
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffective generation of heavy-atom-free triplet photosensitizers containing multiple intersystem crossing mechanisms based on deep learning†
Kepeng Chena,
Xiaoting Zhang b,
Jike Wanga,
Dan Lia,
Tingjun Hou*a,
Wenbo Yang*b and
Yu Kang*a
b,
Jike Wanga,
Dan Lia,
Tingjun Hou*a,
Wenbo Yang*b and
Yu Kang*a
aCollege of Pharmaceutical Sciences, Zhejiang University, Hangzhou, Zhejiang 310058, China. E-mail: yukang@zju.edu.cn
bSchool of Chemistry, State Key Laboratory of Fine Chemicals, Frontier Science Center for Smart Materials, Dalian Key Laboratory of Intelligent Chemistry, Dalian University of Technology, Dalian 116024, China. E-mail: wbyang@dlut.edu.cn
First published on 8th July 2025
Abstract
Photodynamic therapy (PDT) is a clinically approved therapeutic modality that has demonstrated significant potential for cancer treatment, and triplet photosensitizers (PSs) play a key role in its efficacy. Despite deep learning having emerged as a next-generation tool for material discovery, existing methods mainly target a limited subset of triplet PSs, such as thermally activated delayed fluorescence (TADF) materials, neglecting the critical intersystem crossing (ISC) between the high-lying singlet and triplet states (ΔESnTn). To overcome this limitation, we compiled a comprehensive dataset (∼1.90 × 109) of triplet PSs encompassing various ISC mechanisms. Then, we proposed a novel strategy that incorporates two models: a fragment-based model (Frag-MD) and a character-based model (MD), both integrating a conditional transformer, recurrent neural networks, and reinforcement learning. In silico experiments revealed that the Frag-MD model outperforms the MD model in generating larger conjugated motifs with higher average ring numbers and atom counts; while the MD model generates twice as many unique motifs and excels in novelty and diversity, as evaluated by conditional and MOSES metrics. Therefore, our approach is highly effective for modifying conjugated motifs and designing novel triplet PSs. Notably, the recently reported high-efficiency triplet PSs have been re-identified through ablation experiments using our proposed models, which target ΔESnTn and significantly outperform traditional baselines, achieving a prediction accuracy of 73% versus 4%. Our approach holds the potential to establish a new paradigm for discovering novel PSs applicable in PDT.
1 Introduction
Photodynamic therapy (PDT) has been extensively investigated for decades as a treatment for tumors due to its minimally invasive nature, ability to preserve normal tissues, and approval for early-stage cancer treatment with relatively low pain.1,2 Triplet photosensitizers (PSs) play a key role in determining the efficacy of PDT.3,4 However, the traditional trial-and-error efforts to identify successful triplet PSs, which rely on expert knowledge, are time-consuming and intricate processes. This approach involves selecting potential chromophores, introducing reasonable motif derivatization, ensuring molecular synthesizability, achieving long wavelength absorption, and most importantly, securing high intersystem crossing (ISC) efficiency.In recent years, the development of artificial intelligence (AI), especially through machine learning (ML) and deep learning (DL) approaches for molecular design, has significantly transformed the research paradigm in molecular discovery within the medical and materials science communities, sparking considerable interest.5–7 Leveraging advances in computing power and algorithm efficiency, these approaches exhibit an ever-increasing non-linear exploration capability to navigate the vast potential search space and optimize multiple properties simultaneously. They have demonstrated remarkable effectiveness in molecular property prediction, high-throughput virtual screening, and inverse molecular design or molecular generation under given constraints.5,8–14 Therefore, these approaches hold great promise for accelerating the discovery of novel triplet PSs.
Given that the ISC process of triplet PSs is a first-order perturbation process, the ISC rate constant can be described by eqn (1), a derivation of equation from the Fermi golden rule, which inevitably depends on quantum chemistry calculations (e.g., semi-empirical methods or density functional theory (DFT) calculations) to obtain the key parameter of the ISC process (ΔESnTn, n ≥ 1, the excited singlet–triplet energy gap).
 | (1) |
It is obvious that the high computational cost of quantum chemistry limits the availability of labeled data for DL-based molecular design approaches. On the other hand, DL-based inverse molecular design, which generates de novo molecules with specific and desirable properties rather than relying on high-throughput virtual screening by a molecule prediction model, explores a much broader chemical space. However, it remains a highly challenging task to design successful triplet PSs using DL-based inverse molecular design while simultaneously considering numerous factors such as synthetic accessibility (SA), quantitative estimate of drug-likeness (QED), absorption wavelength, ISC efficiency, and so on.15 For triplet PSs, heavy-atom-free triplet PSs offer advantages such as low cost, reduced potential toxicity, easier preparation and longer triplet lifetimes compared to triplet PSs that rely on heavy-atom enhanced ISC effects. Recently, heavy-atom-free triplet PSs have gained significance not only in PDT3,4 but also in photocatalysis,16–20 triplet–triplet-annihilation upconversion (TTA-UC),21,22 thermally activated delayed fluorescence (TADF),23–25 and other applications. Therefore, our work focuses on designing heavy-atom-free triplet PSs.
To address the challenges of insufficient labeled data in high-throughput virtual screening26–28 and inverse molecular design,5,8–10,14,29–33 various strategies have been developed, including constructing chemically reasonable and constrained molecules, selectively labeling a subset of molecules, and employing DFT functionals with lower computational cost. For example, Gómez-Bombarelli et al.5 built a space of 1.6 million molecules by combining 110 donor, 105 acceptor and 7 bridging moieties. More specifically, Buglak et al.30 focused on 70 heavy-atom-free boron-dipyrromethene (BODIPY) dyes in three different solvents to predict singlet oxygen generation using a multiple linear regression model within a DL framework. To improve the model's prediction accuracy of ΔES1T1 and reduce its dependence on labeled data, Wang et al. developed a smart strategy that combines a heuristic algorithm (active learning) with a deep neural network.34 This resulted in an efficient and useful self-improving discovery system from a space of more than 7 million molecules. Similarly, Nigam et al. developed a DL workflow to find novel organic emitters with desired ΔES1T1 and appreciable fluorescence rates,13 using a genetic algorithm as the heuristic algorithm. More recently, Blaschke et al.35 constructed a framework that connects the REINVENT reinforcement learning framework to discover acceptors in TTA-UC.14 However, a key point that has been overlooked is the ISC process arising from higher excited singlet and triplet states (ΔESnTn, n > 1), leading to limited studies on the ΔES1T1 value36 and TADF molecules (ΔES1T1 ≤ 0.2 eV).37 While the ISC process of TADF molecules includes the SOC-ISC and the revised ISC (RISC) process, which extend the triplet state lifetime and enhance their applicability in PDT, TADF molecules represent only a small subset of ideal triplet PSs. Notably, some monomers achieve high ISC efficiency purely through the SOC-ISC mechanism, without requiring RISC. For example, porphyrin derivatives have been used as commercial reagents in PDT.38,39 Similarly, other donor–acceptor systems can be effective candidates through a spin orbital charge transfer ISC (SOCT-ISC)40 or radical pair ISC (RP-ISC) mechanism,41 which are modulated by solvent polarity, electronic coupling, and other related parameters.42
In this study, we first developed a comprehensive dataset (∼1.90 × 109) of triplet PSs with multiple ISC mechanisms by combining linkers, donors and acceptors. The ISC efficiency metric was set within a ΔESnTn range of 0–0.3 eV. Beyond traditional molecular-level metrics, we adopted conjugated-motif-level metrics as a more reasonable evaluation approach. We then proposed a novel strategy for designing heavy-atom-free triplet PSs, which mimics the design process of a chemistry expert and can be divided into two main tasks. Task 1 is the de novo design aimed at discovering new conjugated motifs, and Task 2 employs a conjugated-motif-based method focused on conjugated motif derivation. To accomplish these two tasks, we utilized a character-based molecular generation architecture (MD/GD)43 for Task 1 and a fragment-based molecular generation architecture (Frag-MD/Frag-GD)44 for Task 2 (Fig. 1). The general reinforcement learning (RL) workflow for target-directed molecular generation consists of a recurrent neural network (RNN) and scoring function models.35,45 Frag-GD and GD only consist of general modules, while Frag-MD and MD consist of mixture modules which introduce a conditional transformer for molecule dataset generation, followed by general modules,44 which bias the molecular space towards the desired region. The final label ‘D’ means the scoring function model of the studied models trained from diverse molecule types and multiple ISC mechanisms. In contrast, Frag-MB/Frag-GB and MB/GB are regarded as 4 baseline models, where the scoring function model is trained on data labeled with ΔES1T1. In silico experiments demonstrate the practical viability of our method in accomplishing both tasks. Notably, recently reported high-efficiency triplet PSs have been re-identified through ablation experiments, further demonstrating the molecular design ability of our proposed models. Thus, Frag-MD/MD has the potential to discover novel PSs applicable in PDT.
