
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDistinct oxygen reduction pathways for solar H2O2 production by regulating unsaturated bonds in covalent organic frameworks†
Jie-Yu Yue*a,
Zi-Xian Pana,
Yan Guoa,
Peng Yang *a and
Bo Tang*ab
*a and
Bo Tang*ab
aKey Laboratory of Molecular and Nano Probes, Ministry of Education, Collaborative Innovation Center of Functionalized Probes for Chemical Imaging in Universities of Shandong, College of Chemistry, Chemical Engineering and Materials Science, Shandong Normal University, Jinan, 250014, P. R. China. E-mail: yuejieyu@sdnu.edu.cn; yangpeng@sdnu.edu.cn
bLaoshan Laboratory, Qingdao, 266200, P. R. China. E-mail: tangb@sdnu.edu.cn
First published on 19th June 2025
Abstract
Photocatalytic H2O2 generation via the two-electron oxygen reduction reaction (2e− ORR) is a highly sustainable approach, capable of proceeding via either a one-step or two-step 2e− ORR route. Nonetheless, precise regulation of the 2e− ORR pathways still remains a formidable challenge. Herein, for the first time, we modulate the 2e− ORR pathway through unsaturated bond control in covalent organic frameworks (COFs). We synthesize a pair of isostructural COFs distinguished only by their unsaturated bonds. The alkyne-containing TY-COF favors the two-step 2e− ORR route, whereas the alkene-containing TE-COF follows the one-step 2e− ORR route. Without any sacrificial agents in O2, the TY-COF and TE-COF display impressive H2O2 production rates of 6455 and 4804 μmol g−1 h−1, respectively. Further theoretical results reveal that the regulation of unsaturated bonds alters the electron–hole distribution along the COF skeletons, prompting the reorganization of the catalytic centers for ORR (the benzene ring in TY-COFs and the triazine in TE-COFs), which leads to divergent ORR pathways. Additionally, free-standing TY-COF and TE-COF membranes, fabricated via the interfacial polymerization method, are also able to drive H2O2 photosynthesis. The present work offers a new strategy and valuable inspiration for modulating 2e− ORR pathways via strategic architectural engineering of COFs.
Introduction
H2O2 is a multifaceted and eco-friendly oxidant, known for its broad range of applications in various industries, including chemical synthesis, environmental remediation, and medical sterilization.1–3 The application of photocatalytic techniques, powered by solar energy, enables the generation of H2O2 via the oxygen reduction reaction (ORR) and water oxidation reaction (WOR), establishing a sustainable and cost-efficient strategy.4–6 This method not only reduces energy consumption but also allows for on-site H2O2 generation, eliminating transportation and storage issues associated with concentrated H2O2 solutions.In contrast to the kinetically sluggish WOR route, photosynthetic H2O2 production via the two-electron ORR (2e− ORR) path presents a more promising and readily implementable alternative.7–9 Depending on the electron transfer processes and intermediates involved, the 2e− ORR mechanism for H2O2 production can be classified into two divergent routes: the direct one-step 2e− ORR route (O2 + 2e− + 2H+ → H2O2) and the sequential two-step 2e− ORR route (O2 + e− → O2˙−; O2˙− + e + 2H+ → H2O2).4,5 The key distinction between the one-step and two-step 2e− ORR lies in the formation of the O2˙− intermediate. In numerous practical scenarios, particularly in aqueous environments containing geochemical metals and dissolved organic matter, the O2˙− intermediate is prone to disproportionation,10 which consequently results in diminished H2O2 production efficiency. As such, optimizing the 2e− ORR pathway is essential for enabling in situ H2O2 generation with high efficiency across diverse practical contexts, yet it remains a formidable challenge.11
In the realm of materials explored for photocatalytic H2O2 production, covalent organic frameworks (COFs) have attracted considerable interest since 2020.12–17 COFs are characterized by their crystalline, porous architectures, which are formed from organic building units linked together by covalent bonds. Their highly ordered structures, tunable pore sizes, and versatile functionalities render them ideal candidates for photocatalytic applications.18–20 Furthermore, the precise control over their molecular design facilitates the rational engineering of COFs with enhanced light absorption, efficient charge separation, and optimized catalytic sites. These distinctive features establish COFs as promising platforms for the efficient and selective photogeneration of H2O2.21–27 To date, the majority of reported COFs generate H2O2 via the two-step 2e− ORR route,28–34 while only a few studies have successfully regulated the 2e− ORR route.35–37 For instance, FS-COF with sulfone units on the backbone,35 PyIm-COF featuring pyridyl-imine structures,36 and s-heptazine-based HEP-TAPT-COF can drive H2O2 generation through the one-step 2e− ORR pathway.37 Notwithstanding these advances, the strategic modulation of 2e− ORR pathways within COFs architectures remains a nascent domain, necessitating the exploration of innovative design paradigms and synthetic methodologies.
Unsaturated bonds constitute covalent connections that link two atoms via multiple bonds, including triple and double bonds with hybridized orbitals, such as sp-hybridized alkyne bonds (–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–), sp2-hybridized alkene bonds (–C
C–), sp2-hybridized alkene bonds (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–), and azo bonds (–NN–), which profoundly modulate the electronic states of organic materials. While COFs incorporating –CC– and –NN– have exhibited distinguished electrocatalytic and photocatalytic ORR activities,38,39 the comparative impact of –CC– and –CC– on the photocatalytic 2e− ORR pathways for H2O2 production remains unexplored.
C–), and azo bonds (–NN–), which profoundly modulate the electronic states of organic materials. While COFs incorporating –CC– and –NN– have exhibited distinguished electrocatalytic and photocatalytic ORR activities,38,39 the comparative impact of –CC– and –CC– on the photocatalytic 2e− ORR pathways for H2O2 production remains unexplored.
Herein, for the first time, we modulate the 2e− ORR pathways in COFs by manipulating unsaturated bonds (–CC– and –CC–) on the skeleton. We synthesize two structurally similar COFs (named TY-COF and TE-COF), which differ only in the type of unsaturated bonds (–CC– or –CC–, Fig. 1a). Experimental results exhibit that the TY-COF, with –CC–, prefers the two-step 2e− ORR route, while the TE-COF, with –CC–, undergoes the one-step 2e− ORR pathway. Without sacrificial agents in O2, the TY-COF and TE-COF produce H2O2 at rates of 6455 and 4804 μmol g−1 h−1, respectively, comparable to high-performance COF-based photocatalysts. Furthermore, TY-COF and TE-COF membranes are built via interfacial polymerization, which can also produce H2O2 smoothly. Theoretical calculations reveal that the regulation of unsaturated bonds alters the electron–hole distribution along the COF skeletons, changing the ORR catalytic centers (the benzene ring in TY-COFs and the triazine in TE-COFs), which ultimately leads to divergent 2e− ORR pathways. The present work underscores the critical role of regulating the charge carrier distribution in catalysts to modulate the 2e− ORR pathways.
