
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Promoting electrocatalytic CO2 reduction to n-propanol over ethanol at Cu step sites†
Yuanyuan
Xue
,
Ximeng
Lv
 ,
Chao
Yang
,
Lu
Song
,
Lijuan
Zhang
* and
Gengfeng
Zheng
*
,
Chao
Yang
,
Lu
Song
,
Lijuan
Zhang
* and
Gengfeng
Zheng
*
Laboratory of Advanced Materials, State Key Laboratory of Porous Materials for Separation and Conversion, Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials, Fudan University, Shanghai, 200438, China. E-mail: gfzheng@fudan.edu.cn; zhanglijuan@fudan.edu.cn
First published on 27th June 2025
Abstract
Obtaining valuable C3+ products directly from the electrocatalytic reduction of CO2 or CO is an attractive but challenging task, due to the much more complicated reaction pathways and sluggish kinetics of C3+ products than their C1 and C2 counterparts. As different C3+ products and competitive C2 side-products may share the common rate-determining step (e.g. the carbon–carbon coupling), the regulation of subsequent selectivity-determining step(s) is critical for promoting the selectivity of C3+ products. Herein, we focused on tuning the selectivity competition between n-propanol (n-C3H7OH, an important C3+ alcohol) versus ethanol (C2H5OH, a major C2 side product), based on the constant potential computations on the Cu surface with different step sites. The critical selectivity-determining steps for the n-C3H7OH and C2H5OH pathways have been identified, and the impact of Cu step sites on the competitive relation between n-C3H7OH and C2H5OH has been explored. Moreover, a descriptor related closely to the n-propanol selectivity has been developed, showing that controlling the competitive hydrogenation of C2 intermediates and C1–C2 coupling processes is vital to differentiate the selectivity of n-propanol from ethanol. This work can inspire the screening and rational design of unconventional electrocatalytic sites for generating more value-added C3+ products from the electrocatalytic CO2 reduction.
Introduction
The electrocatalytic CO2 or CO reduction reaction (CO2RR/CORR) using renewable electricity has attractive potential for reducing carbon footprint and energy storage in liquid fuel products like alcohols,1–4 due to their high energy densities, convenient storage, and facile transportation.5,6 C1 and C2 alcohols, i.e., methanol (CH3OH)7–9 and ethanol (C2H5OH),10–12 have relatively high selectivities and activities. In contrast, the selective electroreduction of CO(2) into C3+ alcohols, such as n-propanol (n-C3H7OH), is still challenging versus the competing side reactions of C1 and C2 products. As the CO(2)-to-C3H7OH involves complicated reaction pathways containing both the C1–C1 coupling and subsequent C1–C2 coupling,13,14 most of the reported faradaic efficiencies (FEs) of n-C3H7OH in CO(2) electroreduction are still below 20% to date.15–18A variety of approaches have been investigated to promote the selectivity of the n-propanol product from the CO(2)RR. For instance, doping Au into Cu(100) was reported to decrease the adsorption of CO* (where * refers to the adsorption site) while retaining the intrinsic Cu(100) active sites at the same time, which facilitated the C1–C2 and C1–C1 coupling process and presented a peak FE of 18% for n-C3H7OH.15 Cu co-doped with Ag and Ru was synthesized for the CO electroreduction to n-C3H7OH, with a 37% FE and >100 mA cm−2 of partial current density.19 Nonetheless, the production selectivity and yield of n-C3H7OH by the electrocatalytic CO(2)RR are still much lower than those of the C1 and C2 side products and also far from the commercialization requirements.20–22
The selectivity of C1 and C2 products in the CO(2)RR can be promoted based on the rate-determining step (RDS) regulation,23–29 such as using atomic structure design23 or microenvironmental tuning.24,26 However, as the C3 formation steps (e.g. the C1–C2 coupling and the hydrogenation of C3 intermediates) are far away from the initial reaction stage and unlikely to serve as the RDS,30 different C3+ products and those C2 side products may share the same RDS. Thus, it is hard to improve the selectivity of C3+ products by the RDS tuning strategy. The selectivities of C3+ products should mainly be determined by the selectivity-determining steps (SDSs) for the competitive pathways.31 Ethanol has been proposed as a major competing side product of n-C3H7OH.32–36 Wang and coworkers analyzed the reported CO2RR-relevant studies using the machine learning method and found correlation between FEs/ΔFEs of ethanol and n-propanol, suggesting that ethanol and n-propanol share the common C–C coupling process and compete with each other.36 In addition, according to our previously reported work,2 the pathways to ethanol and n-propanol separate with the acetate/acetic acid pathway at an earlier stage (CH2CO*). Thus, the FE of acetate is generally low under conditions that are advantageous for the n-propanol formation.37,38 Thus, the competitive relationship between ethanol and n-propanol is more critical for determining the n-propanol selectivity in the CO(2)RR. By using differential electrochemical mass spectrometry, it was found that the concentration ratios of acetaldehyde/ethanol and propionaldehyde/n-propanol near the cathode surface are higher than those in the bulk electrolyte during CO2 electroreduction, suggesting that acetaldehyde (CH3CHO) is the bifurcation point of C2H5OH and n-C3H7OH.32 The subsequent coupling of CH3CHO and CO* can lead to the formation of n-C3H7OH, while the further hydrogenation of CH3CHO results in C2H5OH.32 In addition to acetaldehyde, methylcarbonyl (CH3CO*) has also been suggested as another possible branching point for the C2H5OH and n-C3H7OH pathways.34 Nonetheless, despite that they are crucial for the CO(2)RR to n-C3H7OH, the branching intermediates and selectivity-determining steps for the C2H5OH and n-C3H7OH pathways are still ambiguous, precluding the breakthrough of designing efficient electrocatalysts.