 | ||
| Fig. 1 Workflow of Frag-MD with a fragment-based method and MD with a character-based method. (a) Dataset preparation details, (b) ring-cutting method and char-cutting method, (c) transformer decoder, and (d) fragment-based RNN or character-based RNN for reinforcement learning. | ||
2 Methods
The workflow presented here is developed based on a policy gradient model with a modified version of REINVENT35 by introducing a conditional transformer. For de novo molecular design in Task 1, we implemented a conventional char-cutting method,46 referred to as the character-based method, to facilitate free molecular derivation without the constraint of preserving conjugated motifs. For Task 2, which focuses on designing conjugated-motif-based molecules, we split molecules into fragments using a ring-cutting method to retain conjugated motifs (referred to as the fragment-based method).47,48 As shown in Fig. 1, the char-cutting method splits molecules into atoms or functional groups, which serve as tokens for a molecule generation model (e.g., an RNN model). In contrast, the ring-cutting method splits molecules into fragments based on single bonds outside the ring. These fragments include terminal groups (R-groups), conjugated motifs (scaffolds), and linkers. Specifically, the wavy lines represent the cleavage sites, and the star symbols represent the combination sites.2.1 Dataset
To design triplet PSs with multiple ISC mechanisms, a well-curated dataset is crucial. Hence, we constructed our dataset by combining fragments from conventionally studied chromophores,49 custom donors or acceptors in TADF materials,36 and molecules from the QM9 dataset.50 It has the following advantages: (1) conventionally studied chromophores, which are typically large conjugated molecules, exhibit long-wavelength absorption and practical derivatization, aligning well with the desired molecular space; (2) molecular scale and molecular diversity can be further expanded by introducing A–D–A, A–A, and D–D systems to the conventional D–A and D–A–D types (TADF molecules); (3) the incorporation of novel fragments from the QM9 dataset (124 K), which is widely used in quantum chemistry models, significantly enhances molecular diversity while maintaining computational feasibility.Detailed information on dataset construction is provided in ESI Part 1,† and its basic characteristics are summarized in Table 1. The final dataset contains ∼1.90 × 109 molecules, with a subset of 3.5 × 106 molecules (500 K molecules for each molecule type) selected in our study to balance computational costs. To the best of our knowledge, this dataset is substantially larger and contains a broader range of molecular types compared to previously reported datasets (103–105, specifically TADF molecules in the initially studied dataset).1,29,34
| Molecule types | Molecule numbers | Dataset components | Labeled molecules | Descriptions of combination |
|---|---|---|---|---|
| a Con, star, alkyl and aryl stand for conjugated motifs, reaction site, alkyl linker and aryl linker, respectively. The Con recovery rate of dataset is 1.0. | ||||
| Monomers | ∼1.90 × 109 (1898![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 972708) 972708) |
500000 |
∼1.40 × 104 (14005) |
Con* + termi-frag |
| Dimers1 (directly connected) | 500000 |
Con* + Con* | ||
| Dimers2 (alkyl linkers) | 500000 |
2 Con* + alkyl** | ||
| Dimers3 (aryl linkers) | 500000 |
2 Con* + aryl** | ||
| Trimers1 (directly connected) | 500000 |
2 Con* + Con** | ||
| Trimers2 (alkyl linkers) | 500000 |
3 Con* + alkyl*** | ||
| Trimers3 (aryl linkers) | 500000 |
3 Con* + aryl*** | ||
2.2 Agent in reinforcement learning
In RL, the agent for all models adopted an RNN architecture to generate molecules tailored to desired properties through score functions. To constrain the RNN-generated molecules to a reasonable range of SA and QED properties while enabling subsequent target optimization of ΔEST and Eabs and improving the fine-tuning efficiency, a lightweight RNN architecture (MD/Frag-MD) was derived from a heavier transformer decoder model through knowledge distillation.51The conditional transformer decoder was trained on the selected dataset (3.5 × 106). Its architecture is illustrated in Fig. 1, where the next token in a Simplified Molecular Input Line Entry System (SMILES) sequence is generated based on the preceding token. To mitigate the risk of information leakage from tokens within the decoder portion of the sequence, a masked multi-head self-attention layer was implemented in this model. As the core component of the transformer decoder, this layer integrates multiple scaled dot-product attention mechanisms, enhancing the model's ability to effectively extract and prioritize critical information from sequential data. The attention mechanism is concisely represented by eqn (2):
 | (2) |
To generate molecules with desired properties, a conditional character P is incorporated as an input during the molecule generation process. This conditional character P is a Boolean string of length 2, where each bit (1 or 0) indicates whether the molecule meets the QED and SA conditions. Once embedded, the conditional character and the molecular SMILES strings are concatenated, enabling the model to learn from both sources concurrently during training. Specifically, during the training of the conditional transformer decoder model, the following loss function is minimized given a conditional character P, as shown in eqn (3).
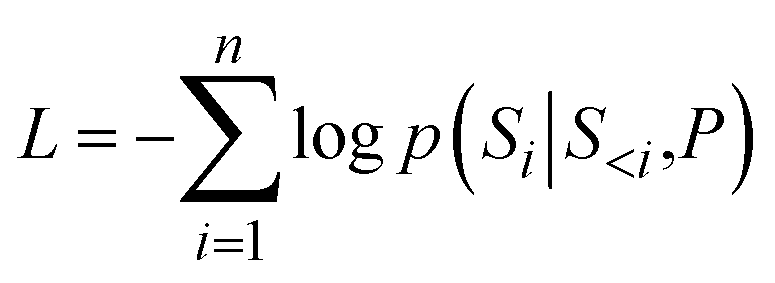 | (3) |
To generate SMILES under a conditional character P, the model learns the conditional distribution p(S|P), as shown in eqn (4).
 | (4) |
Using the trained conditional transformer decoder, constrained desired molecules were generated based on a conditional character P (QED and SA), resulting in a final new dataset of molecules (3.5 × 106). During the training stage of the RNN with this dataset, the loss function aims to minimize the negative log-likelihood or cross-entropy function, as shown in eqn (5).
 | (5) |
In the generation stage of the trained RNN, the autoregressive method was used to generate SMILES strings in units of molecular fragments or characters as shown in eqn (6).