 | ||
| Fig. 1 (a) Chemical structures, (b) calculated electron–hole distributions, electron transfer amount, and electron transfer distances of TY-COFs and TE-COFs in the first excited state by TD-DFT calculations. | ||
Results and discussion
To elucidate the influence of unsaturated bonds on the photocatalytic performance and mechanism for H2O2 generation, we designed a pair of COFs with –CC– and –CC–, respectively. As illustrated in Fig. 1a, the TY-COF was fabricated via the condensation of 4,4′,4′′-(benzene-1,3,5-triyltris(ethyne-2,1-diyl))trianiline (BYA) with 4,4′,4′′-(1,3,5-triazine-2,4,6-triyl)tribenzaldehyde (TTB). The alkyne-containing BYA linker introduced –CC– into the framework, creating a rigid and highly conjugated structure. In parallel, the TE-COF was constructed using 4,4′,4′′–((1E,1′E,1′′E)-benzene-1,3,5-triyltris(ethene-2,1-diyl))trianiline (BEA) in conjunction with the same TTB monomer, thereby integrating –CC– as the pivotal unsaturated bonds.
First of all, we investigated the electronic structure disparities between the TY-COF and TE-COF, emanating from the intrinsic nature of their unsaturated bonds through time-dependent density functional theory (TD-DFT) calculations using CP2K (2023.2). As shown in Fig. 1b, the TY-COF unit cell exhibited a pronounced dipole moment of 5.01 D, whereas the TE-COF unit cell showed a marginally diminished dipole moment of 4.88 D in their first excited state. Typically, enhanced dipole moments correlate with augmented charge separation driving forces.40 To probe their photoinduced charge separation and transfer behaviors, we assessed the differences in charge density that existed between the ground state and the excited state for model compounds of both COFs. The centroid distributions of positive (blue regions) and negative charges (yellow regions) were distinctly localized in their respective segments. The electron transfer amounts (ΔE) of TY-COF and TE-COF were 0.56e and 0.57e, respectively, while the corresponding electron transfer distances (DCT) of TY-COF and TE-COF were 6.75 Å and 7.76 Å. Generally, an elevated absolute value of ΔE/DCT signifies more efficacious charge transfer.41 Compared to the alkene-containing TE-COF, the alkyne-containing TY-COF demonstrated a more substantial dipole moment and pronounced charge transfer characteristics. These predicted subtle yet consequential distinctions suggest that the –CC– and –CC– linkages could generate distinctive electronic environments within the COF architectures, potentially modulating charge transfer dynamics during photocatalytic processes.
To confirm the crystallinity of TY-COF and TE-COF, powder X-ray diffraction (PXRD) analysis was conducted. As shown in Fig. 2a, the TY-COF exhibited a pronounced diffraction peak at 3.50°, accompanied by two weaker peaks at 6.10° and 25.6°, corresponding to the (100), (110), and (001) reflection planes, respectively. The experimental PXRD pattern aligned well with the simulated pattern based on the AA stacking mode. The Pawley-refined cell parameters of TY-COF were a = b = 29.92 Å, c = 3.48 Å, α = β = 90°, and γ = 120° (Rwp = 2.78% and Rp = 2.19%). The experimental PXRD profile of TE-COF (Fig. 2b) illustrated a prominent peak at 3.38° and weaker peaks at 5.90°, 6.86°, 9.10°, and around 25.5°, related to the (100), (110), (200), (210), and (001) planes, respectively. The positions of the experimental PXRD peaks for the TE-COF closely matched the simulated results obtained using the AA stacking mode. The lattice parameters of TE-COF were found to be a = b = 29.99 Å, c = 3.47 Å, α = β = 90°, and γ = 120° (Rwp = 2.64% and Rp = 2.10%). The slight shift in the primary diffraction peak position between the TY-COF and TE-COF (3.50° vs. 3.38°) reflected the structural variations introduced by the –CC– and –CC– modules.
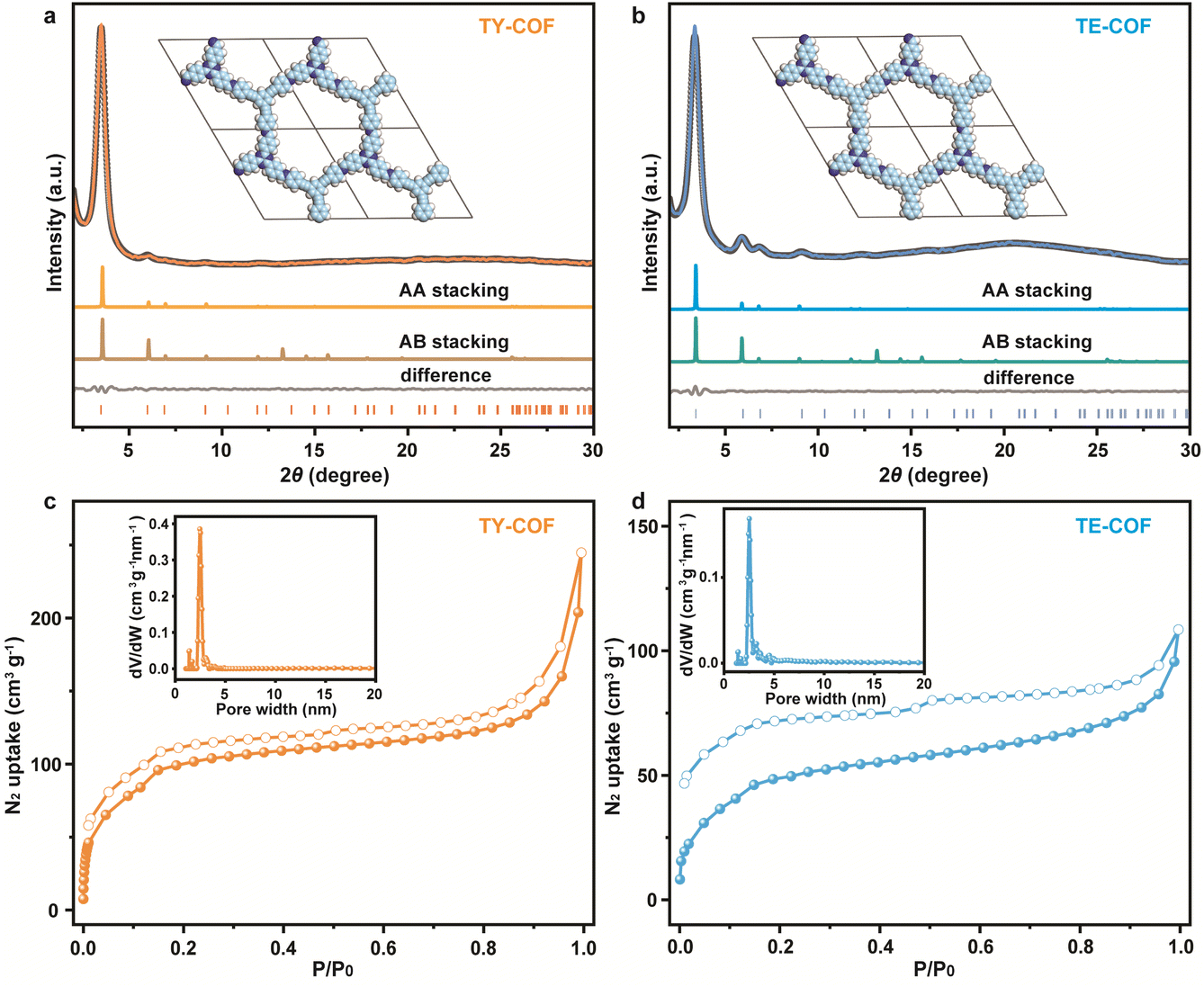 | ||
| Fig. 2 Experimental, Pawley refined, and simulated PXRD patterns of (a) TY-COF and (b) TE-COF. N2 sorption isotherms (insets: pore size distribution curves) of (c) TY-COF and (d) TE-COF. | ||
The permanent porosities of TY-COF and TE-COF were assessed via N2 adsorption–desorption isotherms at 77 K. As depicted in Fig. 2c, the TY-COF featured a type IV isotherm, and the Brunauer–Emmett–Teller (BET) surface area was 350.3 m2 g−1. The pore size distribution analysis (inset of Fig. 2c) revealed a predominant pore width of approximately 2.41 nm for the TY-COF. In contrast, the TE-COF displayed a lower N2 uptake capacity (Fig. 2d), resulting in a BET surface area of 174.8 m2 g−1. This reduced surface area, compared to the TY-COF, can be attributed to the distinct geometric configurations of the alkene linkages, which may introduce slight distortions within the frameworks. Nevertheless, the TE-COF maintained a type IV isotherm with a pore size distribution centered around 2.53 nm (inset of Fig. 2d).
