
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBis(amidophenolate)-supported pnictoranides: Lewis acid-induced electromerism in a bismuth complex†
Simon B. H. Karnbrocka,
Jan F. Köstera,
Isabelle Becker b,
Christopher Golza,
Franc Meyerb,
Martí Gimferrerc and
Manuel Alcarazo*a
b,
Christopher Golza,
Franc Meyerb,
Martí Gimferrerc and
Manuel Alcarazo*a
aInstitute of Organic and Biomolecular Chemistry, University of Göttingen, Tammannstraße 2, 37077 Göttingen, Germany. E-mail: manuel.alcarazo@chemie.uni-goettingen.de
bInstitute of Inorganic Chemistry, University of Göttingen, Tammannstraße 4, 37077 Göttingen, Germany
cInstitute of Physical Chemistry, University of Göttingen, Tammannstraße 6, 37077 Göttingen, Germany
First published on 8th July 2025
Abstract
The synthesis of the complete series of 10-Pn-4 bis(amidophenolate)-supported pnictoranide anions is reported. Coordination of these anions to the cyclopentadienyliron(II) dicarbonyl fragment, [CpFe(CO)2]+, delivers the corresponding 10-Pn-5 complexes for P, As and Sb, while in the case of Bi an electron redistribution takes place, and the corresponding 12-Bi-5 compound is isolated. This case of Lewis acid-induced electromerism has been experimentally confirmed through X-ray diffraction analyses as well as IR, UV-Vis and Mössbauer spectroscopy. Effective Oxidation State analysis of the Fe-complexes assigns an oxidation state +3 to all Pn, while it predicts the Fe-atom to be reduced to Fe(0) in the Bi-derivative. The mixture of the Bi(6p) orbital with the π-system of the bis(amidophenolate) ligand and the efficient electronic donation from this orbital to the Fe-center explain this situation.
Introduction
Electromerism, the process of electron density relocation between redox active sites in a molecule,1 has evolved from being considered a curiosity to a practical tool for the design of magnetic materials, molecular switches, and even active catalysts (Scheme 1a).2 These applications rely on the very different physicochemical properties that electromers exhibit, and the nature of the different stimuli that are able to trigger their interconversion; namely, temperature,3 pressure,4 light5 or constitutional changes, such as the coordination to Lewis acids or bases.6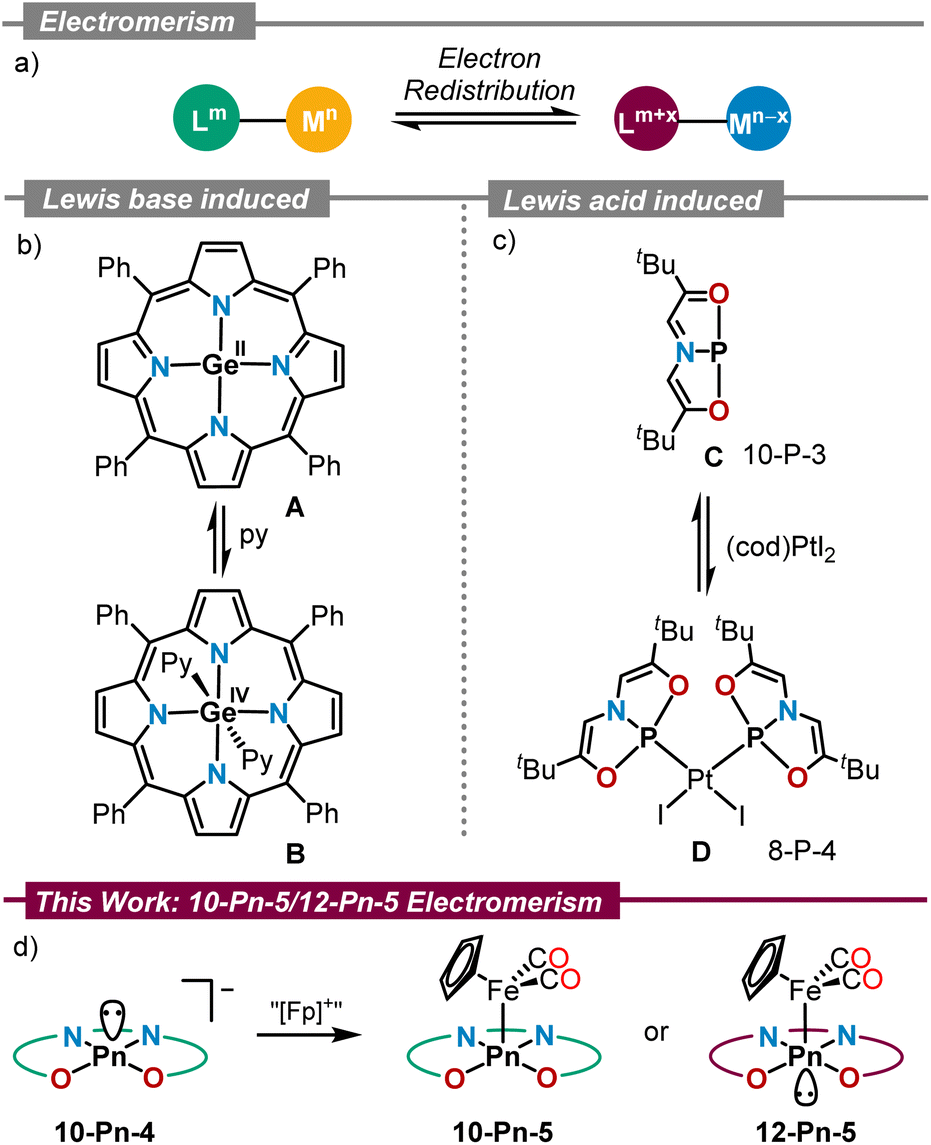 | ||
| Scheme 1 Electromerism in main group chemistry: (a) concept; (b and c) electromerism induced by coordination to Lewis bases and acids; (d) this work. | ||
Ever since the seminal report by Buchanan and Pierpont on electromerism in a cobalt bis(dioxolene)(bipyridyl) complex,7 this field of research has mostly been associated with transition metal chemistry.1 Valence tautomeric p-block element compounds are, in comparison, underrepresented.8 Seminal examples include Ge(II) tetraphenylporphyrin A reported by Vaid, in which Lewis base coordination induces the oxidation of Ge(II) to Ge(IV) with concomitant two-electron reduction of the porphyrin unit to its 20π-electron antiaromatic state (Scheme 1b);9 and the geometrically constrained P(I)-compound C. Interestingly, reaction of C with (cod)PtI2 leads to the formation of the corresponding Pt-complex D, in which the oxidation of the P-center to P(III) has taken place with simultaneous reduction of the supporting ONO scaffold (Scheme 1c).10 More recently, examples of main group electromerism induced by coordination to Lewis acids or Lewis bases have also been found in Si,11 Ge,12 Sn,13 As14 and Sb15 chemistry.
