
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh-efficiency enzymatic biodegradation of polypropylene-based melt-blown fabric debris†
Xiu Huang ab,
Li Huanga,
Qian Wanga,
Qiurong Hea,
Zunzhen Zhanga,
Qian Liu*bc and
Guibin Jiangbc
ab,
Li Huanga,
Qian Wanga,
Qiurong Hea,
Zunzhen Zhanga,
Qian Liu*bc and
Guibin Jiangbc
aWest China School of Public Health and West China Fourth Hospital, Sichuan University, Chengdu 610041, China
bState Key Laboratory of Environmental Chemistry and Ecotoxicology, Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, Beijing 100085, China. E-mail: qianliu@rcees.ac.cn
cCollege of Resources and Environment, University of Chinese Academy of Sciences, Beijing 100190, China
First published on 30th June 2025
Abstract
The coronavirus pandemic (COVID-19) has led to a dramatic increase in the usage and disposal of disposable masks worldwide, most of which are manufactured from melt-blown fabric composed of polypropylene (PP). Biodegradation of PP is considered to be extremely challenging because of its saturated chemical structure and high stability. In this study, we discovered that PP-based melt-blown fabric debris (MBFD; Mw, ∼157 kDa; crystallinity, 40–60%) and PP industrial particles (Mw, 100 kDa; crystallinity, 40–50%) can be degraded by the natural metabolizing enzyme glutathione S-transferase (GST) and digestive enzyme trypsin. The degradation efficiency of MBFD reached 86.7% (trypsin) and 99.2% (GST) with degradation rates of 1.57 and 3.98 g L−1 h−1, respectively, under physiological conditions at 1 atm. Chemical multi-fingerprinting analysis revealed that the mechanisms of MBFD degradation included oxidation and unreported nitridation pathways. Molecular dynamics simulations demonstrated the stability of the enzyme–PP complex system, primarily attributed to hydrophobic interactions. This enzymatic method was also successfully applied to degrade MBFD from disposable activated carbon masks, PP industrial particles, and polyethylene (PE) industrial particles. Finally, we demonstrated the potential of this enzymatic method in biodegradation of MBFD and PP industrial particles in real environmental wastewaters and human serum.
Introduction
The outbreak of the coronavirus pandemic (COVID-19) has led to an unprecedented increase in the consumption of disposable masks, due to their effectiveness in curbing viral transmission.1–6 It is estimated that approximately 340 million disposable medical masks are used daily globally.7 However, the usage and disposal of disposable masks have resulted in a range of environmental and bio-safety issues around the world, such as secondary transmission of pathogens, plastic pollution, and increased health risks to humans.8–12 The production of disposable masks accounts for 1.05% of the global plastic consumption, while the recycling rate for masks is less than 1%.9 More importantly, the mechanical properties of masks deteriorate over time in the environment, becoming more fragile and prone to breaking down, which leads to the generation of micro/nanoplastic particles. Recent reports suggest that microplastics released from face masks pose a biohazard and may increase the mortality risk.13–18 These findings underscore the need to assess the potential environmental and health risks associated with disposable mask materials.The treatment of melt-blown fabrics, the main material in disposable masks, is crucial for reducing the environmental and health risks associated with masks.19 Melt-blown fabrics provide comfort during extended wear due to their lightweight and breathable nature.20 Polypropylene (PP) is the most commonly used material in making protective masks and is extensively used in producing melt-blown fabrics.21 The structural design of PP melt-blown fabrics enhances breathability by optimizing air circulation and moisture release.21 Current treatment methods for melt-blown fabrics include photodegradation, thermal degradation, mechanical degradation, and chemical degradation (including hydrolysis and oxidative degradation).22,23 However, these methods often have low efficiency and limited treatment capacity, potentially leading to secondary pollution risks. In particular, PP materials have significant environmental concerns because they are non-biodegradable.24 Specifically, under various degradation conditions, such as aerobic or anaerobic environments, photo-oxidation, and thermal oxidation, it can take over 10 years for PP-based melt-blown fabrics to fully degrade.25 Consequently, there is an urgent need to develop effective and eco-friendly biodegradation technologies for degrading melt-blown fabrics.
Our previous study unexpectedly discovered that PET plastic can be efficiently degraded by glutathione S-transferase (GST), a natural phase II metabolic isozyme of mammalian origin, under mild conditions.26 This finding highlights the potential of natural enzymes in degrading PET plastic. PET plastic is a thermoplastic polyester composed of monomers terephthalic acid (TPA) and ethylene glycol (EG) with oxidizable and hydrolyzable groups (–COO– and –CO–). Ester bonds in the polymer backbone of PET are more susceptible to biodegradation compared to C–C bonds.27 Chemical depolymerization of PET can be achieved through various reactions like hydrolysis, glycolysis, methanolysis, aminolysis, and ammonolysis.28 PP is much more resistant to stress cracking than PET, due to the fact that PP has too high molecular weight, too much hydrophobicity, too much bonding energy, and too little bioaccessibility, making it difficult to be degraded by biological enzymes in the environment.29 Specifically, (1) the stable chemical structure of PP, mainly consisting of –C–C– and –C–H– covalent bonds, lacks groups prone to oxidation and hydrolysis. Additionally, the absence of enzymes capable of directly breaking the C–C bond in nature impedes their biodegradation; (2) the long-chain structure with a high molecular weight (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–40000 g mol−1) creates spatial site resistance, preventing biological enzymes from accessing active sites for degradation;30,31 (3) PP, being highly hydrophobic and composed of three stereoisomers (isotactic, syndiotactic, and atactic), exhibits a hydrophobic surface that enhances its environmental resistance.32,33 This hydrophobicity makes it challenging for enzymes to interact with it for degradation. Therefore, there are very few reports on the biodegradation of PP plastics. Efficient and highly specific degradation enzymes have not been reported, and the mechanism of degradation remains largely unknown.34 To the best of our knowledge, there have been no reports on the degradation of PP by natural enzymes, particularly those of mammalian and human origin, which play a crucial role in metabolic transformations in living organisms.
000–40000 g mol−1) creates spatial site resistance, preventing biological enzymes from accessing active sites for degradation;30,31 (3) PP, being highly hydrophobic and composed of three stereoisomers (isotactic, syndiotactic, and atactic), exhibits a hydrophobic surface that enhances its environmental resistance.32,33 This hydrophobicity makes it challenging for enzymes to interact with it for degradation. Therefore, there are very few reports on the biodegradation of PP plastics. Efficient and highly specific degradation enzymes have not been reported, and the mechanism of degradation remains largely unknown.34 To the best of our knowledge, there have been no reports on the degradation of PP by natural enzymes, particularly those of mammalian and human origin, which play a crucial role in metabolic transformations in living organisms.
In this study, we tested the feasibility of biodegradation of PP-based melt-blown fabric debris (MBFD) with several typical natural enzymes. Three enzymes—glutathione S-transferase (GST), cytochrome P450 (CYP450), and trypsin—were selected for the biodegradation of MBFD due to their critical roles in biochemical reactions within living organisms, particularly in drug metabolism and conversion of bioactive substances.35–37 Among them, CYP450 and GST are among the most important and representative biometabolic enzymes, making them ideal candidates for studying MBFD degradation in biological systems.35,36 Trypsin, as a key digestive enzyme, serves as the first line of defense following MBFD ingestion, making its role crucial in assessing the fate of MBFD in organisms and subsequent degradation.38 The low cost and ease of production of trypsin also enhance its potential for large-scale industrial applications. Furthermore, previous studies have shown that GST, trypsin, and CYP450 can degrade high-molecular weight polymers, suggesting their potential in breaking down PP-based MBFD as well.26,39–42 We found that GST and trypsin can efficiently degrade MBFD under mild conditions. In contrast, CYP450 showed low efficiency in degradation of MBFD. Small molecule inhibitors and an organic solvent (acetonitrile, ACN) were used to verify the role of enzymes. Multiple characterization techniques, such as scanning electron microscopy (SEM), X-ray photoelectron spectroscopy (XPS), energy-dispersive X-ray spectroscopy (EDX), and laser desorption/ionization time-of-flight mass spectrometry (LDI-TOF MS) were used to further explore the degradation pathways and mechanisms. We also employed molecular docking and molecular dynamics simulations to investigate the molecular interaction between PP and enzymes. Finally, we applied the enzymatic method in the biodegradation of MBFD in real environmental wastewaters, demonstrating the practical applicability of our findings.
Results and discussion
Monitoring of biological degradation of MBFD
Biological enzymes serve as vital factories for the production and transformation of various products in the body, representing a hub of environmental detoxification. To investigate the potential of these enzymes for biodegradation of melt-blown fabrics used in masks, we selected three enzymes, including a commonly used digestive enzyme (trypsin) and two metabolic enzymes (i.e., GST and CYP450).We first characterized the morphology and size of MBFD co-incubated with trypsin. The pristine MBFD after ball milling exhibited a morphology consisting of flakes and strips, with lengths and widths less than 10 μm and 1.2 μm, respectively (Fig. 1A). Upon degradation in the dark, MBFD transformed from fibrous structures to granular materials, with pores of varying sizes appearing at the edges and within the material (Fig. 1A). This result indicated that the degradation occurred simultaneously at both the edges and the interior of the MBFD. After 14 days of incubation with trypsin, the size of the MBFD was significantly reduced to the nanometer level, with degradation products observed at lengths as small as 30 nm (Fig. 1A).
