
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMimicking NADPH oxidase and lipoxygenase by using a biodegradable single-site catalyst via a cascade reaction to trigger tumor-specific ferroptosis†
Xiyang Gea,
Yiyan Yina,
Xiaoni Wanga,
Xiang Liab,
Jin Ouyang c and
Na Na*a
c and
Na Na*a
aKey Laboratory of Radiopharmaceuticals, College of Chemistry, Beijing Normal University, Beijing 100875, China. E-mail: nana@bnu.edu.cn
bState Key Laboratory for Quality Ensurance and Sustainable Use of Dao-di Herbs, National Resource Center for Chinese Materia Medica, China Academy of Chinese Medical Sciences, Beijing 100700, China
cDepartment of Chemistry, Faculty of Arts and Sciences, Beijing Normal University, Zhuhai 519087, China
First published on 5th July 2025
Abstract
Ferroptosis exhibits promising potential in cancer therapy via lipid peroxidation (LPO) accumulation, while its therapeutic efficacy is normally limited by inadequate ROS production and adverse effects on normal tissues. Here, a TME-activated in situ synthesis of a single-site catalyst (Fe(II)–PW11) is reported, which triggers ferroptosis by mimicking natural enzyme activities of NADPH oxidase (NOX) and lipoxygenase (LOX) via cascade reactions. Upon degradation of the nanocarrier by the overexpressed GSH in an acidic TME, Fe(II)–PW11 is obtained through the coordination of Fe2+ into lacunary phosphotungstic acid (PW11). Subsequently, Fe(II)–PW11 catalyzes NADPH depletion and O2˙− generation through a NOX-like process. This facilitates the formation of high-valent Fe(IV)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O–PW11, initiating cascade reactions to generate lipid radicals through hydrogen atom transfer based on LOX-like activity. Thus, Fe(II)–PW11 synergistically accelerates LPO accumulation and antioxidant inhibitions, effectively inducing ferroptosis for cancer therapy. Notably, Fe(II)–PW11 is degraded into low-toxic debris in normal organs, reducing side effects after treatment. Significantly, the whole process is well confirmed by comprehensive characterization studies including online monitoring via ambient mass spectrometry. This work not only reveals a novel ferroptosis-based cancer treatment in a ROS-independent pathway, but also provides a safe therapeutic modality with low toxicity to normal tissues.
O–PW11, initiating cascade reactions to generate lipid radicals through hydrogen atom transfer based on LOX-like activity. Thus, Fe(II)–PW11 synergistically accelerates LPO accumulation and antioxidant inhibitions, effectively inducing ferroptosis for cancer therapy. Notably, Fe(II)–PW11 is degraded into low-toxic debris in normal organs, reducing side effects after treatment. Significantly, the whole process is well confirmed by comprehensive characterization studies including online monitoring via ambient mass spectrometry. This work not only reveals a novel ferroptosis-based cancer treatment in a ROS-independent pathway, but also provides a safe therapeutic modality with low toxicity to normal tissues.
Introduction
Ferroptosis is a unique non-apoptotic-regulated cell death, characterized by lipid peroxidation (LPO) accumulation.1–3 Compared to classical chemotherapeutics, ferroptosis can overcome the high propensities of drug resistance and poor therapeutic efficiencies, exhibiting promising potential in treating therapy-resistant tumors.4 Recently, nano-materials have been developed for catalyzing ROS generation to induce ferroptosis, for example, converting H2O2 to highly active ˙OH via the Fenton reaction.5,6 However, ROS can be easily deactivated due to their short lifetime and diffusion distance (only about 1 ns and 100 nm)7 or be depleted by reductants such as glutathione (GSH).8,9 Although ferroptosis can be enhanced by in situ H2O2 generation and increasing intracellular iron content, the efficacy of ROS-mediated ferroptosis is still limited by hypoxia and overexpression of various REDOX substances in the tumor microenvironment (TME). Therefore, it would be desirable to develop therapeutic agents that induce ferroptosis through a ROS-independent pathway.Natural enzymes such as lipoxygenases (LOXs) and NADPH oxidase (NOX) can facilitate LPO accumulation through enzymatic processes.10,11 As a key enzyme to initiate LPO, LOXs is a non-heme iron-dependent dioxygenase for targeted oxidizing polyunsaturated fatty acid-containing phospholipids (PUFA-PLs).12,13 As another inducer, NOX could oxidize NADPH into NADP+ along with superoxide radical (O2˙−) formation, which inhibits ferroptosis suppression systems.14–16 Therefore, the synergistic mimicking of both natural enzyme activities could be an ideal strategy for effectively inducing ferroptosis-based antitumor therapy. However, there is still a shortage of reports on ferroptosis induced by mimics with both enzymatic activities, which could be challenged by obscure catalytic mechanisms and achieving synergistic activities in cancer cells.
Besides, some challenges still remain in tumor-specific therapy despite the distinct advantages of ferroptosis-based therapy. For instance, the drug's random distribution during the delivery would cause off-target toxicity to normal tissues. In addition, the postoperative side effects of ferroptosis can also lead to serious adverse reactions and damage to normal organs, such as kidney damage and neurodegeneration.17,18 To overcome this limitation, the in situ construction of a ferroptosis-inducing catalyst can be a promising strategy in precise cancer therapy.19,20 However, the complex TME would interfere with the catalyst release and decrease the therapeutic efficiency.21 Moreover, compared with conventional nanoparticles, the single-site catalysts showed great potential in efficient therapy due to their high atomic utilization of isolated catalytic centers with well-defined geometric and electronic structures.22,23 Therefore, developing a therapeutic single-site catalyst for the specific activation of ferroptosis is desirable.