Fourier transform infrared (FT-IR) spectroscopy data were recorded to characterize the chemical structures of TY-COF and TE-COF. Fig. S1† presents the comparative FT-IR spectra of the monomers (BEA, BYA, and TTB) and the resultant COFs (TY-COF and TE-COF), offering crucial insights into their molecular structures and bonding characteristics. The FT-IR spectra of both the TY-COF and TE-COF displayed distinctive –CN stretching vibration bands, serving as definitive evidence for the formation of imine linkages during the condensation reactions. Specifically, the TY-COF exhibited a characteristic –CN stretching band at 1622 cm−1, while the TE-COF showed a similar but slightly shifted band at 1616 cm−1. Solid-state 13C nuclear magnetic resonance (NMR) spectroscopy was carried out on both TY-COF and TE-COF to further validate the successful formation of the intended COFs (Fig. S2†). Signals at 154.9 and 156.3 ppm were attributed to the carbon atoms in the CN bonds of TY-COF and TE-COF, respectively. Signals at 169.7 and 169.5 ppm were identified as the carbon atoms within the triazine unit of TY-COF and TE-COF, respectively. Additionally, the peak at 89.8 ppm was characteristic of carbon atoms in the –CC– linkage, providing direct evidence for the incorporation of the alkyne-containing building blocks into the TY-COF structure. The establishment of TY-COF and TE-COF was further substantiated by X-ray photoelectron spectroscopy (XPS) measurements illustrated in Fig. S3,† along with elemental analysis results provided in Table S1.†
Thermogravimetric analysis (TGA) was utilized to evaluate the thermal robustness of the TY-COF and TE-COF (Fig. S4†). The as-synthesized COFs exhibited a weight loss of less than 5% at 400 °C, which signified their commendable thermal stability. The solvent stability of the TY-COF and TE-COF was assessed through PXRD analysis after treating them in various solvents and under different pH conditions for 8 h. Both TY-COF and TE-COF retained their crystallinity under these conditions, underscoring their potential applicability in diverse settings (Fig. S5†).
To obtain a more comprehensive understanding of the morphological features and nanostructure of TE-COF and TY-COF, both scanning electron microscopy (SEM) and transmission electron microscopy (TEM) analyses were conducted. The TY-COF exhibited an adherent particle stacking morphology (Fig. S6a†). The TE-COF displayed granular structures and coral-like structures in Fig. S6b.† The morphologies of TY-COF and TE-COF observed by TEM (Fig. S6c and d†) were consistent with their SEM images. Besides, we carried out high-resolution transmission electron microscopy (HRTEM) to check the lattice structures of TY-COF and TE-COF. Notably, while lattice fringes were not discernible in the TY-COF, the TE-COF revealed pronounced lattice fringes attributable to the (001) plane (Fig. S6e†).
Afterwards, the optical and electronic properties of TY-COF and TE-COF were investigated. As illustrated in the UV-visible diffuse reflectance spectra (UV-vis DRS) in Fig. 3a, the TY-COF exhibited broad absorption ranges with an edge extending to approximately 480 nm, whereas the TE-COF displayed a slightly red-shifted absorption edge around 540 nm. The corresponding Tauc plots (inset, Fig. 3a) yielded optical bandgaps of 2.54 eV for the TY-COF and 2.29 eV for the TE-COF. The Mott–Schottky plots of TY-COF and TE-COF (Fig. S7†) revealed positive slopes, indicating their n-type semiconductor characteristics, with flat band potentials of −0.99 and −0.89 V (vs. Ag/AgCl). Based on these optical bandgaps and flat band potentials, we constructed the energy band diagrams for TY-COF and TE-COF (Fig. 3b). The TY-COF exhibited a conduction band (CB) potential of −0.79 V (vs. NHE) and a valence band (VB) potential of 1.75 V (vs. NHE). In contrast, the TE-COF exhibited CB potentials of −0.69 V (vs. NHE) and VB potentials of 1.60 V (vs. NHE). Obviously, both TY-COF and TE-COF are thermodynamically favorable for photocatalytic H2O2 production via ORR and WOR pathways.
 | ||
| Fig. 3 (a) UV-vis DRS with Tauc plots, (b) energy band structures, (c) EPR spectra (under dark and light irradiation), (d) PL spectra, (e) time-resolved PL decay curves (excited at 375 nm), and (f) transient photocurrent density of the TY-COF and TE-COF. | ||
To directly observe the unpaired electrons in TY-COF and TE-COF, we conducted electron paramagnetic resonance (EPR) spectroscopy under both dark and illuminated conditions (Fig. 3c). Both COFs showed g values close to 2.003, with significantly enhanced signal intensities under illumination compared to dark conditions. The TY-COF displayed a more obvious EPR signal than the TE-COF, indicating a superior capacity for generating photoinduced electrons at its CB, under light irradiation, compared to TE-COF, beneficial to the photocatalytic reaction.