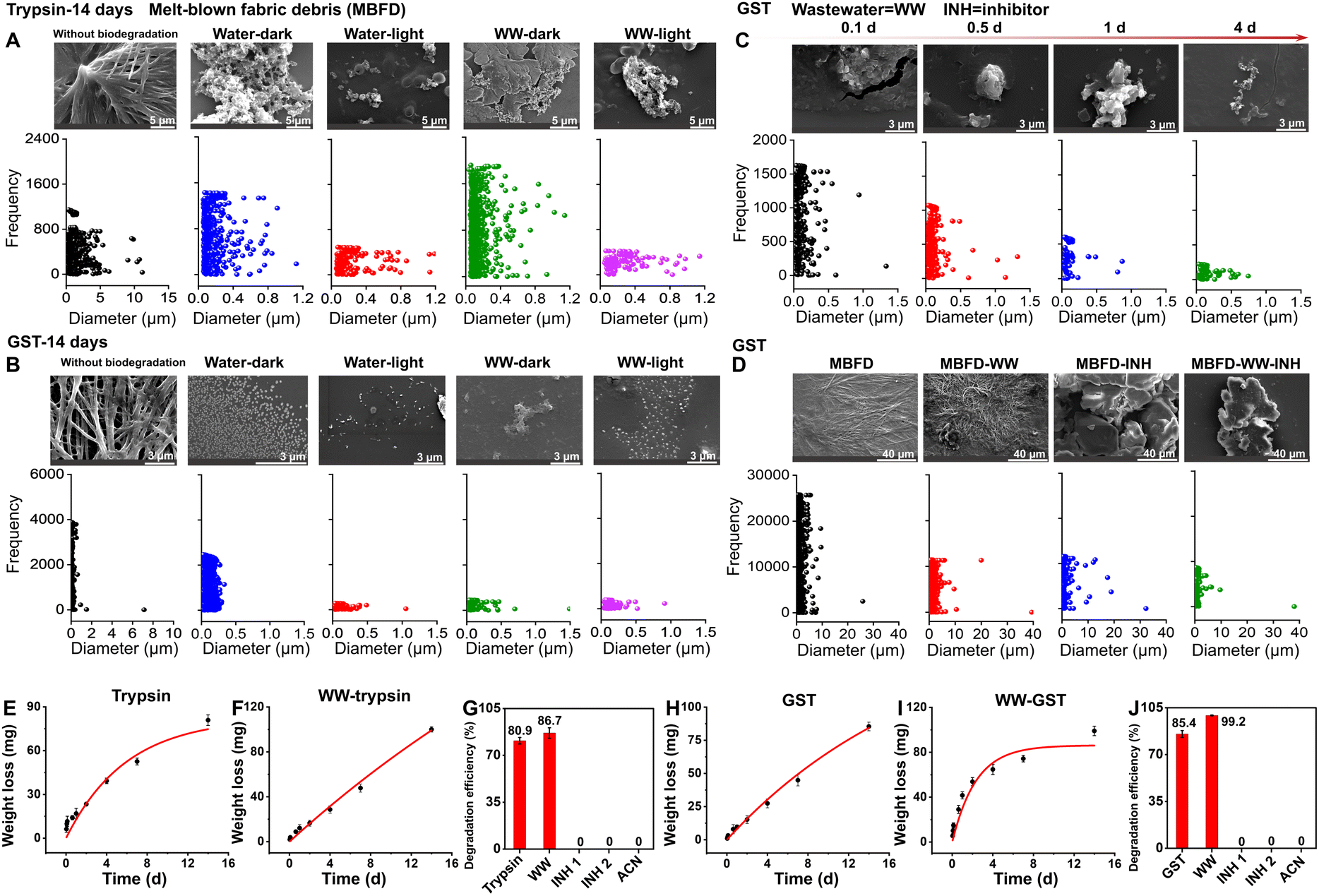 | ||
| Fig. 1 Characterization of morphology, particle size, degradation kinetics, and degradation efficiency during the degradation of MBFD induced by trypsin and GST. (A–D) Characterization of morphology and particle size during the degradation of MBFD induced by trypsin and GST. SEM images and the corresponding size variations under different conditions of treatment with (A) trypsin and (B) GST are shown. (C) Dynamic characterization of MBFD at different incubation times (0.1–4 days), including SEM images and particle size distribution. (D) SEM images and particle size distribution during the degradation of MBFD induced by GST in the presence of small-molecule inhibitors. (E–J) Monitoring the enzymatic depolymerization of MBFD. (E, F, H, and I) Degradation kinetics of MBFD under different conditions. The dynamic curves of MBFD (E and H) in water and (F and I) in wastewater (WW) samples. The degradation efficiency of MBFD treated with (G) trypsin and (J) GST in different ambient environments. Each filled symbol represents the mean ± SD (n = 3). | ||
GST, an important natural metabolic enzyme, was also evaluated. The results obtained with GST were similar to those with trypsin, suggesting that MBFD can also be degraded by GST under mild conditions. As illustrated in Fig. 1B, co-incubation with GST resulted in a granule-like morphology of MBFD. After 14 days of incubation with GST, MBFD was reduced to smaller particles ranging from 16 nm to 500 nm in size (Fig. 1B). SEM images visualized the dynamic process of MBFD degradation over time. As illustrated in Fig. 1C, the enzyme appeared to wrap around the MBFD, gradually breaking it down into smaller particles. After incubation for 14 days in the dark, small particles were observed surrounding the larger ones, indicating the release of degraded materials (Fig. 1B). After just 4 days of incubation, the size distribution of MBFD particles ranged from 6 nm to 750 nm (Fig. 1C). Furthermore, the DLS analysis in Fig. S1† confirms the decreasing trend in particle size variation, transitioning from the micrometer to the nanometer scale. This aligns with the data presented on particle frequency. The use of enzymes from mammals or humans, including metabolic and digestive enzymes, suggests that plastics may be transformed within organisms and humans.26,29,43,44 This transformation can convert plastics into micro/nanoplastics, leading to significant environmental pollution and health risks.45 In human tissues, micro/nanoplastics can cause health issues like inflammatory responses, endocrine disruption, and cell damage.45 It is widely believed that smaller particles can more easily penetrate the human body, causing potential systemic harm.46 At the same time, the degradation of plastics by enzymes is directly linked to their size; generally, smaller particles are easier to degrade.26,47 More research is required to understand how plastics are metabolized and processed in the human body. Specifically, further studies are needed to investigate the degradation process of plastics in living organisms and the human body, focusing on the biotransformation reactions and fate of the resulting degradation products.
Experiments using small-molecule inhibitors and the organic solvent ACN provided compelling evidence for the roles of trypsin and GST in the degradation of MBFD (Fig. 1D, G, J, and S2†). Fig. 1D shows the morphology and size distribution of MBFD in the presence of small-molecule inhibitors and ACN. In the presence of two small-molecule inhibitors (TLK117 and indomethacin), the size of MBFD increased, suggesting aggregation rather than degradation. Further analysis (Fig. 1G and J) shows that in the presence of enzyme inhibitors and ACN, the degradation efficiency of MBFD by trypsin and GST significantly dropped from 80.9% to 0% (TLCK, trypsin inhibitor soybean and ACN) and 85.4% to 0% (TLK117, indomethacin, and ACN). These results strongly support that the degradation of MBFD is enzymatic in nature, as the inhibition of enzyme activity led to a complete cessation of degradation.
Fig. 1E–J show the degradation efficiency and kinetic curves of MBFD, revealing that the degradation follows the first-order kinetics (Fig. 1E–J and Table S1†). When co-incubated with trypsin (Fig. 1E–G) or GST (Fig. 1H–J), MBFD was nearly fully degraded, with the MBFD-to-trypsin ratio ranging from 30.4 to 89.0 kg mg−1 and the MBFD-to-GST ratio ranging from 2.2 to 11.6 kg mg−1. Under the specified incubation time and environmental conditions, the degradation efficiency of MBFD was measured as 80.9% with trypsin (Fig. 1G), which was slightly lower than 85.4% obtained with GST (Fig. 1J). The dynamic degradation curve shows that the degradation rate of MBFD declined over time, eventually reaching a plateau. The maximum degradation rate achieved was 1.57 g L−1 h−1 for trypsin and 3.98 g L−1 h−1 for GST (Table S1†).
We also investigated the potential of CYP450 in MBFD degradation because it is a significant group of hydroxyl-detoxifying enzymes mostly expressed in the liver for phase I metabolic processes. However, the degradation efficiency of MBFD by CYP450 was found to be low at only 0.9% (Fig. S3†). This low efficiency can be attributed to the fact that most CYP450s need NAD(P)H-driven redox partners to function in vivo.39,48 Alternatively, H2O2 or organic surrogates, such as tert-butyl peroxide, cumene hydroperoxide, or iodosylbenzene, can also be used to drive CYP450 catalysis by acting as an oxygen donor to the CYP450 heme and produce catalytically active ferric hydroperoxy species.49,50 Therefore, subsequent experiments focused on GST and trypsin.
Biodegradation of MBFD in real environmental wastewater
To validate the application potential of this method for real-world scenarios, the enzymatic degradation of MBFD was performed in wastewater samples (Fig. 1 and S4†). Since environmental conditions are critical in wastewater applications, we first investigated the effects of environmental factors on the enzymatic degradation performance of MBFD prior to its use in wastewater treatment. Under identical conditions, comparing the degradation in light and dark environments, we found that the particle content and size in light were significantly lower than those in the dark, suggesting that light conditions enhanced the degradation efficiency (Fig. 1A and B). Fig. S2† confirmed the changes in enzyme activity. After 14 days of incubation in the dark, GST and trypsin activity remained stable at around 95% (Fig. S2†). Exposure to light resulted in approximately 5% and 9% increases in GST and trypsin activity, respectively (Fig. S2†), further supporting that light conditions positively influence enzyme efficiency in biodegradation processes. This enhancement may be attributed to the generation of oxygen free radicals through photocatalysis, which, along with increased enzyme activity,51,52 accelerated the degradation of MBFD.53 The underlying mechanism aligns with previous research, demonstrating that UV irradiation or heat treatment can enhance plastic degradation by reducing polymer hydrophobicity and introducing more susceptible functional groups (e.g., C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O or –OH).54–60 Notably, GST plays a pivotal role in managing photo-oxidative stress, serving as a crucial component in exogenous metabolism, oxidative stress response, and cellular signaling pathways.54–60 These biological functions are intrinsically linked to the generation and elimination of reactive oxygen species (ROS), whose production is directly influenced by light exposure, leading to differential enzyme activity in illuminated versus dark environments. The observed increase in GST activity represents an adaptive response to light-induced ROS generation. Furthermore, GST's function as a transducer in UV and red light-regulated signaling pathways suggests that light exposure enhances both its conjugation capacity and catalytic efficiency. This dual mechanism of action – through both ROS- mediated pathways and direct light activation – provides a comprehensive explanation for the enhanced biodegradation observed under illuminated conditions.57
O or –OH).54–60 Notably, GST plays a pivotal role in managing photo-oxidative stress, serving as a crucial component in exogenous metabolism, oxidative stress response, and cellular signaling pathways.54–60 These biological functions are intrinsically linked to the generation and elimination of reactive oxygen species (ROS), whose production is directly influenced by light exposure, leading to differential enzyme activity in illuminated versus dark environments. The observed increase in GST activity represents an adaptive response to light-induced ROS generation. Furthermore, GST's function as a transducer in UV and red light-regulated signaling pathways suggests that light exposure enhances both its conjugation capacity and catalytic efficiency. This dual mechanism of action – through both ROS- mediated pathways and direct light activation – provides a comprehensive explanation for the enhanced biodegradation observed under illuminated conditions.57
Fig. 1F, G, I and J show that both trypsin and GST effectively induced the degradation of MBFD under both dark or light conditions, indicating that the complex matrix of wastewater did no significantly hinder the enzymatic process. We also compared the degradation of MBFD induced by trypsin and GST in wastewater and pure water (Fig. 1). The morphology of MBFD in wastewater was similar with that in pure water (Fig. 1A–D), with both environments showing a transition from irregular to more regular shapes and a decrease in particle size. In wastewater, the particle size of MBFD decreased to a range of 1.2164 nm (Fig. S4†). Remarkably, the degradation efficiency of MBFD in wastewater samples exceeded 99% (Fig. 1G and J), even higher than that in pure water. This result was attributed to the increase of enzyme activity in the wastewater environment, with trypsin activity increasing by ∼3% and GST by ∼11% (Fig. S2†). Furthermore, under light irradiation, the degradation efficiency of MBFD in wastewater was significantly higher than that under dark conditions (Fig. 1A, B and S2†). These findings highlight the potential of natural enzymes to effectively degrade PP plastics in real wastewater samples, presenting a potential eco-friendly strategy for mitigating plastic pollution in wastewater management. To enhance the efficiency and scale of plastic degradation for industrial use, a combination of genetic engineering and microbial technologies can be explored. Additionally, further research is needed to assess the biodegradation of PP in various environmental settings, like soil, and with different types of plastics to better address plastic pollution challenges.
