
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePersonal recollections of a quantum chemist
Frank
Weinhold
 *
*
Theoretical Chemistry Institute and Department of Chemistry, University of Wisconsin-Madison, Madison, Wisconsin 53706, USA. E-mail: weinhold@chem.wisc.edu
First published on 26th June 2025
Abstract
I describe some personal recollections pertaining to the role of quantum mechanics in chemistry, particularly relating to orbital- and resonance-based conceptions of chemical bonding and their role in describing the full range of chemical phenomena from molecular to material scale.
1. Introduction
“Perspective” is gained at a price, whether climbing to higher elevations or enduring larger swatches of time. In this 100 year anniversary of quantum theory, I attempt to recall some personal experiences loosely focused around the rise, fall, and re-rise of Pauling's “resonance” conceptions of chemical bonding. These recollections stretch over the six or so decades I have been privileged to follow such developments, up to the present era of computational chemistry impact in virtually every area of modern chemical research.2. Early contacts with science and scientists
I came from a rural family with little academic background. My father only reached third grade and my mother ninth grade before each was “needed at home” or dispatched westward with other refugees of the depression. They eventually met in the small panhandle town of Gering NE, where I attended high school and was offered my first chance to see a scientist of note.In about 1956, our chemistry instructor, Mr Stanley Bush, took a small science-club group to the “nearby” (ca. 300-mile round-trip!) University at Wyoming at Laramie to hear a lecture on stellar evolution by Hans Bethe, one of the giants of the Manhattan Project. Although I understood not a word and had scarcely heard of “quantum” theories of Nature, I thought it was something so interesting that I should try to learn more.
Mr Bush had studied chemistry at Colorado University in Boulder, and I resolved to do the same. There I had the good fortune of hearing quantum chemistry lectures from Joop de Heer, who had strong European collaborations with Ruben Pauncz1 and Per-Olov Löwdin2 on AMO (alternant molecular orbital) theory of benzene and related aromatic species.3 This was a semi-empirical attempt to adapt Mulliken-style MO methodology4 to Pauling's resonance conception of the chemical bond,5 which by that time was the well-established basis of all undergraduate chemical pedagogy. Such early awareness of Per Löwdin and the circle of quantum experts at his summer (Uppsala) and winter (Sanibel) quantum chemistry schools would soon become a major influence of my scientific life.
At Boulder, I also had opportunity to see early examples of technological advances that were to have powerful effects on chemical research and practice. To help cover college expenses, I had begun work as a freshman undergraduate assistant at the high altitude observatory with Dr Paul Julian. He was first on campus to acquire a general-purpose electronic digital computer –– an early Bendix model of refrigerator-like proportions (Fig. 1, left) that was used to process climate and stream-flow data for court-ordered reallocation of Rocky Mountain irrigation waters to (among others) my father's farm in western Nebraska. In this capacity I was able to gain a first-hand impression of the mysterious “programming language” that governed the contraption and was to become a central focus of my own academic career. Later, I was recruited back to Chemistry as an undergraduate assistant to incoming Prof Melvin Hanna6 and the freshly minted Varian A-60 NMR spectrometer (Fig. 1, right) to manage sample-handling and console operations for this near-magical device. I remember the day when Prof Stan Cristol brought in five test-tubes from the basement repository of synthesis samples, together with the PhD theses describing their structural characterization. Running one sample after another, we saw within minutes that two of the five theses had minor (but distinctive) structural errors in what had been laboriously inferred from pre-NMR methodology. Chemical progress was in the air!
 | ||
| Fig. 1 Promotional materials for first general-purpose Bendix G-15 computer (left) and Varian A60 NMR spectrometer (right) to arrive on the CU-Boulder campus. | ||
3. Grad and postdoc quantum immersion
1962 graduation from CU led to a “vacation” year of study at Freiburg as a Fulbright student. There I completed classwork and requirements for a Scheinprüfung with Georg Karagounis,7 but scientific conditions in the German universities still fell short of their pre-war eminence. However, one had opportunities to freely hear lectures and seminars in any subject area of interest, or to travel on weekends and study breaks to nearby cities and universities. These included “foreign” sites of scientific and cultural interest (Basel, Zürich, Paris, Vienna, Prague,…) as well as German birthplaces of quantum theory [Göttingen, Leipzig, Garching (where Heisenberg still worked), Berlin,…] that provided eye-opening international context for a Nebraska farm boy and his fellow Fulbrighter girl friend.The following year led to beginning of Harvard graduate studies with “E. B.” Wilson, a former student of Pauling and co-author of the best book ever written about how to solve quantum mechanical equations for chemical applications.8 There I immediately fell into comradely fellowship with other Wilson students in his newly formed “theory sub-group,” particularly with Bill Miller,9 who accompanied me to Löwdin's 1964 quantum chemistry school at Abisko, high above the Arctic Circle in far-northern Swedish Lapland. There we met under the midnight sun with many of the former, present, and future leaders of quantum chemistry, gathered together attentively by day for the brilliant schoolroom lectures of Löwdin and his colleagues. Any thought of leaving the lectures was deterred by the unimaginable swarms of blood-thirsty mosquitoes lurking outside, clustering mercilessly around the head of any participant who dared exit for the nearby dormitory without the necessary escape velocity (“Born-Oppenheimer breakdown”).