Herein, a TME-activated in situ synthesis of a single-site catalyst (Fe(II)–PW11) is reported to trigger ferroptosis by mimicking NOX and LOX activity via a cascade reaction. As shown in Scheme 1, a nanocarrier A-MIL-101@PW12 was constructed by loading phosphotungstic acid (PW12) into iron-based MIL-101 and modified with the AS1411 aptamer. This nanocarrier can target cancer cells and prevent the leaking of PW12. Responding to the TME, Fe2+ and lacunary phosphotungstic acid (PW11) were released based on the reduction of A-MIL-101@PW12 by GSH and the spontaneous transformation of PW12 into PW11 under acidic conditions. Thereby, the single-site catalyst of Fe(II)–PW11 was in situ synthesized through the coordination of Fe2+ into PW11. The Fe(II)–PW11 presented NOX-like activity to convert NADPH into NADP+ and catalyzed O2˙− generation. This process facilitated the formation of the Fe(IV)O–PW11 intermediate, which subsequently abstracted a hydrogen atom from lipids to generate lipid radicals based on LOX-like activity. The produced O2˙− reacts with intracellular Fe3+ and catalyzes the conversion of Fe3+ to Fe2+ to promote the intracellular labile iron pool (LIP), synergetically triggering LPO. In addition, the GSH consumption-induced GPX-4 deactivation and NADPH depletion severely inhibited ferroptosis suppression systems. Notably, Fe(II)–PW11 was degraded into low-toxic WO4− in normal tissues. Both in vivo and in vitro experiments verified that Fe(II)–PW11-induced ferroptosis exhibited potent anticancer efficacy with minimal side effects.
 | ||
| Scheme 1 Illustration of in situ synthesis of Fe(II)–PW11 for cancer therapy. (a) Preparation of nanocarriers. (b) Ferroptosis induced by the in situ synthesized Fe(II)–PW11 through mimicking cascade NOX and LOX activities by cascade reactions (in cancer cells) and the postoperative degradation of Fe(II)–PW11 (in normal cells). | ||
Results and discussion
Preparation and characterization of nanocarriers
The nanocarrier of A-MIL-101@PW12 was prepared by a series of procedures (Scheme 1a). First, the iron-based metal–organic framework MIL-101 was synthesized via the solvothermal method using Fe3+ and terephthalic acid as additives. Then, PW12 was loaded into MIL-101 with a porosity of 2.9–3.4 nm (Fig. 1a).24 The obtained MIL-101@PW12 was washed several times to remove surface-absorbed PW12. As characterized by transmission electron microscopy (TEM), both MIL-101 and MIL-101@PW12 exhibited octahedral morphologies (Fig. 1b(i), (ii) and S1†), indicating that the encapsulation did not affect the MIL-101 structure. Meanwhile, small aggregates in MIL-101@PW12 represented the loaded PW12. Besides, X-ray diffraction (XRD) peaks of MIL-101 shifted from 5.2° and 5.8° to 5.8° and 6.5° after encapsulation, indicating that the entry of PW12 affected the crystal face of MIL-101 (Fig. 1c). Therefore, PW12 was confirmed to be successfully loaded onto MIL-101 with a high encapsulation rate of 65.89% (Fig. S2†). To achieve enrichment in the tumor, the AS1411 aptamer was modified onto MIL-101@PW12 to form A-MIL-101@PW12 through electrostatic interaction, which also showed an octahedral morphology (Fig. 1b(iii)). The energy-dispersive spectroscopy (EDS) mapping demonstrated the uniform distribution of C, O, N, W, P, and Fe elements in A-MIL-101@PW12, confirming its successful preparation.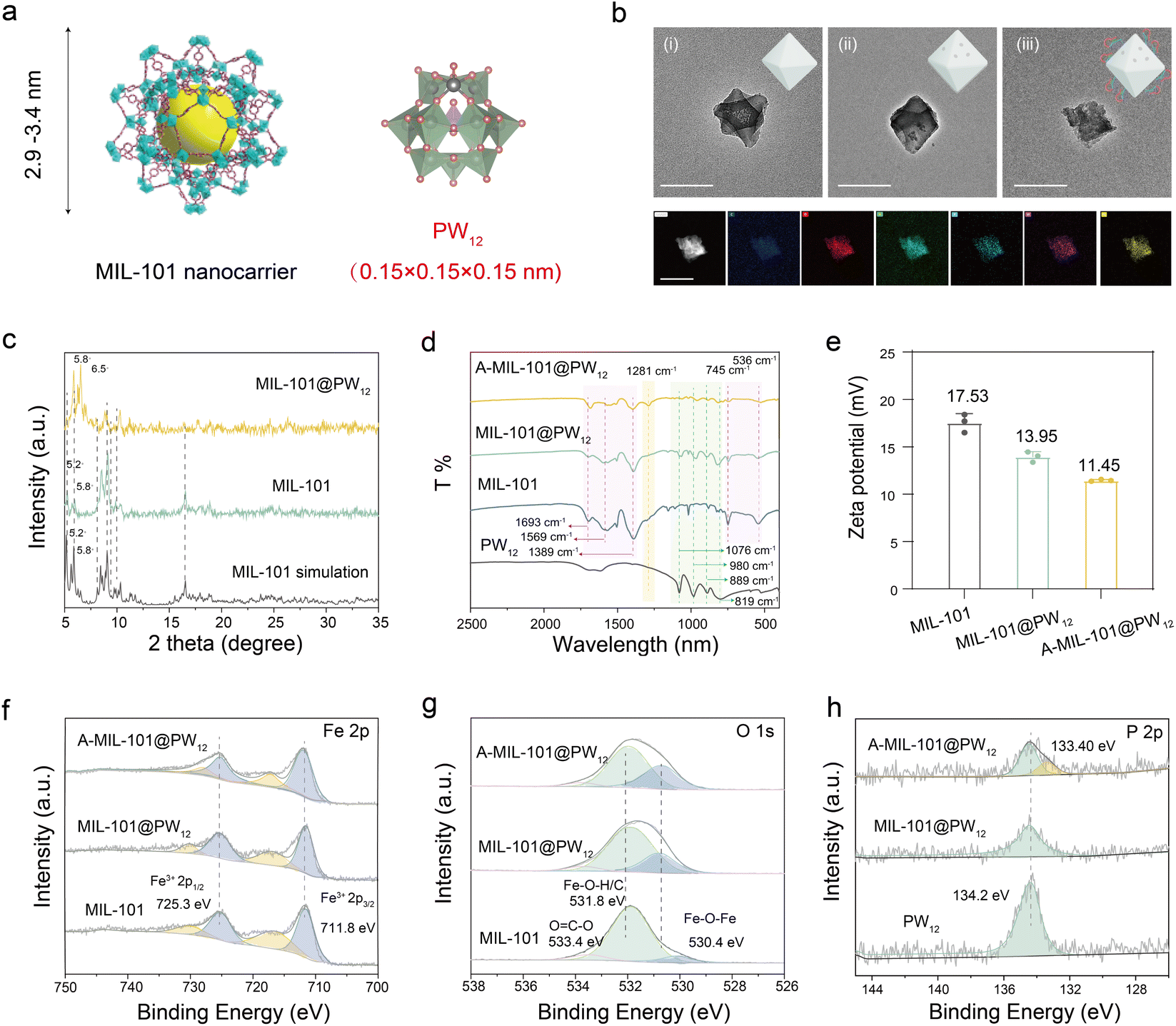 | ||