Elemental fingerprints reveal the molecular degradation pathway
We further characterized the elemental fingerprints of MBFD during the degradation process. Since plastics, enzymes, and small-molecule inhibitors all contain C, O, N, P, and S, we mapped the distribution of these five elements and calculated their relative proportions. As illustrated in Fig. 2A and B, following degradation, the proportion of N and O atoms increased, while the proportion of C atoms decreased. This suggests the formation of nitrogen- and oxygen-containing degradation products. In the presence of an inhibitor, however, the proportion of C, N, and O atoms remained stable, while the ratio of P and S atoms increased (Fig. 2C). Similar trends were observed in wastewater samples (Fig. 2D–F).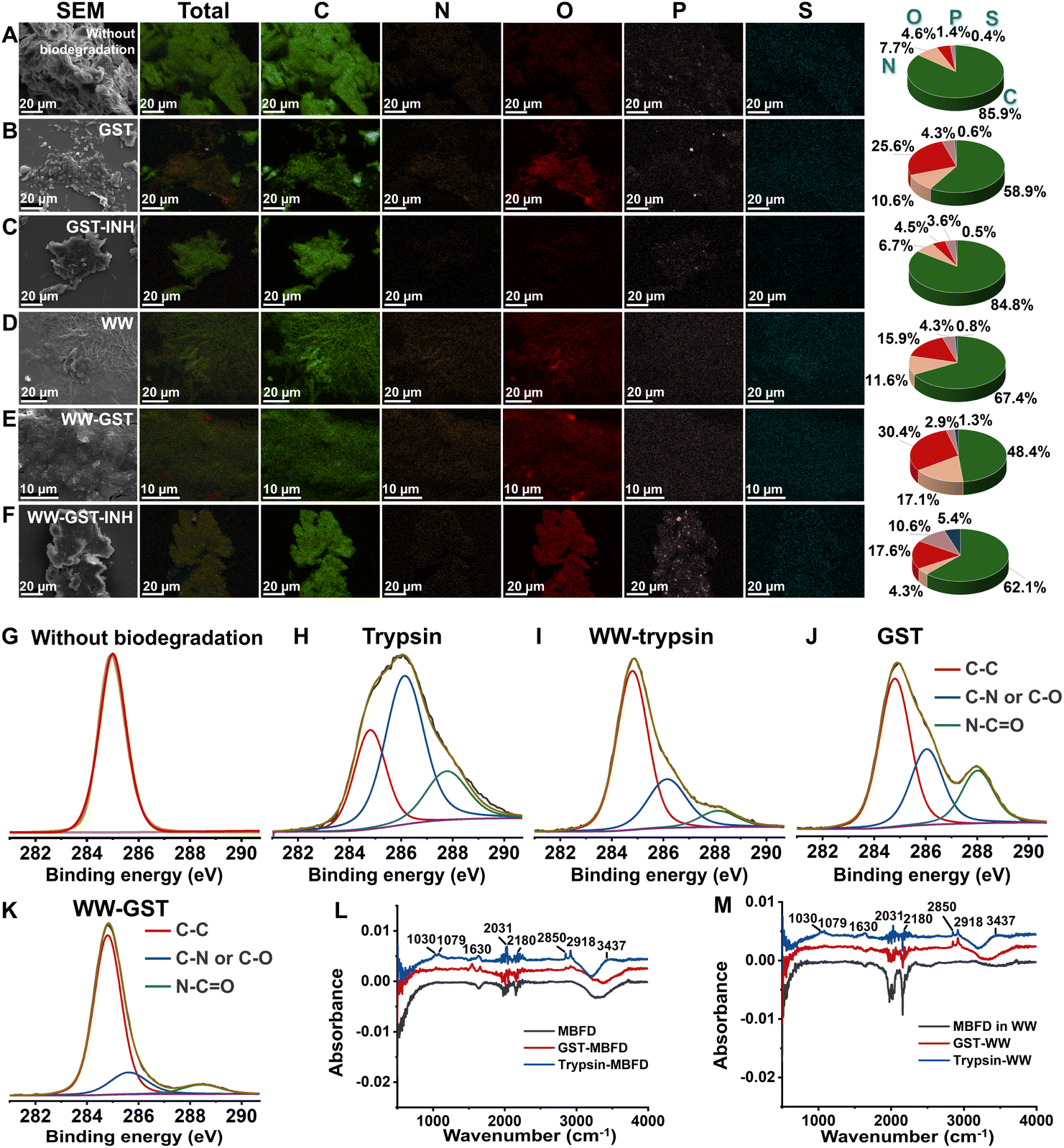 | ||
| Fig. 2 Elemental fingerprints of MBFD generated following treatment with GST and trypsin under different conditions. EDX energy spectrum of MBFD in (A–C) pure water and (D–F) wastewater (WW) samples degraded by trypsin and GST under different conditions. Mapping images of EDX spectra of MBFD during incubation in the (C and F) presence or (B and E) absence of inhibitor. (G–K) The XPS spectra of MBFD treated with enzymes: (G) without biodegradation, (H) degradation under dark conditions (37 °C) by trypsin, (J) degradation under dark conditions (37 °C) by GST, (I) MBFD in wastewater (WW) samples under dark conditions (37 °C) degraded by trypsin, and (K) MBFD in wastewater (WW) samples under dark conditions (37 °C) degraded by GST. FT-IR spectra of MBFD in (L) pure water and (M) wastewater (WW) samples degraded by trypsin and GST, respectively. | ||
XPS analysis further elucidated the elemental fingerprints of MBFD during the degradation process. The pristine MBFD showed an absorption peak at 285.0 eV, corresponding to –C–H– and –C–C– bonds of hydrocarbons (Fig. 2G). Upon mixing MBFD with GST and trypsin, new peaks emerged at 285.8 eV and 287.8 eV, indicating the formation of –C–N– or –C–O– bonds and –CO– or CN– bonds, respectively (Fig. 2H–K). Notably, the intensity of these peaks increased significantly over time. Additionally, FT-IR spectra revealed the appearance of stretching vibration peaks of –C–O–, –CO–, and –CN– bonds (see Fig. 2L–M). Incubation with GST generated –H–N–H– asymmetric and symmetric stretching vibration peaks at 2850 cm−1 and 2918 cm−1, alongside stretching vibration peaks for –CC– and –CO– at 1542 cm−1 and 1656 cm−1, respectively (Fig. 2L–M). In the case of trypsin-induced degradation, several peaks were observed, including those at 2850 and 2918 cm−1 (asymmetric and symmetric stretching vibrations of –H–N–H–), as well as peaks at 1630 cm−1 (stretching vibration of –CO–), 1079 cm−1 (bending vibration of –N–H–), 1030 cm−1 (stretching vibration of –C–O–C–), 2180 cm−1 (stretching vibration of –CN–), and 2031 cm−1 (stretching vibration of –OCN–) (Fig. 2L–M).
Mass spectrometry (MS) fingerprints of MBFD
MALDI-TOF MS provided further validation of the degradation of MBFD by offering detailed molecular structure information. In this study, we found that both MBFD and its degradation products can be directly detected by matrix-free MALDI-TOF MS (Fig. S5†). As shown in Fig. S5,† MBFD exhibited characteristic clusters of molecular peaks in the low-mass region, including the peaks corresponding to C3H6, C5H10, C6H12, C7H14, and C8H16 at m/z 42.0, 70.0, 84.0, 98.0, and 112.0, respectively. The mass-to-charge ratio of MBFD measured by MALDI-TOF MS ranged from 0 to 2000, particularly in the low-mass region (less than 500). Considering the high specificity and low background noise, we selected the low-mass region to effectively monitor the degradation of MBFD. To the best of our knowledge, this also represents the first report of using LDI-TOF MS for the direct analysis of PP without the need for a matrix.This method allowed us to obtain MS fingerprints of MBFD degradation and monitor the degradation process. A series of characteristic peaks of MBFD were generated in both the low-mass and high-mass regions (Fig. S5 and S6†). Extending the incubation time to 14 days, the characteristic peaks of MBFD maintained consistent intensities in the presence of the inhibitor (Fig. S6–S8†), confirming that the degradation was indeed triggered by trypsin and GST. When incubated with trypsin, a gradual shift of peaks to the low-mass region was observed over 2 days, implying a gradual decrease in the size of MBFD induced by trypsin (Fig. S5 and S9A†). As degradation continued, MBFD was broken down into monomers, small organic molecules, or even CO2, leading to a gradual decrease and eventually disappearance of the characteristic MBFD peaks (Fig. S5 and S9A†). During this process, a variety of oxidation products formed, including single-oxygen products (e.g., C3H6O at m/z 58.0) and multi-oxygen products (e.g., C6H12O2 at m/z 116.1 and [C5H10O3]− at m/z 118.1). When trypsin degrades MBFD, it formed amides (e.g., [C7H15NO + Na–2H]− at m/z 150.1 and [C7H16N2O2]− at m/z 160.0) and NO3-containing products ([CH3NO3]− at m/z 77.0). The concentrations of these degradation products increased initially, followed by a decline, suggesting the progressive degradation of MBFD.
Compared with trypsin, the degradation process with GST showed some differences. The MS fingerprints obtained with GST revealed an initial increase followed by a decrease, generating a series of carbon cluster peaks in the low-mass regions (Fig. S5 and S9B†). In addition to carbon clusters, the degradation of MBFD with GST also yielded oxidative and nitridation products. Notably, the degree of nitridation was significantly higher with GST than with trypsin. The MS peaks gradually increased over the first 4 days of incubation with GST, while those with trypsin showed an increase over just 2 days (Fig. S5 and S9†). Afterward, the peaks for both MBFD and its degradation products began to decline (Fig. S5 and S9†), suggesting their further degradation. The changing trend of typical peaks of MBFD in wastewater was similar to that in pure water (Fig. S10 and S11†). To the best of our knowledge, the generation of nitrogen-containing degradation products from PP has been unreported before. This is likely due to dissolved nitrogen in water being introduced into the products during the degradation process.
The MS performance of MBFD in the presence of CYP450 was evaluated over time, comparing both the low-mass and high-mass regions (Fig. S3A–E†). Extending the incubation time from 0 to 14 days, the characteristic peaks of MBFD remained relatively stable in intensity across both regions, indicating a low degradation efficiency of MBFD when exposed to CYP450 (Fig. S3A–E†).
