schrock and P. Müller, Dimers That Contain Unbridged W(IV)/W(IV) Double Bonds, Organometallics, 2006, 25(8), 1978–1986, DOI:10.1021/om050961s.
- R. Toreki, R. R. Schrock and M. G. Vale, Two Rhenium Complexes That Contain an Unsupported Metal-Metal Double Bond in the Presence of Potentially Bridging Ligands, J. Am. Chem. Soc., 1991, 113(9), 3610–3611, DOI:10.1021/ja00009a068.
- J. F. Berry and C. C. Lu, Metal–Metal Bonds: From Fundamentals to Applications, Inorg. Chem., 2017, 56(14), 7577–7581, DOI:10.1021/acs.inorgchem.7b01330.
- J. P. Collman and R. Boulatov, Heterodinuclear Transition-Metal Complexes with Multiple Metal–Metal Bonds, Angew. Chem., Int. Ed., 2002, 41(21), 3948–3961, DOI:10.1002/1521-3773(20021104)41:21<3948::AID-ANIE3948>3.0.CO;2-K.
- S. Lassalle, R. Jabbour, P. Schiltz, P. Berruyer, T. K. Todorova, L. Veyre, D. Gajan, A. Lesage, C. Thieuleux and C. Camp, Metal–Metal Synergy in Well-Defined Surface Tantalum–Iridium Heterobimetallic Catalysts for H/D Exchange Reactions, J. Am. Chem. Soc., 2019, 141(49), 19321–19335, DOI:10.1021/jacs.9b08311.
- M. Oishi, M. Kino, M. Saso, M. Oshima and H. Suzuki, Early–Late Heterobimetallic Complexes with a Ta–Ir Multiple Bond: Bimetallic Oxidative Additions of C–H, N–H, and O–H Bonds, Organometallics, 2012, 31(13), 4658–4661, DOI:10.1021/om300429u.
- J. P. Collman and J. M. Garner, Synthesis and Characterization of a Paramagnetic Osmium-Ruthenium Double Bond, J. Am. Chem. Soc., 1990, 112(1), 166–173, DOI:10.1021/ja00157a027.
- A. Lachguar, A. V. Pichugov, T. Neumann, Z. Dubrawski and C. Camp, Cooperative Activation of Carbon–Hydrogen Bonds by Heterobimetallic Systems, Dalton Trans., 2024, 53(4), 1393–1409, 10.1039/D3DT03571A.
- J. Campos, Bimetallic Cooperation across the Periodic Table, Nat. Rev. Chem., 2020, 4(12), 696–702, DOI:10.1038/s41570-020-00226-5.
- M. Navarro, J. José Moreno, M. Pérez-Jiménez and J. Campos, Small Molecule Activation with Bimetallic Systems: A Landscape of Cooperative Reactivity, Chem. Commun., 2022, 58(80), 11220–11235, 10.1039/D2CC04296G.
- U. Jayarathne, T. J. Mazzacano, S. Bagherzadeh and N. P. Mankad, Heterobimetallic Complexes with Polar, Unsupported Cu–Fe and Zn–Fe Bonds Stabilized by N-Heterocyclic Carbenes, Organometallics, 2013, 32(14), 3986–3992, DOI:10.1021/om400471u.
- C. M. Thomas, J. W. Napoline, G. T. Rowe and B. M. Foxman, Oxidative Addition across Zr/Co Multiple Bonds in Early/Late Heterobimetallic Complexes, Chem. Commun., 2010, 46(31), 5790–5792, 10.1039/C0CC01272F.
- K. M. Gramigna, D. A. Dickie, B. M. Foxman and C. M. Thomas, Cooperative H2 Activation across a Metal–Metal Multiple Bond and Hydrogenation Reactions Catalyzed by a Zr/Co Heterobimetallic Complex, ACS Catal., 2019, 9(4), 3153–3164, DOI:10.1021/acscatal.8b04390.
- B. G. Cooper, J. W. Napoline and C. M. Thomas, Catalytic Applications of Early/Late Heterobimetallic Complexes, Catal. Rev., 2012, 54(1), 1–40, DOI:10.1080/01614940.2012.619931.