| Fig. 1 Characterization of A-MIL-101@PW12. (a) Structure of MIL-101 and PW12. (b) TEM images of MIL-101, MIL-101@PW12 and A-MIL-101@PW12, as well as the HADDF image and element mapping of A-MIL-101@PW12. The scale bar is 200 nm. (c) XRD patterns of MIL-101 simulation, MIL-101 and MIL-101@PW12. (d) IR spectra of MIL-101, PW12, MIL-101@PW12 and A-MIL-101@PW12. (e) Zeta potential of MIL-101, MIL-101@PW12 and A-MIL-101@PW12. (f) Fe 2p XPS and (g) O 1s XPS of MIL-101, MIL-101@PW12 and A-MIL-101@PW12. (h) P 2p XPS of PW12, MIL-101@PW12 and A-MIL-101@PW12. | ||
Moreover, chemical characterization studies were employed to evaluate the construction of A-MIL-101@PW12. By Fourier transform infrared spectroscopy (FT-IR) (Fig. 1d), A-MIL-101@PW12 exhibits peaks of proton vibration and C–H vibration at 1693 cm−1 and 745 cm−1, C–O stretching vibration at 1389 cm−1 and 1569 cm−1 and Fe–O stretching vibration at 536 cm−1, attributed to the MIL-101 framework. In addition, the characteristic peaks of PW12 (P–Oa at 1076 cm−1, WOd at 980 cm−1, W–Ob–W at 889 cm−1 and W–Oc–W at 819 cm−1) were also observed in A-MIL-101@PW12,25 which were slightly red-shifted after encapsulation. This indicated that the symmetry of PW12 was effectively preserved with no decomposition or structural deformation. In addition, the phosphate stretching vibration of the AS1411 aptamer at 1281 cm−1 confirmed the modification of AS1411 onto MIL-101@PW12.26 This was consistent with the approximately negative zeta potential of A-MIL-101@PW12 (Fig. 1e).
The X-ray photoelectron spectroscopy (XPS) analysis verifies the presence of Fe(III) (Fe 2p3/2 711.8 and Fe 2p1/2 725.3 eV) in A-MIL-101@PW12 (Fig. 1f).27 In addition, the distinctive peaks of O 1s at Fe–O–Fe (530.4 eV) and Fe–O–H/O–C (531.8 eV) were detected in MIL-101 (Fig. 1g). Upon loading with PW12, these peaks moved to increased binding energy, indicating the interaction between PW12 and MIL-101. This was consistent with the shift of the W peaks (Fig. S3†). Furthermore, no obvious change of P 2p at 134.2 eV in A-MIL-101@PW12 indicated the structural integrity of PW12 (Fig. 1h). Meanwhile, the XPS peak for N 1s (400.9 eV) and P 2 s (133.40 eV) of the AS1411 aptamer demonstrated the successful modification of AS1411 (Fig. S4†). Therefore, A-MIL-101@PW12 was successfully synthesized and exhibits stability for drug delivery.
Examinations of in situ synthesis of Fe(II)–PW11 in the TME and its biodegradable properties
The TME-activated in situ synthesis of Fe(II)–PW11 was achieved through the A-MIL-101@PW12 degradation (Fig. 2a(i)). Briefly, A-MIL-101@PW12 was reduced by GSH to release PW12 and Fe2+ (eqn (1)). Subsequently, PW12 was spontaneously converted into PW11 under acidic conditions (eqn (2)). Finally, Fe2+ was rapidly coordinated with PW11 to obtain Fe(II)–PW11 (eqn (3)). Notably, this catalyst with a single active site possesses a uniform coordination structure, being convenient to explore molecular changes by MS detections for investigating therapy mechanisms.| A-MIL-101@PW12 + GSH → Fe2+ + PW12 + GSSG | (1) |
 | (2) |
 | (3) |
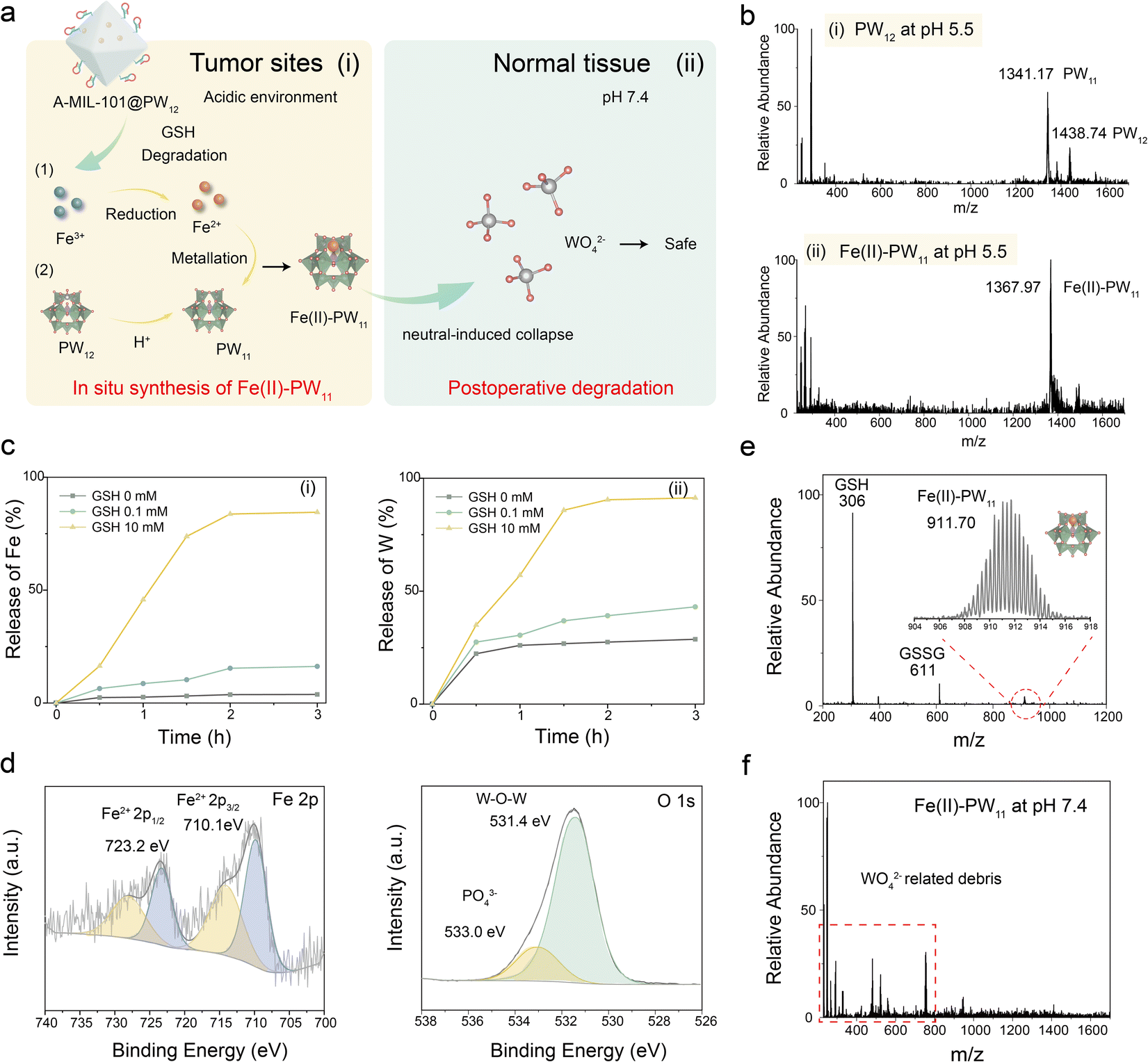 | ||