In this work, we first conducted constant potential computations to identify the selectivity-determining steps and the critical bifurcation intermediate for the n-C3H7OH and C2H5OH pathways. Then we designed a variety of high-index Cu facets with step sites and theoretically investigated for their catalytic performances on the selectivity competition between the n-C3H7OH and C2H5OH pathways. Finally, a critical descriptor was developed to predict the capabilities of different Cu sites for the CO(2)RR to n-C3H7OH, suggesting the potential of developing new electrocatalysts for more value-added products.
Results and discussion
Selectivity mechanism
As Cu(100) has been widely reported for the CO(2)RR to C2+ products (mostly C2 products like ethylene and ethanol though),39 we first conducted constant potential calculations to explore the critical elementary steps regarding the competition between C2H5OH and n-C3H7OH pathways on Cu(100) (computational details in Fig. S1 and Tables S1, S2†). There are two possible bifurcation intermediates (i.e., CH3CO* and CH3CHO*) for the competition pathways between C2H5OH and n-C3H7OH,32–34 and the possible hydrogenation steps and coupling steps of those two intermediates are schematically displayed (Fig. 1a). Although CH2CHO* has also been proposed as a possible precursor to form CH3CHO*,37 the formation of CH3CO is easier than that of CH2CHO* (Fig. S2†). Thus, CH3CO* is chosen as the starting point (Fig. 1a). For the hydrogenation of CH3CO*, the free energy change (ΔG) to CH3CHO* (i.e., CH3CO* + H+ + e− → CH3CHO*) is more negative than that of CH3COH* (i.e., CH3CO* + H+ + e− → CH3COH*) in the whole potential range and pH range (Fig. S3†), indicating that the carbon atom of the carbonyl group in CH3CO* tends to obtain the proton rather than the oxygen atom of the carbonyl group. For the subsequent hydrogenation of CH3CHO*, the carbon atom of the aldehyde group is also easier to obtain the proton (i.e., CH3CH2O*) than the oxygen atom of the aldehyde group (i.e., CH3CHOH*) (Fig. S4†), suggesting that CH3CH2O* is more likely to be the key intermediate toward ethanol than CH3CHOH*. For the n-propanol formation pathway (Fig. S5†), the coupling of CH3CHO* with CO* tends to form CH3COCHO* on Cu(100) within the whole potential and pH ranges, rather than form the CH3CHOCO* intermediate.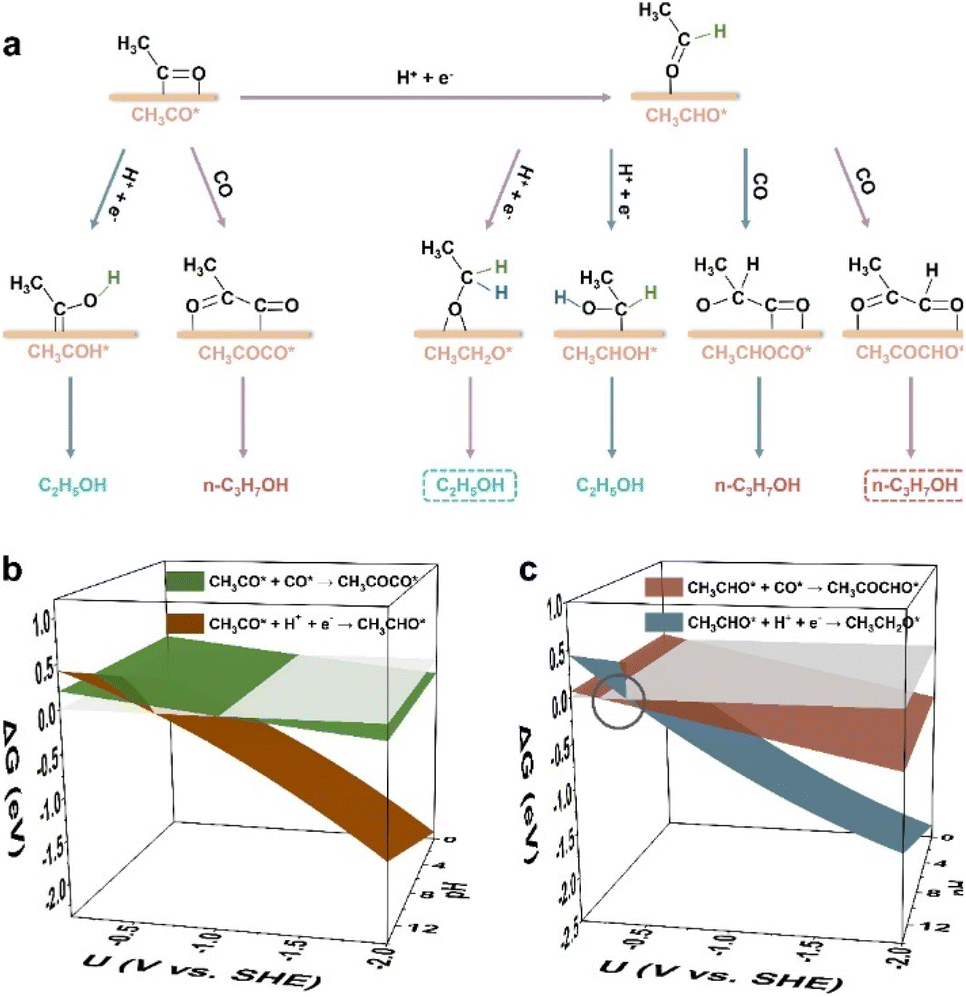 | ||
| Fig. 1 (a) Possible hydrogenation and coupling steps of the two possible branching intermediates (CH3CO* and CH3CHO*) for C2H5OH and n-C3H7OH pathways. The preferable hydrogenation and coupling steps of CH3CO* and CH3CHO* are marked with purple arrows. The hydrogen atoms from the hydrogenation of CH3CO* are shown in green color, and the hydrogen atoms from the hydrogenation of CH3CHO* are shown in blue color. The most possible C2H5OH and n-C3H7OH pathways are highlighted with the dashed boxes. (b) Free energy changes of the hydrogenation and coupling steps of CH3CO* on Cu(100) versus the potential and pH. (c) Free energy changes of the hydrogenation and coupling steps of CH3CHO* on Cu(100) versus the potential and pH. The circle highlights the dominant potential range (at pH 14) where the coupling step proceeds preferably. The grey planes in (b) and (c) are the planes with the function of ΔG = 0 (eV). | ||
