
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotosensitized diradical rearrangement of alkenyl oxime ethers towards amino-featured oxiranes: reaction, mechanism, and structural prediction†
Tu-Ming Liu,
Lin-Yuan Zhu,
Min-Hao Qi,
Si-Jia Li,
Xiao-Jian Wang,
Jia-Rui Xu and
Bing Han *
*
State Key Laboratory of Natural Product Chemistry, College of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou, 730000, China. E-mail: hanb@lzu.edu.cn
First published on 1st July 2025
Abstract
A novel diradical rearrangement protocol of alkenyl ketoxime ethers for the synthesis of amino-featured epoxides under energy-transfer photocatalysis was developed. Mechanism studies revealed that this reaction was triggered by a specific diradical afforded by the second triplet excitation state T2 resulting from the π–π* excited transition of the alkene moiety of the substrate, followed by a cascade radical remote amino group translocation and intramolecular singlet O-, C-diradical cross-coupling after intersystem crossing. As a result, a series of amino-featured epoxides that were previously inaccessible, were synthesized easily and efficiently. Notably, this approach featured mild conditions, good functional group tolerance, excellent stereoselectivity and was very compatible with complex scaffolds (steroids, amino acids, alkaloids). Detailed density functional theory (DFT) calculations provided in-depth insights into the understanding of the reaction as well as structural standards and predictions. This strategy not only enriches the reaction mode of the oxime ethers but also provides a facile synthetic method towards valuable epoxides.
Introduction
As a three-membered oxa-heterocycle with a unique structure, oxirane is found widely in natural products and drug molecules1 (Scheme 1A). In addition, due to its high ring tension and strongly polarized C–O bond, oxirane also serves as a key synthon in organic synthesis by affording various functional group transformations.2,3 Therefore, the efficient and convenient construction of the oxirane structure has been an important focus of organic synthesis. Many strategies have been developed for this purpose: intramolecular nucleophilic substitution of alcohols with a leaving group at the β-position;4 epoxidation of alkenes;5 Corey–Chaykovsky epoxidation of aldehydes and ketones;6 Payne rearrangement7 (Scheme 1B). Despite great achievements, existing methods face several limitations, including poor compatibility with highly active functional groups, unsatisfactory suppression of side reactions caused by the oxidative sensitivity of the substrate, as well as the poor chemo-/site-selectivity of the substrates with multiple olefins and carbonyls. Especially for an amino-containing oxirane, which act as units with specific toxicity against cancer cells,1f,g conventional tactics face problems of inefficiency and product complexity due to the nucleophilicity and oxidative sensitivity of the amine group.8 Thus, the development of novel strategies for the efficient access to oxirane, especially for the amino-containing counterpart, is of great significance and value for the chemistry of organic and medicinal compounds. | ||
| Scheme 1 Synthetic strategy for oxiranes and the representative mode of the alkenyl oxime reaction. | ||
Oximes, as a class of fundamental organic compounds, have been widely used in diverse organic transformations, including traditional ionic conversions and emerging radical reactions. The former are known for the Beckmann rearrangement,9 reductive amination,10 and transition metal-catalysed cross-coupling,11 whereas the latter are represented by iminoxyl radical-mediated O/N-atom dichotomous cyclization12 and iminyl radical-mediated diverse reactions.13 In recent years, visible light-mediated photocatalytic energy transfer (EnT) has provided an opportunity to develop sustainable chemical conversions using triplet-state diradicals.14 Accordingly, some oxime-mediated novel transformations have been undertaken with the help of the EnT tactic.15–17 For example, a series of olefin difunctionalizations has been reported by Glorius and colleagues16 and other research teams (including ours17) independently. In addition, some elegant intra-/intermolecular [2 + 2] cycloadditions of oximes with olefins have been reported by Schindler et al.18 Based on our continuing interest in the research of oximes conversion,19 we wondered if it is possible to use oximes for synthesizing amino group-containing epoxides by taking advantage of their natural structural features.
We were encouraged by our recent work on the radical remote amino translocation of alkenyl oximes by N–O bond cleavage (Scheme 1C).20 Hence, we anticipated that this translocation may also be applicable to a triplet-state diradical counterpart.21 We speculated that if the alkenyl moiety of oxime 1 is excited to its triplet-state T2 through EnT-mediated photocatalysis, the fast 5-exo-trig cyclization of the terminal C-radical to the imine moiety would lead to the formation of a five-membered triplet diradical intermediate INT1. By employing the radical β-scission of a weak N–O bond, a triplet O-, N-diradical intermediate INT2 would be formed, which subsequently undergoes intersystem crossing (ISC) to generate singlet diradical counterpart INT3. Finally, the desired amino-contained oxiranes 2 would be realized via intramolecular cross-coupling between the O-radical and C-radical (Scheme 1D). However, due to the coexistence of imine and alkene moieties in oxime 1, the hypothesized epoxidation reaction faces three challenges: (1) energy transfer sensitization competition between the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond and CC bond with the excited photocatalyst; (2) the supposed reaction begins from the high-energy-level T2 by sensitization of the alkene moiety rather than the low-energy-level T1 by sensitization of the imine moiety, which leads to the deactivation of T2 by an internal conversion to T1; (3) T1 and T2 are inclined to undergo the alternative relaxation, including non-radiation Z–E isomerization of alkenyl oxime 1 and radiative deexcitation.
N bond and CC bond with the excited photocatalyst; (2) the supposed reaction begins from the high-energy-level T2 by sensitization of the alkene moiety rather than the low-energy-level T1 by sensitization of the imine moiety, which leads to the deactivation of T2 by an internal conversion to T1; (3) T1 and T2 are inclined to undergo the alternative relaxation, including non-radiation Z–E isomerization of alkenyl oxime 1 and radiative deexcitation.