| Fig. 2 In situ synthesis of Fe(II)–PW11. (a) Scheme of in situ synthesis of Fe(II)–PW11 at tumor sites and postoperative degradation in normal tissues. (b) The MS spectra of PW12 (i) and Fe(II)–PW11 at pH 5.5 (ii). (c) Accumulated release profiles of Fe (i) and W (ii) of MIL-101@PW12 at different GSH concentrations. (d) XPS peaks of Fe(II)–PW11 for Fe and O elements with 10 mM GSH at pH 5.5. (e) The MS spectra of in situ synthesized Fe(II)–PW11 by mixing A-MIL-101@PW12 with 10 mM GSH at pH 5.5. (f) The MS spectra of Fe(II)–PW11 at pH 7.4. | ||
Initially, the spontaneous conversion of PW12 to PW11 was examined using MS analysis. As demonstrated (Fig. 2b(i) and S5†), PW12 ([PW12O40 + H]2− at m/z 1438.74, calc. 1438.60) was recorded, while it converted to PW11 ([PW11O39 + 5H]2− at m/z 1341.17, calc. 1341.14) at pH 5.5. In addition, Fe(II)–PW11 ([Fe(II)PW11O39 + 3H]2− at m/z 1367.97, calc. 1368.10) at pH 5.5 was also recorded after adding Fe2+ into PW12 (Fig. 2b(ii)), confirming the coordination of Fe2+ to PW11.
Subsequently, to confirm the GSH-induced degradation, the release of Fe2+ and PW12 was evaluated by inductively coupled plasma (ICP)-MS analysis. As exhibited (Fig. 2c), contents of Fe (i) and W (ii) gradually increased with GSH added and became stable after about 2 h. Meanwhile, the increased UV absorption of Fe2+ indicated the reduction of Fe3+ by GSH (Fig. S6†). The biodegradability of A-MIL-101@PW12 was further confirmed by the structural collapse and the final dissolution of A-MIL-101@PW12 after incubating with GSH (Fig. S7†). Besides, XPS confirmed the in situ synthesis of Fe(II)–PW11 after A-MIL101@PW12 degradation (Fig. 2d). As demonstrated, the Fe 2p XPS confirmed the presence of Fe(II) (Fe 2p3/2 710.1 eV and Fe 2p1/2 723.2 eV), indicating that Fe3+ was completely reduced to Fe2+ by excess GSH. The O 1s XPS peaks at 531.4 eV and 533.0 eV correspond to the W–O–W and PO43− of PW11. This is consistent with the rapid conversion of PW12 into PW11 under acidic conditions, contributing to the subsequent synthesis of Fe(II)–PW11 upon coordination of released Fe2+ to PW11.
The in situ synthesis of Fe(II)–PW11 was further evaluated by MS. As demonstrated by MS characterization (Fig. 2e), the ions of Fe(II)–PW11 ([Fe(II)PW11O39 + 2H]3− at m/z 911.70, calc.: 911.73) and the GSH oxidation product ([GSSG–H]− at m/z 611) were recorded by mixing A-MIL-101@PW12 with GSH at pH 5.5, indicating that Fe(II)–PW11 can be synthesized in the TME. While at pH 7.4, no significant signal of Fe(II)–PW11 appeared in the A-MIL-101@PW12 solution (Fig. S8†). This indicated the stability of A-MIL-101@PW12 during delivery. Additionally, this process was monitored with a homemade device of ambient mass spectrometry (Fig. S9†).28 With GSH added into A-MIL-101@PW12, GSSG and PW12 first appeared in 5 min, confirming the efficient degradation of A-MIL-101@PW12 by GSH. Thereafter, Fe(II)–PW11 gradually increased, which was attributed to the generation of PW11 under acidic conditions. In addition, almost no PW11 signals were observed due to the rapid coordination of Fe2+ with PW11. Therefore, based on the reduction of MIL-101 by GSH and the transformation of PW12 into PW11 under acidic conditions, Fe(II)–PW11 was successfully synthesized in the TME.