- Homo- and Heterobimetallic Complexes in Catalysis, Topics in Organometallic Chemistry; Cooperative Catalysis, ed. P. Kalck, Springer International Publishing, Cham, 2016, vol. 59, DOI:10.1007/978-3-319-34184-2.
- A. V. Pichugov, L. Escomel, S. Lassalle, J. Petit, R. Jabbour, D. Gajan, L. Veyre, E. Fonda, A. Lesage, C. Thieuleux and C. Camp, Highly Selective and Efficient Perdeuteration of N-Pentane via H/D Exchange Catalyzed by a Silica-Supported Hafnium-Iridium Bimetallic Complex, Angew. Chem., Int. Ed., 2024, 63(16), e202400992, DOI:10.1002/anie.202400992.
- A. Lachguar, T. Neumann, A. V. Pichugov, E. Jeanneau, L. Veyre, C. Thieuleux and C. Camp, Catalytic H/D Exchange of (Hetero)Arenes with Early–Late Polyhydride Heterobimetallic Complexes: Impact of Transition Metal Pairs, Dalton Trans., 2025, 54(9), 3804–3811, 10.1039/D4DT03171G.
- F. A. Cotton, K. R. Dunbar, B. Hong, C. A. James, J. H. Matonic and J. L. C. Thomas, Complexes Containing Heteronuclear and Homonuclear Quadruple Bonds. Preparation and Characterization of Bis[Bis(Dimethylphosphino)Methane]Tetrachloromolybdenumtungsten and Mo2X4(Dmpm)2 (X = Bromide, Iodide), Inorg. Chem., 1993, 32(23), 5183–5187, DOI:10.1021/ic00075a040.
- M. H. Chisholm, F. A. Cotton, B. A. Frenz, W. W. Reichert, L. W. Shive and B. R. Stults, The Molybdenum-Molybdenum Triple Bond. 1. Hexakis(Dimethylamido)Dimolybdenum and Some Homologs: Preparation, Structure, and Properties, J. Am. Chem. Soc., 1976, 98(15), 4469–4476, DOI:10.1021/ja00431a024.
- J. P. Collman, S. T. Harford, S. Franzen, A. P. Shreve and W. H. Woodruff, Resonance Raman and X-Ray Crystallographic Studies of Intertriad Metal–Metal Bonds. 2. WRu and MoOs Porphyrin Dimers, Inorg. Chem., 1999, 38(9), 2093–2097, DOI:10.1021/ic9810337.
- M. Nippe, G. H. Timmer and J. F. Berry, Remarkable Regioselectivity in the Preparation of the First Heterotrimetallic MoW⋯Cr Chain, Chem. Commun., 2009,(29), 4357–4359, 10.1039/B907402C.
- I. D. Rosal, S. Lassalle, C. Dinoi, C. Thieuleux, L. Maron and C. Camp, Mechanistic Investigations via DFT Support the Cooperative Heterobimetallic C–H and O–H Bond Activation across TaIr Multiple Bonds, Dalton Trans., 2021, 50(2), 504–510, 10.1039/D0DT03818K.
- C. Ehinger, S. Pollitt, J. D. J. Silva, X. Zhou, K. Sakamoto, M. Nachtegaal, O. Safonova and C. Copéret, Alloyed Molybdenum Enables Efficient Alcohol Hydrodeoxygenation with Supported Bimetallic Catalysts, Chem. Sci., 2025, 16(14), 5887–5896, 10.1039/D4SC08532A.
- J. A. Esterhuizen, A. Mathur, B. R. Goldsmith and S. Linic, High-Performance Iridium–Molybdenum Oxide Electrocatalysts for Water Oxidation in Acid: Bayesian Optimization Discovery and Experimental Testing, J. Am. Chem. Soc., 2024, 146(8), 5511–5522, DOI:10.1021/jacs.3c13491.
- L. Fu, Y. Li, N. Yao, F. Yang, G. Cheng and W. Luo, IrMo Nanocatalysts for Efficient Alkaline Hydrogen Electrocatalysis, ACS Catal., 2020, 10(13), 7322–7327, DOI:10.1021/acscatal.0c02254.