To overcome the above-mentioned challenges, we assumed that two factors would facilitate the desired reaction: (1) introducing styrene or the conjugated diene into oxime 1 and using a photocatalyst with high ET to ensure the efficient sensitization of the olefin moiety; (2) increasing the Gibbs free energy gap (ΔGT = GT2 − GT1) between T2 and T1 states to avoid the inactivation of the T2 state by internal conversion; (3) reducing the energy gap (ΔEFO = ELUMO[oxime] − EHSOMO[alkene]) between the highest singly occupied molecular orbital (HSOMO) of the alkenyl diradical and the lowest unoccupied molecular orbital (LUMO) of the CN bond to increase the frontier orbital matching ability of the cyclization process, thereby suppressing the unwanted alternative relaxation. By employing the aforementioned strategy, the desired oxiranes 2 were produced by the photocatalytic epoxidation rearrangement of the readily available alkenyl oximes. This reaction provides a new method for synthesizing amino oxiranes that were previously inaccessible and broadens the boundary of the reaction mode of oximes.
Results and discussion
The present study commenced with the irradiation of a mixture of styrene-tethered oximes 1 and organic photosensitizer 2,4,5,6-tetra(9H-carbazol-9-yl) isophthalonitrile (4CzIPN) in ethyl acetate with blue light (443 nm, 30 W). First, the reaction optimization was carried out with aldoximes. However, benzaldehydoxime E-1-A underwent only Z/E-isomerization to give a mixture of 1-A with a ratio of Z![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :E of 8:1, and the desired oxirane 2-A was not detected. Similarly, benzaldehydoximes E-1-B and E-1-C bearing an electron-donating methoxy group and electron-withdrawing trifluoromethyl group on the para-position of phenyl ring, respectively, only underwent Z/E-isomerization as well (Table 1, entries 2 and 3). In addition, methyl glyoxylatoxime E-1-D was inert under the reaction conditions, and no reaction (even Z–E isomerization) occurred (Table 1, entry 4). Apparently, the aldoxime structure was not suitable for the protocol. Subsequently, we explored the reactivity of ketoximes by varying structural features under these conditions. Neither acetophenonoxime E-1-E nor acetonoxime 1-F could be converted into the desired products. The former underwent only Z–E isomerization, whereas the latter was inert in the reaction (Table 1, entries 5 and 6). Delightedly, diphenylketoxime 1a succeeded in obtaining the desired oxirane 2a in 82% yield (Table 1, entry 7). Next, other photocatalysts, such as thioxanthone (TXT), fac-Ir(ppy)3 (Ir), and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 (Ir–F) with different triplet-state energy (ET), were also tested in the reaction. Although they could also afford 2a, its yield failed to improve, presumably due to inadequate photosensitization under blue light or lower triplet-state energy (Table 1, entries 8–10). Unfortunately, when Ru(bpy)3(PF6)2 [Ru] acted as the photocatalyst, no reaction occurred and 1a was fully recovered, possibly due to its EnT being too low to sensitize 1a effectively (Table 1, entry 11). In addition, solvent screening showed that ethyl acetate was the best one (Table 1, entries 12–14). Both the photocatalyst and light irradiation were indispensable for an efficient reaction to proceed, clearly supporting a photocatalytic mechanism (Table 1, entries 15 and 16).
:E of 8:1, and the desired oxirane 2-A was not detected. Similarly, benzaldehydoximes E-1-B and E-1-C bearing an electron-donating methoxy group and electron-withdrawing trifluoromethyl group on the para-position of phenyl ring, respectively, only underwent Z/E-isomerization as well (Table 1, entries 2 and 3). In addition, methyl glyoxylatoxime E-1-D was inert under the reaction conditions, and no reaction (even Z–E isomerization) occurred (Table 1, entry 4). Apparently, the aldoxime structure was not suitable for the protocol. Subsequently, we explored the reactivity of ketoximes by varying structural features under these conditions. Neither acetophenonoxime E-1-E nor acetonoxime 1-F could be converted into the desired products. The former underwent only Z–E isomerization, whereas the latter was inert in the reaction (Table 1, entries 5 and 6). Delightedly, diphenylketoxime 1a succeeded in obtaining the desired oxirane 2a in 82% yield (Table 1, entry 7). Next, other photocatalysts, such as thioxanthone (TXT), fac-Ir(ppy)3 (Ir), and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 (Ir–F) with different triplet-state energy (ET), were also tested in the reaction. Although they could also afford 2a, its yield failed to improve, presumably due to inadequate photosensitization under blue light or lower triplet-state energy (Table 1, entries 8–10). Unfortunately, when Ru(bpy)3(PF6)2 [Ru] acted as the photocatalyst, no reaction occurred and 1a was fully recovered, possibly due to its EnT being too low to sensitize 1a effectively (Table 1, entry 11). In addition, solvent screening showed that ethyl acetate was the best one (Table 1, entries 12–14). Both the photocatalyst and light irradiation were indispensable for an efficient reaction to proceed, clearly supporting a photocatalytic mechanism (Table 1, entries 15 and 16).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Substrate | Photocatalyst | Solvent | Yielda,b (%) | |||
| R1 | R2 | Z/E | 1 (%, Z/E) | 2 | |||
| a Reaction conditions: 0.2 mmol scale, PC (2 mol%), solvent (0.1 M), 30 W 443 nm blue LEDs, 15 h.b Isolated yields.c No reaction occurred, and the starting material was recovered.d Reaction was conducted in the dark. | |||||||
| 1 | Ph | H | 1A, 0/1 | 4CzIPN | EtOAc | 1A, 80, 8/1 | 2A, 0 |