As continuation to our ongoing efforts to understand the redox-activity of pnictogen compounds supported by the rigid tetradentate bis(amidophenolate) ligand 1,14,16 we now describe the synthesis and characterization of the complete series of pnictide anions derived from that ligand, compounds 3a–d, and their corresponding cyclopentadienyliron dicarbonyl complexes 4a–c and 4d′. X-ray crystallographic analyses evidence that for all anions 3a–d the central pnictogen adopts a 10-Pn-4 configuration from which a Pn(III)-oxidation state is inferred. Upon coordination to [CpFe(CO)2]+ the situation remains identical for the P-, As-, and Sb-complexes 4a–c, respectively; however, in 4d′ a valence shell expansion from 10-Bi-5 to 12-Bi-5 occurs at the Bi-center. An Effective Oxidation State (EOS) analysis of that complex still assigns an oxidation state +3 to Bi, while the bis(amidophenolate) ligand gets oxidised (−2) and Fe reduced from Fe(II) to Fe(0). Thus, the geometrical rearrangement at Bi serves to better transfer the electron density from the bis(amidophenolate) scaffold to Fe.
Results and discussion
Synthesis and structure of 10-Pn-4 anions
Building on our recent studies on bis(amidophenolate)-supported phosphorus and arsenic species 3a,b (ref. 14 and 16) we started our investigation with the syntheses of their higher Sb- and Bi-analogues 3c and 3d, respectively. The necessary stibine precursor 2c was prepared by reaction of SbCl3 and 1 in the presence of triethylamine; yet, the corresponding bismuthine 2d was not accessible following that route. Fortunately, compound 2d could be cleanly prepared by treatment of 1 with Bi(NMe2)3 at −78 °C in Et2O (Scheme 2). Deprotonation of 2a–d was readily achieved by treatment with potassium hexamethyldisilazide (KHMDS) in Et2O employing [2.2.2]cryptand to sequester the potassium cation. Compounds 3a–d precipitate from the reaction mixture, and are obtained in analytically pure form by subsequent washing and drying. Quality crystals for X-ray diffraction analysis were obtained by slowly cooling saturated solutions of 3a–d in an appropriate solvent (see ESI†). | ||
| Scheme 2 Synthesis of 3a–d. (a) 2a: PCl3 (1.05 equiv.), DIPEA (3.15 equiv.) (54%); 2b: AsCl3 (1.00 equiv.), NEt3 (3.00 equiv.) (96%); 2c: SbCl3 (1.00 equiv.), NEt3 (3.00 equiv.) (94%); 2d: Bi(NMe2)3 (1.00 equiv.) (89%). (b) KHMDS (1.00 equiv.), cryptand (1.00 equiv.) 3a (81%), 3b (89%), 3c (92%), 3d (88%). | ||
Interestingly, 3a was found to crystallize as two polymorphs. The first one crystallizes in the hexagonal space group P61 and features a directed interaction between the phosphorus donor atom and the potassium cation. The P1–K1 distance was determined to be 4.3934(13) Å, and is slightly shorter than the sum of van-der-Waals radii (4.55 Å).17 The second polymorph, which crystallizes in the monoclinic space group P21/c, does not feature this interaction (shortest P1–K1 distance: 7.669(3) Å) and is, therefore, better suited for a geometric comparison. In all the higher homologues 3b–d, the ions were also found to be well separated. All 10-Pn-4 anions 3a–d exhibit a geometry between square pyramidal and disphenoidal around the central pnictogen due to the geometric constraint imposed by the tetradentate bis(amidophenolate) scaffold and the lower tendency to hybridize of the orbitals of heavier atoms. The difference in the trans basal angles Δθ around the pnictogen center has been used as quantitative parameter to illustrate the geometric change (Fig. 1a). As expected, the heavier homologues get closer to an ideal square pyramid (3c: Δθ = 4.3°, 3d: Δθ = 5.2° (average value)), while for the lighter congeners the disphenoidal arrangement becomes more favorable (3a: Δθ = 44.8°, 3b: Δθ = 35.5°) (Fig. 1b). Despite of this, the energetic landscape connecting these structures seems to be rather flat and strongly depending of packing effects. Thus, when the potassium cation in phosphoranide 3a is captured with 18-crown-6 ether, the P-atom adopts a perfect square pyramidal geometry and features a short P1–K1 contact (3.5899(12) Å) as reported by Dobrovetsky.18
 | ||
| Fig. 1 (a) Definition of trans basal angles θ around the pnictogen; (b–f) X-ray solid-state structures of 3a–d. Ellipsoids are shown at the 50% probability level. Hydrogens, cations (except for 3a), tert-butyl groups and solvent molecules omitted for clarity. | ||