From the above analysis, the most possible hydrogenation and coupling steps of CH3CO* and CH3CHO* are determined (purple arrows in Fig. 1a), among which CH3CHO* can be obtained from the hydrogenation of CH3CO*. As shown in Fig. 1b, the coupling between CH3CO* and CO* (i.e., CH3CO* + CO* → CH3COCO*) is preferable under alkaline conditions, as the ΔG of the CH3CO* hydrogenation step (i.e., CH3CO* + H+ + e− → CH3CHO*) is more positive in a higher pH environment. However, when the coupling step becomes spontaneous, ΔG of the CH3CO* protonation step is more negative, even at pH 14. Thus, the protonation of CH3CO* to CH3CHO* is generally advantageous during the CO(2)RR. On the other hand, for CH3CHO* in an alkaline environment (Fig. 1c), the coupling step (CH3CHO* + CO* → CH3COCHO*) is more preferable than its protonation step in the potential range of −0.27 to −0.50 V vs. the standard hydrogen electrode (SHE) at pH 14, suggesting that CH3CHO* is more likely to be the branching intermediate for C2H5OH and n-C3H7OH pathways. The corresponding SDS for C2H5OH formation is: CH3CHO* + H+ + e− → CH3CH2O*, and the corresponding SDS for n-C3H7OH formation is: CH3CHO* + CO* → CH3COCHO*. Kastlunger et al. conducted microkinetic simulations based on the constant-potential density functional theory (DFT) to explore the formation of C2 products by the CO2RR on Cu(100)40 and found that the hydrogenation of CH3CHO* to CH3CH2O* led to the formation of C2H5OH, consistent with our results. Recently, the surface reconstruction of Cu(100) during the CO2RR was theoretically explored by the potential-dependent grand canonical Monte Carlo method combined with the environmental kinetic Monte Carlo method and the DFT method, showing that C2H5OH can be produced through the hydrogenation of CH3CHO* to CH3CH2O*.41 This work also supports that the hydrogenation of CH3CHO* is a critical step for the formation of C2H5OH. The free energy profiles of SDSs for both the C2H5OH and n-C3H7OH formation pathways at −0.4 V vs. SHE at pH 14 are displayed (Fig. S6†), indicating the feasibility for the CO(2)RR to n-propanol via the coupling between CH3CHO* and CO*.
When the potential becomes more negative (<−0.50 V vs. SHE, pH 14), the hydrogenation step of *CH3CHO toward ethanol becomes more dominant than the coupling step on Cu(100) (Fig. 1c), indicating that the perfect Cu(100) facet is hard to catalyze the CO(2)RR to n-C3H7OH. In comparison, on Ag-doped Cu, the SDS for the n-C3H7OH pathway becomes dominant in the potential range between 0.22 and −0.96 V vs. SHE at pH 14 (see ΔG(U, pH) and structures in Fig. S7, computational details in Fig. S8 and Table S3†), in accordance with the experimental observation of the enhanced n-C3H7OH selectivity on Ag-doped Cu,42 also confirming the branching intermediate (CH3CHO*) and SDSs for C2H5OH and n-C3H7OH pathways.