Based on MS fingerprinting, we further examined how environmental factors (such as light and environmental matrices), biological factors (like proteins and whole blood), and plastic additives impact plastic degradation. The results showed that MBFD can be degraded by GST and trypsin under different light conditions (in dark and under UV or natural light) and in the presence or absence of plastic additives, humic acid (HA) and bovine serum albumin (BSA) (Fig. S12–S20†). Under natural light, the characteristic peaks of MBFD initially increased and then decreased within 0–7 days (Fig. S12A and S13A†). Conversely, under UV light, the MBFD peaks consistently decreased and nearly vanished after 4 days, suggesting that MBFD can be fully degraded by GST and trypsin (Fig. S12B and S13B†). Comparing the results between UV and visible light, it was found that the degradation efficiency of PP under UV light was significantly higher than that under visible light (Fig. S12 and S13†). These findings are also consistent with our previously obtained results on degradation efficiency and enzyme activity under light conditions (Fig. 1 and S2†). Although the environmental matrix (HA) and biological components (BSA) affected the MBFD degradation efficiency, the MBFD characteristic peaks still showed a tendency of increase and then decrease, indicating that HA and BSA did not affect the degradation ability of GST and trypsin (Fig. S14–S16†). The nitrogenous and oxygenated products produced by degradation were different from those in water, suggesting that the environmental and biological matrices affected the type of degradation products (Fig. S14 and S15†). Additionally, the study investigated the impact of plastic additives on MBFD degradation. The antioxidant 1010 (tetrakis[beta-(3,5-di-tert-butyl-4-hydroxyphenyl)propionic acid] pentaerythritol ester), commonly used in the production of mask melt-blown cloth, was examined. When the incubation time was extended from 0 to 14 days with enzyme and antioxidant 1010, the MBFD peak initially increased and then decreased, similar to the degradation without the antioxidant (Fig. S17 and S18†). This suggests that antioxidant 1010 does not significantly affect the enzyme activity or the degradation process of MBFD. Furthermore, the study assessed the degradation capabilities of trypsin and GST in human serum (Fig. S19 and S20†). The MS peaks showed similar trends to those in water, indicating that these enzymes can effectively degrade MBFD even in the complex environment of human serum.
The enzymatic method was further extended to investigate the degradation of MBFD derived from disposable activated carbon masks, PP industrial particles, and PE particles (Fig. S21–S29†). The experimental results indicated that the characteristic peaks of PP particles, without the addition of enzymes, remained stable throughout the 7-day incubation period (Fig. S21†). This confirms that PP particles are non-degradable under natural conditions. Throughout the incubation period, both PP industrial particles and MBFD from disposable activated charcoal masks exhibited a characteristic trend of initial increase followed by decrease in their peak intensities, similar to the degradation behavior of MBFD (Fig. S22–S24†). These observations further support the broad applicability of the enzymatic approach for degrading various forms of PP plastics. It is particularly noteworthy that under dark conditions, GST was also able to effectively degrade PE particles, resulting in a significant decrease in their characteristic peaks and the production of nitrogen- and oxygen-containing degradation products, which were somewhat different from those degraded from PP particles (Fig. S25–S27†). When tested in real wastewater samples, these plastic particles were successfully degraded by both GST and trypsin (Fig. S28 and S29†). These results collectively demonstrate that GST and trypsin possess the capability to degrade diverse types of PP and PE plastics, including in real wastewater scenarios. Therefore, this study highlights their potential for practical plastic waste treatment for addressing plastic pollution challenges.
Molecular degradation pathways of MBFD
The degradation pathways of MBFD can be elucidated by analyzing the ratios of adjacent fingerprint peaks, as shown in Fig. 3. Generally, the degradation of MBFD involves two main pathways, i.e., oxidation and nitridation (Fig. 4). The peak ratios of oxygen-containing species, such as [CnH2n±1NxOy + Na/K − mH]/[CnH2n±1NxOy−1], [CnH2n±1O2 + Na/K − mH]/[CnH2n±1 + Na/K − mH], [CnH2n±1O + Na/K − mH]/[CnH2n±1 + Na/K − mH], [CnH2n±1O3 + Na/K − mH]/[CnH2n±1O + Na/K − mH], [CnH2n±1O2 + Na/K − mH]/[CnH2n±1O + Na/K − mH], [CnH2n±1O2]/[CnH2n±1], [CnH2n±1NxO + Na − 2H]/[CnH2n±1Nx], and [CnH2n±1O3 + Na/K − mH]/[CnH2n±1O2 + Na/K − mH] with the same C number showed a gradual increase throughout the degradation process (Fig. 3 and Table S2†), indicating a direct oxidation of MBFD. Similarly, the peak ratios of nitrogen-containing species, such as [CnH2n±1NxOy]/[CnH2n±1Nx−1Oy], [CnH2n±1NxOy + Na/K − mH]/[CnH2n±1 + Na/K − mH], [CnH2n±1NOy + Na/K − mH]/[CnH2n±1Oy + Na/K − mH], [CnH2n±1NOy]/[CnH2n±1Oy], [CnH2n−2NxOy + Na − 2H]/[CnH2nOy], and [CnH2n±1NxO2 + Na/K − mH]/[CnH2n±1O2 + Na/K − mH], with the same C number also exhibited increasing trends during the degradation, suggesting that direct nitridation is occurring alongside oxidation. Furthermore, we found that the peak ratios such as [Cn−1H2n−2O + Na − 2H]/[CnH2nO2 + Na − 2H], [Cn−1H2n−2 + Na/K − 2H]/[CnH2nO + Na/K − 2H], [Cn−1H2n−2O + Na − 2H]/[CnH2nO2 + Na − 2H], [Cn−1H2n−2 + Na/K − 2H]/[CnH2nO + Na/K − 2H], [Cn−1H2n−2O + Na − 2H]/[CnH2nO3 + Na − 2H], and [Cn−1H2n−2 + Na − 2H]/[CnH2nO2 + Na − 2H] with a single C difference increased during the degradation (Fig. 3 and Table S2†), corresponding to decarbonization of MBFD (releasing a single CO2 or CO molecule).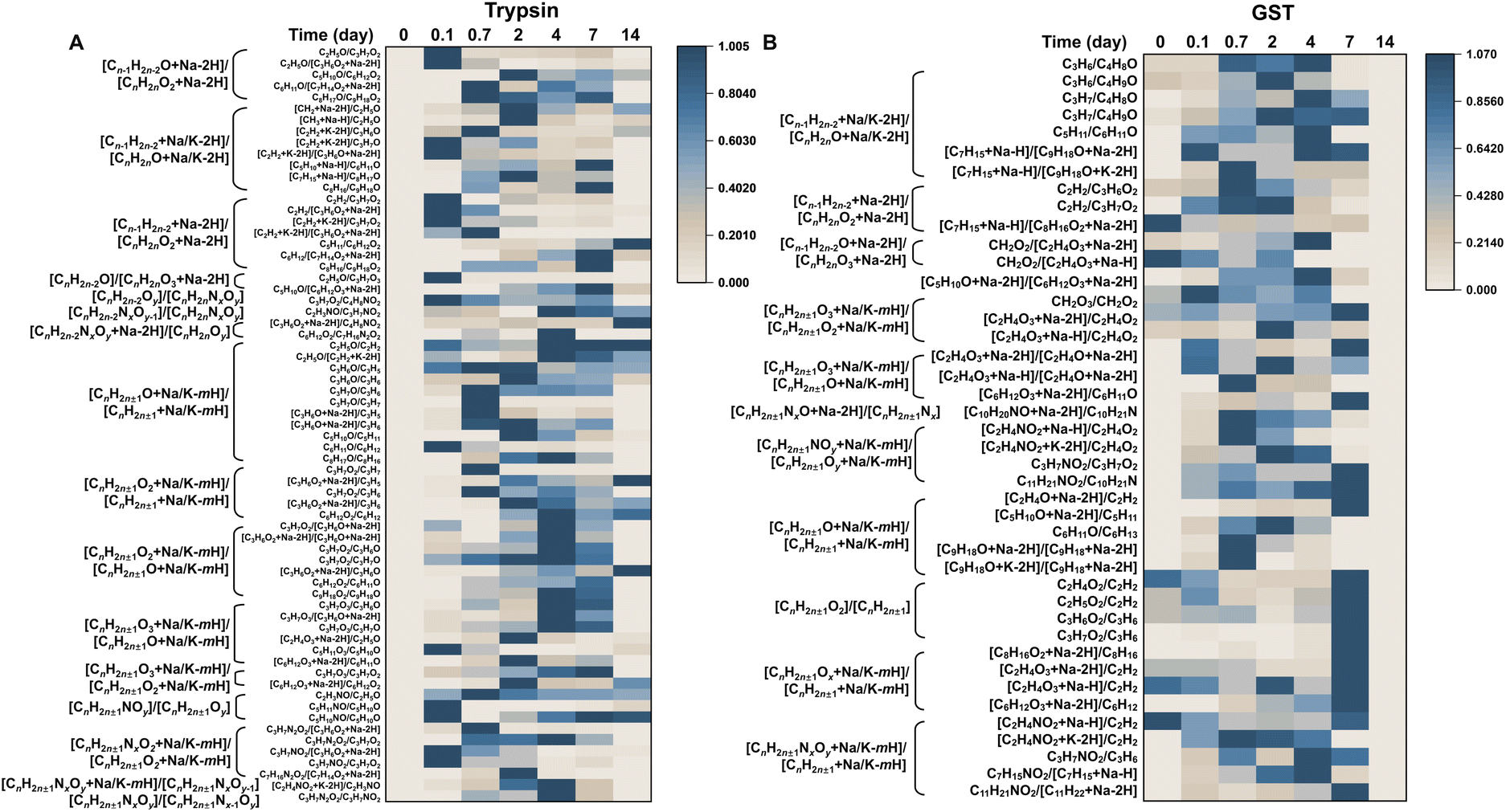 | ||
| Fig. 3 LDI-TOF MS fingerprinting of MBFD degradation induced by trypsin and GST. Heat map representing the ratio of typical fingerprint peaks of MBFD degradation by (A) trypsin and (B) GST at physiological temperature for 14 days. | ||
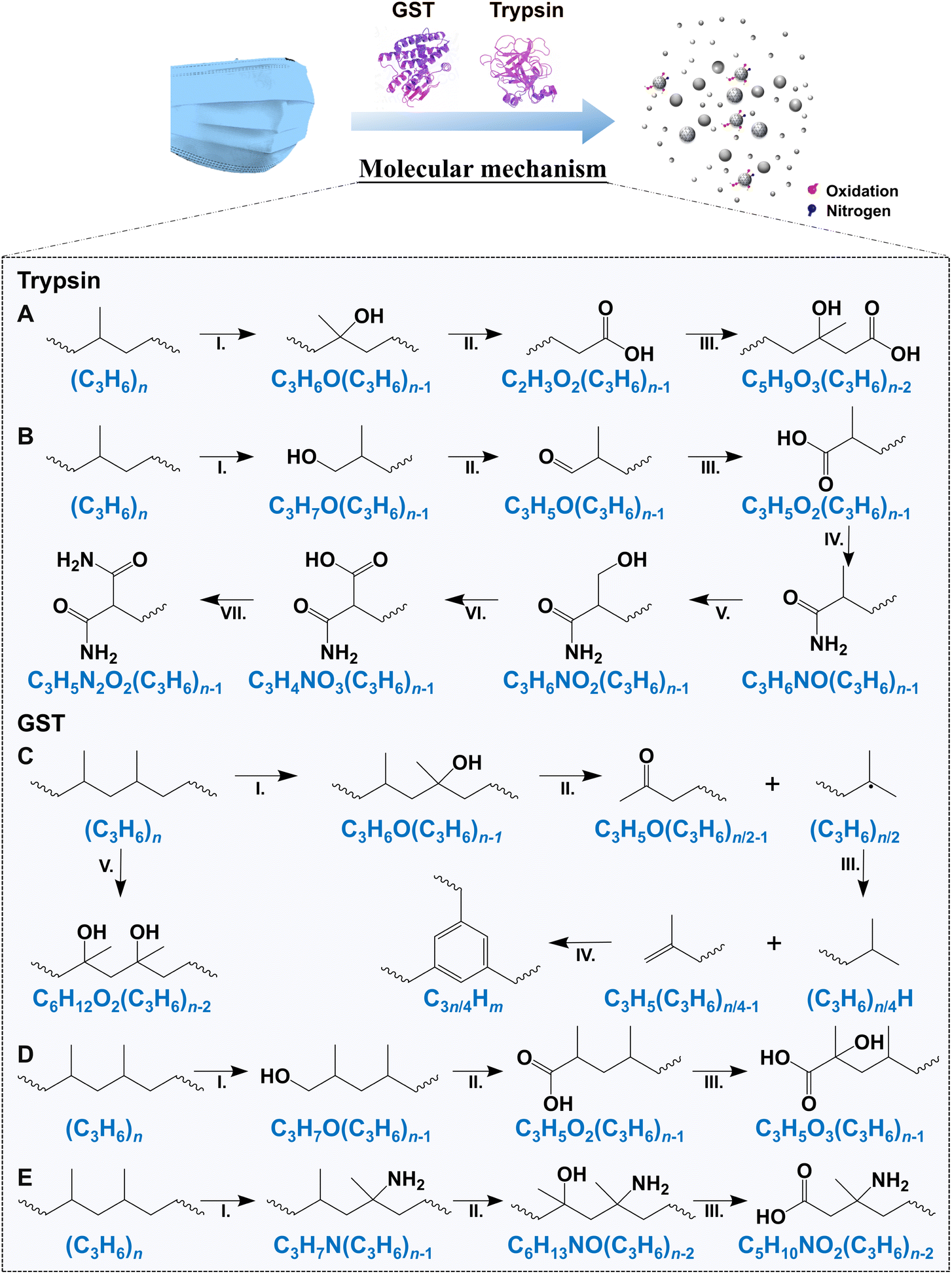 | ||
| Fig. 4 Potential molecular mechanisms of trypsin- and GST-induced degradation of MBFD. The molecular degradation pathway of MBFD by (A and B) trypsin and (C–E) GST based on the results of multiple techniques. | ||