| 2 | p-MeOPh | H | 1B, 0/1 | 4CzIPN | EtOAc | 1B, 85, 6/1 | 2B, 0 |
| 3 | p-CF3Ph | H | 1C, 0/1 | 4CzIPN | EtOAc | 1C, 88, 5/1 | 2C, 0 |
| 4 | CO2Me | H | 1D, 0/1 | 4CzIPN | EtOAc | 1D, 98%, 0/1c | 2D, 0 |
| 5 | Ph | Me | 1E, 0/1 | 4CzIPN | EtOAc | 1E, 88, 5/1 | 2E, 0 |
| 6 | Me | Me | 1F, — | 4CzIPN | EtOAc | 1F,— | 2F, 0 |
| 7 | Ph | Ph | 1a, — | 4CzIPN | EtOAc | 0 | 2a, 82 |
| 8 | Ph | Ph | 1a, — | TXT | EtOAc | 0 | 2a, 42 |
| 9 | Ph | Ph | 1a, — | Ir–F | EtOAc | 0 | 2a, 78 |
| 10 | Ph | Ph | 1a, — | [Ir] | EtOAc | 0 | 2a, 70 |
| 11 | Ph | Ph | 1a, — | [Ru] | EtOAc | 1d, 98, —c | 2a, 0 |
| 12 | Ph | Ph | 1a, — | 4CzIPN | MeCN | 0 | 2a, 63 |
| 13 | Ph | Ph | 1a, — | 4CzIPN | MeOH | 0 | 2a, 60 |
| 14 | Ph | Ph | 1a, — | 4CzIPN | DCM | 0 | 2a, 0 |
| 15 | Ph | Ph | 1a, — | — | EtOAc | 95, —c | 2a, 0 |
| 16 | Ph | Ph | 1a, — | 4CzIPN | EtOAc | 80, —c,d | 2a, 0 |

To evaluate the applicability of the protocol, various alkenyl ketoxime ethers were investigated under the optimal reaction conditions, as shown in Scheme 2. The scope of the alkene moiety was first explored. Styrene moieties with substituents of different electronic properties on the para-position of the phenyl ring, such as OMe, Me, F, Cl, Br, CN, were all compatible with the reaction conditions, and converted smoothly to the corresponding amino-tethered oxiranes 2b–2g in good (70%) to excellent (92%) yields. Accordingly, ortho-/meta-fluoro and meta-methoxy substituted styrenes also reacted very well using this approach, delivering the desired products 2h–2j in good yields. Dimethyl-substituted styrene was also suitable for the reaction, affording the desired product 2k in 92% yield. Both electron-rich and electron-deficient styrenes could be converted in the reaction, indicating that electronic effects did not influence the reaction. In addition to styrenes, other aryl and heteroaryl groups incorporated into ethenes, such as naphthyl, furan, thiophene, and benzothiophene, were all good partners for the reaction, as demonstrated in cases 2l–2o, in which the corresponding oxiranes were formed in moderate (55%) to good (76%) yields. Importantly, the protocol could be extended readily from aryl-substituted alkenes to 1,3-dienes, establishing a novel approach for the regioselective functionalization of conjugated dienes. Acyclic and cyclic conjugated 1,3-dienes were good candidates for this method. Acyclic 1,3-dienes, such as 1,3-butadiene and isoprene, were efficiently transformed in the reaction, yielding products 2p and 2q in 50% yields. Cyclic dienes, such as cyclopentene- and cyclohexene-fused ethenes, proceeded smoothly and provided products 2r and 2s in 67% and 75% yields, respectively. O-atom-embedded and difluoro-substituted cyclohexenes converted smoothly as well, resulting in the corresponding products 2t–2u in excellent yields. When the ketal-containing cyclohexene was employed in the conversion, the desired product 2v was obtained in 82% yield, suggesting the potential for constructing epoxy-ketone architectures. Subsequently, the boundary of the oxime moiety was explored using the tethered styrene as the reaction partner. Symmetric diaryl ketoximes, represented by benzophenonoximes bearing para-methoxy, para-bromo, para-cyano, and meta-trifluoromethyl on the phenyl rings, were good candidates and converted to the corresponding products 2w–2z in good yields, and no significant electronic effects were observed. Furthermore, 9-fluorenonoxime and thienylketoxime also showed good compatibility, giving products 2aa and 2ab in 63% and 61% yields, respectively. Notably, asymmetric diaryl ketoximes, such as pMeO-phenyl-phenyl, phenyl-pyridinyl, and phenyl-thienyl ketoximes, were also very compatible with the protocol, as demonstrated in cases 2ac–2ae. Delightfully, in addition to diaryl ketoximes, monoaryl ketoximes bearing electron-deficient groups, such as CF3, ester, and amide, also performed very well under the conditions, affording the desired products 2af–2ah in 67–80% yields. Importantly, this approach was also compatible with complex natural scaffolds, such as amino acids, steroids, and alkaloids, as in cases 2ai–2ak, showing a promising application for late-stage modification.
 | ||
| Scheme 2 aReaction conditions: 0.2 mmol scale, 4CzIPN (2 mol%), EtOAc (0.1 M), blue LEDs (443 nm, 30 W), 15 h. bIsolated yields. | ||
Having achieved 1,1-disubstituted oxiranes by this method, we hope that it was also suitable for synthesizing the 1,1,2-trisubstituted counterparts using secondary alkenyl alcohol-derived benzopheonnoximes ethers as substrates. To our delight, the protocol was suitable for obtaining 1,1,2-trisubstituted oxiranes and exhibited a single diastereoselectivity for 1,2-substitutions, and the results are summarized in Scheme 3. Ketoxime ethers bearing a series of substituted phenyls with different electronic properties at the α-position of the O-atom reacted very well in the reaction and produced the corresponding products 2al–2ap in good yields as single diastereomers. The structure and configuration of epoxide 2an were confirmed by X-ray crystallography.21 When the aforementioned phenyls were replaced by pyridinyl and thienyl, the conversions were smooth, giving rise to 2aq and 2ar in 72% and 63% yields, respectively. Notably, the substitution of phenyl by alkyl chains or cycloalkyls did not affect the reactivity, as demonstrated in cases 2as–2aw, in which propyl, cyclopropyl, cyclopentyl, cyclohexyl, and piperidinyl were involved. In particular, ketoxime ethers that replaced the phenyl group with alkynyl and alkenyl groups were also converted to the desired products 2ax and 2ay in yields of 55% and 53%, respectively.