Among the 10-Pn-4 anions, 3d is particularly intriguing. While its solid-state structure definitely shows a pyramidal Bi(III) atom, another minimum (3d′) lying only 4.0 kcal mol−1 higher in energy and characterized by a square planar geometry around the Bi-center has been found using DFT calculations at the PBE0-D3(BJ)/def2-TZVP(PCM)//TPSS-D3(BJ)/def2-TZVP level of theory (Fig. 2 and ESI†).19 We assign the weak absorption centred at 980 nm in the UV-Vis spectrum of 3d to traces of conformer 3d′ present in solution (see Fig. S15†). In 3d′, a formally vacant 6p-orbital at Bi is available for mixing with the π-system of the redox active ligand making the assignation of the oxidation state of Bi not straightforward; in fact, the electronic structure of 3d′ is reminiscent to that of a Bi(I)-complex.20 Similar triamide-Bi complexes have been described as Bi(I)/Bi(III) “redox-confused” for this reason.21 This ill-defined situation motivated us to further evaluate the electronic structure in 3d′ by analyzing the effective fragment orbitals (EFOs)22 and resulting effective oxidation states.23
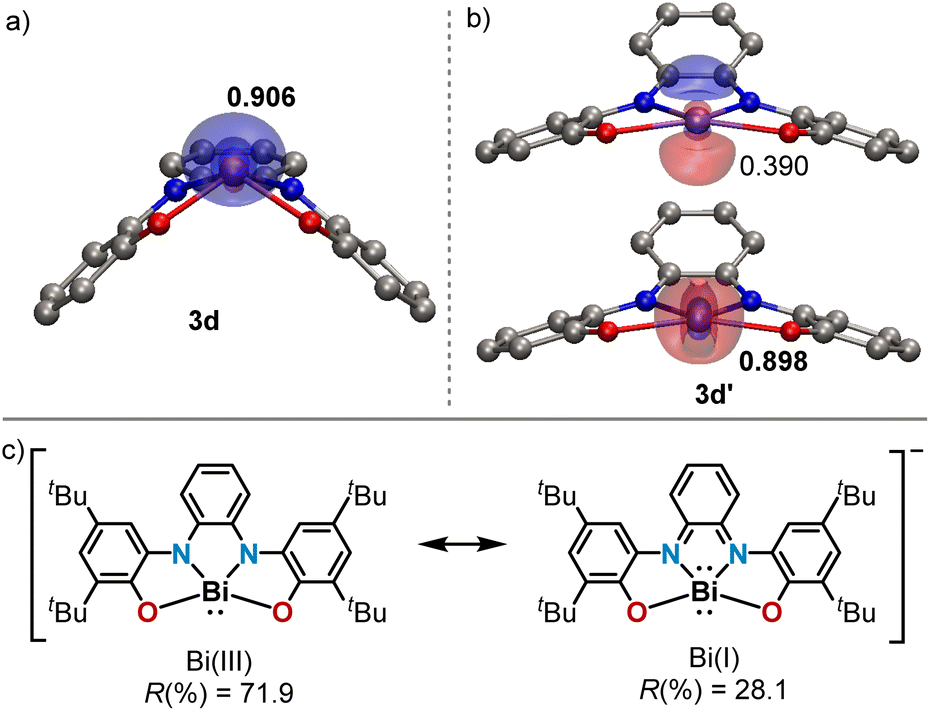 | ||
| Fig. 2 (a and b) Optimized structures of 3d and 3d′, respectively, at the PBE0-D3(BJ)/def2-TZVP(PCM)//TPSS-D3(BJ)/def2-TZVP level of theory including the most relevant effective fragment orbitals (EFOs). Occupation values provided for each EFO, marked in bold the formally occupied. (c) Possible resonance structures of 3d′ including their associated EOS reliability index. | ||
In the experimentally observed pyramidal structure 3d, the bismuth is undoubtedly in an oxidation state of +3 surrounded by a tetraanionic bis(amidophenolate) framework. The reliability index (R(%)) that quantifies how close the overall oxidation state assignment and the actual electronic structure of a molecule are, rises to 98.2% in this case. Yet, in 3d′ two electronic states are conceivable (Fig. 2b and c), which can be classified as Robin–Day Class III electromers.24 According to EOS, the Bi(III) assignment persists as the most appropriate description, although with a lower reliability index (R(%) = 71.9).25 The alternative description of 3d′ as a Bi(I) center within a dianionic tetradentate scaffold can be artificially produced within the EOS framework by switching the formal occupation of the last occupied and first unoccupied EFOs, yielding a non-aufbau electronic assignment with R(%) = 28.1.
Synthesis and characterization of Pn[CpFe(CO)2] complexes
After the initial hints about possible electron redistribution from the surrounding framework to the pnictogen, we envisioned that this process might be fostered using Lewis acid coordination as a trigger. The cyclopentadienyliron(II) dicarbonyl cation was chosen for this task since it serves as probe for both IR and Mössbauer spectroscopy (Scheme 3).26 | ||
| Scheme 3 Synthesis of 10-Pn-5 and 12-Pn-5 Fe-complexes. Reaction conditions: (a) CpFe(CO)2I (1.00 equiv.), THF, rt, 4a (64%).; (b) [CpFe(thf)(CO)2]BF4 (1.00 equiv.), DCM, −78 °C to rt, 4b (47%), 4c (72%), 4d′ (64%). | ||
Phosphoranide 3a was found to readily substitute the iodide in CpFe(CO)2I upon mixture of the reagents in THF; however, the heavier homologues 3b–d only coordinate the Fe-center when the more reactive [CpFe(thf)(CO)2]BF4 was employed. All compounds were obtained in moderate to good yields as air sensitive solids. Crystallization from MeCN solutions delivered monocrystals of sufficient quality for single-crystal X-ray diffraction analysis and the obtained solid-state structures confirm the coordination of 3a–d to Fe through the central pnictogen donor atom, forming 4a–c and 4d′. As expected, the Pn–Fe bond lengths progressively increase when moving down in the pnictogen group, and are in agreement with the reported values for Pn–Fe bonds featuring pnictogen-based X-type ligands Fig. 3a–d.26a,27
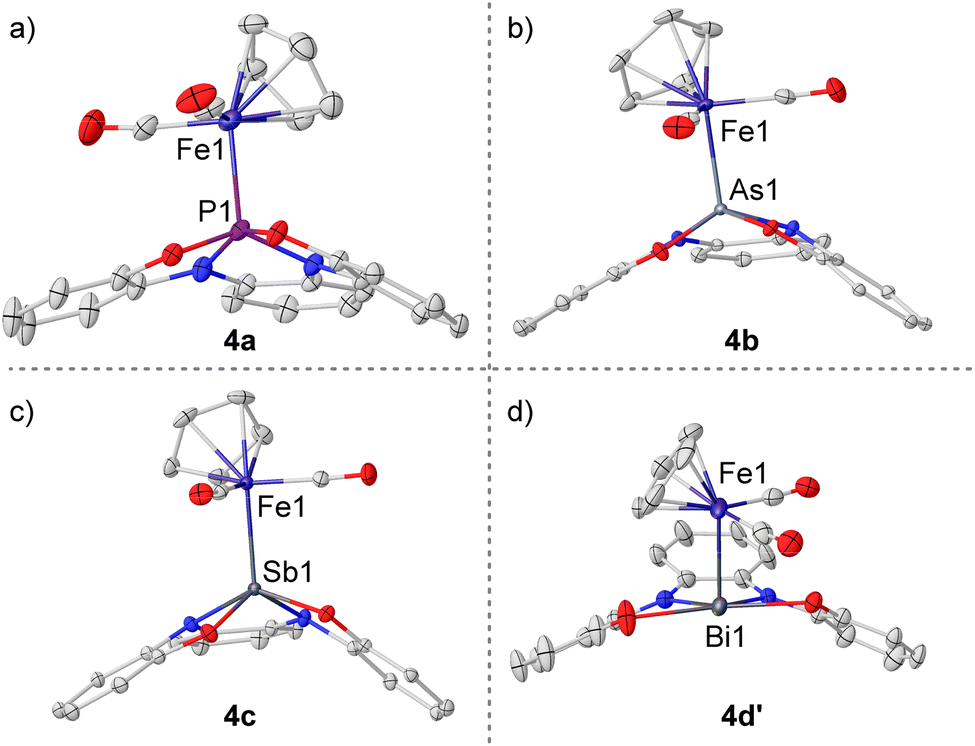 | ||
| Fig. 3 (a) X-ray solid-state structures of 4a–c and 4d′. Ellipsoids are shown at the 50% probability level. Hydrogens, cations, tert-butyl groups and solvent molecules omitted for clarity. Selected bond lengths (Å): 4a, P1–Fe1 2.2540(4); 4b, As1–Fe1 2.3071(3); 4c, Sb1–Fe1 2.4507(3); 4d′, Bi1–Fe1 2.6052(13). | ||
The geometries of complexes 4a–c are very similar, only an increased pyramidalization degree is found for the pnictogen atom when descending in the group (sum of basal angles: 340.4° (4a); 335.7° (4b, average value); 326.9° (4c)). The bismuth congener, however, opposes this trend having the Bi atom perfectly embedded in the plane defined by the oxygen and nitrogen donors (sum of basal angles around Bi = 360.1(11)°). This indicates that, unlike for the lighter pnictogens, in 4d′ the Bi(6p) orbital is used to form the bond with Fe while the Bi atom retains its 6s inert pair; hence, the 12-Bi-5 electromeric state is favored over the 10-Pn-5 observed for 4a–c.