Fig. 4 shows the degradation of MBFD under the action of GST and trypsin. The degradation pathways of MBFD mainly include nitridation and oxidative degradation pathways (Fig. 4). In the presence of trypsin, the oxidation and nitridation degradation tend to induce the formation of hydroxy compounds and amides, respectively (Fig. 4A, B and S9A†). Specifically, trypsin-induced degradation of MBFD is more likely to result in oxidation and subsequent nitration, leading to the formation of amides (Fig. 4B and S9A†). Trypsin can degrade MBFD through the NO–NO3–N2O2 nitridation pathway (Fig. 4B). This may be due to the ability of trypsin to specifically cleave the amide and guanidyl terminus of positively charged glutamine and arginines. On the other hand, GST-induced degradation of MBFD appears to initiate carbonization and carboxylation (Fig. 4C–E and S9B†). The degradation of MBFD induced by GST is more likely to result in the formation of carbon cluster peaks, which are indicative of carbon nanostructures (Fig. 4C and S9B†). For the nitridation pathway, MBFD may undergo direct nitridation followed by oxidation (Fig. 4E and S9B†). Additionally, the degradation of MBFD by these enzymes may also be influenced by its physical properties, such as size, surface properties, and interfacial effects. This is consistent with previously reported mechanisms of plastic biodegradation. This process typically leads to a reduction in carbonyl groups, followed by their conversion into carboxylic acids.61 Eventually, these enzymes further break down the long carbon chains into small hydrocarbons, releasing carbon dioxide as a byproduct.62 This study revealed the degradation mechanism of plastics, which involved depolymerization and monomer fracture. It is crucial to control the biodegradation conditions to achieve maximum depolymerization without damaging PP monomers. This balance is essential for biological recycling and regeneration. Biological recycling is vital for sustainable development due to its adaptability to various raw materials and strong environmental protection. Therefore, this research contributes to the potential use of biorecycling pathways through biodegradation, supporting long-term development in this field.
The proposed degradation pathways of MBFD were substantiated using multiple methods, including FT-IR, EDX, XPS, anaerobic experiments with argon gas, gel permeation chromatography (GPC), gas chromatography-mass spectrometry (GC-MS), and stable isotope patterns (Fig. 2 and S30–S42†). Results from EDX, XPS, and FT-IR experiments provided evidence for the presence of key intermediates and products (Fig. 2 and S30–S33†). Notably, the varying trends of N- and O-containing functional groups observed in the EDX, XPS, and FT-IR spectra supported the proposed degradation pathways (Fig. 2 and S30–S33†). For instance, a comparative analysis of the elemental fingerprints of MBFD after co- incubation with GST and trypsin revealed that the XPS spectra yielded significantly higher proportions of functional groups such as –OC–N–, –OC–, –O–C–, –N–H–, –N–O–, and –O–H– compared to pristine MBFD over time, suggesting that MBFD was effectively biodegraded by enzymes (Fig. 2 and S30–S33†). The presence and progressive increase of the –OC–N–, –N–H–, and –N–O– bonds in the N1 XPS spectra throughout the degradation process further corroborated the formation of important oxidation and nitridation products (Fig. S30 and S31†). Similarly, the O1 XPS spectra showed peaks corresponding to –OC–, –OC–N–, –O–C–, and –O–H– bonds after the biodegradation by trypsin and GST (Fig. S32 and S33†). Additionally, Ar gas was used as a protective gas to verify the N and O addition mechanisms (Fig. S34†). Almost no peaks corresponding to N- and O-containing products were obtained after 2 days of incubation with Ar gas purge (Fig. S34A, B, D and E†). However, in the presence of air, the peaks of N- and O-containing degradation products gradually increased (Fig. S34C and F†). These results evidence that MBFD can be degraded by GST and trypsin, and the N and O in the degradation products originate from the air dissolved in the solution, thus supporting the molecular mechanisms in Fig. 4. Overall, these results verified the proposed degradation pathways.
We conducted further analysis of the degradation products using GPC and GC-MS, which revealed N- and O-containing degradation products, confirming the generation of degradation products. The GPC analysis results only showed peaks for GST and trypsin, with no peaks detected for other proteins (Fig. S35†). This suggests that neither GST nor trypsin have any carry-over proteins affecting their degradation activity. After 1 day of incubation, the molecular weight distribution (MWD) of the treated PP membranes was analyzed using GPC, revealing a trend towards depolymerization, as shown in Fig. S36.† The Mw of GST-treated, trypsin-treated, and enzyme-free MBFD was 140201, 146504, and 157265, respectively, showing a decrease of 10.9% and 6.8% (Fig. S36†). These results indicate that the inclusion of GST and trypsin treatments caused chain breakage in MBFD, leading to a reduction in their molecular weights. To further investigate the degradation of MBFD by GST and trypsin, we analyzed the decomposition products using GC-MS (Fig. S37–S42†). MBFD and PE plastics, which were not degraded by GST and trypsin, exhibited characteristic peaks at retention times of 16.481 and 16.482 minutes, respectively (Fig. S37 and S38†). The close retention times indicate that GC-MS cannot differentiate between PE- and PP-based MBFD because PE and PP have similar characteristics. Following degradation, the PE plastics and PP-based MBFD treated with trypsin and GST showed characteristic peaks at specific retention times of 9.846, 14.352, 16.467, 15.310, 14.989, 11.865, 9.897, 18.172, 9.682, 12.011, 15.929, 10.155, 11.213, 11.729, 12.405, 15.492, 10.888, 13.558, and 11.371 minutes (Fig. S37 and S38†). Mass spectrometry was then employed to analyze the components detected in the aforementioned retention time peaks (Fig. S39–S42†). The results indicated that the degradation products of PE included “oxalic acid, 2-ethylhexyl hexyl ester” (1), “n-hexadecanoic acid” (2), “phenol,2,2′-methylenebis[6-(1,1-dimethylethyl)-4-methyl]” (3), “octadecanoic acid” (4), “eicosyl nonyl ether” (5), “2,4-di-tert-butylphenol” (6), “benzaldehyde” (7), “13-docosenamide” (8), “dodecane” (9), “pentadecane” (10), “tetracosane” (11), “tridecane” (12), “nonadecane” (13), “hexadecane” (14), “heneicosane” (15), “eicosane” (16), “2-dimethyldodecane” (17), “octacosane” (18), and “heptadecane” (19) (see Fig. S39–S42 and Table S3†). All these compounds have high matching scores (over 80%) in the NIST library. The results indicated that the degradation products of GST and trypsin on PP included oxygen-containing compounds, nitrogen-containing compounds, and hydrocarbons (see Fig. S39–S42 and Table S3†). The presence of nitrogenation and oxidation products validated the reliability of our findings.
The stable isotope patterns of carbon, nitrogen, and oxygen further validated the proposed degradation pathways (Fig. S43 and S44†). In the negative-ion LDI-TOF MS, five nitridation and oxidation products were identified: [C2H4O]−, [C5H10O2]−, [C7H15NO + Na–2H]−, [C17H34O]−, and [CH4NO3]−. The naturally occurring stable isotopes of these elements are as follows: oxygen has three isotopes with a ratio of 16O:17O:18O = 99.763%:0.038%:0.200%; nitrogen has two naturally occurring stable isotopes with a ratio of 14N:15N = 99.634%:0.366%; and carbon has two naturally occurring stable isotopes with a ratio of 12C:13C = 98.893%:1.107%. For the nitridation products, i.e., [C7H15NO + Na–2H]− and [CH4NO3]−, the measured stable isotope patterns for C, N, and O closely matched the theoretical values (Fig. S43 and S44†). Similarly, for the oxidation products, including [12C13CH416O]−, [12C413CH1016O2]−, [12C1613C1H3416O]−, [12C1513C21H3416O]−, [12C171H3418O]−, [12C1613C1H332H16O]−, [13C1H414N16O3]−, [12C1H414N16O217O]−, and [12C1H32H14N16O217O]−, the measured stable isotope patterns for C and O were also consistent with the theoretical values, supporting the production of oxidation products (Fig. S43 and S44†).