- J. Fang, H. Wang, Q. Dang, H. Wang, X. Wang, J. Pei, Z. Xu, C. Chen, W. Zhu, H. Li, Y. Yan and Z. Zhuang, Atomically Dispersed Iridium on Mo2C as an Efficient and Stable Alkaline Hydrogen Oxidation Reaction Catalyst, Nat. Commun., 2024, 15(1), 4236, DOI:10.1038/s41467-024-48672-9.
- J. Shi, R. Li, J. Zhang, Y. Wang, W. Ma, Z. Yue, C. Jin, Y. Liu, L. Zheng, J. Bai, X. Li, K. Leng and Y. Qu, N-Coordinated Iridium–Molybdenum Dual-Atom Catalysts Enabling Efficient Bifunctional Hydrogen Electrocatalysis, ACS Appl. Mater. Interfaces, 2024, 16(1), 889–897, DOI:10.1021/acsami.3c16300.
- A. Albinati, A. Togni and L. M. Venanzi, Cationic Molybdenum(IV)- and Tungsten(IV)-Iridium(III) Complexes with Hydride and η5:η1-Cyclopentadienyl Bridging Ligands. Syntheses and x-Ray Crystal Structure of [(η5-C5H5)M(Mu-H)2(Mu-(η5:η1-C5H4))IrH(PMePh2)2][BPh4] (M = Mo and W), Organometallics, 1986, 5(9), 1785–1791, DOI:10.1021/om00140a005.
- L. Dahlenburg, E. Halsch, A. Wolski and M. Moll, Metallorganische Verbindungen Des Iridiums Und Rhodiums: XXXI. Heterobimetallische Zweikern-Komplexe Durch Addition von Cp(CO)3 MH (M = Mo, W) an Ir(CH3)(CO)(PPh3)2, J. Organomet. Chem., 1993, 463(1), 227–233, DOI:10.1016/0022-328X(93)83422-R.
- L. Escomel, I. Del Rosal, L. Maron, E. Jeanneau, L. Veyre, C. Thieuleux and C. Camp, Strongly Polarized Iridiumδ−–Aluminumδ+ Pairs: Unconventional Reactivity Patterns Including CO2 Cooperative Reductive Cleavage, J. Am. Chem. Soc., 2021, 143(12), 4844–4856, DOI:10.1021/jacs.1c01725.
- C. Z. Ye, I. D. Rosal, M. A. Boreen, E. T. Ouellette, D. R. Russo, L. Maron, J. Arnold and C. Camp, A Versatile Strategy for the Formation of Hydride-Bridged Actinide–Iridium Multimetallics, Chem. Sci., 2023, 14(4), 861–868, 10.1039/D2SC04903A.
- J. C. Green, M. L. H. Green and G. Parkin, The Occurrence and Representation of Three-Centre Two-Electron Bonds in Covalent Inorganic Compounds, Chem. Commun., 2012, 48(94), 11481–11503, 10.1039/C2CC35304K.
- M. J. Butler and M. R. Crimmin, Magnesium, Zinc, Aluminium and Gallium Hydride Complexes of the Transition Metals, Chem. Commun., 2017, 53(8), 1348–1365, 10.1039/C6CC05702K.
- N. Curado, M. Carrasco, J. Campos, C. Maya, A. Rodríguez, E. Ruiz, S. Álvarez and E. Carmona, An Unsaturated Four-Coordinate Dimethyl Dimolybdenum Complex with a Molybdenum–Molybdenum Quadruple Bond, Chem.–Eur. J., 2017, 23(1), 194–205, DOI:10.1002/chem.201604618.
- M. Perez-Jimenez, N. Curado, C. Maya, J. Campos, J. Jover, S. Alvarez and E. Carmona, Coordination of LiH Molecules to Mo
![[quadruple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e003.gif) Mo Bonds: Experimental and Computational Studies on Mo2LiH2, Mo2Li2H4, and Mo6Li9H18 Clusters, J. Am. Chem. Soc., 2021, 143(13), 5222–5230, DOI:10.1021/jacs.1c01602.