Closer comparison of the bond lengths and geometries in 4a–c and 4d′ reveals that the extra electron pair surrounding the Bi center stems from the bis(amidophenolate) scaffold. The diagnostic N1–C15 and N2–C20 bond lengths in 4d′ are significantly shortened when compared with those in 4c (ca. −0.06 Å). Moreover, the alternating short and long C–C bond lengths throughout the central carbocycle in 4d′ further suggests the partial dearomatization of this ring and a considerable contribution of a bis(imine) structure for the ligand in 4d′. The bond lengths in the amidophenolate side-arms were also inspected using the metrical oxidation state (MOS) method reported by Brown.28 The values obtained for 4c (−2.02(6), −1.98(6)) are the typical for completely reduced amidophenolate moieties, while in 4d′ the MOS's are significantly increased (−0.99(14), −1.12(10)) revealing a substantial (imino)semiquinone character (Fig. 4).
 | ||
| Fig. 4 Comparative analysis of the bond lengths, angles and metrical oxidation states in 4c (a) and 4d′ (b). | ||
In order to scrutinize electronic changes at the iron center, all four iron complexes were further analyzed by 57Fe Mössbauer spectroscopy (Fig. 5). The obtained parameters for the isomeric shift δ are very similar throughout the series (4a: 0.08 mm s−1, 4b: 0.12 mm s−1, 4c: 0.14 mm s−1, 4d′: 0.14 mm s−1), indicating a similar s-electron density at the iron nuclei and suggesting that differences in Fe(s) character mixing into σ(Fe–Pn) orbitals and in Fe(d) orbital populations are either negligible, or that their effects are compensating. The δ values agree with those of previous examples of Fp-complexes with X-type ligands such as [CpFeII(CO)2X] (X = Me, Cl, Br, I or P(O)(OMe)2),26b,c but also with those found in formal CpFe0(CO)2 compounds.26d The quadrupole splitting ΔEQ is basically equal for the structurally similar complexes 4a–c, reflecting a similar local geometry and electric field gradient at Fe. The ΔEQ value decreases by around 0.1 mm s−1 for the bismuth congener 4d′, which might be related to the altered geometry of the bis(amidophenolato) bismuth fragment; however, no clear relation to bond distances or angles around the Fe-center was found.
 | ||
| Fig. 5 Zero-field 57Fe Mössbauer spectra of 4a–c and 4d′ at 80 K. Isomer shifts are given relative to α-Fe at 298 K. | ||
More precise information on the electronic situation at the Fe-center was obtained from infrared spectroscopic measurements. The carbonyl regions of the IR spectra of 4a–c and 4d′ are depicted in Fig. 6. According to these spectra, the carbonyl stretching bands are higher in energy when descending in the period from 4a to 4c. This suggests lower donor ability for the heavier pnictogens as consequence of a more internal electron pair. However, 4d′ breaks this trend and exhibits the least energetic stretching bands of all the series (![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) sym = 2004, asym = 1962 cm−1), which are indicative of the strongest donor ability of the bis(amidophenolate)Bi moiety and the weakest polarization of the iron–bismuth bond along the pnictogen series.27b
sym = 2004, asym = 1962 cm−1), which are indicative of the strongest donor ability of the bis(amidophenolate)Bi moiety and the weakest polarization of the iron–bismuth bond along the pnictogen series.27b
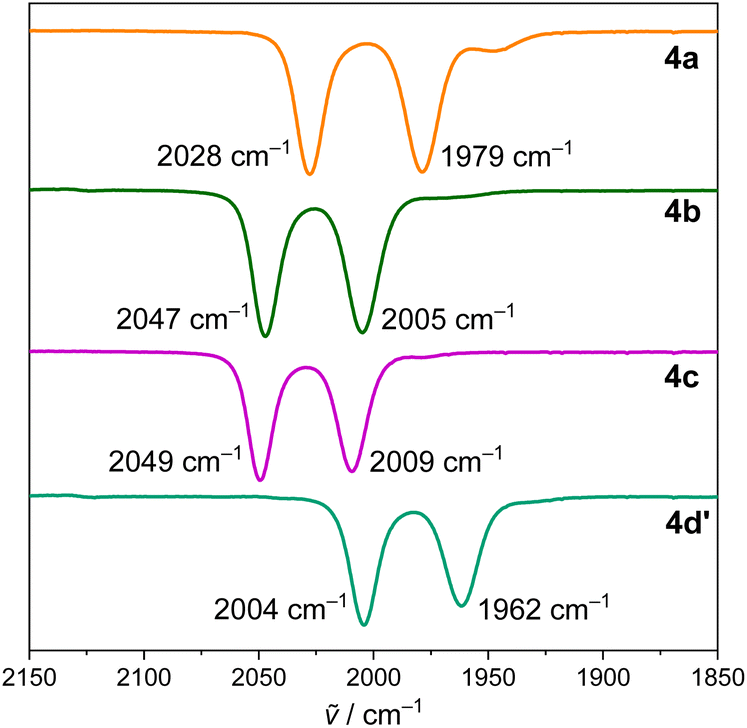 | ||
| Fig. 6 FT-IR spectra of 4a–c and 4d′ in DCM solution. | ||
Compounds 4a–c and 4d′ were further analyzed by UV/vis spectroscopy (Fig. 7). The spectra of 4a–c in DCM are quite similar at first glance and are dominated by intense transitions at around 300 nm. Interestingly, a broad band in the visible region was present with distinct maxima at 556 nm (4b) and 622 nm (4c), this band is also observed in the spectrum of 4a as a shoulder ranging from ca. 400 to 500 nm. Time-dependent DFT calculations aided in the assignment of this band as a HOMO → LUMO transition (see ESI†). The HOMOs of 4a–c are in all cases a π-orbital mainly localized at the bis(amidophenolate) unit. LUMOs are mostly distributed along the Pn–Fe bond axes and have σ*(Pn–Fe) character (see the ESI). Thus, these transitions can be described as charge-transfer bands and the redshift observed from P to Sb is explained by the LUMO energy decrease along the series, which aligns with the electron density reduction at the Fe-center (detected by IR spectroscopy).