Furthermore, the in situ synthesized Fe(II)–PW11 can be decomposed into low-toxic WO42−-related fragments under neutral conditions of normal tissues (Fig. 2a(ii) and eqn (4)). This process avoids the toxicity of Fe(II)–PW11 to healthy tissues in postoperative metabolism. To evaluate postoperative degradation, the MS signal of Fe(II)–PW11 at pH 7.4 was examined. As demonstrated (Fig. 2f), most debris (at m/z < 800) was observed, while no signals of Fe(II)–PW11 appeared, indicating the disintegration of Fe(II)–PW11 in the neutral environment. This was consistent with UV-vis spectra of PW12 and Fe(II)–PW11 under different conditions (Fig. S10†). Besides, the generation of WO42− debris was directly confirmed by the in situ Raman spectroscopy characterization (Fig. S11†), showing the increased characteristic bond of WO42− (931 cm−1) along with the decrease in Fe(II)–PW11 peaks (at 995 and 980 cm−1).29–31 This neutral environment-induced degradation reduced toxicity to normal tissues, showing potential in selective and efficient therapy.
 | (4) |
NOX-like activity of Fe(II)–PW11
The in situ synthesized Fe(II)–PW11 exhibited NOX-like activity by accepting electrons from NADPH, and hence catalyzing the conversion of O2 to O2˙−. First, the NOX-like activity was evaluated by UV-vis spectroscopy analysis. As demonstrated, the absorbance of NADPH decreased at 339 nm after adding Fe(II)–PW11 (Fig. 3a), while no significant change in NADPH was observed in the absence of Fe(II)–PW11, indicating that NADPH oxidation was catalyzed by Fe(II)–PW11 (Fig. S12†). Besides, the ions of [NADP–2H]− at m/z 742 verified the NADP+ generation in the reaction system (Fig. 3b). The electron paramagnetic resonance (EPR) analysis was conducted using 5-(2,2-dimethyl-1,3-propoxycyclo-phosphoryl)-5-methyl-1-pyrroline-N-oxide (CYPMPO) as a spin trap. In the presence of CYPMPO, the carbon-centered NADP˙ radical was detected in the Fe(II)–PW11 and NADPH solution (Fig. 3c).32 Notably, O2˙− was also observed in the reaction, confirmed by the EPR signal of BMPO–OOH with 5-tert-butoxycarbonyl-5-methyl-1-pyrroline N-oxide (BMPO) as a trapping agent (Fig. S13†).33 In addition, H2O2 was detected by peroxide detection strips (Fig. S14†), which were formed by the interaction between O2˙− and H+ along with the electron transfer from the NADP˙ radical to NADP+.34 These results confirmed the superior NOX-like activity of Fe(II)–PW11 for catalyzing O2 into O2˙− and the oxidation of NADPH to NADP+. | ||
| Fig. 3 The NOX-like activity of Fe(II)–PW11. (a) The UV-vis spectra of NADPH after treatment with Fe(II)–PW11 in air at different times. (b) Mass spectra of NADPH before (i) and after (ii) adding Fe(II)–PW11 in air. (c) EPR spectra of CYPMPO–NADP˙. c(CYPMPO) = 100 mM. (d) The CV curves of Fe(II)–PW11 with or without adding NADPH in air and N2. (e) The optimization structure of Fe(III)–PW11–O2−. (f) Mass spectra of Fe(II)–PW11 before (i) and after (ii) adding NADPH in air. (g) EPR spectra of the in situ synthesized Fe(II)–PW11 before and after absorbing with O2. | ||
Furthermore, the cyclic voltammetry (CV) current of Fe(II)–PW11 significantly increased after adding NADPH in air, indicating the occurrence of electron transfer between Fe(II)–PW11 and NADPH (Fig. 3d). However, no obvious change in Fe(II)–PW11 in the N2-treated solution demonstrated the importance of O2 in the Fe(II)–PW11 catalysis process. The electron transfer in the reaction between Fe(II)–PW11 and NADPH may be facilitated by the coordination of O2 with Fe. Thereby, the coordination of O2 by Fe(II)–PW11 was investigated through Density Functional Theory (DFT) calculations.35 As calculated (Fig. 3e), O2 was connected to an Fe atom in an end-on way, exhibiting a stretched O–O bond (1.24 Å) compared with that in O2 (1.20 Å). This indicated that Fe(II)–PW11 can activate O2 via the Fe–O bond to facilitate the catalytic reaction.
The intermediates in the reaction were evaluated through MS detections. As a result (Fig. 3f), the intermediate of Fe(III)–PW11–O2− ([Fe(III)PW11O39–O2− + 3H]2− at m/z 1384.12, calc. 1384.09) was first formed, confirming the ability of Fe(II)–PW11 to absorb O2. After adding NADPH, the intermediate of Fe(II)–PW11–O2˙− ([Fe(II)PW11O39–O2˙− + 4H]2− at m/z 1384.60, calc. 1384.60) was formed during the reaction, which also confirmed the formation of O2˙−. In addition, EPR analysis demonstrated that the high-spin Fe (S = 2, g = 11.58) in the in situ synthesized Fe(II)–PW11 was converted to the low-spin state (S = 1/2, g = 2.00) during O2 coordination (Fig. 3g). This indicated that the change of Fe spin would facilitate the activation of O2, permitting electron transfer from NADPH. To explore the effect of Fe(II)–PW11, the electron properties were calculated using the Multiwfn software package.36 As demonstrated (Fig. S15†), the LUMO energy of O2 in Fe(III)–PW11–O2− was lower than that of the O2 empty orbital  . This indicated that Fe(II)–PW11 can activate O2 to accept the electron from NADPH easily.
. This indicated that Fe(II)–PW11 can activate O2 to accept the electron from NADPH easily.
Consequently, the mechanism of NOX-like activity of Fe(II)–PW11 was confirmed (Fig. S16†). Initially, Fe(II)–PW11 activates O2 by forming the Fe(III)–PW11–O2− intermediate through spin change. With NADPH present, an electron is transferred from NADPH to Fe(III)–PW11–O2− to form Fe(II)–PW11–O2˙− and NADPH+˙, easily forming the NADP˙ radical upon deprotonation. Finally, an electron was transferred from NADP˙ to O2˙− that desorbed from Fe(II)–PW11, facilitating the generation of NADP+ and H2O2. It should be noted that the present single-site catalyst of Fe(II)–PW11 exhibited better NOX-like performance than the conventional catalysts of Fe3O4 nanoparticles (the characterization studies are shown in Fig. S17†), demonstrated by the evaluation of NADPH content after treatment with both species (Fig. S18†).