Mo Bonds: Experimental and Computational Studies on Mo2LiH2, Mo2Li2H4, and Mo6Li9H18 Clusters, J. Am. Chem. Soc., 2021, 143(13), 5222–5230, DOI:10.1021/jacs.1c01602. - F. A. Cotton, Discovering and Understanding Multiple Metal-to-Metal Bonds, Acc. Chem. Res., 1978, 11(6), 225–232, DOI:10.1021/ar50126a001.
- N. T. Lucas, J. P. Blitz, S. Petrie, R. Stranger, M. G. Humphrey, G. A. Heath and V. Otieno-Alego, Mixed-Metal Cluster Chemistry. 19. Crystallographic, Spectroscopic, Electrochemical, Spectroelectrochemical, and Theoretical Studies of Systematically Varied Tetrahedral Group 6−Iridium Clusters, J. Am. Chem. Soc., 2002, 124(18), 5139–5153, DOI:10.1021/ja0173829.
- N. T. Lucas, E. G. A. Notaras, S. Petrie, R. Stranger and M. G. Humphrey, Mixed-Metal Cluster Chemistry. 22. Synthesis and Crystallographic, Electrochemical, and Theoretical Studies of Alkyne-Coordinated Group 6−Iridium Clusters Linked by Phenyleneethynylene Groups, Organometallics, 2003, 22(4), 708–721, DOI:10.1021/om020910k.
- M. D. Randles, P. V. Simpson, V. Gupta, J. Fu, G. J. Moxey, T. Schwich, A. L. Criddle, S. Petrie, J. G. MacLellan, S. R. Batten, R. Stranger, M. P. Cifuentes and M. G. Humphrey, Syntheses of Pentanuclear Group 6 Iridium Clusters by Core Expansion of Tetranuclear Clusters with Ir(CO)2(H5-C5Me4R) (R = H, Me), Inorg. Chem., 2013, 52(19), 11256–11268, DOI:10.1021/ic401502f.
- P. V. Simpson, M. D. Randles, V. Gupta, J. Fu, G. J. Moxey, T. Schwich, M. Morshedi, M. P. Cifuentes and M. G. Humphrey, Phosphine, Isocyanide, and Alkyne Reactivity at Pentanuclear Molybdenum/Tungsten–Iridium Clusters, Dalton Trans., 2015, 44(16), 7292–7304, 10.1039/C5DT00525F.
- J. Fu, M. D. Randles, G. J. Moxey, R. Stranger, M. P. Cifuentes and M. G. Humphrey, Mixed-Metal Cluster Chemistry. 39. Syntheses and X-Ray Structures of Mo3Ir3(M4-H2-CO)(M3-CO)(CO)10(H5-C5H5)3 and Mo3RhIr3(μ-CO)4(CO)7(μ5-C5H5)3(μ5-C5Me5), J. Organomet. Chem., 2017, 829, 66–70, DOI:10.1016/j.jorganchem.2016.10.016.
- T. Takao, S. Matsuzawa and M. Nagaoka, Synthesis of Bimetallic Dinuclear Hydrido Complexes of Re and Rh/Ir Supported by a Direct M–M′ Bond: The Role of the M–M′ Bond in the Site Exchange of Hydrides, Inorg. Chem., 2024, 63(46), 22214–22226, DOI:10.1021/acs.inorgchem.4c03837.
- L. Escomel, N. Soulé, E. Robin, I. Del Rosal, L. Maron, E. Jeanneau, C. Thieuleux and C. Camp, Rational Preparation of Well-Defined Multinuclear Iridium–Aluminum Polyhydride Clusters and Comparative Reactivity, Inorg. Chem., 2022, 61(15), 5715–5730, DOI:10.1021/acs.inorgchem.1c03120.
- S. Lassalle, J. Petit, R. L. Falconer, V. Hérault, E. Jeanneau, C. Thieuleux and C. Camp, Reactivity of Tantalum/Iridium and Hafnium/Iridium Alkyl Hydrides with Alkyl Lithium Reagents: Nucleophilic Addition, Alpha-H Abstraction, or Hydride Deprotonation?, Organometallics, 2022, 41(13), 1675–1687, DOI:10.1021/acs.organomet.2c00158.
- K. Ishihara, Y. Araki, M. Tada, T. Takayama, Y. Sakai, W. M. C. Sameera and Y. Ohki, Synthesis of Dinuclear Mo−Fe Hydride Complexes and Catalytic Silylation of N2, Chem.–Eur. J., 2020, 26(43), 9537–9546, DOI:10.1002/chem.202000104.