Molecular simulations of interaction between enzymes and PP
Molecular docking and molecular dynamics simulations were performed to improve the understanding of the molecular interaction between PP and enzymes. Fig. 5 illustrates the structural conformations of two groups of GST and trypsin complexes before and after simulation under specified conditions. The Root Mean Square Deviation (RMSD) curves for the protein–substrate complexes and substrates were calculated individually over time to evaluate system stability. Fig. S45 and S46† demonstrate the stability of two sets of systems during the simulation. All atoms, including those of the enzyme (e.g., C, O, N, H, S, and P atoms) and the constituent atoms of PP (e.g., C and H atoms), were used to calculate the RMSD. The RMSD values of the protein–substrate complex structures stabilized between 0.3 and 0.5 nm. In the GST–PP system, the RMSD values of the substrate PP stabilized with fluctuations of ∼0.2 nm (Fig. S45A†). The substrate PP in trypsin–PP fluctuated between 0.1 and 0.3 nm (Fig. S46A†). The fluctuations observed in the first 20 ns may be attributed to errors in the periodic processing of the program, which also caused fluctuations in the trypsin–PP composite system before 20 ns (Fig. S46A†). Furthermore, the evolution of the distance over time between the key amino acid of the protein and PP showed that the fluctuation in the distance between GST and PP was relatively small, indicating that they had consistently been in close proximity (Fig. S47 and S48†). The Root Mean Square Fluctuation (RMSF) of the proteins in the two systems was calculated over a 500 ns simulation trajectory within a physiological solvent environment. The smoothness of protein fluctuations during the molecular dynamics simulation at 500 ns was analyzed, as depicted in Fig. S45B and S46B.† The analysis revealed that GST exhibited greater fluctuations in amino acids 80–130 compared to the trypsin groups, which showed relatively consistent fluctuations. By examining the molecular trajectories, it can be observed that trypsin mainly fluctuated with the substrate within the amino acid range of 42–114 (see Videos 2†). | ||
| Fig. 5 Molecular dynamics simulation of GST and trypsin. (A) The potential energy of GST and PP, (B) the Gibbs free energy landscape of GST–PP, and (C) the representative structure of the free energy minimum of the GST–PP complex (hydrophobic interactions are shown as gray dashed lines). (D) The potential energy of trypsin and PP, (E) the Gibbs free energy landscape of trypsin–PP, and (F) the representative structure of the free energy minimum of trypsin–PP complex (hydrophobic interactions are shown as gray dashed lines). | ||
Additionally, we analyzed the secondary structure of the proteins to detect any changes. Fig. S49† shows that the secondary structures of the two protein groups remained stable during the simulation, suggesting that binding to the substrate PP did not lead to significant structural changes in the proteins.
Subsequently, we calculated the binding free energy between the PP and enzyme (i.e., GST and trypsin).63,64 The simulation process was reliable for both two groups of systems, and the protein–substrate complexes exhibited stability. Therefore, we proceeded to analyze the binding of the substrate PP to the proteins. The non-bonding binding energies of the substrates were examined to determine the relative non-bonding binding free energies of GST–PP and trypsin–PP. As depicted in Fig. 5A, the coulombic short-range (Coul-SR) energy and Lennard-Jones short-range (LJ-SR) energy of GST–PP significantly contributed to the overall binding stability, with values of 3 ± 1 kJ mol−1 and −150 ± 18 kJ mol−1. The Coul-SR energy of trypsin–PP was 3 ± 1 kJ mol−1, while the LJ-SR energy was −64 ± 20 kJ mol−1 (Fig. 5D). Analysis of the relative free energy surface maps of the GST–PP complexes in Fig. 5B and C revealed the structure with the lowest relative binding free energy. By analyzing the interaction mode, it is evident that PP was only engaged in hydrophobic interactions with the protein. The relative free energy surface map analysis of the trypsin–PP complexes, shown in Fig. 5E and F, indicated that the structure with the lowest relative binding free energy was obtained. Furthermore, the interaction pattern analysis demonstrated that PP was engaged in hydrophobic interactions of trypsin.
The binding free energies of the substrate PP and the two groups of proteins were further analyzed using the gmx_MMPBSA program.64,65 The Gibbs free energy of GST with PP was −24 ± 4 kcal mol−1, and the Gibbs free energy of trypsin with PP was −11 ± 2 kcal mol−1 (Fig. S50†). The energy contribution of relative amino acid residues at the substrate–protein interface was also analyzed previously (Fig. S50†). To further investigate the factors influencing enzymatic degradation efficiency, molecular dynamics simulation was also performed on the CYP450-PP system (Fig. S51–S54†). The results revealed that both the substrate PP and the HEM group in CYP450-PP exhibited fluctuations of approximately 0.1 nm (Fig. S51†). Analysis of the non-bonding interactions indicated that the relative free energy contributions were 0.3 ± 0.9 kJ mol−1 for Coul-SR interactions and −147 ± 10 kJ mol−1 for LJ-SR interactions, as illustrated in Fig. S53 and S54.† The Gibbs free energy of the CYP450–PP complex was determined to be −10 ± 6 kcal mol−1, which was lower than that of GST and trypsin. This confirms previous findings that its degradation efficiency is lower compared to the other two enzymes (Fig. S54†).
Conclusions
In summary, the use of trypsin, a natural digestive enzyme originated from mammals, and GST, a human-derived natural metabolic enzyme, has shown promise in effective biodegradation of MBFD. The results indicated that the enzymatic degradation rate of MBFD reached 1.57 and 3.98 g L−1 h−1 in water and environmental wastewater samples, respectively. The degradation efficiency of trypsin and GST was 86.7% and 99.2%, respectively. Notably, MBFD can be degraded by GST and trypsin under different conditions (in dark and under UV or natural light) and in the presence or absence of plastic additives, HA, and BSA. Through chemical multi-fingerprint analysis, we proposed the mechanisms of MBFD degradation, including the oxidative pathway as well as the previously unreported nitrogenation pathway. Molecular dynamics simulations confirmed that hydrophobic interactions were the main driving force in the enzyme–PP complex system. Furthermore, this enzymatic method has been successfully applied in degradation of MBFD from disposable activated carbon masks, PP industrial particles, and PE industrial particles, and it is also valid in complex matrices such as actual wastewater and human serum. Our results not only offer valuable insights for selective enzymatic degradation of plastics but also pave the way for new research avenues to address the issue of plastic pollution.Experimental
Chemicals and materials
Disposable medical masks and disposable activated carbon masks were obtained from Youhekang (Guangzhou, China) and DOCTOR MASK (Japan), respectively. Melt-blown fabrics of masks are primarily made of PP plastics with a molecular weight of approximately 157 kDa and a crystallinity of 40–60%. PP industrial particles (Mw: 100 kDa; d50 = 6.5 μm; crystallinity: 40–50%) and PE industrial particles (Mw: 60 kDa; d50 = 6.5 μm; crystallinity: 80–90%) used in this study were obtained from Yize Industry in Zhoukou, China. GST, CYP450 3A4 human, sodium phosphate buffer, Na-benzoyl-L-arginine ethyl ester (BAEE), tetrakis[beta-(3,5-di-tert-butyl-4-hydroxyphenyl)propionic acid] pentaerythritol ester, and hydrochloric acid solution were obtained from Sigma-Aldrich (St. Louis, Missouri, USA). Trypsin (2.5%, no phenol red) were purchased from Thermo Scientific (Waltham, MA). The inhibitors for GST (indomethacin, 99%) and trypsin (trypsin inhibitor soybean) were purchased from Rhawn (Shanghai, China) and Aladdin (Shanghai, China), respectively. L-γ-Glutamyl-S-(phenylmethyl)-L-cysteinyl-2-phenyl (TLK117) was purchased from MedChemExpress (New Jersey, USA). (3S)-1-Chloro-3-tosylamido-7-amino-2-heptanone hydrochloride (TLCK) was obtained from Biolab (Beijing, China). The GST activity assay kit and bovine serum albumin (BSA) were from Solarbio, Beijing, China. Humic acid (HA) was purchased from Shanghai Macklin Biochemical Technology Co. (Shanghai, China). Dichloromethane was from McLean (Shanghai, China). HPLC grade acetonitrile (ACN) was from J. T. Baker (Phillipsburg, NJ, USA). Ar gas was purchased from Hefei Zhengfan Electronic Materials Co. (Hefei, China). Ultrapure water was prepared using the Millipore Milli-Q system (Billerica, MA, USA). All reagents were of analytical grade unless otherwise noted.Characterization of MBFD
SEM images were captured with a Thermo Fisher Quattro environmental scanning electron microscope (Waltham, Massachusetts, USA) and a Hitachi S-3000N scanning electron microscope (Tokyo, Japan). The size of MBFD was analyzed with Image-Pro software. Fourier transform infrared (FT-IR) spectra were obtained using a JASCO FT-IR spectrometer (Victoria, B. C., Canada). XPS spectra were obtained on a Thermo Scientific ESCALAB 250Xi X-ray photoelectron spectrometer (Massachusetts, USA) using Al Kα X-ray radiation as the X-ray source.Enzymatic degradation of MBFD
Melt-blown cloth for masks was cut into small fragments and transferred to a PTFE ball mill tank. The melt-blown cloth was ground into particles under vacuum conditions at a speed of 480 rpm. After approximately 24 hours, samples were removed and floated in water to extract MBFD. It is important to mention that mechanical stirring was necessary during this procedure. Certain concentrations of micro/nanoplastics dispersed in water were obtained. In an ultra-clean bench, all consumables were sterilized with alcohol. The MBFD dispersion (222 mg mL−1) was then mixed with GST (19.2 μg mL−1), trypsin (5 μg mL−1), and CYP450 (3.4 μg mL−1). The mixture was then placed in a xenon weather meter (SN-500, Beijing North Lihui Instrument Equipment Co.) equipped with three air-cooled 2500 W xenon lamps as sources of simulated solar light. The degradation experiments were first performed in the dark at 37 °C to simulate physiological conditions. Then, experiments were also performed under light irradiation (i.e., UV and visible light) to test the degradation potential of masks in real-world environments. The light intensity was maintained at 550 W m−2. The reaction solution was analyzed at specific time intervals (1 hour, 15 hours, 2 days, 4 days, 7 days, and 14 days) during the incubation period to monitor the degradation of MBFD. The mass of the mixed dispersions was weighed at different times to calculate the mass loss. All experiments were performed in triplicate (n = 3). The study also explored the impact of environmental factors (such as light and HA), biological factors (like BSA), and plastic additives (antioxidant 1010) on MBFD degradation under the same conditions. Additionally, to confirm the role of dissolved air, we utilized Ar gas as a protective environment to incubate with MBFD and the enzyme. Throughout this process, non-reactive gases (i.e., argon) were maintained to purge the O2 and N2. It is crucial to homogenize the mixed solutions before and after sampling.LDI-TOF MS analysis
LDI-TOF MS was performed by transferring 2 μL of MBFD dispersion directly into a stainless steel MTP 384 target frame III (Bruker Daltonics). The mixture was dried under air in a fume hood, followed by analysis with a Bruker Daltonics Autoflex III Smartbeam MALDI-TOF mass spectrometer with a 355 nm Nd:YAG laser at a frequency of 200 Hz. The procedure was conducted in negative-ion mode by setting the laser power to 70% with an MS range of 0–3000.Real samples
Wastewater samples were collected from Beijing, China, and then refrigerated at 4 °C. Serum samples were collected from the West China Fourth Hospital of Sichuan University in Chengdu, China, and stored at −20 °C. The remaining procedures were consistent with those previously described for water samples.Enzymatic treatment procedure
Trypsin was stored at 4 °C, while GST lyophilized powder was stored at −20 °C. Before use as a degrading enzyme, the GST lyophilized powder was dissolved in water. The CYP450 solution was stored at −80 °C and was subpackaged after being diluted with water. When enzymes were used to degrade MBFD, the enzyme solutions were mixed with the plastic dispersion.Enzyme activity test
The activity of GST was analyzed using a GST activity assay kit and measured via UV-visible spectrophotometry on a VARIOSKAN FLASH multifunctional enzyme labeling instrument (Thermo Scientific, USA) equipped with a continuous rate spectrometer using 1-chloro-2,4-dinitrobenzene (CDNB) as the substrate. The activity of the enzyme was characterized by measuring the absorption of glutathione and the substrate at 340 nm.The activity of trypsin was tested using Nα-benzoyl-L-arginine ethyl ester (BAEE) as the substrate, followed by measurement of UV-vis spectra. The entire process was conducted at 25 °C and pH 7.6.