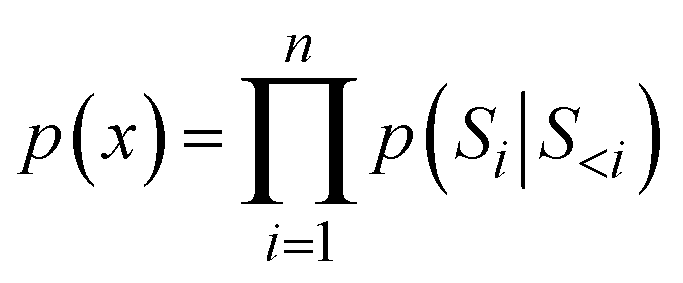 | (6) |
2.3 ΔEST and Eabs prediction models as scoring functions in reinforcement learning
The labeled dataset for the prediction model contains ∼14 K molecules for multiple molecule types that favor multiple ISC mechanisms (Table 1). To determine the energy gap between high excited singlet and triplet states (ΔESnTn, 1 ≤ n ≤ 6) and the energy level of the absorption wavelength (Eabs), ten excited singlets and ten exited triplets were calculated based on the ground state geometries optimized using DFT//B3LYP/6-31G. Including more excited states enhances the accuracy of the first six excited energy levels. These calculations were performed using the Gaussian 16 program package.52 The prediction model employed is a typical molecular graph convolutional neural (GCN) network.53 The molecular graph is an undirected graph G = (V, E), where nodes V and edges E represent atoms and chemical connection bonds between atoms, respectively. The ConvMolFeaturizer function was used to encode the molecular fingerprint into a two-dimensional representation, which facilitates the prediction of quantum properties.54 Hyperparameters such as the number of graph convolutional layers, dense layers, dropout, and learning rate were optimized using a grid search method, which systematically evaluates all possible combinations of specified hyperparameters via cross-validation. An early stopping strategy was implemented during the training stage. The relationship between training epochs and the mean absolute error (MAE) values of ΔEST and Eabs is presented in ESI Fig. S1,† and the MAE of ΔEST in the subset of the dataset was less than 0.3 eV, indicating that our model is accurate enough for RL applications in triplet PS design. Further details about the prediction model are provided in ESI Part 2.†2.4 Loss function in reinforcement learning
In this study, we employed the RL strategy implemented in REINVENT to refine our RNN model (agent). The loss function was optimized using the policy gradient descent optimizer method to fine-tune the agent model. The RL fine-tuning procedure is outlined as follows: (1) copy the trained fragment/character-based RNN model as the prior model, keeping all parameters fixed during fine-tuning, and use the original fragment-based or character-based RNN model as the agent model; (2) sample molecules with the agent model and save the likelihood of each sample molecule as logp(S)Agent, and use the prior model to estimate logp(S)Prior for the generated SMILES strings; (3) calculate the reward score from the prediction models, multiply it by a coefficient σ, and define the loss function as follows:|
Loss = [logp(S)prior + σScore(S) − logp(S)agent]2
| (7) |
2.5 Experiment setting
The objective of our model is to design heavy-atom-free triplet PSs containing multiple ISC mechanisms. A successful triplet PS needs to satisfy the following four constraints: ΔEST ≤ 0.30, Eabs ≤ 2.48, QED ≥ 0.38, and SA ≤ 4.0. The threshold of ΔEST ≤ 0.30 captures not only TADF molecules with the SOC-ISC/RISC mechanism but also more general triplet PSs encompassing multiple ISC mechanisms, such as SOCT-ISC, RP-ISC and SOC. The Eabs value above 0.38 eV signifies an absorption wavelength greater than 500 nm, guiding the model toward molecules with extended conjugated systems for enhanced applicability in PDT. QED ≥ 0.38 was chosen based on the average QED value of the reported triplet PS distribution, and this value of PDT drugs significantly differs from the typical QED of 0.6 for conventional drugs, highlighting the distinct chemical space of triplet PSs. The SA value below 4.0 generally indicates that a molecule is synthetically accessible. Molecules that satisfy these constraints are defined as “desired molecules”, while “successful molecules” are further distinguished by their uniqueness. During the RL process, the batch size for sampling was set to 64. These molecules were labeled with their four scores as the rewards, and the desired molecules were saved during sampling.2.6 Baselines and training setting
Frag-MB/Frag-GB and MB/GB are regarded as four baselines for comparison with the studied generative models. All workflows and datasets remain consistent across models, except that the prediction models in the baseline models were trained on data labeled with ΔES1T1,34 which favors the generation of TADF molecules. For each model, the fine-tuning process of RL was conducted for 5000 iterations. To mitigate experimental variability, each RL fine-tuning procedure was repeated three times. This study employed two distinct sets of evaluation metrics as benchmarks to rigorously assess the performance of the generative models. The first standard metrics were outlined by Jin et al.,55 while the second metrics were adapted from the MOSES benchmarking platform,56 a widely recognized standard in de novo molecular generation. Besides, to evaluate the fragments of generated molecules more effectively, we used conjugate motif diversity, which reflects the hierarchical organization of molecular motifs and better guides models toward generating triplet PSs.3 Results and discussion
3.1 Generation efficiency