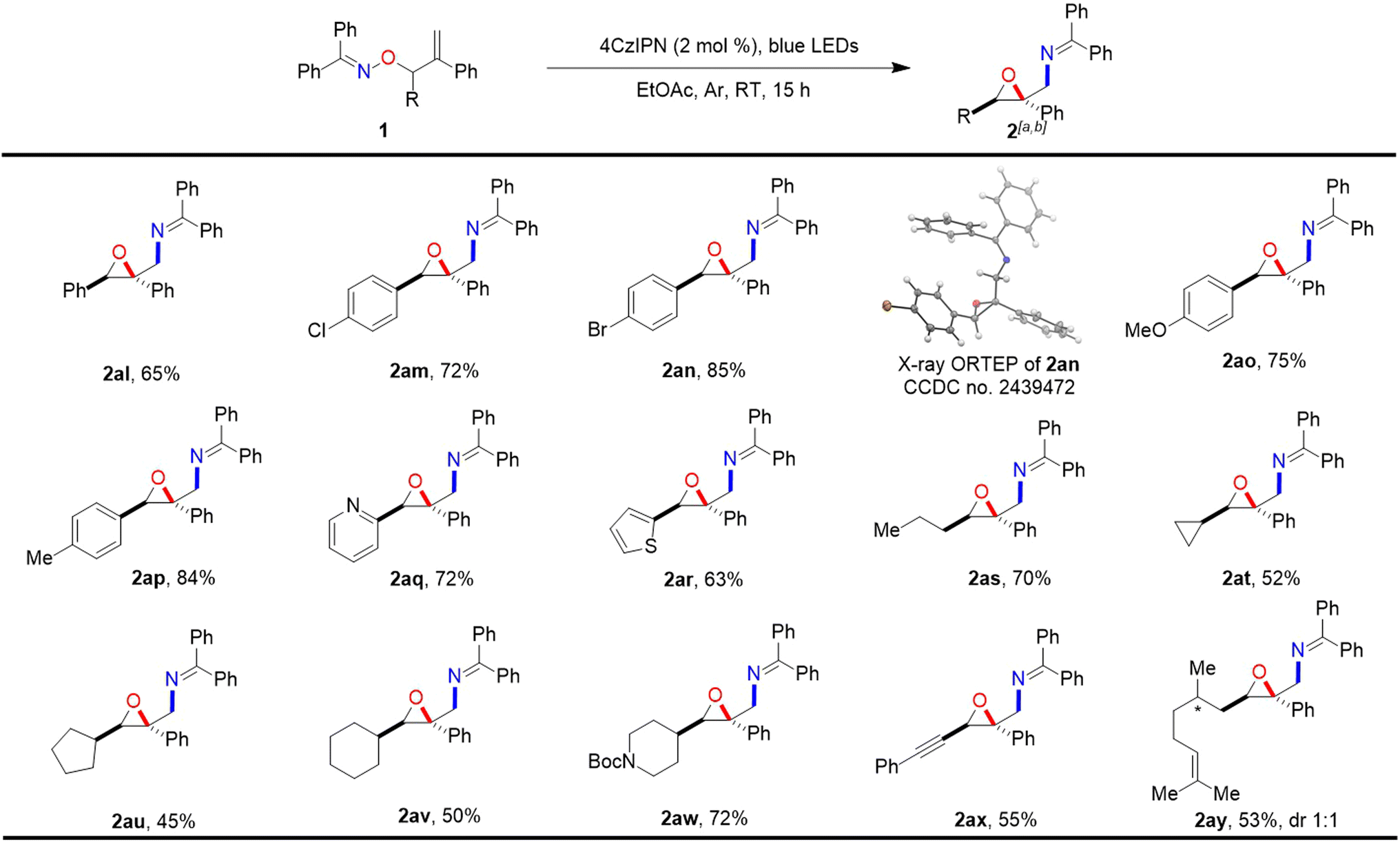 | ||
| Scheme 3 aReaction conditions: 0.2 mmol scale, 4CzIPN (2 mol%), EtOAc (0.1 M), blue LEDs (443 nm, 30 W), 15 h. bIsolated yields. | ||
To further prove the practicability and suitability of our protocol, the gram-scale synthesis and diverse derivation of the amino group-containing oxiranes were carried out, as shown in Scheme 4. The gram-scale synthesis was performed at a 5 mmol scale using substrate 1a, producing oxirane 2a at 1.252 g in 80% yield. Further conversions of 2a were conducted by various transformations of functional groups, such as reduction, hydrolysis, and epoxy ring-opening. The reduction of 2a could be smoothly achieved using NaBH3CN and LiAlH4, respectively. The former provided the diphenylamine oxirane 3 in 62% yield by solely reducing the imine moiety, and the latter produced 2-amino tertiary alcohol 4 in 73% yield by simultaneously reducing imine and epoxy moieties. Using C-, N-and S-nucleophiles, such as phenyl Grignard reagent, imidazole, and 2-mercaptobenzimidazole to react with 2a, tertiary alcohols 5–7 were generated in good yields by the nucleophilic epoxy ring-opening concomitant with the forging of C–C, N–C, and S–C bonds, respectively. Importantly, when 2a was treated with muriatic acid, the hydrolysis of the imine and acidic ring-opening was also achieved, affording the structurally valuable 3-amino-2-phenylpropane-1,2-diol hydrochloride 8 in 62% yield. Acetamide-incorporated 1,2-diol 9 was obtained in 83% yield by a cascade hydrolysis, acylation, and acidic epoxy ring-opening after being treated with acetic acid under heating.