GPC analysis
GPC analysis was conducted using a Waters 1525 system equipped with a Waters 2414 detector and an Agilent 1260 Infinity II system with a VWD detector. MBFD was performed on a high-temperature PL-GPC 20 unit, while enzyme analysis was done at a low temperature. The molecular weight distribution (MWD) of MBFD and enzyme was characterized using a Waters PLgel MIXED-BLS column (300 × 7.5 mm) and a TSKgel G2000SWXL (300 mm × 7.8 mm, 0.5 μm) with a sample concentration of 1 mg mL−1. Trichlorobenzene was used as the mobile phase at a flow rate of 1 mL min−1 and a column temperature of 150 °C for MBFD analysis, while acetonitrile:water:trifluoroacetic acid in a ratio of 45:55:0.1 was used at a flow rate of 0.5 mL min−1 and a column temperature of 30 °C for enzyme analysis. The GPC system was calibrated with a polystyrene (PS) standard (K = 17.5; a = 0.67) and peptide standard solutions (cytochrome C, aprotinase, bacillus enzyme, glycine–glycine–tyrosine–arginine, and glycine–glycine–glycine) to establish a general calibration curve for molecular weight (Mw) determination.
GC-MS analysis
The degradation products of PP and PE were also detected using GC-MS (TQ8050, Shimadzu, Japan). The degradation solution was first centrifuged at 12000 rpm for 30 minutes at 4 °C. The resulting supernatant was freeze-dried and then redissolved in 1 mL of dichloromethane. For GC-MS analysis, 2 μL of the filtered supernatant was injected into the chromatograph at an injection temperature of 300 °C. The column temperature was initially set at 50 °C for 4 minutes and then ramped up to 300 °C at a rate of 20 °C min−1, with a total run time of 15 minutes. The chromatographic column utilized was a DB-5 ms, measuring 30 m in length, with an inner diameter of 0.25 mm and a column thickness of 0.25 μm. Helium served as the carrier gas at a flow rate of 0.8 mL min−1, while the scanning mode monitored ions/fragments within the 30–450 amu range.
Molecular docking and molecular dynamics
Molecular dynamics simulations were conducted using GROMACS 2019.6 to analyze the specific information of three protein complexes over time from kinetic and thermodynamic perspectives.66,67 GST and trypsin structures were obtained from the AlphaFold database,68,69 while the CYP450 structure was retrieved from PDB ID: 1w0e.70 The topological parameters of the receptor proteins were derived from the amber14SB force field in Ambertools23.71,72 The initial molecular dynamics modeling parameters were constructed using the aqueous solvent model and physiological aqueous environment model of TIP3P.71 The substrate, PP (n = 6), was used to establish molecular parameters based on the gaff2 force field in Ambertools,72,73 with molecular atomic charges calculated using AM1-BCC.74 Protein hydrogenation was performed using tleap for autonomous hydrogenation, and protonation was assessed using H++.75 CYP450 was evaluated in conjunction with PyMOL,76 focusing on the catalytic center. Global docking was conducted for GST and trypsin, while CYP450 was docked based on the HEME catalytic center. Autodock Vina 1.2.5 was utilized for docking.77 The most suitable conformations were selected based on docking scoring (affinity in kcal mol−1) and visual inspection in PyMOL.76 Conformations with the lowest binding energy were selected due to the lack of specific pocket information. The substrate was subsequently simulated at a temperature of 300 K. The energy minimization was performed in 50000 steps using the steepest descent and conjugate gradient methods. This was followed by a two-stage pre-equilibrium simulation of the system. Firstly, the system underwent NVT system equilibrium for 500 ps (V-rescale temperature coupled with constant particle number, volume, and temperature).78 Secondly, NPT system equilibrium was conducted for 1000 ps (Berendsen pressure coupled with constant particle number, volume, and temperature and Berendsen pressure coupled with constant particle number, pressure, and temperature).79 All bond lengths and angles were constrained using LINCS.80 Long-range electrostatic interactions were managed using the PME method81 with a cutoff of 1.0 nm for van der Waals interactions. The temperature of the whole system was maintained at 300 K for kinetic simulations over 500 ns, with steps of 2 fs and a time interval of 10 ps to save coordinate files for analytical purposes. After obtaining the trajectory from the all-atom simulation for this system, it was analyzed using the analysis tools provided by GROMACS 2019.6 and the DulvyTools program.82,83
Ethical statement
All experiments were performed in accordance with National Regulations and International Ethical Standards for Human Subject Research and approved by the ethics committee at West China Fourth Hospital of Sichuan University. All participants provided written informed consent prior to study enrollment.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
The contributions of each author to this study are as follows: Q. Liu and X. Huang designed the research; G. Jiang supervised the project; L. Huang, Q. Wang, and Z. Zhang participated in the discussion; Q. He provided serum samples; X. Huang performed the experiments; Q. Liu and X. Huang analyzed the data and wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by Chinese Academy of Sciences Project for Young Scientists in Basic Research (YSBR-086), National Natural Science Foundation of China (No. 22425041, 22188102, 22206136, and 22376145), National Key R&D Program of China (2023YFC3708302), Sichuan University Interdisciplinary Innovation Fund, and Fundamental Research Funds for the Central Universities (YJ202205).Notes and references
- J. Howard, A. Huang, Z. Li, Z. Tufekci, V. Zdimal, H.-M. van der Westhuizen, A. von Delft, A. Price, L. Fridman, L.-H. Tang, V. Tang, G. L. Watson, C. E. Bax, R. Shaikh, F. Questier, D. Hernandez, L. F. Chu, C. M. Ramirez and A. W. Rimoin, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2014564118 CrossRef CAS PubMed.
- T. Mitze, R. Kosfeld, J. Rode and K. Walde, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 32293–32301 CrossRef CAS PubMed.
- S. Kwon, A. D. Joshi, C.-H. Lo, D. A. Drew, L. H. Nguyen, C.-G. Guo, W. Ma, R. S. Mehta, F. M. Shebl, E. T. Warner, C. M. Astley, J. Merino, B. Murray, J. Wolf, S. Ourselin, C. J. Steves, T. D. Spector, J. E. Hart, M. Song, T. VoPham and A. T. Chan, Nat. Commun., 2021, 12, 3737 CrossRef CAS PubMed.
- Y. Wang, H. Tian, L. Zhang, M. Zhang, D. Guo, W. Wu, X. Zhang, G. L. Kan, L. Jia, D. Huo, B. Liu, X. Wang, Y. Sun, Q. Wang, P. Yang and C. R. MacIntyre, BMJ Glob. Health, 2020, 5, e002794 CrossRef PubMed.
- N. Principi, G. Autore, G. Ramundo and S. Esposito, Viruses, 2023, 15, 1160 CrossRef PubMed.
- N. Wu, K. Joyal-Desmarais, P. A. B. Ribeiro, A. M. Vieira, J. Stojanovic, C. Sanuade, D. Yip and S. L. Bacon, Lancet Respir. Med., 2023, 11, 439–452 CrossRef PubMed.
- N. U. Benson, D. E. Bassey and T. Palanisami, Heliyon, 2021, 7, e06343 CrossRef CAS PubMed.
- E. G. Xu and Z. J. Ren, Front. Environ. Sci. Eng., 2021, 15, 125 CrossRef CAS PubMed.
- N. Singh, Y. Tang and O. A. Ogunseitan, Environ. Sci. Technol., 2020, 54, 8500–8502 CrossRef CAS PubMed.
- M. G. Carenbauer, Geoforum, 2021, 120, 79–81 CrossRef PubMed.
- J. C. Prata, A. L. P. Silva, A. C. Duarte and T. Rocha-Santos, Environments, 2021, 8, 31 CrossRef.
- G. L. Sullivan, J. Delgado-Gallardo, T. M. Watson and S. Sarp, Water Res., 2021, 196, 117033 CrossRef CAS PubMed.
- K. Jabeen, B. Li, Q. Chen, L. Su, C. Wu, H. Hollert and H. Shi, Chemosphere, 2018, 213, 323–332 CrossRef CAS PubMed.
- E. E. Burns and A. B. A. Boxall, Environ. Toxicol. Chem., 2018, 37, 2776–2796 CrossRef CAS PubMed.
- K. Ziani, C. B. Ionita-Mindrican, M. Mititelu, S. M. Neacsu, C. Negrei, E. Morosan, D. Draganescu and O. T. Preda, Nutrients, 2023, 15, 617 CrossRef CAS PubMed.
- O. S. Chowdhury, P. J. Schmidt, W. B. Anderson and M. B. Emelko, Environ. Health, 2024, 2, 441–452 CAS.
- C. Fang, O. S. Awoyemi, Y. Luo and R. Naidu, Environ. Health, 2023, 1, 63–71 CAS.