- J. L. Petersen, L. F. Dahl and J. M. Williams, Neutron Diffraction Studies of the Metal-Hydrogen-Metal Bond. I. Symmetric, Bent, Three-Center, Two-Electron Molybdenum-Hydrogen-Molybdenum Bond in μ-Hydrido-μ-Dimethylphosphido-Bis(η.5-Cyclopentadienyldicarbonylmolybdenum), J. Am. Chem. Soc., 1974, 96(21), 6610–6620, DOI:10.1021/ja00828a012.
- N. Queyriaux, J. J. Cabrera-Trujillo, N. Durvin, L. Vendier, K. Miqueu and A. Simonneau, Reactivity of Metal Hydrides with CO2: Going beyond Formate with a High-Valent Cationic Pentahydride Mo(VI) Complex, Chem. Sci., 2024, 15(48), 20582–20589, 10.1039/D4SC04345F.
- W. H. Bernskoetter and N. A. Hazari, Computational Investigation of the Insertion of Carbon Dioxide into Four- and Five-Coordinate Iridium Hydrides, Eur. J. Inorg. Chem., 2013, 2013(22–23), 4032–4041, DOI:10.1002/ejic.201300170.
- E. A. LaPierre, W. E. Piers and C. Gendy, Divergent Reactivity of CO2, CO, and Related Substrates at the Nickel Carbon Double Bond of (PCcarbeneP)Ni(II) Pincer Complexes, Organometallics, 2018, 37(20), 3394–3398, DOI:10.1021/acs.organomet.8b00440.
- F. Bertini, F. Hoffmann, C. Appelt, W. Uhl, A. W. Ehlers, J. C. Slootweg and K. Lammertsma, Reactivity of Dimeric P/Al-Based Lewis Pairs toward Carbon Dioxide and Tert-Butyl Isocyanate, Organometallics, 2013, 32(22), 6764–6769, DOI:10.1021/om3011382.
- B. Stadler, N. Gorgas, S. J. Elliott and M. R. Crimmin, Dyotropic Rearrangement of an Iron–Aluminium Complex, Angew. Chem., Int. Ed., 2024, 63(42), e202408257, DOI:10.1002/anie.202408257.
- V. C. Gibson, C. Redshaw, W. Clegg and M. R. J. Elsegood, Isocyanate versus Isothiocyanate Insertion into Alkoxo and Imido Ligands, J. Chem. Soc. Chem. Commun., 1994, 2635–2636, 10.1039/C39940002635.
- H.-W. Lam, G. Wilkinson, B. Hussain-Bates and M. B. Hursthouse, Reactions of Tert-Butyl Isocyanate and Trimethylsilyl Azide with Imidoamido Compounds of Chromium, Molybdenum and Tungsten, J. Chem. Soc., Dalton Trans., 1993,(5), 781–788, 10.1039/DT9930000781.
- H. E. Dyer, S. Huijser, N. Susperregui, F. Bonnet, A. D. Schwarz, R. Duchateau, L. Maron and P. Mountford, Ring-Opening Polymerization of Rac-Lactide by Bis(Phenolate)Amine-Supported Samarium Borohydride Complexes: An Experimental and DFT Study, Organometallics, 2010, 29(16), 3602–3621, DOI:10.1021/om100513j.
- D. Schöne, D. Gerlach, C. Wiltzsch, E. Brendler, T. Heine, E. Kroke and J. Wagler, A Distorted Trigonal Antiprismatic Cationic Silicon Complex with Ureato Ligands: Syntheses, Crystal Structures and Solid State 29Si NMR Properties, Eur. J. Inorg. Chem., 2010, 2010(3), 461–467, DOI:10.1002/ejic.200900784.
- A. Lachguar, C. Z. Ye, S. N. Kelly, E. Jeanneau, I. D. Rosal, L. Maron, L. Veyre, C. Thieuleux, J. Arnold and C. Camp, CO2 Cleavage by Tantalum/M (M = Iridium, Osmium) Heterobimetallic Complexes, Chem. Commun., 2024, 60(61), 7878–7881, 10.1039/D4CC02207F.