To assess the generation efficiency of each model, we analyzed the relationship between the number of steps and the cumulative number of unique desired molecules (referred to as successful molecules for simplicity). As shown in Tasks 1 and 2 of Fig. 2, the number of successful molecules increased with the generation steps, indicating that the model had effectively learned the association between conditional inputs and generated molecules. However, the generation efficiency differed across models. In Task 1 (left panel of Fig. 2), MD/MB exhibited a faster generation rate of successful molecules compared to GD/GB. Similarly, in Task 2 (right panel of Fig. 2), Frag-MD/Frag-MB demonstrated a faster generation rate than Frag-GD/Frag-GB. This suggests that, the RNN, derived from knowledge distillation of the conditional transformer decoder, enhanced the generation efficiency of successful molecules. More specifically, in Task 1 (left panel of Fig. 2), MD significantly outperformed MB, whereas GD showed a marginal improvement over GB. This finding demonstrated that our scoring function, as a more accurate prediction tool, also contributed to enhancement of generation efficiency in the knowledge distillation-based RNN model. A similar result was observed in Task 2, as shown in the right panel of Fig. 2. When comparing the number of successful molecules generated by MD (the top model in Task 1) with those generated by Frag-MD (the top model in Task 2), we found that MD produced substantially more successful molecules. Additionally, the average number of tokens for MD (88.9) was significantly higher than that for Frag-MD (7.6), indicating a more diverse range of modification for MD. In conclusion, the efficient generation and diverse modification for the character-based model significantly facilitate the exploration of novel molecules. | ||
| Fig. 2 Relationship between the number of successfully generated molecules and the steps in RL of Task 1 (left) and Task 2 (right). | ||
3.2 Diversity and size of conjugated motifs
To further assess the diversity of conjugated motif level (rather than molecular level) generation by the MD model in Task 1, we collected the conjugated motifs in the generated molecules. This involved splitting the generated motifs, selecting unique instances, and removing duplicates. Encouragingly, the MD model exhibited 18836 unique conjugated motifs, nearly twice the number (9754) observed in Frag-MD, further indicating that MD is well-suited for de novo triplet PS design in Task 1 by exploring novel conjugated motifs rather than relying solely on molecule modification or derivation. To examine the size of conjugated motifs, the distributions of ring numbers and atoms numbers of conjugated motifs for the successfully generated molecules were analyzed (Fig. 3). Frag-MD exhibited slightly higher average ring numbers (1.53 vs. 1.28) and atom counts (13.42 vs. 11.16), suggesting that the fragment-based generation method is more advantageous for creating larger motifs, which may exhibit longer wavelength absorption and demonstrate better applicability in PDT. The results for other models are provided in ESI Table S2.†
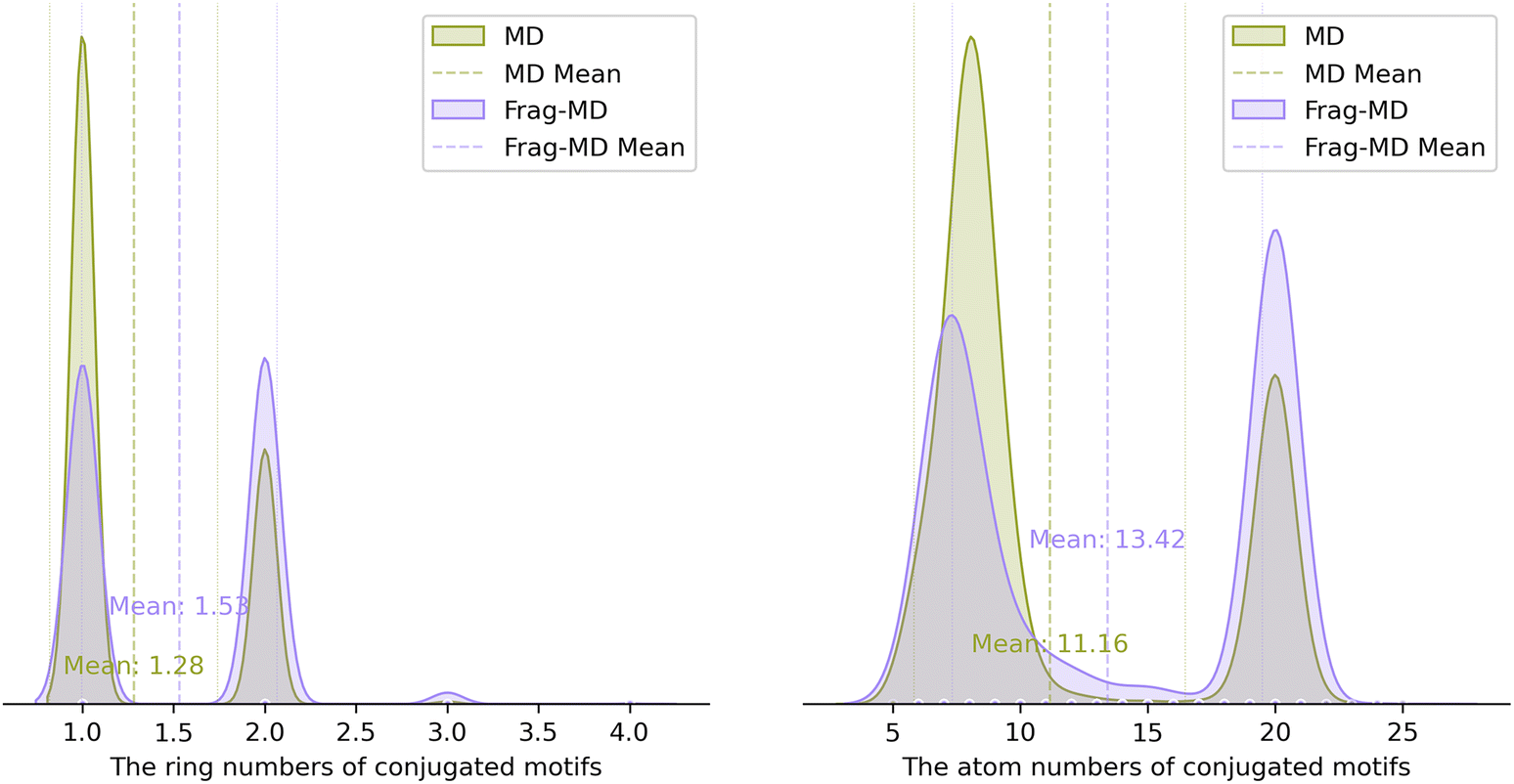 | ||
| Fig. 3 Distribution of the ring numbers of conjugated motifs (left) as well as the atom numbers of conjugated motifs (right) for the successful molecules. | ||
3.3 Benchmark performance
As part of this analysis, we collected the total number of molecules generated in each cycle to evaluate the performance of all models against the benchmarks. The average values are summarized in ESI Table S2.† Regarding conditional metrics, the MD model performed the best in terms of Novelty, whereas the Frag-MD model ranked the last. Additionally, all character-based methods exhibited superior performance compared to their fragment-based counterparts across the Novelty and Div metrics.This observation further demonstrated that character-based molecule generation models are more likely to yield diverse derivatives, consistent with the findings presented in Fig. 2. However, the MG model exhibited the highest diversity, slightly surpassing MD. This result can be attributed to the constraints imposed by the chemical space limitations of the transformer decoder.44 With respect to the MOSES metrics, Frag-MD performed the best in terms of Unique and Frag, despite the overall poor performance of all models. Hence, Frag-MD is proficient in fragment combination and fragment derivation for identifying desired triplet PSs. The MOSES metrics, including SNN and IntDiv, showed similar trends to Novelty and Div observed in the conditional metrics. Although the metrics for the four studied models were compared with their baseline counterparts in both Task 1 and Task2, not all models showed better performance. For example, Frag-MD performed notably worse than its baseline, Frag-MB, in Novelty metrics, and MD exhibited substantially lower diversity compared to its baseline, MB. All studied models consistently showed improved diversity and larger conjugated motifs, as detailed in ESI Table S2.†
We visualized the property distributions of the total successfully generated molecules across all models in both Task 1 and Task 2. As depicted in Fig. 4 and 5 for Task 1 and Task 2, the distributions of ΔEST and Eabs varied among four models, and a uniform distribution was observed across all models. Notably, the left-skewed distribution curves of Eabs, generated by the MD/GD and Frag-MD/Frag-GD models, when compared to their respective baselines, exhibited their capability to generate larger conjugated molecules (Fig. 5). The underlying logic is that a lower Eabs energy level corresponds to longer absorption wavelengths, making these molecules better candidates for triplet PSs. However, the desired left-skewed distribution curves of ΔEST are not observed in Fig. 4, indicating a limitation in the models' ability to accurately reproduce the ΔEST property. To explore potential reasons behind this discrepancy, we performed DFT calculations to obtain ground-truth data.