Step effects
After determining the critical branching intermediate and corresponding SDSs for the C2H5OH and n-C3H7OH pathways, we further investigated the roles of surface step sites in the competition between C2H5OH and n-C3H7OH. The explicit functions of step sites on the n-C3H7OH selectivity were first surveyed by constructing surface steps with different upper terrace widths and lower terrace widths based on the Cu(100) facet (Fig. 2a–h). The step surfaces were constructed by removing the different numbers of atom row on the top layer of Cu(100), and the width of one row is the diameter of Cu (1.8 Å). The formed step surfaces are designated as “Step_u(x)d(y)”, where “u(x)d(y)” refers to the step site unit comprising x rows at the upper terrace and y rows at the lower terrace. It was found that the adsorption of CH3CHO* and CO* competes with each other,33 while the adsorption of CH3CHO* on Cu(100) is always weaker than that of CO* in the whole potential range of the CO(2)RR (Fig. 3a, computational details in Fig. S9 and Table S4†). For the coupling of CH3CHO* and CO* (i.e., the SDS for the n-propanol pathway), the adsorption of both CO* and CH3CHO* should be optimized. Thus, the ΔEads(CH3CHO*)/ΔEads(CO*) ratio is used to evaluate the priority of the n-C3H7OH pathway, from which the ratio close to 1 suggests an optimal match of both CH3CHO* and CO* adsorption. The ΔEads(CH3CHO*)/ΔEads(CO*) ratio reaches the highest value of 0.84 when the width of the lower terrace of the Cu(100) step is 3.6 Å (Fig. 3b, computational details in Table S5†). On the other hand, for the protonation of CH3CHO* to CH3CH2O* (i.e., the SDS of the C2H5OH pathway), when hydrogenated CH3CH2O* is more stable, the possibility for the formation of C2H5OH increases. Thus, the ΔEads(CH3CH2O*)/ΔEads(CO*) ratio is used to represent the protonation capability of the catalyst for C2+ intermediates, from which the smaller ratio represents that the hydrogenation step is less likely to occur. The ΔEads(CH3CH2O*)/ΔEads(CO*) ratio reaches the lowest value (2.45) when the width of the upper terrace is 1.8 Å (Fig. 3c, computational details in Table S5†). Based on the two indicators above, the optimal Cu(100) step is the Step_u1d2, with a lower terrace width of 3.6 Å and an upper terrace width of 1.8 Å (Fig. S10†). | ||
| Fig. 2 (a–h) The side views and top views of different step surfaces constructed based on the Cu(100) facet, including (a) Step_u5d1, (b) Step_u4d2, (c) Step_u3d3, (d) Step_u2d4, (e) Step_u2d2, (f) Step_u1d5, (g) Step_u1d3, and (h) Step_u1d1. The Cu atoms of the uppermost layer are presented with a brown color to clearly display the step sites. These step surfaces were denoted as “Step_u(x)d(y)”, which means that the upper terrace width of the step unit is “x” times the diameter of the Cu atom, and the lower terrace width of the step unit is “y” times the diameter of the Cu atom. The diameter of the Cu atom is 1.8 Å. | ||
 | ||
| Fig. 3 (a) The adsorption energies (ΔEads) of CH3CHO* and CO* on Cu(100) against the potential. (b) The adsorption energy ratios between CH3CHO* and CO* of the step surfaces constructed based on Cu(100) against the width of the lower terrace. (c) The adsorption energy ratios between CH3CH2O* and CO* of the step surfaces constructed based on Cu(100) against the width of the upper terrace. The data in (b) and (c) are from Cu(100), Step_u5d1, Step_u4d2, Step_u3d3, Step_u2d4, and Step_u1d5. (d) The relation of the switching trend (defined as KC2+CO/KC2+H) against the descriptor ΔEads(CH3CH2O*)/ΔEads(CO*). (e) The free energy changes of three reaction steps including the protonation of CH3CHO* (C2 + H), the protonation of CH3COCHO* (C3 + H), and the coupling between CH3CHO* and CO* (C2 + CO), and n-propanol relative selectivity of Cu(100) and step surfaces constructed based on Cu(100). (f) The free energy changes of the SDSs for n-propanol and ethanol pathways on Step_u1d2 against the potential and pH. The grey plane is the plane with the function of ΔG = 0 (eV). The highlighted region with blue color shows the potential range at pH = 14 where the n-propanol is preferably produced. (g) The adsorption configurations of CH3CH2O* on Cu(100) and Step_u1d2 (top), and the atomic charge coloring diagrams of CH3CH2O* on Cu(100) and Step_u1d2 (bottom), the numbers of electron transferred from the surface adsorption sites to CH3CH2O* are marked. | ||
To evaluate the n-C3H7OH selectivity of different step sites, we set the Cu(100) surface as the benchmark, and the n-C3H7OH relative selectivity compared to the Cu(100) surface is defined as: (KC2+CO/KC2+H × KC3+H), where K = kstep/kCu(100), k refers to the rate constant of an elementary reaction, “step” refers to the step surfaces, and “C2 + CO”, “C2 + H”, and “C3 + H” represent the coupling of CH3CHO* and CO* to CH3COCHO*, the hydrogenation of CH3CHO* to CH3CH2O*, and the hydrogenation of CH3COCHO* to CH3COCHOH* (Fig. S11†), respectively. The relative n-C3H7OH selectivity of the Cu(100) surface is set as 1. “KC2+CO/KC2+H” represents the switching trend of the C2H5OH and n-C3H7OH pathways, which shows a linear correlation with the ΔEads(CH3CH2O*)/ΔEads(CO*) value (Fig. 3d). Then the relative selectivity of n-C3H7OH on different Cu(100) steps was calculated, among which the Step_u1d2 sites show the highest n-C3H7OH relative selectivity of 5.7 × 1010 (Fig. 3e, right y-axis). The SDS of the n-C3H7OH pathway on Step_u1d2 is more dominant than the ethanol pathway in the potential range of −0.41 to −1.03 V vs. SHE at pH 14 (Fig. 3f, computational details in Fig. S12 and Table S6†), wider than that of the perfect Cu(100) surface (Fig. 1c, −0.27 to −0.50 V vs. SHE). By comparing ΔG values of the hydrogenation and coupling steps of CH3CHO* and the hydrogenation step of CH3COCHO* on Step_u1d2 and Cu(100) (Fig. 3e, left y-axis), the suppression of the CH3CHO* protonation contributes the most to the enhanced n-C3H7OH relative selectivity of Step_u1d2. The adsorption of CH3CH2O* is switched from a bridged-adsorption mode on the Cu(100) surface, to a top-adsorption mode on the Step_u1d2 sites due to the confined surface structure (Fig. 3g). This top-adsorption mode leads to the less electron transfer from Cu atoms to CH3CH2O* according to the Bader charge and differential charge density analysis (Fig. 3g and S13†), thus decreasing the binding strength of CH3CH2O* on Step_u1d2 (Fig. S14a, computational details in Fig. S12 and Table S6†). On the other hand, the adsorption of CH3CHO* on Step_u1d2 is stronger than that on Cu(100) (Fig. S14b, computational details in Fig. S12 and Table S6†). The angle between the Cu–O bond (the O atom from CH3CHO*) and the surface plane of Step_u1d2 is 64° (Fig. S15†), smaller than that of CH3CHO* on Cu(100) (82°), indicating a geometric affinity of Step_u1d2 for the CH3CHO* adsorption. Thus, the weak adsorption of CH3CH2O* and the strong adsorption of CH3CHO* on Step_u1d2 together contribute to the inhibited protonation of CH3CHO* and enhanced n-C3H7OH relative selectivity.