 | ||
| Fig. 7 Electronic absorption spectra of 4a–c and 4d′. | ||
The spectrum of 4d′ is starkly different and its most diagnostic feature is the strong low-energy absorption band with a maximum at 1090 nm (22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 M−1 cm−1). This band corresponds to a π→π* transition that has been observed in numerous two electron-oxidized derivatives of the bis(amidophenolate) ligand 1.14,29 Thereby, the spectral data nicely corroborate the change of electronic structure as a result of the 10-Pn-5/12-Pn-5 electromerism.
900 M−1 cm−1). This band corresponds to a π→π* transition that has been observed in numerous two electron-oxidized derivatives of the bis(amidophenolate) ligand 1.14,29 Thereby, the spectral data nicely corroborate the change of electronic structure as a result of the 10-Pn-5/12-Pn-5 electromerism.
Iron–pnictogen bonding and oxidation state determination
In order to elucidate the nature of the pnictogen–iron bonding interactions, we evaluated the electronic structure and performed chemical bonding analysis on the experimentally synthesized systems 4a–c and 4d′, together with the computationally modelled electromers 4b′ and 4c′, in which the Pn-atom depicts square planar geometry and the ligand is formally oxidized. The computed relative Gibbs free energies agree with the experimentally observed electromeric forms. In particular, 4b is more stable than 4b′ by 24.0 kcal mol−1; for 4c and 4c′ the energy difference decreases to 8.2 kcal mol−1. All attempts to find the conceivable structures 4a′ and 4d as local minima in the potential energy surface failed. Notably, our calculations predict the lowest electronic state of 4c′ and 4d′ to be an open-shell singlet.The topology of the computed electron densities was investigated within the quantum theory of atoms in molecules (QTAIM) framework to shed light on the Pn–Fe bonding situation.30 As illustrative example, Fig. 8 shows the contour maps of the electron density Laplacian for As-electromers 4b and 4b′. The Fe–As bond critical point lies in a region of charge depletion for both electromers, a common feature for bonding between atoms having more than three atomic shells.31 For these relatively heavy atoms, Bader's classification of bonding interactions based on the sign of the Laplacian cannot simply be extrapolated,32 and other parameters need to be considered.31 Thus, structures 4b and 4b′ exhibit negative relative energy densities H/ρ at the bond critical point, fulfilling the Cremer–Kraka criteria for covalent bonding (Table 1).33
 | ||
| Fig. 8 Contour plots of the Laplacian of the electron density ∇2ρ for 4b and 4b′ calculated at the PBE0-D3(BJ)/def2-TZVP(PCM)//TPSS-D3(BJ)/def2-TZVP level of theory. Blue: charge accumulation (∇2ρ < 0). Red: charge depletion (∇2ρ > 0). | ||
| ρ/a.u. | ∇2ρ/a.u. | H/ρ/a.u. | DI | d/Å | |
|---|---|---|---|---|---|
| 4a | 0.094 | 0.066 | −0.40 | 0.72 | 2.286 |
| 4b | 0.083 | 0.073 | −0.39 | 0.70 | 2.355 |
| 4c | 0.069 | 0.056 | −0.35 | 0.68 | 2.506 |
| 4b′ | 0.073 | 0.070 | −0.36 | 0.64 | 2.407 |
| 4c′ | 0.062 | 0.034 | −0.32 | 0.66 | 2.585 |
| 4d′ | 0.057 | 0.043 | −0.28 | 0.69 | 2.665 |
The electron density at the bond critical point decreases quite uniformly throughout the 4a–c series as result of the longer bond distances and the more diffuse nature of the electron density for the heavier pnictogens. The calculated delocalization indices (DI) are in agreement with the formulation of covalent Pn–Fe single bonds for all compounds; yet, an intriguing trend is revealed. For the 10-Pn-5 compounds 4a–c, the DI gradually decreases for the heavier pnictogens, while the opposite trend is observed for the 12-Pn-5 isomers 4b′–d′. Since the DI is directly related with the extent of delocalization of electron pairs between two atomic basins31 and has been used to quantify covalent bond orders,32 it seems that the covalency of the Pn–Fe bond reaches two maxima, one for the lighter 10-P-5 system in 4a and a second one for the heavier 12-Bi-5 electromer in 4d′.