Steady-state photoluminescence (PL) spectra (Fig. 3d) revealed distinct emission profiles for the as-synthesized COFs. The TY-COF showed an emission maximum at approximately 515 nm, while the TE-COF exhibited a red-shifted emission peak centered at 550 nm. The variation in emission wavelengths reflected their distinctive excited-state energy landscapes, influenced by the nature of their unsaturated bonds. Notably, the TY-COF displayed lower PL intensity compared to the TE-COF, indicating reduced radiative recombination of photogenerated charge carriers. This observation implied that the –CC– linkage in TY-COF facilitated more efficient charge separation and transfer, potentially enhancing its photocatalytic performance.
Time-resolved photoluminescence decay measurements (Fig. 3e) provided further insights into charge carrier dynamics. Both TY-COF and TE-COF displayed similar bi-exponential decay profiles, with average fluorescence lifetimes of 2.80 and 2.71 ns, respectively. The slightly longer lifetime observed in the TY-COF may contribute to enhanced charge separation efficiency, allowing photogenerated electrons additional time to engage in catalytic reactions prior to recombination.
Moreover, under intermittent illumination, the TY-COF generated a substantially higher photocurrent density than the TE-COF (Fig. 3f). This nearly two-fold enhancement in photocurrent for the TY-COF indicated superior charge separation and transport properties, consistent with the findings from EPR and PL analyses. Besides, electrochemical impedance spectroscopy (EIS) was applied to elucidate the charge transfer properties and electrical conductivity of TY-COF and TE-COF (Fig. S8†). The smaller radius in the Nyquist plots of the TY-COF indicated a substantially lower charge transfer resistance compared to the TE-COF. All the above characterization studies proved that the TY-COF featured more efficient charge transfer capabilities than the TE-COF, in accordance with the theoretically predicted results shown in Fig. 1.
Next, we systematically investigated the photocatalytic H2O2 generation performance of the TY-COF and TE-COF to elucidate how their distinct unsaturated bonds influence the reaction pathways and efficiency. All H2O2 concentrations were quantified using the titanium sulphate colorimetric method (Fig. S9†). The time-dependent H2O2 production of TY-COF and TE-COF under visible light irradiation (λ > 420 nm) in air and pure water is depicted in Fig. 4a, and TY-COF and TE-COF displayed H2O2 photogeneration rates of 5772 and 4429 μmol g−1 h−1. The superior performance of the TY-COF aligned with its enhanced charge separation and transfer capabilities.
 | ||
| Fig. 4 (a) Time-dependent H2O2 photogeneration amount over TY-COF and TE-COF in air and pure water under visible light irradiation (λ > 420 nm, Xe lamp). (b) The H2O2 photogeneration rate of TY-COF and TE-COF in O2, air, and Ar atmospheres, respectively. (c) H2O2 yields of TY-COF and TE-COF in the presence of TEMPOL (1 mM), BQ (0.01 mM), and KBrO3 (1 mM) in air and pure water under illumination for 1 h, respectively. (d) Detection of O2˙− using NBT in TY-COF and TE-COF photocatalytic systems. (e) DMPO spin-trapping signals of O2˙− of TY-COF and TE-COF. (f) H2O2 selectivity and electron transfer numbers of TY-COF and TE-COF tested by RRDE (KOH, 0.1 M). In situ DRIFT spectra of (g) TY-COF and (h) TE-COF during the H2O2 photoproduction process. (i) Quantity of H2O2 generated over TY-COF and TE-COF in Ar when KBrO3 (1 mM) and CH3OH (10%) were present. | ||
Furthermore, the kinetic parameters governing H2O2 production, specifically the formation (kf) and decomposition (kd) rate constants, were systematically determined for both TY-COF and TE-COF (Fig. S10†).31 The kf values for TY-COF and TE-COF were ascertained to be 0.84 and 0.77 mM h−1, respectively, while their corresponding kd values were 0.52 and 0.49 h−1, respectively. Throughout the 12 h photocatalytic process, the H2O2 photosynthetic activity of the TY-COF consistently outperformed that of the TE-COF (Fig. S11†). Furthermore, the H2O2 production rates of TY-COF and TE-COF did not decrease significantly over five consecutive photocatalytic cycles (Fig. S12†). After 12 h of photocatalytic H2O2 experiments and 5 photocatalytic cycles, both TY-COF and TE-COF maintained their crystallinity and chemical structures, as confirmed by the PXRD curves (Fig. S13†), FT-IR spectra (Fig. S14†), and XPS spectra (Fig. S15†).
To elucidate the reaction pathways involved in H2O2 generation, we conducted experiments under varying atmospheric conditions (Fig. 4b). Both TY-COF and TE-COF exhibited the highest H2O2 production rates in a pure O2 atmosphere (6455 μmol g−1 h−1 for the TY-COF, and 4804 μmol g−1 h−1 for the TE-COF), with solar-to-chemical conversion (SCC) efficiencies of 0.67% and 0.45%, respectively, among the highest reported for COF-based photocatalysts for H2O2 production (Tab. S2). The apparent quantum efficiencies (AQE) of TY-COF and TE-COF were measured at different wavelengths (420, 450, and 520 nm), respectively. As illustrated in Fig. S16,† both TY-COF and TE-COF exhibited the highest AQE at 450 nm (9.5% for TY-COF, 6.0% for TE-COF). Since TY-COF and TE-COF can drive H2O2 synthesis via the 2e− ORR and 2e− WOR pathways, the turnover number (TON) was calculated by dividing the amount of H2O2 generated by the amount of photocatalyst.23,42 In O2 and pure water, the TON values of TY-COF and TE-COF for H2O2 photogeneration were determined to be 4.92 and 3.69, respectively.
Besides, with the addition of sacrificial agents, such as EtOH and BnOH (10%), the H2O2 photogeneration rates of TY-COF (8121 and 39![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 060 μmol g−1 h−1) and TE-COF (6023 and 34351 μmol g−1 h−1) were all enhanced (Fig. S17†). Exceptionally, TY-COF and TE-COF still showed measurable H2O2 production even in an Ar atmosphere, albeit at significantly reduced rates (370 μmol g−1 h−1 for TY-COF, and 44 μmol g−1 h−1 for TE-COF). This observation showed that TY-COF and TE-COF can photosynthesize H2O2 via the ORR and WOR dual pathways, with the ORR path playing a predominant role.43 The TY-COF and TE-COF showed weak H2O2 decomposition efficiency in Ar (Fig. S18†).