- S. Lassalle, R. Jabbour, I. Del Rosal, L. Maron, E. Fonda, L. Veyre, D. Gajan, A. Lesage, C. Thieuleux and C. Camp, Stepwise Construction of Silica-Supported Tantalum/Iridium Heteropolymetallic Catalysts Using Surface Organometallic Chemistry, J. Catal., 2020, 392, 287–301, DOI:10.1016/j.jcat.2020.10.016.
- CRC Handbook of Chemistry and Physics: A Ready-Reference Book of Chemical and Physical Data, ed. D. R. Lide, CRC Press, Boca Raton, Fla, 2008, vol. 88 Search PubMed.
- L. S. Bartell, E. A. Roth, C. D. Hollowell, K. Kuchitsu and J. E. Young Jr, Electron-Diffraction Study of the Structures of C2H4 and C2D4, J. Chem. Phys., 1965, 42(8), 2683–2686, DOI:10.1063/1.1703223.
- J. Chatt and L. A. Duncanson, 586. Olefin Co-Ordination Compounds. Part III. Infra-Red Spectra and Structure: Attempted Preparation of Acetylene Complexes, J. Chem. Soc., 1953, 2939–2947, 10.1039/JR9530002939.
- M. J. Bezdek and P. J. Chirik, Proton-Coupled Electron Transfer to a Molybdenum Ethylene Complex Yields a β-Agostic Ethyl: Structure, Dynamics and Mechanism, J. Am. Chem. Soc., 2018, 140(42), 13817–13826, DOI:10.1021/jacs.8b08460.
- K. S. Cook, W. E. Piers, P. G. Hayes and M. Parvez, Reactions of a Borataalkene Ligand at Tantalocene Centers: Isonitrile Insertion into the B−C Bond of the [CH2B(C6F5)2] Ligand via the H1 Bonding Mode, Organometallics, 2002, 21(12), 2422–2425, DOI:10.1021/om020145+.
- M. Baltrun, F. A. Watt, R. Schoch and S. Hohloch, Dioxo-, Oxo-Imido-, and Bis-Imido-Molybdenum(VI) Complexes with a Bis-Phenolate-NHC Ligand, Organometallics, 2019, 38(19), 3719–3729, DOI:10.1021/acs.organomet.9b00472.
- D. C. H. Do, N. Đorđević and H. V. Huynh, Diverse Post-Metathesis Reactivities of Mixed Amido-Isothiocyanato Molybdenum Complexes, Inorg. Chem., 2023, 62(45), 18583–18590, DOI:10.1021/acs.inorgchem.3c02885.
- G. D. Forster and G. Hogarth, Reversible Insertion of an Isocyanate into an Imido Moiety at a Dimolybdenum Centre. Synthesis and X-Ray Crystal Structure of [Mo2(NPh)2(S2CNEt2)2(μ-NPh){μ-PhNC(O)NPh}], J. Chem. Soc., Dalton Trans., 1993,(16), 2539–2540, 10.1039/DT9930002539.
- R. B. King, R. M. Murray, R. E. Davis and P. K. Ross, Organonitrogen Derivatives of Metal Carbonyls: XIII. Reactions of Dialkylaminotrimethylsilylacetylenes with Metal Carbonyls, J. Organomet. Chem., 1987, 330(1), 115–132, DOI:10.1016/0022-328X(87)80282-6.
- W.-S. Ojo, E. Paugam, F. Y. Pétillon, P. Schollhammer, J. Talarmin and K. W. Muir, Formation of C−C, C−N, and C−O Links between Isonitrile, Cyclopentadienyl, and Hydroxide Ligands Bound to Molybdenum(III): Syntheses and Crystal Structures of μ-Aminocarbyne and μ-Amino-Oxycarbene Dimolybdenum Complexes, Organometallics, 2006, 25(16), 4009–4018, DOI:10.1021/om060383v.
- W.-S. Ojo, F. Y. Pétillon, P. Schollhammer and J. Talarmin, C−C, C−S, and C−N Coupling versus Dealkylation Processes in the Cationic Tris(Thiolato)Dimolybdenum(III) Complexes [Mo2Cp2(μ-SMe)3L2]+ (L = XylNC, t-BuNC, CO, MeCN), Organometallics, 2008, 27(16), 4207–4222, DOI:10.1021/om8003702.