The shape and occupation values of the EFOs involved in the σ-type Pn–Fe bond are shown in Fig. 9. The EFO/EOS analysis was performed for all systems considering each ligand coordinated at the Fe and Pn atoms as fragments. This allows the disentangling of the Fe–Pn interaction from the rest of the ligands. For the pyramidal-shaped species 4a–c (Fig. 9 top), the Fe–Pn bond is formed by a d-type EFO sitting on Fe, with the s-type EFO from the pnictogen. The latter shows an increase of occupation value from 0.516 (4a) to 0.630 (4a) and in 0.636 (4c). Hence, the heavier the central element, the more polarized the Fe–Pn bond towards Pn. This is well in agreement with the lower tendency of the higher pnictogens to involve their s-electrons in bonding, and also supported by our IR data. The difference in occupation between these two EFOs also hints at the degree of covalency of the bond. The lower the difference is, the greater the covalent character of the bond. Therefore, the Fe–P (4a) bond is more covalent than both Fe–As (4b) and Fe–Sb (4c), agreeing with the topological analysis of the electron densities (see ESI†).
 | ||
| Fig. 9 Frontier EFOs and their associated occupation values for the reduced and oxidized species studied. For open-shell systems, beta EFO occupancies are reported in parentheses. Marked in bold are the EFOs formally occupied according to the EOS analysis. | ||
For the 12-Pn-5 species 4b′–d′ (Fig. 9 bottom), the Fe–Pn bond is formed by a d-type Fe EFO interacting with a properly oriented p-type EFO from the pnictogen, which is formally occupied as consequence of the bis(amidophenolate) oxidation. Interestingly, for this species the pnictogen s-type EFO does not participate in bonding to the Fe.
Finally, the oxidation states of 4a–c and 4d′ have been calculated. In 4a–c, the Cp, CO and ONNO scaffolds are characterized by oxidation states −1, 0 and −4, respectively. For the central Pn an oxidation state +3 is assigned, and subsequently, the Fe atom is in oxidation state +2. This electronic distribution is more evident for As and Sb (R(%) = 70.0 and 68.9, respectively); yet, for 4a the reliability index is significantly reduced (R(%) = 56.9) due to the large covalency of the P–Fe bond. The same analysis in Bi complex 4d′ assigns an oxidation state of −2 to the ONNO ligand, +3 to Bi, and 0 to Fe, instead of the more intuitive Bi(I)/Fe(II) oxidation states. It should be noted that this solution shows a lower reliability index (R(%) = 56.5), denoting how difficult the electron pair assignment in the Bi–Fe interaction is (see Fig. 9). This discrepancy suggests that in 4d′ the planarized Bi-atom simply serves as transmission belt to transfer electron density for the bis(amidophenolate) ligand to the Fe-center.
Conclusions
A series of geometrically constrained bis(amidophenolate)-supported pnictoranide anions has been synthesized and structurally characterized. An Effective Oxidation State analysis of these anions unambiguously assigns an oxidation state +3 to the P-, As- and Sb-complexes, while it suggests some +1 character to the Bi-derivative due to the efficient mixture of the Bi(6p) orbital with the π-system of the redox-active supporting ligand. Coordination of these species to the [CpFe(CO)2]+ moiety was achieved. For the P-, As- and Sb-derivatives the pnictogen atom remains pyramidal but the Bi-atom planarizes. The Effective Oxidation State analysis of that complex still assigns an oxidation state +3 to Bi while the Fe atom is reduced from +2 to 0. The mixture of the Bi(6p)-orbital with the π-system of the bis(amidophenolate) ligand in the square planar geometry, and the efficient electronic donation from that orbital to the Fe-center explain this non-intuitive situation.Data availability
All data associated with this article are available from the ESI.†Author contributions
S. B. H. K. and M. A. conceived the project and designed the experiments. S. B. H. K. and J. F. K. performed the experiments and analysed the results. S. B. H. K. and M. G. carried out the computational studies. C. G. performed the X-ray crystallographic analysis. I. B. and F. M. collected and analysed the Mössbauer spectroscopy data. All authors discussed the results, and S. B. H. K., M. G. and M. A. prepared the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the DFG through the projects INST 186/1237-1, INST 186/1318-1 and INST 186/1324-1 is gratefully acknowledged. We also thank the NMR and MS services, as well as the analytical laboratory at the Faculty of Chemistry (University of Göttingen) for technical assistance, and Prof. Ricardo A. Mata (University of Göttingen) for insightful discussions. I. B. is grateful for a Kekulé PhD fellowship from the Fonds der Chemischen Industrie (FCI). M. G. acknowledges support by the DFG – 217133147/SFB 1073.Notes and references
- (a) M. A. Dunstan and K. S. Pedersen, Chem. Commun., 2025, 61, 627–638 RSC; (b) J. R. Reimers and L. K. McKemmish, in Mixed Valence Systems:Fundamentals, Synthesis, Electron Transfer, and Applications, ed. Y.-W. Zhong, C. Y. Liu and J. R. Reimers, Wiley-VCH GmbH, Weinheim, 2023, ch. 2 Search PubMed; (c) T. Tezgerevska, K. G. Alley and C. Boskovic, Coord. Chem. Rev., 2014, 268, 23–40 CrossRef CAS; (d) E. Evangelio and D. Ruiz-Molina, Eur. J. Inorg. Chem., 2005, 15, 2957–2971 CrossRef; (e) P. Gütlich and A. Dei, Angew. Chem., Int. Ed., 1997, 36, 2734–2736 CrossRef; (f) L. W. Jones, Science, 1917, 46, 493–502 CrossRef CAS PubMed.
- (a) K. S. Kumar and M. Ruben, Coord. Chem. Rev., 2017, 346, 176–205 CrossRef; (b) M. D. Manrique-Juarez, S. Rat, L. Salmon, G. Molnar, C. M. Quintero, L. Nicu, H. J. Shepherd and A. Bousseksou, Coord. Chem. Rev., 2016, 308, 395–408 CrossRef CAS; (c) O. Sato, J. Tao and Y.-Z. Zhang, Angew. Chem., Int. Ed., 2007, 46, 2152–2187 CrossRef CAS PubMed; (d) J.-F. Létard, P. Guionneau and L. Goux-Capes, Top. Curr. Chem., 2004, 235, 221–249 CrossRef; (e) O. Kahn and C. J. Martinez, Science, 1998, 279, 44–48 CrossRef CAS.