LOX-like activity of Fe(II)–PW11
Natural LOX catalyzes the peroxidation of PUFA through hydrogen atom transfer (HAT) in the presence of an active site of Fe(III) (Fig. 4a). During the catalytic reaction in an Fe(II)–PW11 system, the O2-activated generation of high-valent Fe species can catalyze the LPO process through HAT, enabling cascade NOX-like and LOX-like activities of Fe(II)–PW11. Initially, linoleic acid (LA), a kind of PUFA, was selected as the model for examinations. As a result (Fig. 4b), only [LA–H]− at m/z 279 was recorded after adding Fe(II)–PW11 into LA. While with NADPH added, LA peroxidation was confirmed by recording ions of [LA–OH–H]− (m/z 295) and [LA–OOH–H]− (m/z 311) (Fig. 4c). Meanwhile, [NADP–2H]− (m/z 742) demonstrated the formation of NADP+ in the reaction system, confirming enzymatic catalysis of NADPH and LA oxidation upon the cascade reaction catalyzed by Fe(II)–PW11. As the reaction progressed, various oxidation products and peroxidation products of LA at m/z 309 ([LAO–OH–H]−), 325 ([LAO–OH–OH–H]−) and 327 ([LA–OH–OOH–H]−) were observed at the 20 min reaction, respectively (Fig. S19†). Furthermore, other oxidation products and LPO markers of 4-hydroxynonenal (4-HNE) (m/z 171 and 155) were detected at 1 h (Fig. S20†), indicating the deep oxidation and bond breakage of LA. The corresponding structures were confirmed by collision-induced dissociation (CID) MS (Fig. S21†).
 | ||
| Fig. 4 The LOX-like activity of Fe(II)–PW11. (a) The HAT mechanism of natural LOXs. (b) The MS signal of a mixture of LA and Fe(II)–PW11. (c) The MS signal of LA oxidation and peroxidation products in the Fe(II)–PW11–NADPH system for 5 min. (d) The MS spectra of Fe(II)–PW11 with SA, OA and LA added. (e) The MS spectra of the OA–NADPH system with Fe(II)–PW11 added. (f) EPR spectra of the LA peroxidation process with BMPO as the trapping agent. (g) The MS spectra of the Fe(IV)–PW11O intermediate during the reaction. (h) Proposed Fe(II)–PW11 catalytic mechanism of enzymatic NOX–LOX-like activities upon cascade reactions. | ||
To confirm the specific peroxidation of PUFA, saturated fatty acids (stearic acid, SA) and monounsaturated fatty acids (oleic acid, OA) were selected as the models. The adsorption of SA, OA and LA on the catalytic site of Fe(II)–PW11 was evaluated by mixing Fe(II)–PW11 with LA, OA and SA, respectively (Fig. 4d). As result, the ions of [Fe(II)PW11O39–OA + H2O + 3H]2− (m/z 1527.22, calc. 1527.19), [Fe(II)PW11O39–OA–2H2O + K + 2H]2− (m/z 1546.68, calc. 1546.72), [Fe(II)PW11O39–LA–H2O + 3H]2− (m/z 1526.20, calc. 1525.23) and [Fe(II)PW11O39–LA–2H2O + K + 2H]2− (m/z 1545.70, calc. 1545.72) were observed, while no signal of Fe(II)–PW11–SA was recorded. This indicated that Fe(II)–PW11 could adsorb OA and LA through double bonds for subsequent peroxidation. Furthermore, compared to LA, OA cannot be oxidized in air after adding NADPH (Fig. 4e), further indicating the selective catalytic peroxidation of PUFA by Fe(II)–PW11.
Besides, the peroxidation of PUFA-PLs catalyzed by Fe(II)–PW11 was explored. With lecithin (PC) and cuorin (CL) as substrates (the most abundant phospholipids in the body), the peroxidation product ions of [PC–OOH + H]+ (m/z 790) and [CL–OOH–2H]− (m/z 739) were recorded after adding NADPH into the Fe(II)–PW11 system (Fig. S22 and S23†). This indicated that the phospholipid chain in cells have no effect on the Fe(II)–PW11 catalyzed PUFA-PL peroxidation.
To explore the mechanism of LA peroxidation by Fe(II)–PW11, EPR and MS characterization studies were carried out. In the reaction system (Fig. 4f), the O2˙− and ˙LA radicals were confirmed by the peaks of BMPO–OOH and BMPO–LA, respectively. The signal of BMPO–OOH (AN and AβH at 14.1 G and 9.9 G, respectively) was attributed to O2˙− generation catalyzed by Fe(II)–PW11 through NOX-like activity. In addition, the peaks of BMPO–LA (AN and AβH at 16.0 G and 23.0 G) indicated the formation of the carbon-central ˙LA radical. Notably, the 2AN + AβH was calculated to be 55.0 G, attributed to the radical of bisallyllic carbon rather than olefinic carbon (>56 G) and saturated carbon (∼52 G). Furthermore, the obvious MS signal of the Fe intermediate of Fe(IV)–PW11O ([Fe(IV)PW11O39O + 3H]2− at m/z 1376.06, calc. 1376.06) was observed after adding NADPH and LA solution (Fig. 4g), which was the most important intermediate in the LPO process. Based on the low-lying unoccupied antibonding orbital, Fe(IV)–PW11O can easily abstract a hydrogen atom from LA to generate ˙LA, facilitating LA–OO˙ production through autooxidation. In addition, the activation energy of the transient state (TS) of the HAT process was calculated to be 125.5 kcal mol−1 with cis-2, cis-5-heptadiene as a model (Fig. S24†). Therefore, the HAT-based generation of ˙LA was demonstrated, which would support the deduced mechanism of NADPH and LA oxidation upon the cascade reaction.