 | ||
| Fig. 4 Distribution of ΔEST for the successful molecules generated by all models for Task 1 based on the character-based method (left) and Task 2 based on the fragment-based model (right). | ||
 | ||
| Fig. 5 Distribution of Eabs for the successful molecules generated by all models for Task 1 (left) and Task 2 (right). | ||
3.4 Theoretical validation by DFT calculation
We first collected two datasets of the successful molecules generated by all studied models (dataset 1) and their baseline models (dataset 2), respectively. One key difference between these datasets lies in the ground truth used in the prediction model: ΔESnTn for the studied models and ΔES1T1 for the baseline models. Initially, each generated dataset was labeled exclusively with its own prediction function, implying that each molecule just has one prediction value. Subsequently, we incorporated the counterpart's prediction model to add a secondary prediction value. Ultimately, two sub-datasets (sub-dataset 1 and sub-dataset 2) were formed by selecting molecules with prediction values for both ΔESnTn and ΔES1T1 below 0.3 eV, thereby classifying these molecules as successful molecules in both the baseline and studied models. To further validate these molecules, the ground truth of the two sub-datasets was obtained by DFT calculation. Encouragingly, the ground truth distribution of ΔESnTn predominantly fell below 0.3 eV, whereas the ground truth distribution of ΔES1T1 largely exceeded 0.3 eV, with some values even surpassing 1.0 eV (Fig. 6). This observation highlights the superior prediction accuracy of the investigated models compared to the baselines. According to the ground truth distribution of ΔEST in sub-dataset 1 (left panel in Fig. 6), we observed that the baseline models (ΔES1T1 as ground truth) exhibited a low accuracy rate of 4%, implying a high error rate of 96% in generating successful molecules. Conversely, the studied models (ΔESnTn as groundtruth) exhibited a high accuracy of 73%, resulting in a low error rate of 27% in generating successful molecules. More interestingly, the ground truth distribution of ΔEST below 0.3 eV exhibited a Gaussian-like pattern rather than being concentrated around zero, which is conducive to multiple ISC mechanisms. Similar results were observed in the distribution of ΔEST in sub-dataset 2 (right panel in Fig. 6).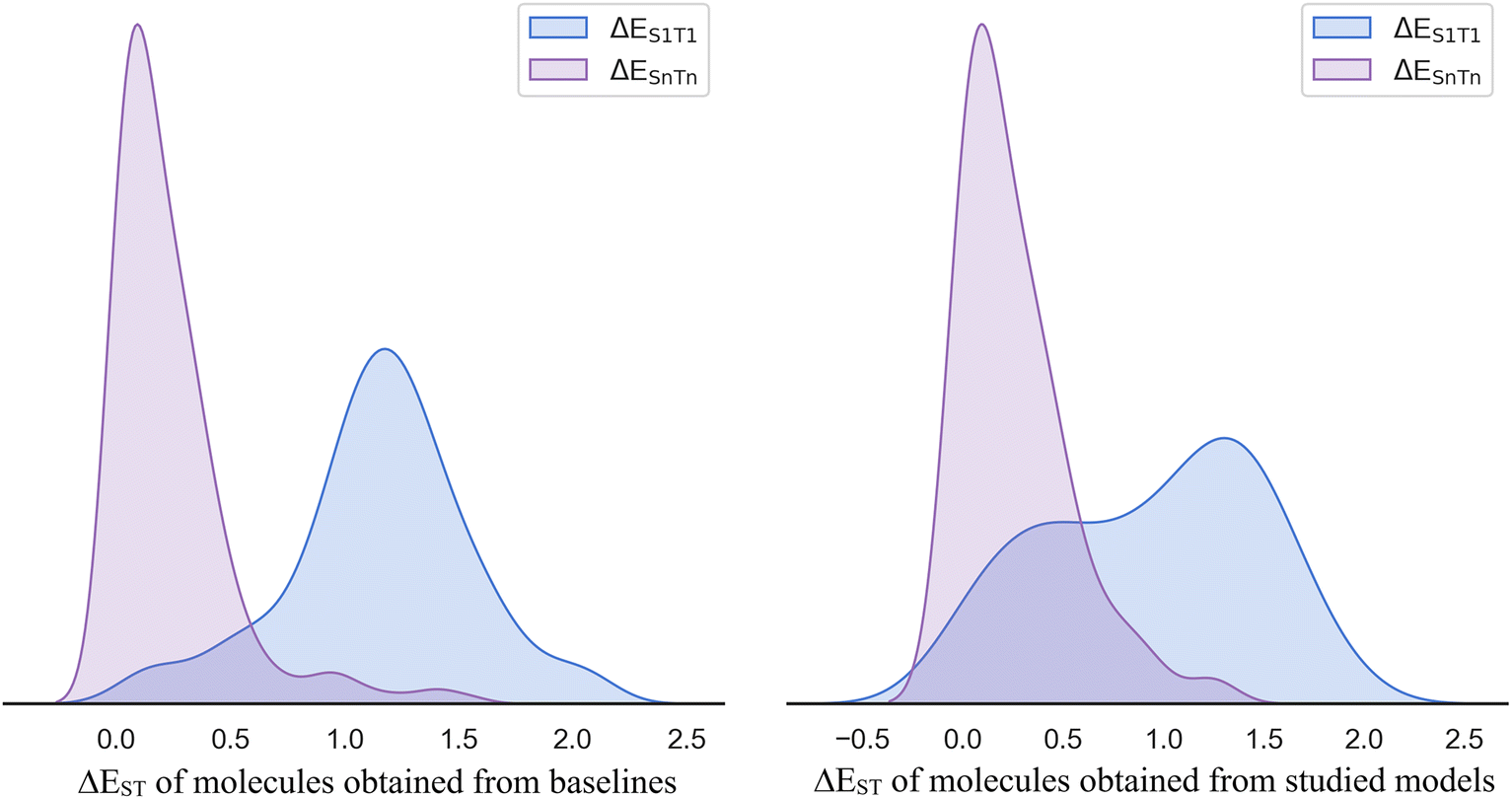 | ||
| Fig. 6 The ground truth distributions of ΔEST of sub-dataset 1 (left) and sub-dataset 2 (right); ΔESnTn is the ground truth for the studied models and ΔES1T1 is the ground truth for the baseline models. | ||
3.5 Ablation studies
Boron dipyrromethene (BODIPY) compounds have been recognized as one of the most promising types of PSs for PDT, according to their excellent photophysical properties, including high extinction coefficients of visible light, superior photostability, and robust chemical stability, as well as practical and efficient derivatization capabilities. Novel BODIPY derivatives functioning as triplet PSs are continuously discovered based on expert knowledge.57 To demonstrate the effectiveness of our DL-based methods to discover heavy-atom-free triplet PSs, we conducted ablation studies by systematically excluding the SMILES representations of BODIPY derivatives from the training and validation datasets, evaluated by Tanimoto similarity (Tanimoto coefficient > 0.4).58To generate BODIPY-constrained molecules, we employed a scaffold-constrained method59 for both the de novo design scenario and the fragment-based molecular generation scenario. In the de novo design scenario, Fig. 7a demonstrates that our character-based method is capable of not only generating structure-similar published compounds but also discovering novel, unpublished compounds. Specifically, M2, a structure-similar published compound, is a fluorescein probe,60 while M1, a derivative molecule of M2, probably exhibits strong absorption in the visible region and shows different triplet properties, which indicates M1 is a potential triplet PS. On the other hand, M3 and M4 can represent de novo compounds, with the chrysene motif in M3 and the phenanthrene motif in M4 that never exist in the training and validation datasets, which are different from the following unpublished molecules by the fragment-based method. Thus, the character-based method demonstrates robust capability to design novel triplet PSs, showing its potential for advancing PDT. In the fragment-based molecular generation scenario, Fig. 7b illustrates that, in addition to structure-similar compounds (M5 as a derivatization of M6 (ref. 61) and M7 as a de-derivatization of M8,62 represented as potential triplet PSs) and unpublished compounds (M11 and M12, represented as a novel fragment combination), known published molecules (M9 (ref. 63) and M10 (ref. 64)) were successfully designed. The ISC process of M10 has been meticulously investigated, and its underlying mechanism is elucidated as the SOCT-ISC mechanism. Similarly, the high triplet quantum yield (ΦT = 0.60) of M9 also comes from the SOCT-ISC mechanism. Furthermore, M9 shows red-shifted spin-allowed charge transfer absorption. These two published molecules highlight the ability of our fragment-based method in fragment combination and fragment derivation in searching for desired triplet PSs. Similarly, other case studies about naphthalimide (NI) derivatives and perylenemonoimide (PMI) derivatives are presented in ESI Fig. S8 and S9.† Our fragment-based method successfully re-identified four well-known published molecules, including two TADF and two non-TADF molecules for NI derivatives. Moreover, published perylene (Pery)-phenothiazine (PTZ) triplet photosensitizers (PSs) were obtained through both Pery-constrained and PTZ-constrained case studies, as detailed in ESI Fig. S10 and S11.†
 | ||