060 μmol g−1 h−1) and TE-COF (6023 and 34351 μmol g−1 h−1) were all enhanced (Fig. S17†). Exceptionally, TY-COF and TE-COF still showed measurable H2O2 production even in an Ar atmosphere, albeit at significantly reduced rates (370 μmol g−1 h−1 for TY-COF, and 44 μmol g−1 h−1 for TE-COF). This observation showed that TY-COF and TE-COF can photosynthesize H2O2 via the ORR and WOR dual pathways, with the ORR path playing a predominant role.43 The TY-COF and TE-COF showed weak H2O2 decomposition efficiency in Ar (Fig. S18†).
To further explore the H2O2 photogeneration mechanisms via the 2e− ORR channel in TY-COF and TE-COF, we conducted scavenger experiments utilizing 4-hydroxy-2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPOL, for O2˙−), benzoquinone (BQ for O2˙−), and KBrO3 (for electrons) in Fig. 4c. The introduction of KBrO3 resulted in a significant decrease in H2O2 production for both TY-COF and TE-COF, suggesting that electrons played a crucial role in their H2O2 synthesis processes. For the TY-COF, the addition of TEMPOL and BQ dramatically suppressed H2O2 production, whereas the TE-COF exhibited minimal changes in H2O2 amount upon the addition of these scavengers. These distinct results confirmed that O2˙− is a critical intermediate in the 2e− ORR path of TY-COF, while it has a negligible impact on the H2O2 generation rate of TE-COF. Furthermore, the formation of O2˙− during the photocatalytic processes of TY-COF and TE-COF was investigated using nitro blue tetrazolium (NBT) as a probe (Fig. 4d). The TY-COF exhibited a significant and continuous decrease in the absorbance intensity ratio (At/A0) at a wavelength of 259 nm over time, reaching approximately 64% after 90 min, indicating substantial O2˙− generation, whereas that of TE-COF showed almost no changes, suggesting minimal O2˙− production. These findings were further corroborated by EPR spectra using 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as a spin-trapping agent for O2˙− (Fig. 4e). Under light irradiation, the TY-COF produced strong characteristic signals of the DMPO-O2˙− adduct, while the TE-COF showed only weak signals, confirming the significantly higher O2˙− generation capacity of the TY-COF. Together with the O2˙− scavenger experiments, the NBT and EPR characterization of O2˙− in the TY-COF and TE-COF photocatalytic systems, it can be deduced that TY-COF and TE-COF underwent divergent 2e− ORR pathways. The TY-COF followed the two-step 2e− ORR pathway, while the TE-COF adopted the one-step 2e− ORR pathway for H2O2 photoproduction. By modulating the unsaturated bonds (–CC– and –CC–) in the TY-COF and TE-COF, the 2e− ORR pathways can be effectively regulated.
Additionally, the H2O2 selectivity and electron transfer numbers during the ORR of TY-COF and TE-COF were quantitatively assessed by rotating ring-disk electrode (RRDE) measurements (Fig. 4f). The TY-COF exhibited high H2O2 selectivity (85–98%) across the potential range of 0.2–0.6 V (vs. RHE), with electron transfer numbers approaching 2, indicating a predominant 2e− ORR pathway. The TE-COF showed H2O2 selectivity of 65–78% with electron transfer numbers close to 2.6. These electrochemical results further supported the conclusion that the TY-COF and TE-COF mainly adopted 2e− ORR pathways for H2O2 production.
To gain deeper insights into the reaction intermediates and pathways during the photocatalytic H2O2 evolution processes of TY-COF and TE-COF, in situ diffuse reflectance FT-IR (DRIFT) spectra were collected. For TY-COF (Fig. 4g), characteristic peaks at 1018 cm−1 (*OH), 1185 cm−1 (O2˙−), and 1243 cm−1 (*OOH) were observed during light irradiation, with signal intensities increasing over time. These spectral features indicated that the TY-COF predominantly followed a sequential O2–O2˙−–*OOH–H2O2 pathway, consistent with its two-step 2e− ORR mechanism. In contrast, the TE-COF (Fig. 4h) showed a strong peak at 1020 cm−1 (*OH) and 1239 cm−1 (endoperoxide),35,36 The absence of O2˙− signals suggested that the TE-COF operated through a different mechanism involving the formation of endoperoxide between O2 and the TE-COF backbone via the one-step 2e− ORR path.
To further distinguish the WOR pathways (2e− WOR or 4e− WOR) of the as-synthesized COFs, we conducted control experiments in an Ar atmosphere with the addition of KBrO3 and CH3OH (a hole scavenger), as shown in Fig. 4i, respectively. In both TY-COF and TE-COF reaction systems, KBrO3 significantly enhanced H2O2 production, while CH3OH suppressed it. Moreover, even with the addition of AgNO3, no O2 could be detected by online gas chromatography (5A molecular sieve column), as shown in Fig. S19.† Combined with the scavenger experiments in Ar and the VB position, it can be concluded that both TY-COF and TE-COF underwent the direct 2e− WOR pathway.
Based on the charge distribution observed in the first excited state of TY-COF and TE-COF illustrated in Fig. 1b, it was evident that the photoinduced electrons were predominantly localized within the units derived from the amine precursors, while the holes were primarily associated with the modules originating from the aldehyde monomers. To gain a more profound mechanistic understanding of the photocatalytic H2O2 generation processes in TY-COF and TE-COF, we further performed theoretical calculations to analyze the charge distribution on the representative fragments of the TY-COF and TE-COF using the transition density matrix (TDM) method.44,45 As illustrated in Fig. 5a and b, the TDM heat maps unveiled distinct charge distribution patterns within TY-COF and TE-COF. In TY-COF (Fig. 5a), photogenerated holes predominantly accumulated at fragment 4 (34.2%), corresponding to the phenyl ring. Meanwhile, electrons were primarily localized at fragment 2 (43.3%), associated with the phenyl ring adjacent to the triazine unit. Conversely, the TE-COF (Fig. 5b) displayed hole distribution at fragment 4 (phenyl ring, 33.6%) and fragment 5 (alkene bond, 21.6%). The electron distribution in TE-COF was more dispersed between fragment 1 (triazine, 22.2%) and fragment 2 (phenyl ring, 29.3%), resulting in less pronounced electron–hole separation compared to TY-COF.