Furthermore, the n-C3H7OH relative selectivity of Cu(100) and step sites shows a volcano trend with the ΔEads(CH3CH2O*)/ΔEads(CO*) value (Fig. 4a), as the adsorption energies of different reaction intermediates are correlated during the reactions.43 When ΔEads(CH3CH2O*)/ΔEads(CO*) decreases at the right side of the volcano, the hydrogenation step of CH3CHO* (i.e., SDS for the C2H5OH pathway) is inhibited as the adsorbed CH3CH2O* becomes unstable. This SDS suppression of the C2H5OH pathway is beneficial for the n-C3H7OH production. When ΔEads(CH3CH2O*)/ΔEads(CO*) further decreases at the left side of the volcano, not only the protonation of CH3CHO* is suppressed, but also the protonation of C3 intermediates, like CH3COCHO*, is also suppressed. Thus, the n-C3H7OH relative selectivity decreases as the ΔEads(CH3CH2O*)/ΔEads(CO*) further decreases (at the left side of the volcano).
 | ||
| Fig. 4 (a) The volcano plot of n-propanol relative selectivity (defined as KC2+CO/KC2+H × KC3+H) versus the descriptor ΔEads(CH3CH2O*)/ΔEads(CO*). (b) The contour map showing the ΔEads(CH3CH2O*)/ΔEads(CO*) values of different Cu facets. (c) The n-propanol relative selectivity and ΔEads(CH3CH2O*)/ΔEads(CO*) values of three efficient Cu facets for the CO(2)RR to n-propanol. (d) The free energy changes of the SDSs for n-propanol and ethanol pathways on Cu (433) against the potential and pH. The grey plane is the plane with the function of ΔG = 0 (eV). The highlighted region with orange color shows the potential range at pH = 14 where the n-propanol is preferably produced. (e) The adsorption energies of CH3CHO* (top) and CH3CH2O* on Cu(100) and Cu(433) against the potential. The potential range from −0.8 to −1.4 V vs. SHE is where the formation of n-propanol is preferable on Cu(433). (f) The adsorption configurations of CH3CHO* on Cu(433) and Cu(100). The angles between the Cu–O bond and the surface are marked. (g) The adsorption configurations of CH3CH2O* on Cu(100) and Cu(433) (top) and the atomic charge coloring diagrams of CH3CH2O* on Cu(100) and Cu(433) (bottom), the numbers of electron transferred from the surface adsorption sites to CH3CH2O* are marked. (h) The volcano plot of the n-propanol relative selectivity versus the descriptor ΔEads(CH3CH2O*)/ΔEads(CO*), including the data of the step surfaces (the Step_u(x)d(y) surfaces and Cu facets), the Cu-based bimetals, and other metals. | ||
Facet prediction
As the high-index facets of Cu show characteristics of different step sites, we further screened the potential facets for the electroreduction of CO(2) toward n-C3H7OH using the ΔEads(CH3CH2O*)/ΔEads(CO*) descriptor (Fig. 4b). The ΔEads(CH3CH2O*)/ΔEads(CO*) values of (433), (321), and (310) are located in the optimal range (2.0−3.0 eV). In our work, the high-index facets have been constructed from the primitive cell of Cu, to control the suitable model size for DFT computations. For instance, Cu(321) studied in this work corresponds to Cu(210) (Fig. S16†), and a distinct experiment performance of the Cu(210) facets for the CO2RR to n-propanol was previously reported,44 further confirming the practicability of the selectivity descriptor.Compared to different facets, Cu(433) exhibits the highest relative selectivity (∼109) of n-propanol (Fig. 4c). The potential range for n-propanol production on Cu(433) was calculated to be −0.40 to −1.49 V vs. SHE at pH 14 (Fig. 4d, computational details in Fig. S17 and Table S7†), which covers the experimentally observed potential range (−1.20 to −1.50 V vs. SHE, at pH 14) for n-propanol production,15,20 further indicating the great potential of Cu(433) in the CO(2)RR to n-propanol. Compared to Cu(100), Cu(433) shows a stronger adsorption for CH3CHO* and a weaker adsorption for CH3CH2O* in the potential range for n-propanol production (Fig. 4e, computational details in Fig. S17 and Table S7†). Thus, the hydrogenation of CH3CHO* on Cu(433) becomes difficult and the ethanol pathway is inhibited. The strong adsorption of CH3CHO* on Cu(433) is attributed to the geometric effect from the step sites. Compared to Cu(100), CH3CHO* adsorbed on Cu(433) is closer to the surface (Fig. 4f), allowing a strong interaction between the CH3CHO* and the Cu(433) surface. On the other hand, CH3CH2O* is adsorbed at the bridged-sites on Cu(100), and at the top-sites on Cu(433) (Fig. 4g). The less electron transfer from Cu(433) to the adsorbed CH3CH2O* results in the weak adsorption of CH3CH2O* based on the Bader charge and differential charge density analysis (Fig. 4g and S18†).