| Fig. 7 Sampled generated molecules by different methods. (a) De novo design method and (b) fragment-based molecule generation method. Note: M1, M5, and M7 represent designed compounds, while M2, M6, and M8 correspond to structure-similar published compounds. | ||
4 Conclusion
In this study, we have successfully developed DL-based methods for de novo design and motif optimization of heavy-atom-free triplet photosensitizers (PSs), which incorporate multiple intersystem crossing (ISC) mechanisms. In particular, we developed a comprehensive dataset (∼1.90 × 109) of triplet PSs for the first time, which included a variety of molecular types, favoring multiple ISC mechanisms. Subsequently, we trained a prediction model with the high excited singlet–triplet gap (ΔESnTn ≤ 0.3 eV as the ground truth, n ≥ 1) for the scoring function. To search de novo conjugate motifs (Task 1) as well as perform a conjugated-motif-based modification (Task 2) for desired triplet PSs, a reinforcement learning (RL) network was employed. The silicon experimental results revealed that the Frag-MD model outperformed the MD model in generating larger conjugated motifs, with higher average ring numbers (1.53 vs. 1.28) and atom counts (13.42 vs. 11.16), which illustrated the advantages of PS modification in Task 2 by creating larger motifs with longer wavelength absorption. In contrast, the MD model generated twice as many unique conjugated motifs and performed better in terms of the conditional and MOSES metrics for novelty and diversity, which showed the de novo design ability of the character-based method in Task 1. Impressively, theoretical calculations indicated that our proposed models (Frag-MD/MD and Frag-GD/GD) outperformed their baselines (Frag-MB/MB and Frag-GB/GB) in which the label data of prediction model based on ΔES1T1 has a significantly higher prediction accuracy (73% vs. 4%). High-efficiency triplet PSs reported in recent studies were re-identified through ablation experiments. Thus, our method proves to be highly practical and effective, holding the promise of establishing a new paradigm for the discovery of novel PSs applicable in PDT.Data availability
The code and datasets used in the study are publicly available from the GitHub repository: https://github.com/Kepeng2019/Fragment_and_char_based_PSs_generation. The detailed procedures for constructing the dataset of photosensitizers, prediction models for score function in reinforcement learning, conjugated motif diversity for all models, distribution of QED and SA properties for the unique desired molecules generated by all models for Task 1 and Task 2, as well as DFT/TDDFT calculation results of selected compounds.Author contributions
Yu Kang and Tingjun Hou organized and supervised the research and guided the manuscript writing and experimental design. Kepeng Chen and Jike Wang conceived the overall framework of the character-based and fragment-based models for triplet photosensitizer design. Kepeng Chen constructed the DFT-labeled dataset and performed model validation. Xiaoting Zhang and Wenbo Yang evaluated the generated molecules and proposed practical optimization strategies. Kepeng Chen drafted the manuscript, with critical revisions by Dan Li, Tingjun Hou, Wenbo Yang, and Yu Kang. Yu Kang and Tingjun Hou contributed their expertise in computational chemistry to improve the paper in terms of triplet photosensitizer design. Jike Wang provided specialized guidance on neural network adaptation for this task.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Fundamental Research Funds for the Central Universities (DUT25Z2722), National Natural Science Foundation of China (92370130, 22220102001, 82373791, and 22272017), Natural Science Foundation of Zhejiang Province (LD22H300001) and Excellent Youth Fund of Liaoning Province (2024JH3/10200005).References
- R. Pollice, G. dos Passos Gomes, M. Aldeghi, R. J. Hickman, M. Krenn, C. Lavigne, M. Lindner-D'Addario, A. Nigam, C. T. Ser, Z. Yao and A. Aspuru-Guzik, Data-driven strategies for accelerated materials design, Acc. Chem. Res., 2021, 54, 849–860 CrossRef CAS PubMed.
- R. Baskaran, J. Lee and S.-G. Yang, Clinical development of photodynamic agents and therapeutic applications, Biomater. Res., 2018, 22, 25 CrossRef PubMed.
- X. Zhao, J. Liu, J. Fan, H. Chao and X. Peng, Recent progress in photosensitizers for overcoming the challenges of photodynamic therapy: from molecular design to application, Chem. Soc. Rev., 2021, 50, 4185–4219 RSC.
- J. Xie, Y. Wang, W. Choi, P. Jangili, Y. Ge, Y. Xu, J. Kang, L. Liu, B. Zhang, Z. Xie, J. He, N. Xie, G. Nie, H. Zhang and J. S. Kim, Overcoming barriers in photodynamic therapy harnessing nano-formulation strategies, Chem. Soc. Rev., 2021, 50, 9152–9201 RSC.
- R. Gómez-Bombarelli, J. Aguilera-Iparraguirre, T. D. Hirzel, D. Duvenaud, D. Maclaurin, M. A. Blood-Forsythe, H. S. Chae, M. Einzinger, D.-G. Ha, T. Wu, G. Markopoulos, S. Jeon, H. Kang, H. Miyazaki, M. Numata, S. Kim, W. Huang, S. I. Hong, M. Baldo, R. P. Adams and A. Aspuru-Guzik, Design of efficient molecular organic light-emitting diodes by a high-throughput virtual screening and experimental approach, Nat. Mater., 2016, 15, 1120–1127 CrossRef PubMed.
- H. Mai, T. C. Le, D. Chen, D. A. Winkler and R. A. Caruso, Machine learning for electrocatalyst and photocatalyst design and discovery, Chem. Rev., 2022, 122, 13478–13515 CrossRef CAS PubMed.
- J. Westermayr and P. Marquetand, Machine learning for electronically excited states of molecules, Chem. Rev., 2021, 121, 9873–9926 CrossRef CAS PubMed.
- P. Li, Z. Wang, W. Li, J. Yuan and R. Chen, Design of thermally activated delayed fluorescence materials with high intersystem crossing efficiencies by machine learning-assisted virtual screening, J. Phys. Chem. Lett., 2022, 13, 9910–9918 CrossRef CAS PubMed.
- J.-M. Kim, K. H. Lee and J. Y. Lee, Extracting polaron recombination from electroluminescence in organic light-emitting diodes by artificial intelligence, Adv. Mater., 2023, 35, 2209953 CrossRef CAS PubMed.