My own studies with Wilson10 dealt largely with mathematical N-representability conditions for valid 1st- and 2nd-order reduced density matrices, relating to the foundations of modern density functional theory. However, a focus for all theoretical students, including those of Martin Karplus, Bill Lipscomb, and other faculty, was the rapid progress then underway in Roald Hoffmann's collaborations with synthetic demi-god Robert A. Woodward, employing simple FMO (frontier molecular orbital) symmetry concepts to rationalize and predict chemical reaction mechanisms for a broad range of organic substrates of synthetic interest. This was probably the greatest advance outside the time of Pauling when improved quantum-chemical understanding propelled rapid advances in practical chemical applications. The entire department gathered for celebration when a new IBM 1620 minicomputer arrived in Mallinckrodt to serve as workhorse for quantum calculations that were now being pursued in many research groups, too important to be left to the then-available university computer facilities at Harvard.
My graduate studies included Summer 1966 appointment as “Visiting Research Associate” with Darwin Smith at Gainesville on formal density matrix theory and reuniting with the Florida Quantum Theory Group contingent of Per Löwdin's annual migrations between Uppsala and Gainesville. The Florida group focused ever more strongly on ab initio theory and methods, such as the highly accurate coupled-cluster studies later led by Rod Bartlett.
My postdoctoral studies included an NSF Fellowship (1967-68) with Charles Coulson's group in the Maths Institute at Oxford University, where Coulson's important early contributions to valence theory11 were pursued. This time was largely focused on methods for obtaining mathematically rigorous upper and lower bounds for quantum–mechanical properties, carried out independently during Coulson's extended absences for service with the World Council of Churches and chairmanship of Oxfam during my time there.
Conditions for quantum-chemical programming and computations at Oxford were well behind those to which I was accustomed at Harvard. The algol-oriented English Electric computer then available was accessible to fortran users for only one midnight run per day, with the job to be submitted by late afternoon as punched paper tape (a la Bendix days!) in a plastic bag that was returned (if successful, with printed output) the following morning. The paper tape, if containing an error or otherwise damaged by crimping or tearing during the computer run, had to be replaced by an entirely new tape that required hours of error-free concentration for the following day's run. My exasperation was complete one day when the plastic bag was returned with torn tape, no output page, and a cigarette butt!
A second year (1968-69) of postdoctoral study brought me to Berkeley as an independent Miller Fellow. There I interacted most strongly with George Pimentel, whose work on H-bonding12 I much admired. Other contacts were with key applied mathematicians then involved in IBM System/360 mainframe design, where the increasingly high-precision needs of quantum chemists were being accommodated by the double-precision 64 bit representation of floating-point numbers, well beyond those needed for common “business machine” applications. Even as I was completing the year in preparation for the move across the bay to my first academic appointment at Stanford, the Berkeley faculty was recruiting Bill Miller and Fritz Schaefer as dual appointments in the burgeoning field of computational quantum chemistry.