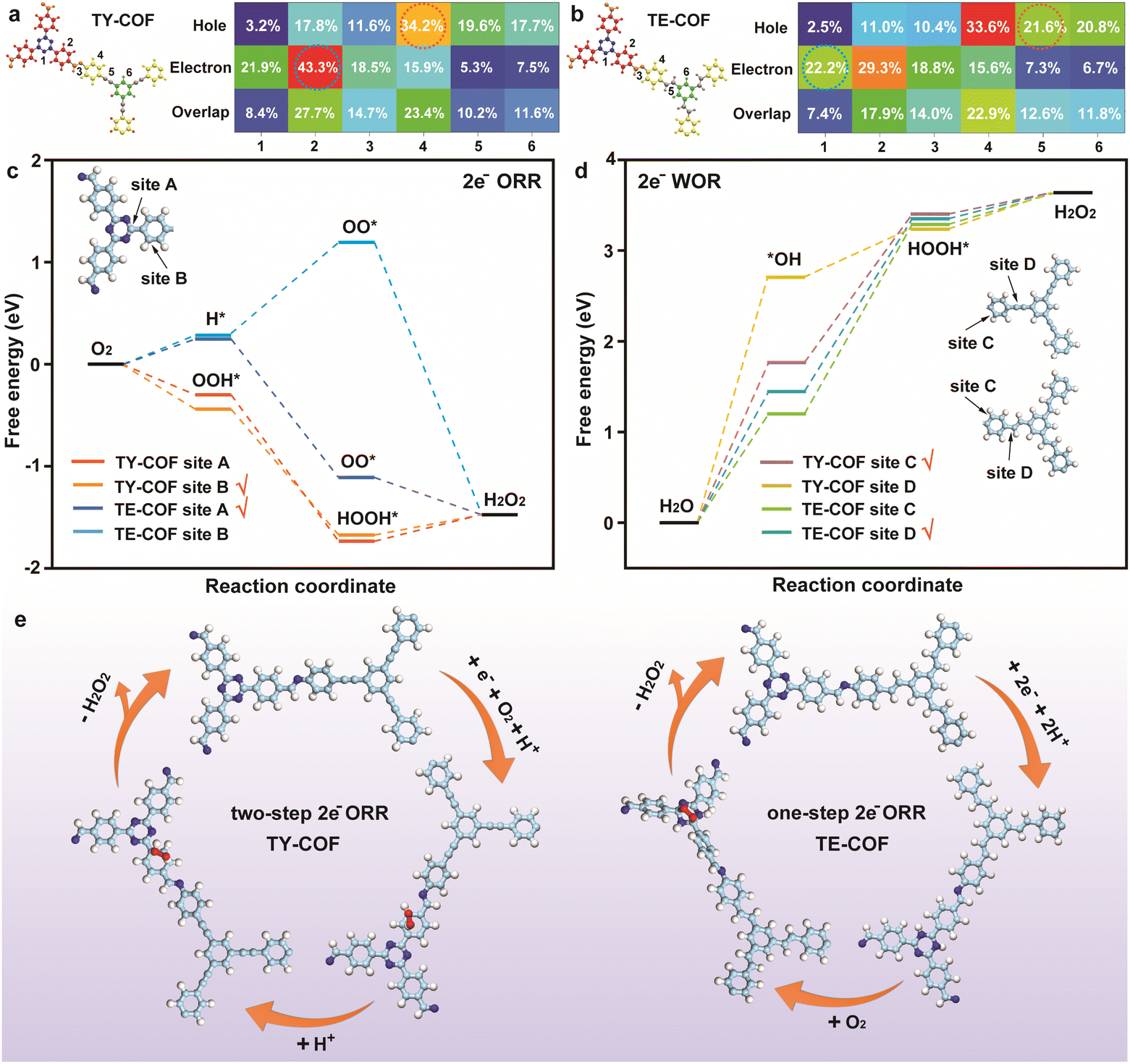 | ||
| Fig. 5 Fragments chosen for TDM analysis and the heat map of hole–electron distributions of (a) TY-COF and (b) TE-COF. Free energy diagrams of photocatalytic H2O2 generation over TY-COF and TE-COF via (c) 2e− ORR paths and (d) 2e− WOR paths. (e) Proposed 2e− ORR mechanism for H2O2 photoproduction via TY-COF and TE-COF. | ||
According to the TDM analysis, the identification of the probable active sites for the 2e− ORR was achieved by evaluating the O2 adsorption energies in fragment 1 and fragment 2 (Fig. S20†) of TY-COF and TE-COF, while those for the 2e− WOR were acquired by evaluating the H2O adsorption energies in fragment 4 and fragment 5 (Fig. S21†). After that, site A and site B were chosen to calculate the free energy diagrams for the 2e− ORR of TY-COF and TE-COF. Site C and site D were chosen to calculate the free energy diagrams for the 2e− WOR of TY-COF and TE-COF. As depicted in Fig. 5c, at site A and site B of the TY-COF, the formation of the OOH* intermediate was exergonic (−0.30 eV and −0.44 eV), and the subsequent conversion to HOOH* (−1.74 eV and −1.62 eV) was also thermodynamically favorable. During the two-step 2e− ORR of TY-COF, the desorption of H2O2 was the rate determining step (RDS), with energy barriers of 0.27 eV at site A and 0.15 eV at site B. Consequently, for TY-COF, the ORR active center was identified as site B (the carbon atom in the phenyl ring in fragment 2). Since the TE-COF followed the one-step 2e− ORR pathway, H* and OO* intermediates were involved.11,37 For the TE-COF, in contrast to site B, site A (the carbon atom in the triazine unit) with lower energy barriers (0.25 eV) in the RDS (the formation of *H) was the most likely ORR active center. The modulation of unsaturated bonds resulted in differing electron distributions between TY-COF and TE-COF, ultimately altering the ORR active centers.
Regarding the 2e− WOR pathway (Fig. 5d), both COFs showed considerable uphill energy profiles, with the formation of *OH and *HOOH intermediates. For TY-COF, the generation of *OH constituted the RDS, exhibiting an activation energy barrier of 1.76 eV at site C, markedly lower than the corresponding barrier observed at site D (2.70 eV). Conversely, for TE-COF, the formation of *HOOH represented the RDS, exhibiting an energy barrier of 1.89 eV at site D, substantially reduced compared to the analogous barrier encountered at site C (2.13 eV). Apparently, based on the energy barriers, site C (the carbon atom in the phenyl ring) in the TY-COF and site D (the carbon atom in the alkene) in the TE-COF represented the most active sites for their 2e− WOR pathways. This thermodynamic limitation explained the limited H2O2 production observed under Ar, in accordance with our experimental results.