- N. Noto, A. Yada, T. Yanai and S. Saito, Machine-learning classification for the prediction of catalytic activity of organic photosensitizers in the nickel(II)-salt-induced synthesis of phenols, Angew. Chem., Int. Ed., 2023, 62, e202219107 CrossRef CAS PubMed.
- D. M. Anstine and O. Isayev, Generative models as an emerging paradigm in the chemical sciences, J. Am. Chem. Soc., 2023, 145, 8736–8750 CrossRef CAS PubMed.
- R. L. Greenaway and K. E. Jelfs, Integrating computational and experimental workflows for accelerated organic materials discovery, Adv. Mater., 2021, 33, 2004831 CrossRef CAS PubMed.
- A. Nigam, R. Pollice, P. Friederich and A. Aspuru-Guzik, Artificial design of organic emitters via a genetic algorithm enhanced by a deep neural network, Chem. Sci., 2024, 15, 2618–2639 RSC.
- C.-H. Li and D. P. Tabor, Generative organic electronic molecular design via reinforcement learning integration with quantum chemistry: tuning singlet and triplet energy energy levels, ChemRxiv, preprint, 2023, DOI:10.26434/chemrxiv-2023-bgcjg-v2.
- S. Kang and K. Cho, Conditional molecular design with deep generative models, J. Chem. Inf. Model., 2019, 59, 43–52 CrossRef CAS PubMed.
- L. Shi and W. Xia, Photoredox functionalization of C-H bonds adjacent to a nitrogen atom, Chem. Soc. Rev., 2012, 41, 7687 RSC.
- J. Xuan and W. Xiao, Visible-light photoredox catalysis, Angew. Chem., Int. Ed., 2012, 51, 6828–6838 CrossRef CAS PubMed.
- D. P. Hari and B. König, Die photokatalytische Meerwein-Arylierung: Eine klassische Aryldiazoniumsalz-Reaktion in neuem Licht, Angew. Chem., 2013, 125, 4832–4842 CrossRef.
- D. Ravelli, M. Fagnoni and A. Albini, Photoorganocatalysis. What for?, Chem. Soc. Rev., 2013, 42, 97–113 RSC.
- A. F. Zahrt, J. J. Henle, B. T. Rose, Y. Wang, W. T. Darrow and S. E. Denmark, Prediction of higher-selectivity catalysts by computer-driven workflow and machine learning, Science, 2019, 363, eaau5631 CrossRef CAS PubMed.
- T. N. Singh-Rachford and F. N. Castellano, Photon upconversion based on sensitized triplet-triplet annihilation, Coord. Chem. Rev., 2010, 254, 2560–2573 CrossRef CAS.
- M. Nakashima, K. Iizuka, M. Karasawa, K. Ishii and Y. Kubo, Selenium-containing BODIPY dyes as photosensitizers for triplet-triplet annihilation upconversion, J. Mater. Chem. C, 2018, 6, 6208–6215 RSC.
- X. Cao, D. Zhang, S. Zhang, Y. Tao and W. Huang, CN-containing donor-acceptor-type small-molecule materials for thermally activated delayed fluorescence OLEDs, J. Mater. Chem. C, 2017, 5, 7699–7714 RSC.
- X. Xiong, F. Song, J. Wang, Y. Zhang, Y. Xue, L. Sun, N. Jiang, P. Gao, L. Tian and X. Peng, Thermally activated delayed fluorescence of fluorescein derivative for time-resolved and confocal fluorescence imaging, J. Am. Chem. Soc., 2014, 136, 9590–9597 CrossRef CAS PubMed.
- M. Li, Y. Liu, R. Duan, X. Wei, Y. Yi, Y. Wang and C. Chen, Aromatic-imide-based thermally activated delayed fluorescence materials for highly efficient organic light-emitting diodes, Angew. Chem., Int. Ed., 2017, 56, 8818–8822 CrossRef CAS PubMed.
- P. Chebotaev, A. Buglak, A. Sheehan and A. Filatov, Predicting fluorescence to singlet oxygen generation quantum yield ratio for Bodipy dyes using QSPR and machine learning, Phys. Chem. Chem. Phys., 2024, 26, 25131–25142 RSC.
- L. He, J. Dong, Y. Yang, Z. Huang, S. Ye, X. Ke, Y. Zhou, A. Li, Z. Zhang, S. Wu, Y. Wang, S. Cai, X. Liu and Y. He, Accelerating the discovery of type II photosensitizer: experimentally validated machine learning models for predicting the singlet oxygen quantum yield of photosensitive molecule, J. Mol. Struct., 2025, 1321, 139850 CrossRef CAS.
- Z. Liao, J. Lu, K. Xie, Y. Wang and Y. Yuan, Prediction of Photochemical Properties of Dissolved Organic Matter Using Machine Learning, Environ. Sci. Technol., 2023, 57, 17971–17980 CrossRef CAS PubMed.
- C. Tu, W. Huang, S. Liang, K. Wang, Q. Tisssan and W. Yan, Combining machine learning and quantum chemical calculations for high-throughput virtual screening of thermally activated delayed fluorescence molecular materials: the impact of selection strategy and structural mutations, RSC Adv., 2022, 12, 30962–30975 RSC.
- A. A. Buglak, A. Charisiadis, A. Sheehan, C. J. Kingsbury, M. O. Senge and M. A. Filatov, Quantitative structure-property relationship modelling for the prediction of singlet oxygen generation by heavy-atom-free BODIPY photosensitizers, Chem.–Eur. J., 2021, 27, 9934–9947 CrossRef CAS PubMed.
- A. A. Buglak, M. A. Filatov, M. A. Hussain and M. Sugimoto, Singlet oxygen generation by porphyrins and metalloporphyrins revisited: a quantitative structure-property relationship (QSPR) study, J. Photochem. Photobiol. A, 2020, 403, 112833 CrossRef CAS.
- Q. Zhang, Y. J. Zheng, W. Sun, Z. Ou, O. Odunmbaku, M. Li, S. Chen, Y. Zhou, J. Li, B. Qin and K. Sun, High-efficiency non-fullerene acceptors developed by machine learning and quantum chemistry, Adv. Sci., 2022, 9, 2104742 CrossRef CAS PubMed.
- A. G. Hofmann and A. Agibetov, Artificial intelligence-based molecular property prediction of photosensitizing effects of drugs, J. Drug Target., 2023, 33, 556–561 CrossRef PubMed.
- S. Xu, J. Li, P. Cai, X. Liu, B. Liu and X. Wang, Self-improving photosensitizer discovery system via Bayesian search with first-principle simulations, J. Am. Chem. Soc., 2021, 143, 19769–19777 CrossRef CAS PubMed.
- T. Blaschke, J. Arús-Pous, H. Chen, C. Margreitter, C. Tyrchan, O. Engkvist, K. Papadopoulos and A. Patronov, REINVENT 2.0: an AI tool for de novo drug design, J. Chem. Inf. Model., 2020, 60, 5918–5922 CrossRef CAS PubMed.
- Z. Tan, Y. Li, Z. Zhang, X. Wu, T. Penfold, W. Shi and S. Yang, Efficient adversarial generation of thermally activated delayed fluorescence molecules, ACS Omega, 2022, 7, 18179–18188 CrossRef CAS PubMed.