To more clearly show the practicability of the selectivity descriptor ΔEads(CH3CH2O*)/ΔEads(CO*), the experimentally reported Cu(321) facet was compared with the Cu(100) and Cu(433) facets. As shown in Fig. S19,† the high n-propanol relative selectivity of Cu(321) is also mainly from its capability for inhibiting the hydrogenation of CH3CHO*. The binding strength of Cu(321) for CH3CHO* is stronger than that of Cu(100) and weaker than that of Cu(433) (Fig. S20†). The adsorption configuration of CH3CHO* adsorbed on Cu(321) was analyzed (Fig. S21†). The angle between the Cu–O bond and the surface plane is smaller than that of Cu(100) (82°) and larger than that of Cu(433) (55°), suggesting that the capability of Cu(321) to stabilize the CH3CHO* intermediate is superior to that of Cu(100) and inferior to that of Cu(433). On the other hand, the adsorption of CH3CH2O* on Cu(321) is weaker than that on Cu(100) and stronger than that on Cu(433) (Fig. S22†). Furthermore, CH3CH2O* is also adsorbed on Cu(321) in a top-adsorption way, and the charge transfer of Cu(321) to the CH3CH2O* intermediate is less than that of Cu(100) and more than that of Cu(433) (Fig. S23†), confirming that the capability of Cu(321) to adsorb CH3CH2O* is between that of Cu(100) and Cu(433). Therefore, the n-propanol relative selectivity of Cu(321) is higher than that of Cu(100) and lower than that of Cu(433) (Fig. 4c). On Cu(321), the preferable potential range (at pH 14) for the coupling of CH3CHO* with CO* is 0 to −0.75 V vs. SHE according to the constant potential calculations (Fig. S24, computational details in Fig. S25, and Table S8†). The overall selectivities of Cu(100), Cu(321), and Cu(433) for n-propanol were further calculated by considering the mainly competitive carbon-containing products (methane, methanol, ethylene, and ethanol) in the CO(2)RR to n-propanol (Fig. S26†). Cu(100) was also used as a reference in those calculations. The n-propanol overall selectivities (by considering all the possible carbon-containing products) on Cu(433) and Cu(321) are calculated to be ∼109 and ∼106, respectively (Fig. S27†), which are close to the n-propanol relative selectivities of the two facets (Fig. 4c). This result confirms that the n-propanol relative selectivity is a reasonable metric to evaluate the n-propanol selectivity of different structures.
Finally, the relative selectivities of n-propanol of all step surfaces (including the step surfaces based on Cu(100) and different Cu facets), the Cu-based bimetals (structures in Fig. S28†), and other metals (structures in Fig. S29†), with respect to the descriptor ΔEads(CH3CH2O*)/ΔEads(CO*), exhibit a volcano correlation (Fig. 4h). This result suggests that the selectivity descriptor ΔEads(CH3CH2O*)/ΔEads(CO*) is universal in finding the various catalysts for the CO(2)RR to n-propanol. The Step_u1d2 sites and Cu(433) are located at the top of the volcano plot, suggesting that the capability of those surface Cu catalytic sites toward higher CO(2)-to-n-propanol conversion selectivities. Although the step surfaces may experience reconstruction during the CO(2)RR due to the high surface energies and the harsh reaction conditions, there have been some reports those have successfully synthesized the high-index Cu-based facets and retained good reaction stability.44–46 For example, by utilizing OH− anions as the controlling reagents and the ascorbic acid for the slow growth of the nanocrystals, the Cu2O(211) facets were synthesized, showing a FEC2H4 of 87% in the CO2RR after being stored in 1 M KOH for one month.45 In addition, it has been found that the presence of the low-index facets can help to stabilize the high-index facets under electroreduction conditions.46 Those studies can inspire the synthesis of high-index Cu-based facets for the CO(2)RR catalysis.
Conclusions
In summary, this work represents a rational theoretical design for the electrocatalytic sites for efficient CO(2)-to-C3+ products based on the constant potential computations. For the formation of n-propanol, ethanol shares the common RDS and is a main side product. In our work, CH3CHO* has been identified as the critical intermediate for the bifurcation of n-propanol and ethanol pathways, and ΔEads(CH3CH2O*)/ΔEads(CO*) has been proposed as a key descriptor for the formation of n-propanol. Based on this descriptor, different step sites have been screened to select the optimal catalytic sites, and Cu(433) facets have been suggested as the most promising facets for the electrochemical CO(2)-to-n-propanol conversion. Our work highlights the significance of SDS regulation in the CO(2)RR and allows understanding the competition mechanism between the C2 and C3+ products.Data availability
All data supporting this work are included in the ESI.†Author contributions
Y. Xue and G. Zheng designed the research. L. Zhang and G. Zheng supervised the research. Y. Xue, X. Lv, C. Yang, and L. Song performed the research and analyzed the data. Y. Xue and G. Zheng wrote the manuscript. All the authors discussed, commented on and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the following funding agencies for supporting this work: the National Key Research and Development Program of China (2024YFB4106400 and 2024YFB4106401), the National Natural Science Foundation of China (22025502, U23A20552, and 22379026), and the Natural Science Foundation of Shanghai (23ZR1407000). The computations in this research were performed using the CFFF platform of Fudan University.Notes and references
- A. Shayesteh Zeraati, F. Li, T. Alkayyali, R. Dorakhan, E. Shirzadi, F. Arabyarmohammadi, C. P. O’Brien, C. M. Gabardo, J. Kong, A. Ozden, M. Zargartalebi, Y. Zhao, L. Fan, P. Papangelakis, D. Kim, S. Park, R. K. Miao, J. P. Edwards, D. Young, A. H. Ip, E. H. Sargent and D. Sinton, Nat. Synth., 2025, 4, 75–83 CrossRef CAS.
- Y. Ji, Z. Chen, R. Wei, C. Yang, Y. Wang, J. Xu, H. Zhang, A. Guan, J. Chen, T.-K. Sham, J. Luo, Y. Yang, X. Xu and G. Zheng, Nat. Catal., 2022, 5, 251–258 CrossRef CAS.