These theoretical insights corroborated our experimental findings, confirming that the 2e− ORR pathway was the dominant contributor to H2O2 production in both COFs, with TY-COF exhibiting superior performance due to its more efficient charge separation, heightened electron density at the active center, and more favorable reaction energetics. Intriguingly, the distinct electronic structures arising from the differing unsaturated bonds (–CC– in TY-COF and –CC– in TE-COF) fundamentally transformed the catalytic centers, resulting in divergent 2e− ORR pathways, as depicted in Fig. 5e. In addition, the 2e− WOR mechanism for H2O2 photoproduction by TY-COF and TE-COF is illustrated in Fig. S22.†
In practical applications of H2O2 photogeneration, it is crucial and convenient that H2O2 can be easily separated. To this end, we fabricated free-standing TY-COF and TE-COF membranes via the interfacial polymerization method,46,47 The PXRD patterns and FT-IR spectra shown in Fig. S23† confirmed the crystallinity and chemical structures of the TY-COF and TE-COF membranes. Both TY-COF and TE-COF membranes displayed surface morphologies with occasional bumps (Fig. S24†). The optical photographs and cross-sectional SEM images of TY-COF and TE-COF membranes are exhibited in Fig. 6a and b. Under air and pure water conditions, the TY-COF membrane (thickness: 19.4 μm) and the TE-COF membrane (thickness: 84.3 μm) can produce H2O2 smoothly at rates of 3100 and 2503 μmol g−1 h−1, respectively (Fig. 6c).
 | ||
| Fig. 6 Optical photographs and cross-sectional SEM images of the (a) TY-COF membrane and (b) TE-COF membrane. (c) H2O2 generation rates of the TY-COF membrane and TE-COF membrane in air and pure water. (d) H2O2 photoproduction rates of TY-COF and TE-COF in different waters. | ||
Moreover, to ascertain the practical viability of solar-driven H2O2 production utilizing TY-COF and TE-COF, photocatalytic experiments were conducted across diverse aqueous matrices, including natural seawater, spring water, municipal tap water, acidic solution, and alkaline solution. As illustrated in Fig. 6d, both TY-COF and TE-COF demonstrated robust H2O2 generation capabilities across various aqueous environments, emphasizing their potential applicability in real world water treatment scenarios.
Conclusions
In conclusion, by rationally designing –CC– and –CC– linkages into the skeletons of COFs, we successfully modulated the 2e− ORR pathways. Scavenger experiments, NBT probing, EPR spectra, and in situ DRIFT spectra confirmed that the –CC– incorporated TY-COF favored the two-step 2e− ORR pathway, while the –CC– incorporated TE-COF followed the one-step 2e− ORR pathway. In the absence of sacrificial agents, the H2O2 evolution rates of TY-COF and TE-COF (6455 and 4804 μmol g−1 h−1, respectively) were equivalent to those of high-performance COF-based photocatalysts. The SCC efficiencies of TY-COF and TE-COF were 0.67% and 0.45%, respectively. The outcomes of theoretical calculations suggested that the manipulation of unsaturated bonds induced a shift in the distribution of electrons and holes along the COF skeletons. This alteration led to a modification of the 2e− ORR catalytic centers (the benzene ring in the TY-COF and triazine in the TE-COF), ultimately resulting in the emergence of two distinct 2e− ORR pathways in TY-COF and TE-COF. Additionally, TY-COF and TE-COF membranes were constructed through an interfacial polymerization strategy, which can drive the H2O2 photosynthesis smoothly. This work highlights the critical role of manipulating charge carrier distribution in catalysts through structural design to modulate 2e− ORR pathways.
Data availability
All the data supporting this article have been included in the main text and the ESI.†Author contributions
J. Y. Y. designed the research, and wrote and revised the manuscript; Z. X. P. and Y. G. performed the experiments; P. Y. and B. T. provided financial support.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22134004, 21804081, and 22278249), the Natural Science Foundation of Shandong Province (ZR2023MB063 and ZR2021YQ13), the Taishan Scholars Program of Shandong Province (tsqn202306151), and the Project of Shandong Provincial Center for Fundamental Science Research (YDZX2024150). The authors thank Suzhou Deyo Bot Advanced Materials Co., Ltd (https://www.dy-test.com) for support with material characterization.Notes and references
- Z. Chen, D. Yao, C. Chu and S. Mao, Chem. Eng. J., 2023, 451, 138489 CrossRef CAS.
- R. Liu, M. Zhang, F. Zhang, B. Zeng, X. Li, Z. Guo and X. Lang, Small, 2025, 21, 2411625 CrossRef CAS.
- J.-Y. Yue, L.-P. Song, Y.-F. Fan, Z.-X. Pan, P. Yang, Y. Ma, Q. Xu and B. Tang, Angew. Chem., Int. Ed., 2023, 62, e202309624 Search PubMed.
- X. Zeng, Y. Liu, X. Hu and X. Zhang, Green Chem., 2021, 23, 1466–1494 RSC.
- H. Cheng, J. Cheng, L. Wang and H. Xu, Chem. Mater., 2022, 34, 4259–4273 Search PubMed.
- X. Wang, Z.-Y. Yi, Y.-Q. Wang and D. Wang, Angew. Chem., Int. Ed., 2025, 64, e202413673 Search PubMed.
- J.-Y. Yue, J.-X. Luo, Z.-X. Pan, Q. Xu, P. Yang and B. Tang, Angew. Chem., Int. Ed., 2025, 64, e202417115 Search PubMed.
- J.-Y. Yue, L.-P. Song, Z.-X. Pan, P. Yang, Y. Ma, Q. Xu and B. Tang, ACS Catal., 2024, 14, 4728–4737 CrossRef CAS.
- Y. Liu, L. Li, Z. Sang, H. Tan, N. Ye, C. Sun, Z. Sun, M. Luo and S. Guo, Nat. Synth., 2025, 4, 134–141 CrossRef CAS.
- Z. Luo, Y. Yan, R. Spinney, D. D. Dionysiou, F. A. Villamena, R. Xiao and D. Vione, Water Res., 2024, 261, 122023 CrossRef CAS.
- H. Cheng, H. Lv, J. Cheng, L. Wang, X. Wu and H. Xu, Adv. Mater., 2022, 34, 2107480 CrossRef CAS.
- C. Krishnaraj, H. Sekhar Jena, L. Bourda, A. Laemont, P. Pachfule, J. Roeser, C. V. Chandran, S. Borgmans, S. M. J. Rogge, K. Leus, C. V. Stevens, J. A. Martens, V. Van Speybroeck, E. Breynaert, A. Thomas and P. Van Der Voort, J. Am. Chem. Soc., 2020, 142, 20107–20116 Search PubMed.
- J. Yang, X. Zeng, M. Tebyetekerwa, Z. Wang, C. Bie, X. Sun, I. Marriam and X. Zhang, Adv. Energy Mater., 2024, 14, 2400740 Search PubMed.
- J.-Y. Yue, L.-P. Song, Z.-X. Pan, M. Cheng, X. Wang, Q. Xu and P. Yang, Chem. Eng. J., 2025, 504, 158983 Search PubMed.
- E. Zhou, F. Wang, X. Zhang, Y. Hui and Y. Wang, Angew. Chem., Int. Ed., 2024, 63, e202400999 CrossRef CAS.
- Y. Zhang, J. Qiu, B. Zhu, G. Sun, B. Cheng and L. Wang, Chin. J. Catal., 2024, 57, 143–153 CrossRef CAS.