4. Inception of natural bond orbital methods
At Stanford, my major research focus continued to be on mathematically rigorous variational bounds, quantifying limits of error as inequalities between “truth” and present best-estimate in quantum chemical calculations.13 However, the limitations of such methods for practical chemical applications was increasingly apparent. In many cases, the underlying mathematical basis of such rigorous “bounding” properties can be recognized as the elementary geometry of the Hilbert space in which all formulations of quantum mechanics reside, as first recognized by Göttingen mathematician David Hilbert. Thus, I was acutely aware of the axiomatic requirements for such geometrically-shaped formal structure of a physical theory.Such awareness led me inevitably toward a still more esoteric consideration, namely, whether other specialized areas of physical theory might also find their common foundation in such geometry-shaped mathematical structure. I had excellent training in the fundamentals of Gibbsian thermodynamics from Joop de Heer at Boulder, so was well aware of the most famous inequality in all science, the inexorable increase of entropy in natural events as expressed in the second law of thermodynamics. Such inherent non-negativity often leads to combining rules of convex type, “weighted averages” with non-negative weightings {wL} summing to unity,
| ΣLwL = 1, each wL ≥ 0 | (1) |
If an experimental measurement (Ê) can lead to an enumerable (L) list of distinguishable alternative outcomes (EL), their weighted-average 〈Ê〉 can be written as the convex combination,
| 〈Ê〉 = ΣLwL × EL | (2) |
As the thermodynamic geometry papers were first reaching publication in 1975, attention was already turning in a new direction that was to have broader impact on chemical research and pedagogy. Although the term “natural bond orbital” (or NBO acronym) did not appear in the first “Principle of Maximum Overlap” paper with student Terry Brunck,21 one can clearly see the seeds of future NBO developments that were to dominate our research efforts from that point forward.
The year 1975 also marked a time of transition for my group and my family. When I was first hired to Stanford in 1969 by department chairman Bill Johnson (brought in from Madison to hire a raft of Nobel-worthy senior faculty, including Linus Pauling), it was made clear to me and other junior faculty that there would be no realistic prospect for tenure promotions in the foreseeable future. Accordingly, I had accepted the invitation to come to the University of Wisconsin in 1976 to join Joe Hirschfelder and colleagues in the Theoretical Chemistry Institute, home of the famous Molecular Theory of Gases and Liquids treatise22 and one of the leading centers of theoretical chemistry research from wartime on.
As will be seen, the maximum overlap paper employed convexity concepts that were closely related both to the geometry of thermodynamics as well as to Pauling's original resonance formulation of localized orbital and bond-order measures. To see any such measure of commonality between intrinsically quantal and intrinsically thermodynamic properties is at first glance quite surprising. However, it is also instructive to recall that the only parcel of 19th-century science that passed unscathed through the quantum revolution of 1925-26 was that of Gibbs's chemical thermodynamics, which required not an iota of change from what Gibbs carefully wrote down in 1876. Any such measure of quantal/thermodynamic commonality implies that there is a connected conceptual highway that somehow spans the perceived abyss (ca. 1023 in magnitude) between “molecular” and “material” levels of description. How can this be?
5. NBO terminology and conceptions of covalent bonding
It is inappropriate to attempt any detailed mathematical description of NBO algorithms for this perspective overview. Suffice it to say that NBO analysis has no role in selecting or altering the form of wavefunction, basis functions or other computational minutiae that the user has employed (for whatever reason) to describe the electronic system of interest. Nor does it matter what specific electronic-structure package among the many available choices (Gaussian, GAMESS, Orca, Q-Chem, Molpro,…)23 was used to perform the quantum-chemical calculation (up to the exact solution of Schrödinger's equation) that the interactive NBO7 program may be requested to analyze. The only mission of the NBO7 program is to find the optimal forms of localized Lewis/Pauling-type descriptors that can best describe the calculated e-density as provided by the MYPROG component of a tandem MYPROG/NBO7 (interactive binary-pair) installation on your computer system.In general, NBO7 output makes it fairly easy to “read” how the printed NBOs relate to the elementary Lewis-structure diagram of e-pairs in a closed-shell molecule, either as a “lone pair” (LP) on a single-atom (1c) site or a 2c “bond” (BD) between atoms. The “shapes” of NBOs can be graphically displayed in the NBO7@Jmol24 or other orbital viewer, or alternatively described numerically in terms of conventional Pauling-type spn “natural” hybrid orbitals (NHOs) hA, hB and their polarization coefficients cA, cB.
As an example, Fig. 2–4 illustrate simple NBO features of carbon monoxide, the strongest known example of binding between two atoms. CO also serves as a simple example of the close connection of NBOs to the early concepts of G. N. Lewis, following the path from Lewis's shared e-pair concept to the logic and terminology of quantization as pioneered by Linus Pauling. Fig. 2 shows the Lewis-structure diagram of CO, with one lone pair (in parentheses) on each atom and three bond pairs (sticks) connecting them, a triple-bonded molecule. Fig. 3 shows orbital imagery of the σCO sigma-bond [from elementary DFT-level B3LYP-D3/cc-pVTZ description], and Fig. 4 shows the corresponding imagery for one of the two equivalent πCO pi-bonds. Table 1 summarizes numerical descriptors of the hybrid and bond orbitals for this species, all recognizable to those trained in Lewis-structural concepts.