 | ||
| Scheme 4 Gram-scale synthesis and derivatization of oxiranes. | ||
To understand the reaction mechanism of this protocol, control experiments, cyclic voltammetry, UV-visible absorption, and Stern–Volmer experiments were carried out, as shown in Scheme 5. When the triplet reaction inhibitor 2,5-dimethylhexa-2,4-diene was added, the desired reaction was completely inhibited in the sample reaction. In contrast, the reaction proceeded smoothly, and the product 2a was obtained in 75% yield when the singlet reaction inhibitor acrylonitrile was added. In addition, the sample reaction still occurred under the direct-irradiation with violet LEDs (365 nm, 20 W) in the absence of a photocatalyst, despite delivering 2a in 50% yield under a conversion rate of 70%. These results indicated that the protocol experienced a triplet photoreaction by energy-transfer catalysis. The influence of temperature on the reaction was also investigated. The reaction could proceed effectively between −40 °C and +60 °C, and the yield of 2a was almost unaffected. However, the reaction was completely inhibited at −60 °C, probably because sufficient activation energy could not be provided for the reaction at such a low temperature. Moreover, crossover experiments demonstrated that under standard reaction conditions, the concurrent addition of substrates 1c and 1w afforded epoxides 2c (78% yield) and 2w (88% yield) exclusively, with no detectable formation of other epoxides. These results indicated that the reaction experienced an intramolecular cleavage and recombination of the N–O bond. UV-Vis absorption spectra clearly showed that ketoxime 1a did not have obvious absorption above 400 nm, and weak absorption at 350 nm. These data indicated that it could not be excited by blue light, but could be excited by UV light, which was completely consistent with our experimental results. The measured oxidation potential Eox of ketoxime 1a was 2.1 V, while its reduction potential Ered was lower than −2.0 V, implying that the effective REDOX reaction could not occur between 1a and the excited photocatalyst 4CzIPN and was more likely to follow an energy-transfer process. Moreover, Stern–Volmer quenching experiments clearly showed that ketoxime 1a could be quenched by the excited 4CzIPN. Compound 1a had the CN double bond of the oxime moiety and the CC double bond of olefin moiety that could both be sensitized by a photosensitizer to trigger the reaction. Hence, we tried to determine which site of the reaction was triggered. Thus, compounds 1a-1 and 1a-2 as analogues of 1a were also investigated under Stern–Volmer quenching experiments. Apparently, both the alkene moiety and oxime moiety could be quenched by the excited 4CzIPN.
 | ||
| Scheme 5 Mechanism studies. | ||
To further confirm the initiation site and the mechanism of this diradical rearrangement reaction as well as to account for the excellent diastereoselectivity, density functional theory (DFT) calculations were conducted using 1al as the model. The calculated energy profiles are illustrated in Scheme 6A. The obtained T1 triplet state with an energy of 41.9 kcal mol−1 was caused by sensitization of the CN bond, whereas the T2 triplet state with an energy of 49.5 kcal mol−1 was caused by sensitization of the CC bond. Apparently, the iminyl moiety had lower sensitization energy and was more easily sensitized than olefin. Theoretically, T1 and T2 states can form the same triplet diradical intermediate INT1 by N-radical-mediated 5-endo-trig cyclization to olefin via TS1′ (ΔG‡ = 26.5 kcal mol−1) and C-radical-mediated 5-exo-trig cyclization to oxime via TS1 (ΔG‡ = 9.1 kcal mol−1), respectively. However, the activation energy of TS1′ was much higher than that of TS1 (ΔΔG‡ = 17.4 kcal mol−1), and therefore, it is more reasonable to initiate the reaction from the higher energy T2 state despite the T1 state being readily produced. This view was also consistent with the experimental result showing that the low-energy photosensitizer Ru(bpy)3(PF6)2 (ET = 46.0 kcal mol−1) failed to trigger the reaction. Next, we calculated two possible paths from INT1 to product 2al.
 | ||
| Scheme 6 (A) DFT-calculated energy profiles for the reaction and rationalization of the excellent diastereoselectivity of compound 2al. (B) Analysis of the atomic spin population and spin density visualization (blue isosurfaces indicate regions of positive spin density, and green isosurfaces indicate regions of negative spin density, at an isovalue of 0.01 a. u.). All calculations were performed at the B3LYP/def2-TZVP, SMD(EA)//B3LYP/6-31G(d,p) level with Gaussian 16. Gibbs free energies are given in kcal mol−1. For details, see the ESI.† | ||
One was a stepwise path that experiences the β-scission of the O–N bond to produce the O-radical and subsequent intramolecular cross-coupling of the O-radical and C-radical to yield 2al. The other was a concerted path in which cleavage of the N–O bond and formation of the C–O bond were carried out simultaneously without formation of the O-radical. In the first path, the calculated energy barrier for the cleavage of the O–N bond to produce the triplet diradical INT2 through TS2 was only 1.1 kcal mol−1, indicating that formation of the O-radical was very easy due to the weak N–O bond, as we reported previously.20 INT2 could be transformed further into two configurations of singlet diradicals INT3 and INT3′ by ISC via the minimal energy crossing point (MECP). The intramolecular O- and C-radicals cross-coupling of the singlet diradicals INT3 and INT3′ finally produced the products trans-2al and cis-2al, respectively. This formation of the O–C bond by radical cross-coupling was a barrierless process. Hence, the distribution of the products trans-2al and cis-2al could be determined by the energies of INT3 and INT3′ according to the Curtin–Hammett principle. Apparently, INT3 had 1.0 kcal mol−1 lower energy than INT3′, resulting in the formation of only trans-2al without cis-2al, which was consistent with the experimental observation. In the second path, INT1 first underwent ISC via MECP2 to produce singlet diradicals INT4 and INT4′. The latter would also produce cis-2al and trans-2al via the bicyclic transition states TS3′ (ΔG‡ = 2.2 kcal mol−1) and TS3 (ΔG‡ = 3.8 kcal mol−1), respectively. However, the conversion of INT4′ to product 10 in the form of cross-coupling of two C-radicals via TS4 looked impossible because of a higher energy barrier of 9.1 kcal mol−1 compared with that for the conversion of INT4′ to cis-2al via TS3′ with a lower activation energy. The concerted process provided an alternative path to access 2al. However, considering that the ISC of this pathway requires more energy (ΔΔG = 9.6 and 12.0 kcal mol−1, respectively) and that the activation energies of TS3 and TS3′ are both higher than that of TS2, we believe that the stepwise path is more favorable than the concerted pathway.