- M. Park, H. S. Kim, H. Yoon, J. Kim, S. Lee, S. Yoo and S. Jeon, Controllable singlet-triplet energy splitting of graphene quantum dots through oxidation: from phosphorescence to TADF, Adv. Mater., 2020, 32, 2000936 CrossRef CAS PubMed.
- J. Kou, D. Dou and L. Yang, Porphyrin photosensitizers in photodynamic therapy and its applications, Oncotarget, 2017, 8, 81591–81603 CrossRef PubMed.
- G. Gunaydin, M. E. Gedik and S. Ayan, Photodynamic therapy for the treatment and diagnosis of cancer-a review of the current clinical status, Front. Chem., 2021, 9, 686303 CrossRef CAS PubMed.
- V.-N. Nguyen, Y. Yan, J. Zhao and J. Yoon, Heavy-atom-free photosensitizers: from molecular design to applications in the photodynamic therapy of cancer, Acc. Chem. Res., 2021, 54, 207–220 CrossRef CAS PubMed.
- M. Imran, D. Liu, K. Ye, X. Zhang and J. Zhao, The rhodamine-perylene compact electron donor-acceptor dyad: spin-orbit charge-transfer intersystem crossing and the energy balance of the triplet excited states, Photochem, 2024, 4, 40–56 CrossRef CAS.
- K. Chen, Y. Dong, X. Zhao, M. Imran, G. Tang, J. Zhao and Q. Liu, Bodipy derivatives as triplet photosensitizers and the related intersystem crossing mechanisms, Front. Chem., 2019, 7, 821 CrossRef CAS PubMed.
- J. Wang, C.-Y. Hsieh, M. Wang, X. Wang, Z. Wu, D. Jiang, B. Liao, X. Zhang, B. Yang, Q. He, D. Cao, X. Chen and T. Hou, Multi-constraint molecular generation based on conditional transformer, knowledge distillation and reinforcement learning, Nat. Mach. Intell., 2021, 3, 914–922 CrossRef.
- J. Wang, Y. Zeng, H. Sun, J. Wang, X. Wang, R. Jin, M. Wang, X. Zhang, D. Cao, X. Chen, C.-Y. Hsieh and T. Hou, Molecular generation with reduced labeling through constraint architecture, J. Chem. Inf. Model., 2023, 63, 3319–3327 CrossRef CAS PubMed.
- M. Olivecrona, T. Blaschke, O. Engkvist and H. Chen, Molecular de-novo design through deep reinforcement learning, J. Cheminform., 2017, 9, 48 CrossRef PubMed.
- K. Kawai, K. Yoshimaru and Y. Takahashi, Generation of target-selective drug candidate structures using molecular evolutionary algorithm with SVM classifiers, J. Comput. Chem. Jpn., 2011, 10, 79–87 CrossRef CAS.
- N. Ståhl, G. Falkman, A. Karlsson, G. Mathiason and J. Boström, Deep reinforcement learning for multiparameter optimization in de novo drug design, J. Chem. Inf. Model., 2019, 59, 3166–3176 CrossRef PubMed.
- K. Kawai, N. Nagata and Y. Takahashi, De novo design of drug-like molecules by a fragment-based molecular evolutionary approach, J. Chem. Inf. Model., 2014, 54, 49–56 CrossRef CAS PubMed.
- J. F. Joung, M. Han, M. Jeong and S. Park, Experimental database of optical properties of organic compounds, Sci. Data, 2020, 7, 295 CrossRef CAS PubMed.
- R. Ramakrishnan, P. O. Dral, M. Rupp and O. A. Von Lilienfeld, Quantum chemistry structures and properties of 134 kilo molecules, Sci. Data, 2014, 1, 140022 CrossRef CAS PubMed.
- J. Gou, B. Yu, S. J. Maybank and D. Tao, Knowledge distillation: a survey, Int. J. Comput. Vis., 2021, 129, 1789–1819 CrossRef.
- K. Chen, J. Zhao, X. Li and G. G. Gurzadyan, Anthracene-naphthalenediimide compact electron donor/acceptor dyads: electronic coupling, electron transfer, and intersystem crossing, J. Phys. Chem. A, 2019, 123, 2503–2516 CrossRef CAS PubMed.
- F. Scarselli, M. Gori, A. C. Tsoi, M. Hagenbuchner and G. Monfardini, The graph neural network model, IEEE Trans. Neural Netw., 2009, 20, 61–80 Search PubMed.
- S. Liu, M. Chen, X. Yao and H. Liu, Fingerprint-enhanced hierarchical molecular graph neural networks for property prediction, J. Pharm. Anal., 2025, 15, 101242 CrossRef PubMed.
- W. Jin, R. Barzilay and T. Jaakkola, Multi-objective molecule generation using interpretable substructures, Proc. Mach. Learn. Res., 2020, 119, 4849–4859 Search PubMed.
- D. Polykovskiy, A. Zhebrak, B. Sanchez-Lengeling, S. Golovanov, O. Tatanov, S. Belyaev, R. Kurbanov, A. Artamonov, V. Aladinskiy, M. Veselov, A. Kadurin, S. Johansson, H. Chen, S. Nikolenko, A. Aspuru-Guzik and A. Zhavoronkov, Molecular sets (MOSES): a benchmarking platform for molecular generation models, Front. Pharmacol, 2020, 11, 565644 CrossRef CAS PubMed.
- J. Zhao, K. Xu, W. Yang, Z. Wang and F. Zhong, The triplet excited state of Bodipy: formation, modulation and application, Chem. Soc. Rev., 2015, 44, 8904–8939 RSC.
- D. Bajusz, A. Rácz and K. Héberger, Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations?, J. Cheminform., 2015, 7, 20 CrossRef PubMed.
- Z. Liu, Z. Guo, Y. Yao, Z. Cen, W. Yu, T. Zhang and D. Zhao, Constrained decision transformer for offline safe reinforcement learning, Proc. Mach. Learn. Res, 2023, 202, 21611–21630 Search PubMed.
- Z. Zhou and T. Maki, Ratiometric fluorescence acid probes based on a tetrad structure including a single BODIPY chromophore, J. Org. Chem., 2021, 86, 17560–17566 CrossRef CAS PubMed.
- M. Tobisu, Y. Masuya, K. Baba and N. Chatani, Palladium(II)-catalyzed synthesis of dibenzothiophene derivatives via the cleavage of carbon-sulfur and carbon-hydrogen bonds, Chem. Sci., 2016, 7, 2587–2591 RSC.
- L. Wang and Y. Qian, A SOCT-ISC type photosensitizer coumarin-BODIPY promoted by AIE effect: mechanism of singlet oxygen generation, simulated PDT in A-549 cells and fluorescence imaging in zebrafish, Dyes Pigm., 2021, 195, 109711 CrossRef CAS.
- Z. Wang, J. Zhao, M. Di Donato and G. Mazzone, Increasing the anti-Stokes shift in TTA upconversion with photosensitizers showing red-shifted spin-allowed charge transfer absorption but a non-compromised triplet state energy level, Chem. Commun., 2019, 55, 1510–1513 RSC.
- M. Kosaka, K. Miyokawa and Y. Kurashige, Molecular mechanism of the triplet states formation in Bodipy-phenoxazine photosensitizer dyads confirmed by ab initio prediction of the spin polarization, Phys. Chem. Chem. Phys., 2024, 26, 29449–29456 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc03192c |
| This journal is © The Royal Society of Chemistry 2025 |