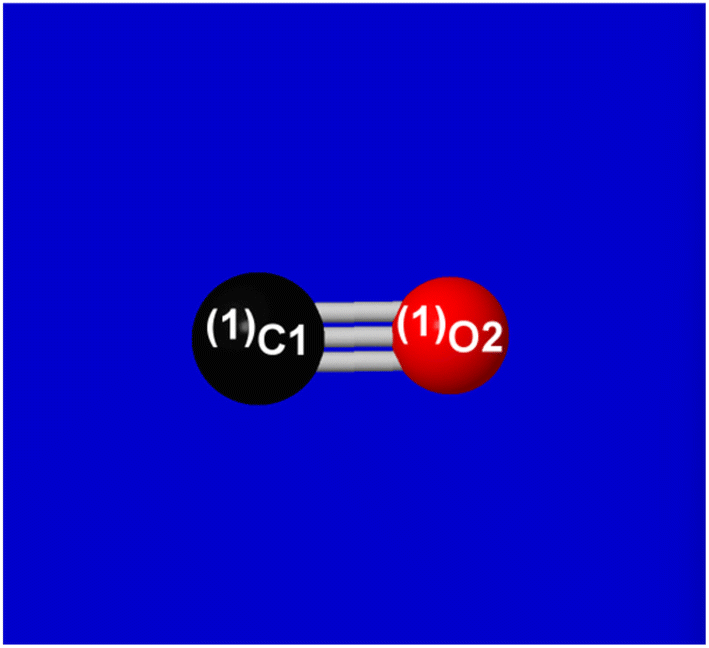 | ||
| Fig. 2 Natural Lewis structure (NLS) ball-and-stick diagram for carbon monoxide. | ||
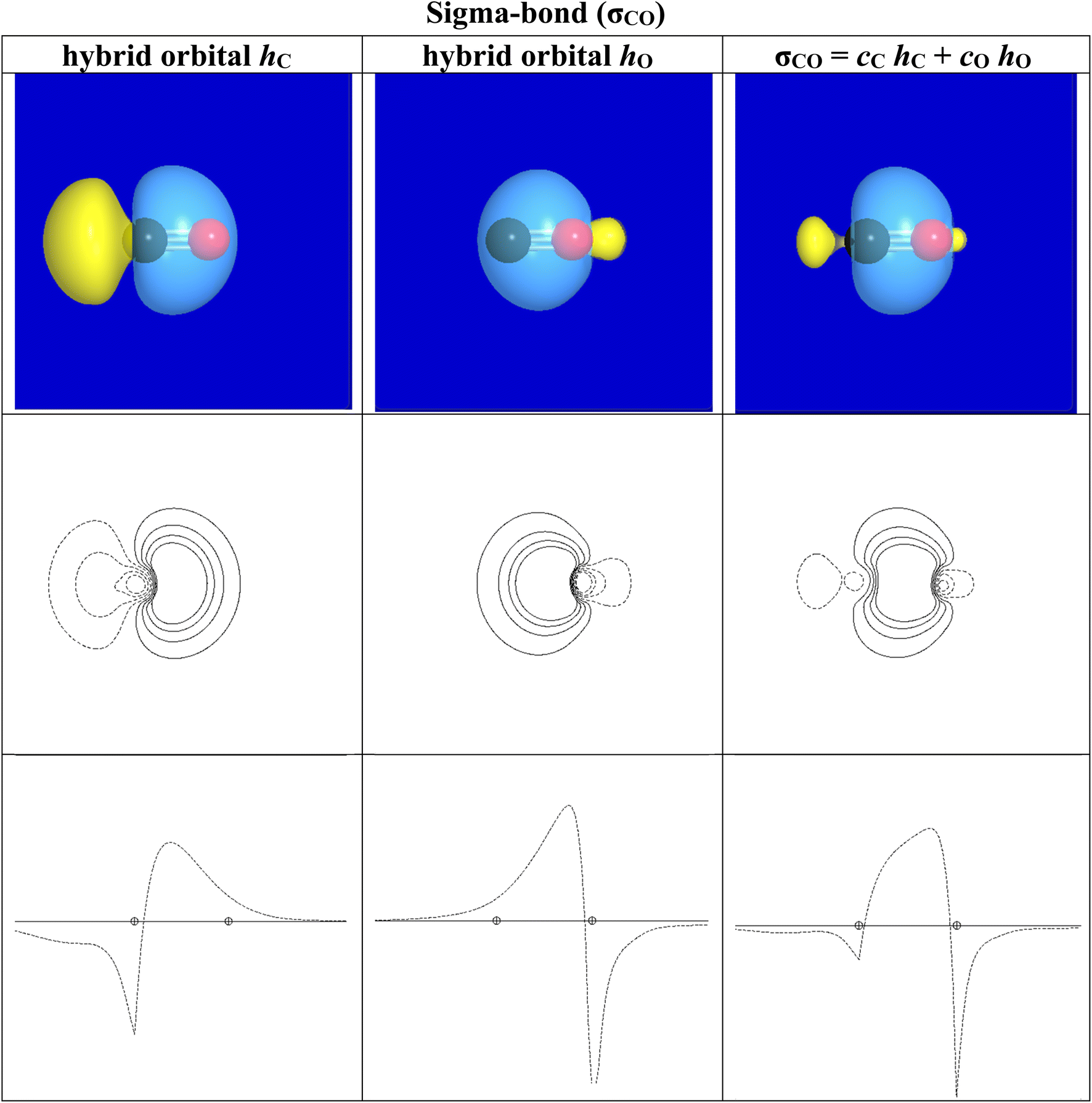 | ||
| Fig. 3 Orbital imagery for the sigma-bond (σCO) of CO, showing carbon hybrid orbital (hC, left), oxygen hybrid orbital (hO, center), and sigma-bond orbital (σCO, right) in 3D view (upper), 2D contour (middle), and 1D profile (lower) plots. Phase differences are denoted by solid (positive) or dashed (negative) lines in the 2D plots. | ||
 | ||
| Fig. 4 Similar to Fig. 3, for the CO pi-bond (πCO) (the 1D profile plot along the bond axis is everywhere zero for a pi-bond). | ||
| Hybrid | h C | h O |
|---|---|---|
| spn | sp2.66 | sp1.20 |
| %-s; %-p | 27%-s; 73%-p | 45%-s; 54%-p |
| Coeff | 0.4976 | 0.8674 |
How “good” is the simple DFT-level Lewis-structure and Pauling hybridization picture in depicting the full electron density of CO? Theoreticians were sometimes prone to denigrate the accuracy of such Pauling-type depictions as “oversimplified,” but they are actually quite good when the numerical descriptors of hybridization and bonding are properly optimized by the NBO algorithms. The accuracy of the Pauling-type description can be quantified as %-ρNLS, the percentage of total electon density (ρ) that is properly captured in the natural Lewis-structure (NLS) description, with numerical value
| %-ρNLS = 99.924%. | (3) |
Furthermore, the simple DFT-level quantum chemical description we are using is able to predict the actual measurable properties of CO quite respectably. Table 2 summarizes some comparisons of DFT-theory vs. experiment for bond-length (RCO), dipole moment (dCO),25 and vibrational frequency (νCO) of this interesting molecule. Again, not bad! It is clear that ever-improving DFT methods have led to ever-better NBOs.
| Property | DFT | Experiment |
|---|---|---|
| R CO (Å) | 1.126 | 1.128 |
| d CO (D) | 0.126 | 0.122 |
| ν CO (cm−1) | 2213 | 2143 |
The intellectual debt to G. N. Lewis's pre-quantal conceptions and their quantal-“translation” by Linus Pauling extends further to Lewis acid-base interactions. In modern terminology, a Lewis acid is the “e-acceptor” and Lewis base the “e-donor” in the give-and-take of electronic donor–acceptor interactions. All such “non-Lewis” (NL) corrections to the elementary Lewis-structure picture are inherently incorporated in Pauling's conception of resonance, to whose NBO-based implementation we now turn.