- K. Qi, Y. Zhang, N. Onofrio, E. Petit, X. Cui, J. Ma, J. Fan, H. Wu, W. Wang, J. Li, J. Liu, Y. Zhang, Y. Wang, G. Jia, J. Wu, L. Lajaunie, C. Salameh and D. Voiry, Nat. Catal., 2023, 6, 319–331 CrossRef CAS.
- X. He, L. Lin, X. Li, M. Zhu, Q. Zhang, S. Xie, B. Mei, F. Sun, Z. Jiang, J. Cheng and Y. Wang, Nat. Commun., 2024, 15, 9923 CrossRef CAS.
- C. F. Shih, T. Zhang, J. Li and C. Bai, Joule, 2018, 2, 1925–1949 CrossRef CAS.
- J. M. Spurgeon and B. Kumar, Energy Environ. Sci., 2018, 11, 1536–1551 RSC.
- S. Kong, X. Lv, X. Wang, Z. Liu, Z. Li, B. Jia, D. Sun, C. Yang, L. Liu, A. Guan, J. Wang, G. Zheng and F. Huang, Nat. Catal., 2023, 6, 6–15 CrossRef CAS.
- J. Zhang, P. Yu, C. Peng, X. Lv, Z. Liu, T. Cheng and G. Zheng, ACS Catal., 2023, 13, 7170–7177 CrossRef CAS.
- Y. Xin, C. B. Musgrave III, J. Su, J. Li, P. Xiong, M. Meng-Jung Li, Y. Song, Q. Gu, Q. Zhang, Y. Liu, W. Guo, L. Cheng, X. Tan, Q. Jiang, C. Xia, B. Zhong Tang, W. A. Goddard III and R. Ye, Angew. Chem., Int. Ed., 2025, 64, 202420286 CrossRef.
- Z. Liu, L. Song, X. Lv, M. Liu, Q. Wen, L. Qian, H. Wang, M. Wang, Q. Han and G. Zheng, J. Am. Chem. Soc., 2024, 146, 14260–14266 CrossRef CAS PubMed.
- C. Peng, J. Ma, G. Luo, S. Yan, J. Zhang, Y. Chen, N. Chen, Z. Wang, W. Wei, T.-K. Sham, Y. Zheng, M. Kuang and G. Zheng, Angew. Chem., Int. Ed., 2024, 63, 202316907 CrossRef PubMed.
- C. Peng, S. Yang, G. Luo, S. Yan, M. Shakouri, J. Zhang, Y. Chen, Z. Wang, W. Wei, T.-K. Sham and G. Zheng, Small, 2023, 19, 2207374 CrossRef CAS.
- M. Sun and B. Huang, Adv. Energy Mater., 2024, 14, 2400152 CrossRef CAS.
- T. Yan, X. Chen, L. Kumari, J. Lin, M. Li, Q. Fan, H. Chi, T. J. Meyer, S. Zhang and X. Ma, Chem. Rev., 2023, 123, 10530–10583 CrossRef CAS PubMed.
- S. Jeong, C. Huang, Z. Levell, R. X. Skalla, W. Hong, N. J. Escorcia, Y. Losovyj, B. Zhu, A. N. Butrum-Griffith, Y. Liu, C. W. Li, D. Reifsnyder Hickey, Y. Liu and X. Ye, J. Am. Chem. Soc., 2024, 146, 4508–4520 CrossRef CAS PubMed.
- N. Sakamoto, K. Sekizawa, S. Shirai, T. Nonaka, T. Arai, S. Sato and T. Morikawa, Nat. Catal., 2024, 7, 574–584 CrossRef CAS.
- C. Peng, G. Luo, J. Zhang, M. Chen, Z. Wang, T.-K. Sham, L. Zhang, Y. Li and G. Zheng, Nat. Commun., 2021, 12, 1580 CrossRef CAS.
- B. Yang, L. Chen, S. Xue, H. Sun, K. Feng, Y. Chen, X. Zhang, L. Xiao, Y. Qin, J. Zhong, Z. Deng, Y. Jiao and Y. Peng, Nat. Commun., 2022, 13, 5122 CrossRef CAS PubMed.
- X. Wang, P. Ou, A. Ozden, S.-F. Hung, J. Tam, C. M. Gabardo, J. Y. Howe, J. Sisler, K. Bertens, F. P. García de Arquer, R. K. Miao, C. P. O'Brien, Z. Wang, J. Abed, A. S. Rasouli, M. Sun, A. H. Ip, D. Sinton and E. H. Sargent, Nat. Energy, 2022, 7, 170–176 CrossRef CAS.
- W. Niu, Z. Chen, W. Guo, W. Mao, Y. Liu, Y. Guo, J. Chen, R. Huang, L. Kang, Y. Ma, Q. Yan, J. Ye, C. Cui, L. Zhang, P. Wang, X. Xu and B. Zhang, Nat. Commun., 2023, 14, 4882 CrossRef CAS PubMed.
- J. Li, F. Che, Y. Pang, C. Zou, J. Y. Howe, T. Burdyny, J. P. Edwards, Y. Wang, F. Li, Z. Wang, P. De Luna, C. T. Dinh, T. T. Zhuang, M. I. Saidaminov, S. Cheng, T. Wu, Y. Z. Finfrock, L. Ma, S. H. Hsieh, Y. S. Liu, G. A. Botton, W. F. Pong, X. Du, J. Guo, T. K. Sham, E. H. Sargent and D. Sinton, Nat. Commun., 2018, 9, 4614 CrossRef.