Next, we tried to elucidate the features of alkenyl oximes 1 that enabled the epoxidation rearrangement to occur smoothly because aldoxime and some ketoximes did not undergo the reaction (as shown in the optimization). Initially, we thought that in addition to the efficient sensitization of alkenes, there were two important factors that would synergistically affect the epoxidation reaction. One factor was that the energy difference (ΔGT = GT2 − GT1) of the excited triplet states T1 and T2 must be large enough to hinder the decay of T2 by internal conversion, thus ensuring its smooth cyclization. The other factor was that matching the frontier orbital energy levels (ΔEFO = ELUMO[oxime] − EHSOMO[alkene], serving as a measure of frontier orbital energy) of the alkene diradical and oxime would lead to a favorable 5-exo-trig cyclization by reducing the activation energy ΔG‡ and ultimately promoting reactivity. To verify this hypothesis, we computed ΔGT and ΔEFO across a series of alkenyl oximes by DFT calculations, and the results are summarized in Scheme 7A by means of quadrant analyses. The reactive substrates were concentrated in the second quadrant of the diagram. That is, the reaction could proceed smoothly if the frontier orbitals were matched (ΔEFO < 4 eV) and the energy gap between T1 and T2 was large enough (ΔGT >7.8 kcal mol−1). However, the substrates that met only one of these two factors failed to achieve the desired conversion. For instance, alkenyl oximes 1A, 1B, 1C, and 1E that satisfied the former factor (ΔEFO < 4 eV) but not the latter, only underwent Z–E isomerization of the oxime without the formation of the desired products (probably due to the fast internal conversion resulting in T2 state inactivation). For alkenyl oximes 1D and 1F, neither factor was satisfied, resulting in no reaction. Importantly, higher ΔEFO was negatively correlated with higher ΔGT (Pearson correlation coefficient of −0.75), indicating that the better matched the reaction orbitals were, the larger the ΔGT was, thus the easier the reaction proceeded. Obviously, this correlation was based on the fact that the internal conversion of the T2 state and 5-exo-trig cyclization in this reaction were two competing paths, the former making it easy for T2 to decay to T1 and the latter to covert it to the product. In this way, replacing the cumbersome ΔG‡ with ΔEFO can be used to establish the aforementioned criterion to rapidly assess the reactivity of substrate structures. To further confirm the accuracy of the developed criterion for structural prediction, the reactivity of a new structure of alkenyl oxime ether 1G was predicted on this criterion (Scheme 7B). DFT evaluations showed that it was located in the first quadrant and far from the average value of ΔGT and ΔEFO, indicating that this structure was not suitable for the diradical epoxidation rearrangement reaction. Subsequently, we synthesized 1G and carried out the reaction under standard reaction conditions. The desired rearrangement reaction did not occur, which fully verified the accuracy of the established criterion. During preparation of our manuscript, a related study was reported by Huang et al.22 Our protocol surpasses the reported one in terms of efficacy and scope, and provides new insights and an in-depth understanding of reaction mechanism and the structural requirements of the substrates through DFT calculations.
 | ||
| Scheme 7 Core criteria for the structural requirements of alkenyl oxime ethers in epoxidation rearrangement reactions and application of the structural prediction. | ||
Conclusions
We developed a novel photosensitized diradical rearrangement protocol of alkenyl ketoxime ethers for synthesizing amino-featured oxiranes under the visible light-facilitated energy-transfer photocatalysis. This protocol features mild conditions, good tolerance of functional groups, wide scope of substrates, excellent diastereoselectivity and is compatible with complex scaffolds (steroids, amino acids, alkaloids). Mechanism studies revealed that the reaction initiated from excitation of the alkenyl moiety of alkenyl ketoxime ethers to triplet-state diradical by photocatalytic energy transfer. These actions were followed by a tandem sequence of alkenyl diradical-mediated 5-exo-trig cyclization onto a ketoxime moiety, amino translocation, and formation of intramolecular C–O bonds. DFT calculations confirmed the proposed reaction mechanism and rationalized the excellent diastereoselectivity. A criterion for rapid assessment of reaction occurrence was also established by DFT calculations. That is, the substrate must meet both the large ΔGT (ΔGT = GT2[alkene] − GT1[oxime]) and small ΔEFO (ΔEFO = ELUMO[oxime] − EHSOMO[alkene]). This protocol provides a new method for the synthesis of valuable amino-featured oxiranes that were previously inaccessible, but also broadens the boundary of oxime-mediated reaction modes.Data availability
Detailed data are in the ESI.†Author contributions
B. H. and T. L. conceived and directed the project. T. L., L. Z., M. Q., S. L., X. W. and J. X. performed experiments. T. L., L. Z. and X. W. prepared the ESI.† B. H. wrote the paper. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (22471109 and 22171118) and Science and Technology Major Program of Gansu Province of China (22ZD6FA006, 23ZDFA015, and 24ZD13FA017) for financial support.References
- (a) D.-H. Wei, B.-L. Lei, M.-S. Tang and C.-G. Zhan, J. Am. Chem. Soc., 2012, 134, 10436–10450 CrossRef CAS PubMed; (b) L.-H. Meng, R. Mohan, B. H. B. Kwok, M. Elofsson, N. Sin and C. M. Crews, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 10403–10408 CrossRef CAS PubMed; (c) J. Lee, J.-L. Liu, X.-D. Feng, M. A. S. Hernández, P. Mucka, D. Ibi, J. W. Choi and U. Ozcan, Nat. Med., 2016, 22, 1023–1032 CrossRef CAS; (d) J.-S. Wang, L.-Q. Zhao, X.-W. Liu and Z.-Y. He, Food Chem., 2023, 405, 134962 CrossRef CAS; (e) J. Wu, X.-Q. Wang, Y.-L. Hou and P. Gong, Med. Chem. Res., 2024, 33, 221–238 CrossRef CAS; (f) Z. Y. Subeh, C. J. Pearce and N. H. Oberlies, J. Nat. Prod., 2023, 86, 596–603 CrossRef; (g) Y. Liu, H. Yang, Y. Liu, J. Liu, Z. Li and W. Zhang, Preparation method and application of chiral cyclopentadienyl rhodium complex, CN118580245A, 2024.