- L. Biancalana and F. Marchetti, Aminocarbyne Ligands in Organometallic Chemistry, Coord. Chem. Rev., 2021, 449, 214203, DOI:10.1016/j.ccr.2021.214203.
- N. Cabon, E. Paugam, F. Y. Pétillon, P. Schollhammer, J. Talarmin and K. W. Muir, Unexpected Coupling of Cp and Two RNC Ligands at a {Mo2(μ-SMe)3} Nucleus, Organometallics, 2003, 22(21), 4178–4180, DOI:10.1021/om030447r.
- K. Fujita, H. Nakaguma, F. Hanasaka and R. Yamaguchi, Synthesis of a DMPM and Hydrido-Bridged Diiridium Complex, [(Cp*Ir)2(μ-Dmpm)(μ-H)2][OTf]2, and Its Reactivity toward Alkynes and Isocyanides, Organometallics, 2002, 21(18), 3749–3757, DOI:10.1021/om020266+.
- K. S. Ratliff, D. L. DeLaet, J. Gao, P. E. Fanwick and C. P. Kubiak, Roles of Bridging Ligand -Acidity in Determining the Nature of the Lowest Excited States of Binuclear Complexes of Nickel(0). Ligand-Centered (M2 μ-LCT), Metal-Centered (μ-L M2CT), and Intraligand (IL) Lowest Excited States for the Series of Complexes 2(μ-)(CNMe)2(PPh2CH2PPh2)2 (L = CNR, CNMe(R)+, NO+), Inorg. Chem., 1990, 29(20), 4022–4027, DOI:10.1021/ic00345a023.
- L. Busetto, F. Marchetti, S. Zacchini and V. Zanotti, Synthesis, Characterization and Reactivity of New (μ-Aminocarbyne)Diruthenium Complexes Containing Alkynylimino Ligands, Eur. J. Inorg. Chem., 2004, 2004(7), 1494–1504, DOI:10.1002/ejic.200300772.
- K. Hüttinger, C. Förster, T. Bund, D. Hinderberger and K. Heinze, Stereochemical Consequences of Oxygen Atom Transfer and Electron Transfer in Imido/Oxido Molybdenum(IV, V, VI) Complexes with Two Unsymmetric Bidentate Ligands, Inorg. Chem., 2012, 51(7), 4180–4192, DOI:10.1021/ic202588u.
- H. Y. Cho, S.-G. Roh and J. H. Jeong, Syntheses and Reactivities of New Bis(Imido)Mo(VI) Complexes of Hydrotris(3,5-Dimethyl-1-Pyrazolyl)Borate and X-Ray Molecular Structures of [{HB(3,5-Me2C3N2H)3}Mo(Nmes)2X] (X=Cl or OH; Mes=2,4,6-Trimethylphenyl), Polyhedron, 2002, 21(12), 1211–1215, DOI:10.1016/S0277-5387(02)01008-2.
- A. Wlodarczyk, J. P. Maher, S. Coles, D. E. Hibbs, M. H. B. Hursthouse and K. M. A. Malik, Oxo-Bridged Binuclear Molybdenum Nitrosyl Halides: Structural and Redoxstudies, Mixed-Valence Behaviour, and Characterisation of Mononuclearhydroxo Precursors, J. Chem. Soc., Dalton Trans., 1997,(15), 2597–2606, 10.1039/A703279J.
- D. A. Dobbs and R. G. Bergman, Synthesis and Reactivity of Bridging Imido and Imido-Oxo Complexes of Iridium. Water-Catalyzed and -Uncatalyzed Dimerization of Terminal Iridium Imido Complexes, Organometallics, 1994, 13(11), 4594–4605, DOI:10.1021/om00023a072.
- D. A. Dobbs and R. G. Bergman, Synthesis of Bridging Iridium Bis(Imido) and Imido-Oxo Complexes. Imide and Oxygen Transfer Reactions and Hydrogenation of an Imido Ligand, J. Am. Chem. Soc., 1993, 115(9), 3836–3837, DOI:10.1021/ja00062a084.