- Y. Wang, F. Wang and M. Song, Environ. Health, 2024, 2, 514–516 CAS.
- Z. Chen, W. Zhang, H. Yang, K. Min, J. Jiang, D. Lu, X. Huang, G. Qu, Q. Liu and G. Jiang, Environ. Sci.:Processes Impacts, 2022, 24, 649–674 RSC.
- H. Memon, S. Liao, R. Maryam, A. Patrucco and C. Riccardi, Mater. Adv., 2024, 5, 8333–8350 RSC.
- L. Chena, Y. Kailua, C. Mingxinga, W. Xinyaa and Z. Weia, Adv. Text. Biotechnol., 2024, 32, 116 Search PubMed.
- L. Lyu, M. Bagchi, N. Markoglou, C. An, H. Peng, H. Bi, X. Yang and H. Sun, J. Hazard. Mater., 2024, 461, 132566 CrossRef CAS PubMed.
- A. M. Oliveira, A. L. Patrício Silva, A. M. V. M. Soares, D. Barceló, A. C. Duarte and T. Rocha-Santos, J. Environ. Chem. Eng., 2023, 11, 109308 CrossRef CAS PubMed.
- S. Razzaq, S. Shahid and Y. Nawab, Int. J. Biol. Macromol., 2024, 282, 136791 CrossRef CAS PubMed.
- A. L. Andrady, P. W. Barnes, J. F. Bornman, T. Gouin, S. Madronich, C. C. White, R. G. Zepp and M. A. K. Jansen, Sci. Total Environ., 2022, 851, 158022 CrossRef CAS PubMed.
- X. Huang, Y. Li, Z. Shu, L. Huang, Q. Liu and G. Jiang, Environ. Sci. Technol., 2024, 58, 13358–13369 CAS.
- I. Taniguchi, S. Yoshida, K. Hiraga, K. Miyamoto, Y. Kimura and K. Oda, ACS Catal., 2019, 9, 4089–4105 CrossRef CAS.
- E. Barnard, J. J. R. Arias and W. Thielemans, Green Chem., 2021, 23, 3765–3789 RSC.
- V. Tournier, S. Duquesne, F. Guillamot, H. Cramail, D. Taton, A. Marty and I. André, Chem. Rev., 2023, 123, 5612–5701 CrossRef CAS PubMed.
- V. Busico and R. Cipullo, Prog. Polym. Sci., 2001, 26, 443–533 CrossRef CAS.
- J.-W. Housmans, R. J. Steenbakkers, P. C. Roozemond, G. W. Peters and H. E. Meijer, Macromolecules, 2009, 42, 5728–5740 CrossRef CAS.
- A. Khoironi, H. Hadiyanto, S. Anggoro and S. Sudarno, Mar. Pollut. Bull., 2020, 151, 110868 CrossRef CAS PubMed.
- A. R. Othman, H. A. Hasan, M. H. Muhamad, N. I. Ismail and S. R. S. Abdullah, Environ. Chem. Lett., 2021, 19, 3057–3073 CrossRef CAS.
- S. Bahl, J. Dolma, J. J. Singh and S. Sehgal, Mater. Today, 2021, 39, 31–34 CAS.
- C. Aloke, O. O. Onisuru and I. Achilonu, Biochem. Biophys. Res. Commun., 2024, 734, 150774 CrossRef CAS PubMed.
- F. P. Guengerich, Pharmacol. Rev., 2024, 76, 1104–1132 CrossRef CAS PubMed.
- J. Kaur and P. K. Singh, Crit. Rev. Anal. Chem., 2022, 52, 949–967 CrossRef CAS PubMed.
- C. M. Shuford and R. P. Grant, J. Mass Spectrom. Adv. Clin. Lab, 2023, 30, 74–82 CrossRef CAS PubMed.
- S.-J. Yeom, T.-K. Le and C.-H. Yun, Trends Biotechnol., 2022, 40, 166–179 CrossRef CAS PubMed.
- M. Wang, Q. Li, C. Shi, J. Lv, Y. Xu, J. Yang, S. L. Chua, L. Jia, H. Chen, Q. Liu, C. Huang, Y. Huang, J. Chen and M. Fang, Nat. Nanotechnol., 2023, 18, 403–411 CrossRef CAS PubMed.
- R. Smith, C. Oliver and D. Williams, J. Biomed. Mater. Res., 1987, 21, 991–1003 CrossRef CAS PubMed.
- Q. Cai, G. Shi, J. Bei and S. Wang, Biomaterials, 2003, 24, 629–638 CrossRef CAS PubMed.
- N. Mohanan, Z. Montazer, P. K. Sharma and D. B. Levin, Front. Microbiol., 2020, 11, 580709 CrossRef PubMed.
- S. Yoshida, K. Hiraga, T. Takehana, I. Taniguchi, H. Yamaji, Y. Maeda, K. Toyohara, K. Miyamoto, Y. Kimura and K. Oda, Science, 2016, 351, 1196–1199 CrossRef CAS PubMed.
- P. Li and J. Liu, Environ. Sci. Technol., 2024, 58, 3065–3078 CAS.
- W. Wang, Y. Lin, H. Yang, W. Ling, L. Liu, W. Zhang, D. Lu, Q. Liu and G. Jiang, Environ. Sci. Technol., 2022, 56, 6857–6869 CrossRef CAS PubMed.
- X. Jiang, P. Zhang, S. Li, X. Tan, Z. Hu, B. Deng, K. Wang, C. Li, M. A. Sullivan and E. Li, Eur. Polym. J., 2016, 82, 175–180 CrossRef CAS.
- T.-K. Le, J. H. Park, G.-Y. Lee, W. S. Choi, K. J. Jeong, C. B. Park and C.-H. Yun, Green Chem., 2019, 21, 515–525 RSC.
- H.-Y. Jo, S.-H. Park, T.-K. Le, S. H. Ma, D. Kim, T. Ahn, Y. H. Joung and C.-H. Yun, Biotechnol. Lett., 2017, 39, 1245–1252 CrossRef CAS PubMed.
- T. H. H. Nguyen, S.-J. Yeom and C.-H. Yun, Appl. Sci., 2021, 11, 603 CrossRef CAS.
- K. Zhu, H. Jia, S. Zhao, T. Xia, X. Guo, T. Wang and L. Zhu, Environ. Sci. Technol., 2019, 53, 8177–8186 CrossRef CAS PubMed.
- T. De Jager, A. Cockrell and S. Du Plessis, Ultraviolet Light in Human Health, Diseases and Environment, 2017, pp. 15–23 Search PubMed.
- P. Pospíšil, Front. Plant Sci., 2016, 7, 232551 Search PubMed.
- N. Inoue, Xenobiotica, 1999, 29, 43–51 CrossRef CAS PubMed.
- X. Kammerscheit, F. Chauvat and C. Cassier-Chauvat, Front. Microbiol., 2019, 10, 471843 Search PubMed.
- A. Galle, Z. Czekus, K. Bela, E. Horvath, A. Ordog, J. Csiszar and P. Poor, Front. Plant Sci., 2019, 9, 427522 Search PubMed.
- Á. Gallé, M. Farkas, A. Pelsőczi, Z. Czékus, A. Kukri, Z. Dorner, A. Ördög, J. Csiszár, K. Bela and P. Poór, Agronomy, 2023, 13, 2053 CrossRef.
- C.-Y. Huang, M.-L. Roan, M.-C. Kuo and W.-L. Lu, Polym. Degrad. Stab., 2005, 90, 95–105 CrossRef CAS.
- H. Kaczmarek, D. Ołdak, P. Malanowski and H. Chaberska, Polym. Degrad. Stab., 2005, 88, 189–198 CrossRef CAS.
- X. Ramis, A. Cadenato, J. Salla, J. Morancho, A. Valles, L. Contat and A. Ribes, Polym. Degrad. Stab., 2004, 86, 483–491 CrossRef CAS.
- S. S. Ali, T. Elsamahy, E. Koutra, M. Kornaros, M. El-Sheekh, E. A. Abdelkarim, D. Zhu and J. Sun, Sci. Total Environ., 2021, 771, 144719 CrossRef CAS PubMed.
- N. Zhang, M. Ding and Y. Yuan, Microorganisms, 2022, 10, 1537 CrossRef CAS PubMed.
- D. A. Case, H. M. Aktulga, K. Belfon, I. Ben-Shalom, S. R. Brozell, D. S. Cerutti, T. E. Cheatham III, V. W. D. Cruzeiro, T. A. Darden and R. E. Duke, Amber 2021, University of California, San Francisco, 2021 Search PubMed.
- B. R. Miller III, T. D. McGee Jr, J. M. Swails, N. Homeyer, H. Gohlke and A. E. Roitberg, J. Chem. Theory Comput., 2012, 8, 3314–3321 CrossRef PubMed.
- M. S. Valdés-Tresanco, M. E. Valdés-Tresanco, P. A. Valiente and E. Moreno, J. Chem. Theory Comput., 2021, 17, 6281–6291 CrossRef PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. Berendsen, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek and A. Potapenko, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- M. Varadi, D. Bertoni, P. Magana, U. Paramval, I. Pidruchna, M. Radhakrishnan, M. Tsenkov, S. Nair, M. Mirdita and J. Yeo, Nucleic Acids Res., 2024, 52, D368–D375 CrossRef CAS PubMed.
- P. A. Williams, J. Cosme, D. M. Vinkovic, A. Ward, H. C. Angove, P. J. Day, C. Vonrhein, I. J. Tickle and H. Jhoti, Science, 2004, 305, 683–686 CrossRef CAS PubMed.
- P. Mark and L. Nilsson, J. Phys. Chem. A, 2001, 105, 9954–9960 CrossRef CAS.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- J. Wang, W. Wang, P. A. Kollman and D. A. Case, J. Mol. Graphics, 2006, 25, 247–260 CrossRef CAS PubMed.
- A. Jakalian, D. B. Jack and C. I. Bayly, J. Comput. Chem., 2002, 23, 1623–1641 CrossRef CAS.
- R. Anandakrishnan, B. Aguilar and A. V. Onufriev, Nucleic Acids Res., 2012, 40, W537–W541 CrossRef CAS PubMed.
- W. L. DeLano, Prot. Cryst., 2002, 40, 82–92 Search PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CrossRef CAS PubMed.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- H. J. Berendsen, J. v. Postma, W. F. Van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- B. Hess, H. Bekker, H. J. Berendsen and J. G. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- Adasme et al., PLIP 2021: expanding the scope of the protein-ligand interaction profiler to DNA and RNA. NAR 2021, https://plip-tool.biotec.tu-dresden.de/plip-web/plip/index.
- CharlesHahn, CharlesHahn/DuIvyTools: DuIvyTools (v0.3.0).Zenodo, 2022, DOI:10.5281/zenodo.6340263.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc03097h |
| This journal is © The Royal Society of Chemistry 2025 |