- For reviews, see: (a) J. G. Smith, Synthesis, 1984, 1984, 629–656 CrossRef; (b) J. He, J. Ling and P. Chiu, Chem. Rev., 2014, 114, 8037–8128 CrossRef CAS; (c) C. Wang, L. Luo and H. Yamamoto, Acc. Chem. Res., 2016, 49, 193–204 CrossRef CAS PubMed; (d) Y. Zhang, B. Hu, Y. Chen and Z. Wang, Chem.–Eur. J., 2024, 30, e202402469 CrossRef CAS PubMed; (e) Z. Liu, D. Wang, H. Cheng and Q. Zhou, ChemCatChem, 2024, 16, e202301764 CrossRef CAS; (f) A. Brandolese and A. W. Kleij, Acc. Chem. Res., 2022, 55, 1634–1645 CrossRef CAS PubMed; (g) M. I. Childers, J. M. Longo, N. J. Van Zee, A. M. LaPointe and G. W. Coates, Chem. Rev., 2014, 114, 8129–8152 CrossRef CAS.
- For recent examples, see: (a) J. Liu, Y. Wei and P. Tang, J. Am. Chem. Soc., 2018, 140, 15194–15199 CrossRef CAS; (b) A. Potrząsaj, M. Musiejuk, W. Chaładaj, M. Giedyk and D. Gryko, J. Am. Chem. Soc., 2021, 143, 9368–9376 CrossRef; (c) S. H. Lau, M. A. Borden, T. J. Steiman, L. S. Wang, M. Parasram and A. G. Doyle, J. Am. Chem. Soc., 2021, 143, 15873–15881 CrossRef CAS PubMed; (d) Y. Wang, S. Tang, G. Yang, S. Wang, D. Ma and Y. Qiu, Angew. Chem., Int. Ed., 2022, 61, e202207746 CrossRef CAS PubMed; (e) L. Li, S. Yang, Z. Xu, S. Li, J. Jiang and Y.-Q. Zhang, J. Am. Chem. Soc., 2024, 146, 13546–13557 CrossRef CAS; (f) L. Li, P. Sivaguru, D. Wei, M. Liu, Q. Zhu, S. Dong, E. Casali, N. Li, G. Zanoni and X. Bi, Nat. Commun., 2024, 15, 1951 CrossRef CAS; (g) W. Zhang, S. Chen, Y. Zhang, S. Liu and X. Shen, Angew. Chem., Int. Ed., 2025, 64, e202422020 CrossRef CAS PubMed.
- (a) Y. Usami, Y. Mizobuchi, M. Ijuin, T. Yamada, M. Morita, K. Mizuki, H. Yoneyama and S. Harusawa, Mar. Drugs, 2022, 20, 438 CrossRef CAS PubMed; (b) Y. Zheng, S.-H. Zhang, K. H. Low, W.-W. Zi and Z.-X. Huang, J. Am. Chem. Soc., 2022, 144, 1951–1961 CrossRef CAS; (c) K. Choudhary, R. G. Biswas, A. Manna and V. K. Singh, J. Org. Chem., 2023, 88, 12041–12053 CrossRef CAS PubMed.
- (a) R. Goswami, J. Am. Chem. Soc., 1980, 102, 5974–5976 CrossRef; (b) Y. Gao, R. M. Hanson, J. M. Klunder, S. Y. Ko, H. Masamune and K. B. Sharpless, J. Am. Chem. Soc., 1987, 109, 5765–5780 CrossRef CAS; (c) F. Severin, G. M. Fusi, C. Wartmann, J. M. Neudörfl and A. Berkessel, Angew. Chem., Int. Ed., 2022, 61, e202201790 CrossRef CAS PubMed.
- (a) E. J. Corey and M. Chaykovsky, J. Am. Chem. Soc., 1962, 84, 867 CrossRef CAS; (b) E. J. Corey and M. Chaykovsky, J. Am. Chem. Soc., 1965, 87, 1353 CrossRef CAS; (c) M. Szostak and J. Aubé, J. Am. Chem. Soc., 2009, 131, 13246–13247 CrossRef CAS PubMed.
- T. Ibuka, Chem. Soc. Rev., 1998, 27, 145–154 RSC.
- A. Boucherif, Q.-Q. Yang, Q. Wang, J.-R. Chen, L.-Q. Lu and W.-J. Xiao, J. Org. Chem., 2014, 79, 3924–3929 CrossRef CAS PubMed.
- (a) E. Beckmann, Ber. Dtsch. Chem. Ges., 1886, 19, 988 CrossRef; (b) Y. Furuya, K. Ishihara and H. Yamamoto, J. Am. Chem. Soc., 2005, 127, 11240–11241 CrossRef CAS; (c) X.-B. Mo, T. D. R. Morgan, H. T. Ang and D. G. Hall, J. Am. Chem. Soc., 2018, 140, 5264–5271 CrossRef CAS.
- (a) J. Mas-Rosello and N. Cramer, Chem.–Eur. J., 2022, 28, e202103683 CrossRef CAS; (b) B. Li, J.-Z. Chen, D. Liu, I. D. Gridnev and W.-B. Zhang, Nat. Chem., 2022, 14, 920–927 CrossRef CAS PubMed; (c) E. D. Heafner, A. L. Smith, C. V. Craescu, K. N. Raymond, R. G. Bergman and F. D. Toste, Chem, 2025, 11, 102368 CrossRef CAS.
- (a) D. S. Bolotin, N. A. Bokach, M. Y. Demakova and V. Yu. Kukushkin, Chem. Rev., 2017, 117, 13039–13122 CrossRef CAS PubMed; (b) R. Lavernhe, R. O. Torres-Ochoa, Q. Wang and J.-P. Zhu, Angew. Chem., Int. Ed., 2021, 60, 24028–24033 CrossRef CAS PubMed.