6. On to resonance!
The essence of Pauling's powerful advancement of chemical theory is its Lewis-like focus on localized electronic interactions, elevating the shared e-pair “chemical bond” to primacy as the logical unit of chemical structure and reactivity. This viewpoint naturally came into conflict with the more physical perspective of what later became known as molecular orbital (MO) theory, also called mean-field or self-consistent field (SCF) theory. Important developers of SCF-MO theory (including Hartree, Fock, Hückel, Slater, Hund, Mulliken) were all physicists by training and departmental association, and their primary focus was atomic physics rather than chemistry.26 Only Hund's variational reformulation of SCF theory in terms of Slater-determinant trial functions made it feasible for Mulliken (around 1940) to re-shape the theory for “molecular orbital” studies toward the way we know it today. SCF-MO theory is indisputably more suited for automated computation, but intrinsically ignores important e-correlation effects such as London dispersion. The path-breaking developments of Walter Kohn and others remove some of these restrictions and underlie the current DFT theory of this genre. Ever deeper connections to Pauling's deeply “chemical” conceptions are obtained in these developments.27The NBO7 (https://nbo6.chem.wisc.edu/)-analysis program that is interfaced to many modern electronic-structure programs23 first saw the extension to more general “natural resonance theory” (NRT) analysis in the work of Eric Glendening in the early 1990s.28 In effect, the NRT algorithms generalize and subsume the NBO functionality of its namesake host program. The original variational algorithms to extract optimized Pauling-type resonance structures, weightings, and bond-orders from the input wavefunction or density were subsequently upgraded to the far more efficient convex-programming techniques of Steve Wright,29 which are intimately related to the form (1) of the resonance expansion itself in NBO7.30
The fully generalized NRT implementation now available in NBO7 is able in principle to extract Pauling-type weightings and interatomic bond orders {bAB} for any wavefunctions or density that is presented to it for analysis, up to and including exact solution of the Schrödinger equation. Thus, the algorithms are indifferent to whatever assumptions, approximations, or “biases” may underlie the method selected by the user for analysis. The only criterion of success or failure in the analysis is whether the output NRT weightings and bond orders can achieve the same type of virtually magical correlation with chemical observations that appeared in virtually every page of Pauling's Nature of the Chemical Bond.
The NRT method not only brings Pauling's early conceptions into the framework of modern computational quantum chemistry, but opens the door to extending the quantum-chemical conceptions of the molecular domain seamlessly to the thermodynamic and kinetic conceptions of the macrosopic “materials” domain. Such a smooth pathway between microscopic- and macroscopic-level descriptions is made possible by Fukui's intrinsic reaction coordinate (IRC) concept31 and recognition32 that the intrinsic unit of “chemical change” is the elementary chemical reaction that converts the pattern of bond orders {bAB(R)} of initial reactant species to those {bAB(P)} of final product species. The sequence of mechanistic reaction steps that underlie an overall chemical transformation can therefore be continuously followed along the IRC pathway, spanning not only the stationary equilibrium states that terminate each reaction step but also the intermediate dynamical (non-equilibrium) {bAB(IRC)} values that smoothly connect these termini. As shown by Fukui, the high-energy transition-state stationary feature defines and shapes all features of the IRC. The NRT description provides the detailed {bAB(IRC)} bond-order descriptors that literally quantify mechanistic progress along the IRC pathway, both for static and reactive IRC values.
As a simple example, we may take the proton-transfer step of nitrosamine degradation to diazonium cation that obstructs proper G–C pairing in DNA. The upper panels of Fig. 5 show the overall reaction (left) and transition-state geometry (right; taken from ref. 32) for this step, while the lower panels show the corresponding E(IRC) energy profile (left) and mechanistic “NRT portrait” (right) of bond-order shifts in this reaction, with color-coded bond-order pairs matched to the atom numberings in the upper panel. Unlike Pauling's long-held opposition to “no-bond resonance” of the intermolecular regime, the NRT descriptions extends uniformly over intra- and intermolecular interactions and exhibits the beautiful NRT symmetries between bond-shifts in the sub-integer and supra-integer range.
 | ||
| Fig. 5 Schematic reaction (upper left), transition-state geometry (upper right), E (IRC) energy profile (lower left), and mechanistic NRT profile of bond-order shifts (lower right) for carcinogenic nitrosamine degradation.32 | ||
It is also evident that NRT's intrinsic inclusion of “no-bond” resonance implies a greatly expanded role in the analysis of weaker “soft matter” interactions, particularly near transition-states for reactive bond-shifting. An equilibrium example of such soft–matter interactions is provided by diberyllocene,33 a sandwich-type complex in which two beryllium atoms (notoriously averse to bonding) are found to be positioned in D5h-like geometry between near-eclipsed C5H5 rings. Fig. 6 displays the calculated NRT bond orders for this species,34 which exhibit sub-integer values for all but the ring bCC bonds, ranging down to bCBe = 0.014. The lesson of this example, and many others likely to follow, is that the importance of resonance-type corrections is increasing in the intermolecular domain, where their existence was long denied by Pauling.