- H. Yu, X. Zhang, Q. Chen, P.-K. Zhou, F. Xu, H. Wang and X. Chen, Chem. Res. Chin. Univ., 2024 DOI:10.1007/s40242-024-4213-3.
- Y.-N. Gong, X. Guan and H.-L. Jiang, Coord. Chem. Rev., 2023, 475, 214889 CrossRef CAS.
- B. Mishra, A. Alam, A. Chakraborty, B. Kumbhakar, S. Ghosh, P. Pachfule and A. Thomas, Adv. Mater., 2024, 2413118 Search PubMed.
- M. Zhang, M. Lu, M.-Y. Yang, J.-P. Liao, Y.-F. Liu, H.-J. Yan, J.-N. Chang, T.-Y. Yu, S.-L. Li and Y.-Q. Lan, eScience, 2023, 3, 100116 CrossRef.
- D. Tan, R. Zhuang, R. Chen, M. Ban, W. Feng, F. Xu, X. Chen and Q. Wang, Adv. Funct. Mater., 2024, 34, 2311655 CrossRef CAS.
- J. Hao, Y. Tang, J. Qu, Y. Cai, X. Yang and J. Hu, Small, 2024, 20, 2404139 CrossRef CAS.
- R. Liu, Y. Chen, H. Yu, M. Položij, Y. Guo, T. C. Sum, T. Heine and D. Jiang, Nat. Catal., 2024, 7, 195–206 Search PubMed.
- Z. Gong, Y. Gao, J. Li, Z. Cai, N. Liu and J. Jiang, Angew. Chem., Int. Ed., 2025, e202423205 CAS.
- L. Zhang, C. Wang, Q. Jiang, P. Lyu and Y. Xu, J. Am. Chem. Soc., 2024, 146, 29943–29954 Search PubMed.
- W. Zhang, M. Sun, J. Cheng, X. Wu and H. Xu, Adv. Mater., 2025, 37, 2500913 CrossRef CAS.
- F. Zhang, X. Lv, H. Wang, J. Cai, H. Wang, S. Bi, R. Wei, C. Yang, G. Zheng and Q. Han, Adv. Mater., 2025, 37, 2502220 CrossRef CAS.
- Z. Yong and T. Ma, Angew. Chem., Int. Ed., 2023, 62, e202308980 Search PubMed.
- Q. Xue, H. Li, P. Jin, X. Zhou and F. Wang, Angew. Chem., Int. Ed., 2025, e202423368 CAS.
- J. Cheng, Y. Wu, W. Zhang, L. Wang, X. Wu and H. Xu, Adv. Mater., 2025, 37, 2410247 CrossRef CAS.
- J.-Y. Yue, Z.-X. Pan, R.-Z. Zhang, Q. Xu, P. Yang and B. Tang, Adv. Funct. Mater., 2025, 35, 2421514 Search PubMed.
- Z. Xie, X. Chen, W. Wang, X. Ke, X. Zhang, S. Wang, X. Wu, J. C. Yu and X. Wang, Angew. Chem., Int. Ed., 2024, 63, e202410179 CrossRef CAS.
- H. Xu, S. Xia, C. Li, Y. Li, W. Xing, Y. Jiang and X. Chen, Angew. Chem., Int. Ed., 2024, 63, e202405476 Search PubMed.
- H. Yu, F. Zhang, Q. Chen, P.-K. Zhou, W. Xing, S. Wang, G. Zhang, Y. Jiang and X. Chen, Angew. Chem., Int. Ed., 2024, 63, e202402297 CrossRef CAS.
- Y. Luo, B. Zhang, C. Liu, D. Xia, X. Ou, Y. Cai, Y. Zhou, J. Jiang and B. Han, Angew. Chem., Int. Ed., 2023, 62, e202305355 CrossRef CAS.
- W. Wu, Z. Li, S. Liu, D. Zhang, B. Cai, Y. Liang, M. Wu, Y. Liao and X. Zhao, Angew. Chem., Int. Ed., 2024, 63, e202404563 CrossRef CAS.
- D. Chen, W. Chen, Y. Wu, L. Wang, X. Wu, H. Xu and L. Chen, Angew. Chem., Int. Ed., 2023, 62, e202217479 CrossRef CAS.
- X. Yan, B. Wang, J. Ren, X. Long and D. Yang, Angew. Chem., Int. Ed., 2022, 61, e202209583 Search PubMed.
- H.-H. Sun, Z.-B. Zhou, Y. Fu, Q.-Y. Qi, Z.-X. Wang, S. Xu and X. Zhao, Angew. Chem., Int. Ed., 2024, 63, e202409250 CrossRef CAS.
- C. Wu, Z. Teng, C. Yang, F. Chen, H. B. Yang, L. Wang, H. Xu, B. Liu, G. Zheng and Q. Han, Adv. Mater., 2022, 34, 2110266 CrossRef CAS.
- Q. Niu, W. Deng, Y. Chen, Q. Lin, L. Li, Z. Liu, J. Bi and Y. Yu, ACS Energy Lett., 2024, 9, 5830–5835 CrossRef CAS.
- M. Bonchio, J. Bonin, O. Ishitani, T.-B. Lu, T. Morikawa, A. J. Morris, E. Reisner, D. Sarkar, F. M. Toma and M. Robert, Nat. Catal., 2023, 6, 657–665 CrossRef.
- L. Guan, Z. Li, K. Wang, L. Gong, Y. Fang, G. Yu, M. Zhu and S. Jin, Angew. Chem., Int. Ed., 2025, 64, e202419867 CrossRef CAS.
- H. Yan, Y. Peng, Y. Huang, M. Shen, X. Wei, W. Zou, Q. Tong, N. Zhou, J. Xu, Y. Zhang, Y.-X. Ye and G. Ouyang, Adv. Mater., 2024, 36, 2311535 CrossRef CAS.
- Y. Yang, Y. Dong, W. Chi, X. Sun, J. Su, X. Chen, H. Zhao and Y. Zhu, Chem. Eng. J., 2025, 503, 158646 CrossRef CAS.
- X. Liu, R. Huang, L. Peng, J. Yang, J. Yan, B. Zhai, Y. Luo, C. Zhang, S. Tan, X. Liu, L. Ding and Y. Fang, Angew. Chem., Int. Ed., 2025, 64, e202414472 CrossRef CAS.
- Q. Shen, Q. Song, Z. Mai, K.-R. Lee, T. Yoshioka, K. Guan, R. R. Gonzales and H. Matsuyama, Sci. Adv., 2023, 9, eadf6122 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Materials, measurements, experimental details, and supplementary figures. See DOI: https://doi.org/10.1039/d5sc03270a |
| This journal is © The Royal Society of Chemistry 2025 |