Consequently, the mechanism of cascade NOX-like and LOX-like activity catalyzed by Fe(II)–PW11 can be proposed as shown in Fig. 4h. Initially, Fe(II)–PW11 would effectively bind and activate O2 to form the Fe(III)–PW11–O2˙− intermediate upon the donation of an electron by NADPH. During the oxidation, Fe(II)–PW11–O2˙− is rapidly protonated into an intermediate of Fe(III)–PW11–OOH under acidic conditions. Thereafter, the intermediate of Fe(IV)O–PW11 is formed upon protonation and hetero-cleavage of the O–O bond, along with losing one water. This intermediate subsequently initiated HAT for ˙LA production. Meanwhile, ˙LA can be oxidized into LAOO˙ radicals in the presence of O2, promoting the formation of lipid peroxides. The absorbed hydroxyl group can recombine with ˙LA to form Fe(III)–PW11–LA–OH, followed by the release of LA–OH and Fe(III)–PW11 unit. Finally, Fe(III)–PW11 would accept an electron from NADPH to form Fe(II)–PW11, initiating the next catalytic cycle. It should be noted that the present single-site catalyst of Fe(II)–PW11 exhibited better LOX-like performance than the conventional catalysts of Fe3O4 nanoparticles, as demonstrated by the monitoring of LA peroxidation by mass spectrometry after treatment with both species (Fig. S25†).
In situ synthesis of Fe(II)–PW11 for cancer therapy
As designed, A-MIL-101@PW12 can be recognized by receptors of cancer cells for cellular uptake and degraded by upregulated GSH for the in situ synthesis of Fe(II)–PW11. Herein, HeLa cells were selected as models to investigate TME-activated in situ synthesis. First, the stability of A-MIL-101@PW12 was evaluated using hydrodynamic size distributions (DLS) analysis (Fig. S26†), without significant change of the A-MIL-101@PW12 size in water, PBS and DMEM cell culture medium at different dispersion times. This indicated that A-MIL-101@PW12 exhibited excellent stability under physiological conditions for cancer therapy. To explore the nanomedicine uptake, the fluorescent Cyanine 5 (Cy5) was labeled on the AS1411 aptamer to form Cy5-A-MIL-101@PW12 with red fluorescence.37 As exhibited in both 2D and 3D Z-stack images of HeLa cells (Fig. 5a), the uptake of A-MIL-101@PW12 into the cytoplasm was demonstrated by the significantly increased red FL signals. Furthermore, the GSH consumption in HeLa cells was recorded in all groups and much higher consumptions were exhibited in MIL-101@PW12 and A-MIL-101@PW12 groups (Fig. 5b). This confirmed the GSH-initiated decomposition of MIL-101 nanocarriers and the NADPH-regulated GSH depletion catalyzed by Fe(II)–PW11. Thereafter, in situ synthesis of Fe(II)–PW11 was confirmed by the detection of Fe(II)–PW11 at m/z 911.75 in the lysate of HeLa cells incubated with A-MIL-101@PW12 (Fig. 5c). | ||
| Fig. 5 Investigation on TME-induced in situ synthesis of Fe(II)–PW11 for cancer therapy. (a) Merged 2D and 3D Z-stack images of HeLa cells incubated with Cy5-A-MIL-101@PW12 for 0 h, 2 h, 4 h and 8 h. Red channel: Cy5-A-MIL-101@PW12; blue channel: nuclei stained by Hoechst 33342. The scan bar is 10 μm. (b) The degradation of A-MIL-101@PW12 in HeLa cells. (i) The illustration of A-MIL-101@PW12 degradation and (ii) the GSH consumption after treatment with different groups. (c) The MS spectra of HeLa cell lysates (i) and HUVEC lysates (ii) after incubation with A-MIL-101@PW12. (d) MTT assay of HeLa and HUVEC cells incubated with different groups. (e) MTT assay of HeLa cells incubated with different concentrations of A-MIL-101@PW12. (f) CLSM images of HeLa cells and HUVECs with different treatments. The cells were stained with calcein-AM (green signals indicated living cells) and propidium iodide (PI) (red signals indicated cell death). The scan bar is 80 μm. Bars represent mean ± SD (n = 3 independent samples) calculated by the one-way ANOVA multiple comparison test (ns (p > 0.05), *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001). | ||
To evaluate the therapeutic effect of cancer therapy, the methylthiazole tetrazole (MTT) assay was performed for HeLa cells and HUVECs cultured with different groups. As demonstrated (Fig. 5d), MIL-101, Fe(II)–PW11, MIL-101@PW12, and A-MIL-101@PW12 showed hypotoxicity to HUVEC cells, indicating the good biocompatibility of these nanomedicines. Notably, Fe(II)–PW11 exhibited high toxicity to HeLa cells but low toxicity to HUVECs, attributed to the degradation of Fe(II)–PW11 into low-toxic species under neutral conditions. Besides, A-MIL-101@PW12 exhibited high cell lethality in HeLa cells based on the in situ synthesized Fe(II)–PW11 (Fig. 5e). Furthermore, live and dead cell staining experiments demonstrated the significant damage of HeLa cells treated by Fe(II)–PW11, MIL-101@PW12 and A-MIL-101@PW12 groups (red signals represented cell death) (Fig. 5f). Therefore, Fe(II)–PW11 can achieve tumor-specific and efficient therapy through TME-activated in situ synthesis in cancer cells, which exhibits low toxicity to normal tissues upon postoperative degradation under neutral conditions.