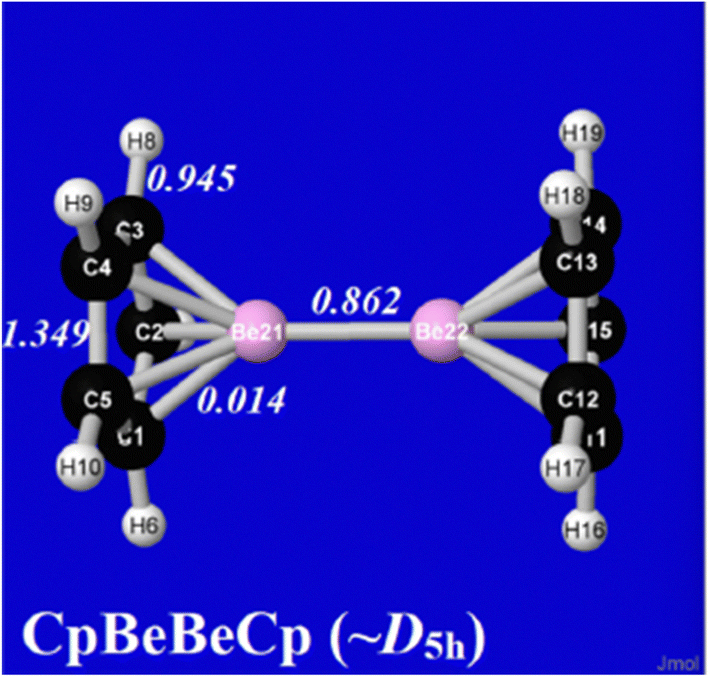 | ||
| Fig. 6 NRT bond orders of diberyllocene. | ||
The extension of resonance conceptions into the intermolecular domain of soft–matter interactions has other consequences and implications for the future of quantum chemistry. As the number of possible contributing resonance structures continues to increase in the long-range limit, the dominance of any particular Lewis-structural bonding pattern continues to wane. Inevitably, the “parent” NLS that was initially chosen for “correction” may become intermingled in weighting with many others, leading to situations where the bonding pattern of the leading “natural resonance-type Lewis-structure” (NLRS) may differ from that of the original NLS from which the NRT search was initiated. In this manner, the NRT “extension” of NBO theory can lead to the “resonance primacy” limit where no sensible vestige of the “dominant” localized Lewis-structure picture (on which NBO theory was built) survives. This limit serves to suggest how a conceptual picture of chemical bonding, like that of its developers, may have only a transient lifetime of usefulness in the longer-range development of chemical science.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this perspective.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Among many other collaborators, I thank Eric Glendening and Clark Landis for their many contributions to the work described herein.References
- R. Pauncz, ACS Cent. Sci., 2017, 3, 515–523 CrossRef CAS PubMed.
- E. J. Brändas, Mol. Phys., 2017, 115, 1995–2024 CrossRef.
- R. Pauncz, J. de Heer and P. O. Löwdin, J. Chem. Phys., 1962, 36, 2257–2265 CrossRef CAS.
- R. S. Mulliken, J. Pure App. Chem., 1974, 2, 203–215 Search PubMed.
- L. Pauling, The Nature of the Chemical Bond, Cornell U. Press, Ithaca, NY, 1st edn, 1939 Search PubMed.
- M. H. Hanna, Quantum Mechanics in Chemistry, W. A. Benjamin, NY, 1957 Search PubMed.
- G. Karagounis, Einführung in die Elektronentheorie Organischer Verbindungen, Springer Verlag, Berlin, 1959 Search PubMed.
- L. Pauling and E. B. Wilson Jr, Introduction to Quantum Mechanics, McGraw-Hill, NY, 1935 Search PubMed.
- W. H. Miller, J. Phys. Chem. A, 2001, 105, 5487–5489 Search PubMed.
- F. Weinhold and E. B. Wilson Jr, N-representability and the Pauli exclusion principle for fermion pair distributions, in Reduced Density Matrices with Applications to Physical and Chemical Systems, Queen's Papers on Pure and Applied Mathematics, ed. A. J. Coleman and R. M. Erdahl, Queen’s University, Kingston, Ontario, Canada, 1968, vol. 11, pp. 71–82 Search PubMed.
- C. A. Coulson, Valence, Oxford U. Press, Oxford UK, 1961 Search PubMed.