- A. L. Casalnuovo, J. C. Calabrese and D. Milstein, Rational Design in Homogeneous Catalysis. Iridium(I)-Catalyzed Addition of Aniline to Norbornylene via Nitrogen-Hydrogen Activation, J. Am. Chem. Soc., 1988, 110(20), 6738–6744, DOI:10.1021/ja00228a022.
- M. Schau-Magnussen, P. Malcho, K. Herbst, M. Brorson and J. Bendix, Synthesis and X-Ray Crystal Structure of a Novel Organometallic (M3-Oxido)(M3-Imido) Trinuclear Iridium Complex, Dalton Trans., 2011, 40(16), 4212–4216, 10.1039/C0DT01791D.
- P. L. Holland, R. A. Andersen and R. G. Bergman, Cyclopentadienyl and Imide Ligand Transfer from Zirconium to Iridium: Can Early Transition Metal Imido Compounds Be Used as Imide Transfer Reagents?, Organometallics, 1998, 17(3), 433–437, DOI:10.1021/om970827a.
- A. M. Baranger and R. G. Bergman, Cooperative Reactivity in the Interactions of X-H Bonds with a Zirconium-Iridium Bridging Imido Complex, J. Am. Chem. Soc., 1994, 116(9), 3822–3835, DOI:10.1021/ja00088a019.
- A. A. Danopoulos, G. Wilkinson, T. K. N. Sweet and M. B. Hursthouse, Reactions of Imido Complexes of Iridium, Rhodium and Ruthenium, J. Chem. Soc., Dalton Trans., 1996,(19), 3771–3778, 10.1039/DT9960003771.
- F. Albert Cotton and R. Poli, Iridium(I) Dimers with Bridging N,N′-Di-p-Tolylformamidine. X-Ray Molecular Structure of Ir2(μ-p-CH3C6H4NCHN-p-C6H4CH3)(μ-NH-p-C6H4CH3)(C8H14)2, Inorg. Chim. Acta, 1986, 122(2), 243–248, DOI:10.1016/S0020-1693(00)81646-1.
- S. Anderson, T. E. Berridge, A. F. Hill, Y. T. Ng, A. J. P. White and D. J. Williams, Dihapto Carbamoyl (Carboxamide) Complexes of Iron(II), Organometallics, 2004, 23(11), 2686–2693, DOI:10.1021/om0306933.
- A. Müller, U. Seyer and W. Eltzner, The Carbamido Moiety as Fixed and Side-on Bonded Ligand. X-Ray Crystal and Molecular Structure of [(C6H5)4P]2 [Mo(NO)(OCN(CH3)2(NCS)4)], Inorg. Chim. Acta, 1979, 32, L65–L66, DOI:10.1016/S0020-1693(00)91616-5.
- J. N. Coalter, J. C. Huffman and K. G. Caulton, Cleavage of H−C(Sp2) and C(Sp2)−X Bonds (X = Alkyl, Aryl, OR, NR2): Facile Decarbonylation, Isonitrile Abstraction, or Dehydrogenation of Aldehydes, Esters, Amides, Amines, and Imines by [RuHCl(PiPr3)2]2, Organometallics, 2000, 19(18), 3569–3578, DOI:10.1021/om000390y.
- M. H. Chisholm, J. A. Heppert, J. C. Huffman and W. E. Streib, Attack of Isocyanide and Carbon Monoxide on a Bridging Alkylidyne Ligand at a Ditungsten Centre. Cleavage of the C–N and C–O Triple Bonds with Respective Formation of Tungsten–Imido and -Oxo Groups and Concomitant Addition of the Carbon Atom to the Bridging Alkylidyne Carbon Yielding a Bridging σ-W1,H2-W2 Alkynyl Ligand, J. Chem. Soc., Chem. Commun., 1985,(24), 1771–1773, 10.1039/C39850001771.
Footnote |
| † Electronic supplementary information (ESI) available: Data for this article, including chemical syntheses, NMR, infrared, UV-vis spectra, X-ray and computational data. CCDC 2410538–2410544. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc03465e |
| This journal is © The Royal Society of Chemistry 2025 |