- Selected examples: (a) J. T. Janetzki, D. S. Brown, F. Daumann, I. H. Ismail, R. W. Gable, M. A. Hay, R. J. Mulder, A. A. Starikova, B. Weber, M. J. Giansiracusa and C. Boskovic, Chem. Sci., 2025, 16, 5857–5871 RSC; (b) D. A. Lukina, A. A. Skatova, R. V. Rumyntcev, S. V. Demeshko, F. Meyer and I. L. Fedushkin, Dalton Trans., 2024, 53, 8850–8856 RSC; (c) C. Metzger, R. Dolai, S. Reh, H. Kelm, M. Schmitz, B. Oelkers, M. Sawall, K. Neymeyr and H.-J. Krüger, Chem.–Eur. J., 2023, 29, e202300091 CrossRef CAS; (d) K. K. C., T. Woods and L. Olshansky, Angew. Chem., Int. Ed., 2023, 62, e202311790 CrossRef PubMed; (e) C. Lecourt, Y. Izumi, L. Khrouz, F. Toche, R. Chiriac, N. Bélanger-Desmarais, C. Reber, O. Fabelo, K. Inoue, C. Desroches and D. Luneau, Dalton Trans., 2020, 49, 15646–156632 RSC; (f) G. K. Gransbury, B. N. Livesay, J. T. Janetzki, M. A. Hay, R. W. Gable, M. P. Shores, A. Starikova and C. Boskovice, J. Am. Chem. Soc., 2020, 142, 10692–10704 CrossRef CAS; (g) J. Chen, Y. Sekine, Y. Komatsumaru, S. Hayami and H. Miyasaka, Angew. Chem., Int. Ed., 2018, 57, 12043–12047 CrossRef CAS.
- Selected examples: (a) A. Summers, F. Z. M. Zahir, G. F. Turner, M. A. Hay, A. Riboldi-Tunnicliffe, R. l. Williamson, S. Bird, L. Goerigk, C. Boskovic and S. A. Moggach, Nat. Commun., 2024, 15, 8922 CrossRef CAS PubMed; (b) A. Caneschi, A. Die, F. Fabrizi de Biani, P. Gütlich, V. Ksenofontov, G. Levchenko, A. Hoefer and F. Renz, Chem.–Eur. J., 2001, 7, 3926–3930 CrossRef CAS; (c) C. Roux, D. M. Adams, J. P. Itié, A. Polian, D. N. Hendrickson and M. Verdaguer, Inorg. Chem., 1996, 35, 2846–2852 CrossRef CAS.
- Selected examples: (a) W.-H. Xu, Y.-B. Huang, W.-W. Zheng, S.-Q. Su, S. Kanegawa, S.-Q. Wu and O. Sato, Chem. Sci., 2024, 53, 2512–2516 CAS; (b) A. Witt, F. W. Heinemann and M. M. Khusniyarov, Chem. Sci., 2015, 6, 4599–4609 RSC; (c) O. Sato, A. Cui, R. Matsuda, J. Tao and S. Hayami, Acc. Chem. Res., 2007, 40, 361–369 CrossRef CAS PubMed.
- Selected examples: (a) N. Ansmann, M. Kerscher and L. Greb, Angew. Chem., Int. Ed., 2025, 64, e202417581 CrossRef CAS PubMed; (b) A. Witt, F. W. Heinemann, S. Sproules and M. M. Khusniyarov, Chem.–Eur. J., 2014, 20, 11149–11162 CrossRef CAS; (c) T. Storr, E. C. Wasinger, R. C. Pratt and T. D. P. Stack, Angew. Chem., Int. Ed., 2007, 46, 5198–5201 CrossRef CAS PubMed.
- R. M. Buchanan and C. G. Pierpont, J. Am. Chem. Soc., 1980, 102, 4951–4957 CrossRef CAS.
- (a) S. B. H. Karnbrock and M. Alcarazo, Chem.–Eur. J., 2024, 30, e202302879 CrossRef CAS PubMed; (b) L. Greb, Eur. J. Inorg. Chem., 2022, 2022, e202100871 CrossRef CAS.
- J. A. Cissell, T. P. Vaid and G. P. A. Yap, J. Am. Chem. Soc., 2007, 129, 7841–7847 CrossRef CAS.
- A. J. Arduengo, C. A. Stewart and F. Davidson, J. Am. Chem. Soc., 1986, 108, 322–323 CrossRef CAS.
- R. Yadav, X. Sun, R. Köppe, M. T. Gamer, F. Weigend and P. W. Roesky, Angew. Chem., Int. Ed., 2022, 61, e202211115 CrossRef CAS.
- (a) S. Yao, A. Kostenko, Y. Xiong, C. Lorent, A. Ruzicka and M. Driess, Angew. Chem., Int. Ed., 2021, 60, 14864–14868 CrossRef CAS; (b) S. Yao, A. Kostenko, Y. Xiong, A. Ruzicka and M. Driess, J. Am. Chem. Soc., 2020, 142, 12608–12612 CrossRef CAS PubMed.
- M. G. Chegerev, A. V. Piskunov, A. A. Starikova, S. P. Kubrin, G. K. Fukin, V. K. Cherkasov and G. A. Abakumov, Eur. J. Inorg. Chem., 2018, 2018, 1087–1092 CrossRef CAS.
- S. B. H. Karnbrock, J. F. Köster, C. Golz, R. A. Mata and M. Alcarazo, Angew. Chem., Int. Ed., 2025, e202501439 CAS.
- M. Schorpp, R. Yadav, D. Roth and L. Greb, Angew. Chem., Int. Ed., 2022, 61, e202207963 CrossRef CAS PubMed.
- (a) S. B. H. Karnbrock, C. Golz, R. A. Mata and M. Alcarazo, Angew. Chem., Int. Ed., 2022, 61, e202207450 Search PubMed; (b) S. B. H. Karnbrock, C. Golz and M. Alcarazo, Chem. Commun., 2024, 60, 6745–6748 RSC.
- (a) M. Mantina, A. C. Chamberlin, R. Valero, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. A, 2009, 113, 5806–5812 Search PubMed; (b) A. Bondi, J. Phys. Chem., 1964, 68, 441–451 Search PubMed.
- S. Volodarsky, I. Malahov, D. Bawari, M. Diab, N. Malik, B. Tumanskii and R. Dobrovetsky, Chem. Sci., 2022, 13, 5957–5963 RSC.
- (a) C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158 Search PubMed; (b) S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456 CrossRef CAS PubMed; (c) G. Scalmani and M. J. Frisch, J. Chem. Phys., 2010, 132, 114110 CrossRef; (d) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC; (e) J. M. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef.