- Y. Pang, J. Li, Z. Wang, C.-S. Tan, P.-L. Hsieh, T.-T. Zhuang, Z.-Q. Liang, C. Zou, X. Wang, P. De Luna, J. P. Edwards, Y. Xu, F. Li, C.-T. Dinh, M. Zhong, Y. Lou, D. Wu, L.-J. Chen, E. H. Sargent and D. Sinton, Nat. Catal., 2019, 2, 251–258 CrossRef CAS.
- S. Yan, Z. Chen, Y. Chen, C. Peng, X. Ma, X. Lv, Z. Qiu, Y. Yang, Y. Yang, M. Kuang, X. Xu and G. Zheng, J. Am. Chem. Soc., 2023, 145, 26374–26382 CrossRef CAS.
- Z. Liu, X. Lv, S. Kong, M. Liu, K. Liu, J. Zhang, B. Wu, Q. Zhang, Y. Tang, L. Qian, L. Zhang and G. Zheng, Angew. Chem., Int. Ed., 2023, 62, 202309319 CrossRef PubMed.
- M. Kuang and G. Zheng, Chem Catal., 2023, 3, 100565 CrossRef CAS.
- L. Yang, X. Lv, C. Peng, S. Kong, F. Huang, Y. Tang, L. Zhang and G. Zheng, ACS Cent. Sci., 2023, 9, 1905–1912 CrossRef CAS PubMed.
- Y. Xue, L. Zhang, M. Kuang and G. Zheng, ACS Appl. Mater., 2025, 17, 11375–11388 CrossRef PubMed.
- Y. Jiang, L. Huang, C. Chen, Y. Zheng and S.-Z. Qiao, Energy Environ. Sci., 2025, 18, 2025–2049 RSC.
- Z. Zhang, W. Gee, P. Sautet and A. N. Alexandrova, J. Am. Chem. Soc., 2024, 146, 16119–16127 CrossRef CAS.
- K. S. Exner, ACS Catal., 2020, 10, 12607–12617 CrossRef CAS.
- C. Lucky, S. Jiang, C.-R. Shih, V. M. Zavala and M. Schreier, Nat. Catal., 2024, 7, 1021–1031 CrossRef CAS.
- E. L. Clark and A. T. Bell, J. Am. Chem. Soc., 2018, 140, 7012–7020 CrossRef CAS PubMed.
- A. H. M. da Silva, Q. Lenne, R. E. Vos and M. T. M. Koper, ACS Catal., 2023, 13, 4339–4347 CrossRef CAS PubMed.
- X. Chang, A. Malkani, X. Yang and B. Xu, J. Am. Chem. Soc., 2020, 142, 2975–2983 CrossRef CAS PubMed.
- J. Li, C. Li, J. Hou, W. Gao, X. Chang, Q. Lu and B. Xu, J. Am. Chem. Soc., 2022, 144, 20495–20506 CrossRef CAS.
- X. He, Y. Su, J. Zhu, N. Fang, Y. Chen, H. Liu, D. Zhou and C. Wang, J. Mater. Chem. A, 2023, 11, 18106–18114 RSC.
- Q. Li, J. Wu, C. Yang, S. Li, C. Long, Z. Zhuang, Q. Li, Z. Guo, X. Huang and Z. Tang, J. Am. Chem. Soc., 2025, 147, 6688–6697 CrossRef CAS PubMed.
- R. Zhang, J. Zhang, S. Wang, Z. Tan, Y. Yang, Y. Song, M. Li, Y. Zhao, H. Wang and B. Han, Angew. Chem., Int. Ed., 2024, 63, 202405733 CrossRef PubMed.
- G. L. De Gregorio, T. Burdyny, A. Loiudice, P. Iyengar, W. A. Smith and R. Buonsanti, ACS Catal., 2020, 10, 4854–4862 CrossRef CAS PubMed.
- G. Kastlunger, H. H. Heenen and N. Govindarajan, ACS Catal., 2023, 13, 5062–5072 CrossRef CAS.
- S. Zhang, Q. Tang, B. Zhu and Y. Gao, ACS Catal., 2025, 15, 6497–6506 CrossRef CAS.
- X. Wang, Z. Wang, T.-T. Zhuang, C.-T. Dinh, J. Li, D.-H. Nam, F. Li, C.-W. Huang, C.-S. Tan, Z. Chen, M. Chi, C. M. Gabardo, A. Seifitokaldani, P. Todorović, A. Proppe, Y. Pang, A. R. Kirmani, Y. Wang, A. H. Ip, L. J. Richter, B. Scheffel, A. Xu, S.-C. Lo, S. O. Kelley, D. Sinton and E. H. Sargent, Nat. Commun., 2019, 10, 5186 CrossRef PubMed.
- M. M. Montemore and J. W. Medlin, Catal. Sci. Technol., 2014, 4, 3748–3761 RSC.
- D. Zhong, Z.-J. Zhao, Q. Zhao, D. Cheng, B. Liu, G. Zhang, W. Deng, H. Dong, L. Zhang, J. Li, J. Li and J. Gong, Angew. Chem., Int. Ed., 2021, 60, 4879–4885 CrossRef CAS PubMed.
- A. M. Harzandi, S. P. Amouzesh, J. Xu, T. Baghban-Ronaghi, S. Shadman, F. Collins, G. Kim, W. Kaminsky, L. A. Curtiss and C. Liu, Appl. Catal., B, 2025, 366, 125053 CrossRef CAS.
- J. Zhang, X. Xu, L. Luo, T. Peng, B. Liu, L. Jiang, M. Jin, R. Wang, H. Yi and W. Wu, Chem. Eng. J., 2025, 503, 158187 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc02562a |
| This journal is © The Royal Society of Chemistry 2025 |