- (a) M. KitamuraI and K. Narasaka, Chem. Rec., 2002, 2, 268–277 CrossRef; (b) H.-W. Huang, X.-C. Ji, W.-Q. Wu and H.-F. Jiang, Chem. Soc. Rev., 2015, 44, 1155–1171 RSC; (c) J. Li, Y.-T. Hu, D.-P. Zhang, Q. Liu, Y.-H. Dong and H. Liu, Adv. Synth. Catal., 2017, 359, 710–771 CrossRef CAS; (d) J. Bajohr, S.-Y. Li, B. Mirabi, C. E. Johnson and M. Lautens, Angew. Chem., Int. Ed., 2025, 64, e202420479 CrossRef CAS.
- (a) J. Davies, S. P. Morcillo, J. J. Douglas and D. Leonori, Chem.–Eur. J., 2018, 24, 12154–12163 CrossRef CAS PubMed; (b) X.-Y. Yu, Q.-Q. Zhao, J. Chen, W.-J. Xiao and J.-R. Chen, Acc. Chem. Res., 2020, 53, 1066–1083 CrossRef CAS PubMed; (c) K. A. Rykaczewski, E. R. Wearing, D. E. Blackmun and C. S. Schindler, Nat. Synth., 2022, 1, 24–36 CrossRef CAS; (d) X.-Y. Duan, N.-N. Zhou, R. Fang, X.-L. Yang, W. Yu and B. Han, Angew. Chem., Int. Ed., 2014, 53, 3158–3162 CrossRef CAS PubMed; (e) E. M. Dauncey, S. P. Morcillo, J. J. Douglas, N. S. Sheikh and D. Leonori, Angew. Chem., Int. Ed., 2018, 57, 744–748 CrossRef CAS PubMed.
- (a) Q.-Q. Zhou, Y.-Q. Zou, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 1586–1604 CrossRef CAS; (b) F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Chem. Soc. Rev., 2018, 47, 7190–7202 RSC; (c) S.-Y. Wen, J.-J. Chen, Y. Zheng, J.-X. Han and H.-M. Huang, Angew. Chem., Int. Ed., 2025, 64, e202415495 CrossRef CAS PubMed.
- (a) D. Lee, V. Soni and E. Cho, N−O Bond Activation by Energy Transfer Photocatalysis, Acc. Chem. Res., 2022, 55, 2526–2541 CrossRef CAS; (b) Q. Sun, S.-P. Wang, Y. Xu, A. Yin, L. Yang, J. Zhu, C.-L. Zheng, G. Wang, Z. Fang, S. Sui, D. Wang, Y. Dong, D. Zhang, C.-S. Wang and K. Guo, ACS Catal., 2025, 15, 1854–1941 CrossRef CAS.
- (a) T. Patra, P. Bellotti, F. Strieth-Kalthoff and F. Glorius, Angew. Chem., Int. Ed., 2020, 59, 3172–3177 CrossRef CAS PubMed; (b) T. Patra, M. Das, C. G. Daniliuc and F. Glorius, Nat. Catal., 2021, 4, 54–61 CrossRef CAS; (c) G. Tan, M. Das, R. Kleinmans, F. Katzenburg, C. Daniliuc and F. Glorius, Nat. Catal., 2022, 5, 1120–1130 CrossRef CAS; (d) J. E. Erchinger, R. Hoogesteger, R. Laskar, S. Dutta, C. Hümpel, D. Rana, C. G. Daniliuc and F. Glorius, J. Am. Chem. Soc., 2023, 145, 2364–2374 CrossRef CAS PubMed; (e) R. Laskar, S. Dutta, J. C. Spies, P. Mukherjee, Á. Rentería-Gómez, R. E. Thielemann, C. G. Daniliuc, O. Gutierrez and F. Glorius, J. Am. Chem. Soc., 2024, 146, 10899–11090 CrossRef CAS; (f) X.-X. Zhang, S.-T. Xu, X.-T. Li, T.-T. Song, D.-W. Ji and Q.-A. Chen, J. Am. Chem. Soc., 2025, 147, 11533–11542 CrossRef CAS.
- S.-Q. Lai, B.-Y. Wei, J.-W. Wang, W. Yu and B. Han, Angew. Chem., Int. Ed., 2021, 60, 21997–22003 CrossRef CAS PubMed.
- (a) M. R. Becker, A. D. Richardson and C. S. Schindler, Nat. Commun., 2019, 10, 5095–5510 CrossRef PubMed; (b) E. R. Wearing, Y.-C. Yeh, G. G. Terrones, S. G. Parikh, I. Kevlishvili, H. J. Kulik and C. S. Schindler, Science, 2024, 384, 1468–1476 CrossRef CAS PubMed.
- (a) B. Han, X.-L. Yang, R. Fang, W. Yu, C. Wang, X.-Y. Duan and S. Liu, Angew. Chem., Int. Ed., 2012, 51, 8816–8820 (Angew. Chem., 2012, 124, 8946–8950) CrossRef CAS PubMed; (b) R.-H. Liu, D. Wei, B. Han and W. Yu, ACS Catal., 2016, 6, 6525–6530 CrossRef CAS; (c) X.-X. Peng, D. Wei, W.-J. Han, F. Chen, W. Yu and B. Han, ACS Catal., 2017, 7, 7830–7834 CrossRef CAS.
- D. Wei, T. M. Liu, Y. H. He, B. Y. Wei, J. H. Pan, J. W. Zhang, N. Jiao and B. Han, Angew. Chem., Int. Ed., 2021, 60, 26308–26313 CrossRef CAS.
- Deposition number 2439472 (for 2an) contains the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- Q.-Z. Wang, Y. Zheng, W.-T. Wu and H.-M. Huang, J. Am. Chem. Soc., 2025, 147, 16248–16254 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2439472. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc03379a |
| This journal is © The Royal Society of Chemistry 2025 |