- G. C. Pimentel and A. L. McClellan, The Hydrogen Bond, W. H. Freeman & Co., San Francisco, 1960 Search PubMed.
- F. Weinhold, Upper and lower bounds to quantum-mechanical properties, in Advances in Quantum Chemistry, ed. P.-O. Löwdin, Academic Press, New York, 1972, vol. 6, pp. 299–331 Search PubMed.
- F. Weinhold, J. Chem. Phys., 1975, 63, 2479–2483 CrossRef CAS , and ensuing papers..
- F. Weinhold, Phys. Today, 1976, 29, 23–29 CrossRef CAS.
- F. Weinhold, Geometrical aspects of equilibrium thermodynamics, in Theoretical Chemistry: Advances and Perspectives, ed. H. Eyring and D. Henderson, Academic Press, New York, 1978, vol. 3, pp. 15–54 Search PubMed.
- F. Weinhold, Classical and Geometrical Theory of Chemical and Phase Thermodynamics, John Wiley, Hoboken NJ, 2009 Search PubMed.
- J. W. Gibbs, Trans. Conn. Acad., 1876, 3, 108–248 Search PubMed ; 1878, 343–524..
- https://en.wikipedia.org/wiki/Riemannian_geometry .
- https://sciety.org/articles/activity/10.20944/preprints202504.0685.v2 .
- F. Weinhold and T. K. Brunck, J. Am. Chem. Soc., 1976, 98, 3745–3749 CrossRef CAS.
- J. O. Hirschfelder, C. F. Curtiss and R. B. Bird, Molecular Theory of Gases and Liquids, Wiley, NY, 1954 Search PubMed.
- For a listing of NBO7-affiliated electronic-structure programs and their degree of interactivity, https://nbo6.chem.wisc.edu/affil_css.htm.
- https://nbo6.chem.wisc.edu/nbopro_adv.htm .
- Note that the agreement extends to the sign, as well as the magnitude, of the CO dipole moment, which has the curious reversal from naïve electrostatic conceptions; A. Schwarz, A. Köhler, J. Grenz and R. Wiesendanger, Appl. Phys. Lett., 2014, 105, 011606 CrossRef . Note also that CO shows no further need for resonance-type “corrections,” such as the dative-type donor-acceptor interactions described by natural resonance theory (Section 6). This specifically refutes Sidgwick’s early suggestion of dative-type σ-bonding in CO [N. V. Sidgwick, Chem. Rev., 1931, 9, 77–88], which was also discounted by Pauling (Ref. [5], p. 9)..
- Key papers in this development include: (a) D. R. Hartree, Math. Proc. Cambridge Philos. Soc., 1928, 24, 111–132 CrossRef CAS; (b) J. C. Slater, Phys. Rev., 1928, 32, 339–348 CrossRef; (c) V. A. Fock, Z. Phys., 1930, 61, 126–148 CrossRef ; Ref. [4].
- E. D. Glendening and F. Weinhold, Molecules, 2021, 26, 4110 CrossRef CAS PubMed.
- E. D. Glendening and F. Weinhold, J. Comput. Chem., 1998, 19, 593–609 CrossRef CAS , and ensuing papers..
- (a) E. D. Glendening, S. J. Wright and F. Weinhold, J. Comput. Chem., 2019, 40, 2028–2035 CrossRef CAS PubMed; (b) E. D. Glendening, C. R. Landis and F. Weinhold, J. Am. Chem. Soc., 2019, 141, 4156–4166 CrossRef CAS.
- E. D. Glendening, C. R. Landis and F. Weinhold, J. Comput. Chem., 2019, 40, 2234–2241 CrossRef CAS PubMed.
- K. Fukui, J. Phys. Chem., 1970, 74, 4161–4163 CrossRef CAS.
- F. Weinhold, Entropy, 2025, 27, 390 CrossRef CAS PubMed.
- (a) Y. Xie, H. F. Schaefer III and E. E. Jemnis, Chem. Phys. Lett., 2005, 402, 414–421 CrossRef CAS; (b) J. T. Boronski, A. E. Crumpton, L. L. Wales and S. Aldridge, Science, 2023, 380, 1147–1149 CrossRef CAS; (c) J. L. Dutton, Science, 2023, 380, 1106–1107 CrossRef CAS.
- F. Weinhold and E. D. Glendening, Phys. Chem. Chem. Phys., 2024, 26, 2815–2822 RSC.
| This journal is © The Royal Society of Chemistry 2025 |