- (a) M. Kořenková, V. Kremláček, M. Erben, R. Jirásko, F. De Proft, J. Turek, R. Jambor, A. Růžička, I. Císařova and L. Dostál, Dalton Trans., 2018, 47, 14503–14514 Search PubMed; (b) I. Vránová, T. Dušková, M. Erben, R. Jambor, A. Růžička and L. Dostál, J. Organomet. Chem., 2018, 863, 15–20 CrossRef; (c) M. Kořenková, V. Kremláček, M. Erben, R. Jambor, Z. Růžičková and L. Dostál, J. Organomet. Chem., 2017, 845, 49–54 CrossRef; (d) I. Vránová, V. Kremláček, M. Erben, J. Tzrek, R. Jambor, A. Růžička, M. Alonso and L. Dostál, Dalton Trans., 2017, 46, 3556–3568 RSC.
- (a) P. Coburger, A. G. Buzanich, F. Emmerling and J. Abbenseth, Chem. Sci., 2024, 15, 6036–6043 RSC; (b) M. B. Kindervater, K. M. Marczenko, U. Werner-Zwanziger and S. S. Chitnis, Angew. Chem., Int. Ed., 2019, 58, 7850–7855 CrossRef CAS PubMed; (c) K. M. Marczenko, J. A. Zurakowski, M. B. Kindervater, S. Jee, T. Hynes, N. Roberts, S. Park, U. Werner-Zwanziger, M. Lumsden, D. N. Langelaan and S. S. Chitnis, Chem.–Eur. J., 2019, 25, 16414–16424 CrossRef CAS PubMed.
- E. Ramos-Cordoba, P. Salvador and I. Mayer, J. Chem. Phys., 2013, 138, 214107 CrossRef PubMed.
- (a) E. Ramos-Cordoba, V. Postils and P. Salvador, J. Chem. Theory Comput., 2015, 11, 1501–1508 CrossRef CAS PubMed; (b) P. Salvador, E. Ramos-Cordoba, M. Montilla, L. Pujal and M. Gimferrer, J. Chem. Phys., 2024, 160, 172502 CrossRef CAS PubMed; (c) In the EOS framework, the difference in occupation between the last occupied (LO) and first unoccupied (FU) EFOs is used to evaluate the reliability of the assignment (R(%) index) as R(%) = 100(λLO − λFU + 1/2). By definition, when the difference between occupations of the frontier EFOs exceeds half an electron, the oxidation state (OS) assignment is considered indisputable (R(%) = 100). An R% value of 50 implies that the two possible effective electronic configurations are equally plausible..
- M. B. Robin and P. Day, in Advances in Inorganic Chemistry and Radiochemistry, ed. H. J. Emeléus and A. G. Sharpe, Academic Press, 1968, vol. 10, 247–422 Search PubMed.
- The same analysis for the T-shape planar triamide bismuthine reported by Chitnis (ref. 21b M. Gimferrer, S. Danés, D. M. Andrada and P. Salvador, Inorg. Chem., 2021, 60, 17657 CrossRef CAS PubMed ) suggest a Bi(III) center and a formal trianionic NNN ligand with R(%) = 65.0.
- (a) K. Kubo, H. Nakazawa, T. Mizuta and K. Miyoshi, Organometallics, 1998, 17, 3522–3531 CrossRef CAS; (b) H. Nakazawa, S. Ichimura, Y. Nishihara, K. Miyoshi, S. Nakashima and H. Sakai, Organometallics, 1998, 17, 5061–5067 CrossRef CAS; (c) M. K. Karunananda, F. X. Vazquez, E. E. Alp, W. Bi, S. Chattopadhyay, T. Shibata and N. P. Mankad, Dalton Trans., 2014, 43, 13661 Search PubMed; (d) D. Kim, D. W. N. Wilson, M. S. Fataftah, B. W. Mercado and P. L. Holland, Chem.–Eur. J., 2022, 28, e202104431 Search PubMed.
- (a) J. Ramler, F. Geist, C. Mihm, L. Lubczyk, S. Reith, C. Herok, F. Fantuzzi and C. Lichtenberg, Chem.–Eur. J., 2025, 31, e202403253 CrossRef CAS PubMed; (b) J. Ramler and C. Lichtenberg, Dalton Trans., 2021, 50, 7120–7138 Search PubMed; (c) J. Ramler, I. Krummenacher and C. Lichtenberg, Angew. Chem., Int. Ed., 2019, 58, 12924–12929 Search PubMed; (d) K. Wójcik, P. Ecorchard, D. Schaarschmidt, T. Rüffer, H. Lang and M. Mehring, Z. Anorg. Allg. Chem., 2012, 638, 1723–1730 CrossRef; (e) Y. Yamamoto, M. Okazaki, Y. Wakisaka and K. Akiba, Organometallics, 1995, 14, 3364–3369 CrossRef CAS; (f) S. K. Chopra and J. C. Martin, Heteroat. Chem., 1991, 2, 71–79 CrossRef CAS; (g) M. Wieber, D. Wirth and C. Burschka, Z. Naturforsch., B: J. Chem. Sci., 1985, 40, 258–262 Search PubMed.
- S. N. Brown, Inorg. Chem., 2012, 51, 1251–1260 Search PubMed.
- (a) J. Underhill, E. S. Yang, T. Schmidt-Räntsch, W. K. Myers, J. M. Goicoechea and J. Abbenseth, Chem.–Eur. J., 2023, 29, e202203266 CrossRef CAS; (b) A. J. Pistner, H. W. Moon, A. Silakov, H. P. Yennawar and A. T. Radosevich, Inorg. Chem., 2017, 56, 8661–8668 CrossRef CAS PubMed; (c) F. Lu, R. A. Zarkesh and A. F. Heyduk, Eur. J. Inorg. Chem., 2012, 2012, 467–470 CrossRef CAS; (d) K. J. Blackmore, N. Lal, J. W. Ziller and A. F. Heyduk, Eur. J. Inorg. Chem., 2009, 2009, 735–743 CrossRef; (e) P. Chaudhuri, M. Hess, J. Müller, K. Hildenbrand, E. Bill, T. Weyhermüller and K. Wieghardt, J. Am. Chem. Soc., 1999, 121, 9599–9610 CrossRef CAS.
- R. F. W. Bader, Atoms in molecules. A quantum theory, Clarendon Press, Oxford, 2003 Search PubMed.
- C. Gatti, Z. Kristallogr. – Cryst. Mater., 2005, 220, 399–457 Search PubMed.
- R. F. W. Bader and H. Essén, J. Chem. Phys., 1984, 80, 1943–1960 Search PubMed.
- D. Cremer and E. Kraka, Angew Chem. Int. Ed. Engl., 1984, 23, 627–628 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2448026–2448035. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc03374h |
| This journal is © The Royal Society of Chemistry 2025 |