Intracellular evaluation of ferroptosis induced by in situ synthesized Fe(II)–PW11
The in situ synthesized Fe(II)–PW11 exhibits NOX-like and LOX-like activities to initiate ferroptosis at tumor sites. As illustrated (Fig. 6a), Fe(II)–PW11 facilitates LPO accumulation through PUFA-PL peroxidation and NADPH consumption. To confirm Fe(II)–PW11-induced ferroptosis for cancer treatment, HeLa cells were incubated with different inhibitors of cell death, including the caspase inhibitor (Z-VAD-FMK and Boc-D-FMK),38 necroptosis inhibitor (Nec-1),39 ferroptosis inhibitor (Fer-1)40 and ROS inhibitor (NAC).41 As shown in Fig. 6b, significantly increased cell viability was recorded after treatment with the Fer-1 inhibitor, indicating that cell death was induced through ferroptosis, rather than apoptosis or necroptosis. In addition, cell viability was slightly restored after being treated with NAC, due to the recovery of GPX-4 activity for ferroptosis suppression. | ||
| Fig. 6 Investigation of ferroptosis induced by in situ synthesized Fe(II)–PW11 in cancer cells. (a) Illustration of NADPH depletion and LPO accumulation for ferroptosis. (b) The cell viability of HeLa cells incubated with A-MIL-101@PW12 and different inhibitors. (c) The LPO verification in HeLa cells. (i) Cell imaging after treatment with different groups. (ii) The quantitative statistics of green fluorescence intensity of (i). The scan bar is 40 μm. (d) The ROS and O2˙− generation. (i) The cell imaging treated with A-MIL-101@PW12 with NAC and without NAC. (ii) The quantitative statistics of fluorescence intensity of (i). The scan bar is 40 μm. (e) MDA content of HeLa cells (i) without NAC and (ii) with NAC after treatment with PBS, Fe(II)–PW11 and A-MIL-101@PW12. (f) NADPH consumption, (g) CoQ10 content and (h) BH4 content of HeLa cells after treatment with different groups. (i(i)) Western blotting assay of the GPX4, DHFR and FSP1 protein levels in cancer cells treated with PBS, Fe(II)–PW11 and A-MIL-101@PW12. (ii) Quantitative analysis of GPX-4, DHFR and FSP1 protein expression based on the western blot data (n = 3). (j) The BioTEM image of HeLa cells treated with A-MIL-101@PW12. The scar bar is 2 μm. Bars represent mean ± SD (n = 3 independent samples) calculated by the one-way ANOVA multiple comparison test (ns (p > 0.05), *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001). | ||
First, the LPO accumulation induced by the in situ synthesized Fe(II)–PW11 was confirmed by the increased green fluorescence upon BODIPY 581/591 C11 oxidation in HeLa cells (Fig. 6c). Interestingly, this LPO accumulation is a ROS-independent process, demonstrated by the absence of ROS but significant LPO signals in cells after treating with A-MIL-101@PW12 for 12 h (Fig. S27†). while obvious ROS signals of O2˙− were observed after 24 h (Fig. S28†) along with cell death, indicating the generation of ROS and O2˙− upon stimulation of oxidative stress after cell death. Furthermore, with the ROS inhibitor NAC added, no significant ROS and O2˙− (Fig. 6d) was observed but LPO accumulation was observed (Fig. S29†). This is in accordance with the ROS-independent LPO accumulation, exhibiting increased content of the LPO product (MDA) with or without the ROS inhibitor NAC added (Fig. 6e).
Additionally, the intracellular NADPH level in HeLa cells significantly decreased after treatment with Fe(II)–PW11, MIL-101@PW12 and A-MIL-101@PW12 without (Fig. 6f) or with (Fig. S30†) the addition of NAC, indicating that in situ synthesized Fe(II)–PW11 facilitated the NADPH oxidation through the ROS-independent NOX/LOX cascade process. The decreased intracellular levels of BH4 (Fig. 6g) and CoQ10 (Fig. 6h) in the A-MIL-101@PW12 group further confirm the inhibition of antioxidants regulated by NADPH. Furthermore, the decreased GPX4 and DHFR protein levels in the A-MIL-101@PW12 and Fe(II)–PW11 groups (Fig. 6i and S31–S33†) indicated the deactivation of GPX-4 and DHFR protein due to the decrease in GSH and BH4 in HeLa cells.42 Notably, the expression of the FSP1 protein remained unchanged, indicating that CoQ10 could have no effect on the activity of FSP1. Consequently, the in situ synthesized Fe(II)–PW11 can induce ferroptosis by reducing intracellular ferroptosis inhibitors and causing related protein deactivation.
Thereafter, intracellular Fe2+ content was also confirmed by the increased red fluorescence after treatment with A-MIL-101@PW12 (Fig. S34†). This demonstrated the promotion of the LIP by the generated O2˙−, which would promote the subsequent LPO process for the cancer therapy The efficient therapy has been confirmed by the distinct mitochondrial shrinkage and the increase in mitochondria membrane density in A-MIL-101@PW12-treated HeLa cells (Fig. 6j).43 Consequently, A-MIL-101@PW12 is effective for cancer therapy through ferroptosis induced by the in situ synthesized Fe(II)–PW11.
In vivo evaluation of tumor suppression
To evaluate the in vivo therapeutic performance of the in situ synthesized Fe(II)–PW11, HeLa-tumor-bearing mice were selected as models. Initially, for tracking the distribution of nanodrugs, fluorescent Cy5 was labeled on the AS1411 aptamer to prepare A-Cy5-MIL-101@PW12. The HeLa-tumor-bearing mice were intravenously administered with A-Cy5-MIL-101@PW12 and the real-time fluorescence images were collected using an in vivo imaging system (IVIS) (Fig. S35†). As demonstrated, tumor sites showed stronger fluorescent signals, while normal tissues showed weaker ones. This was attributed to the enhanced permeability and retention effect of the nanocarrier as well as the tumor-targeting upon AS1411 recognition for in vivo therapeutic applications. Thereafter, the mice were subsequently sacrificed to obtain the main organs for in vivo imaging to identify the distribution of the nanomedicine. As demonstrated, A-Cy5-MIL-101@PW12 was mainly accumulated in tumor and liver tissue, demonstrating that nanocarriers in tumors would be excreted through metabolism after treatment.Subsequently, the HeLa-tumor-bearing mice were injected with nanomedicines, which were randomly divided into five groups: saline (I), MIL-101 (II), Fe(II)–PW11 (III), MIL-101@PW12 (IV), and A-MIL-101@PW12 (V) (1.20 mg kg−1 with the reference LD50 studies) (Fig. 7a and S36†). These nanomedicines were injected into mice every 3 days and the tumor sizes and body weights were monitored. Initially, the hemocompatibility assay of A-MIL-101@PW12 was confirmed by the low hemolysis rates and no significant heme release from the damaged erythrocytes (Fig. S37†). This indicated the negligible hemolytic toxicity, further confirmed by stable levels of biochemical indices in different groups (Fig. S38†).44 Therefore, A-MIL-101@PW12 satisfied biosafety for the subsequent therapeutic applications. As monitored, the tumor growth was inhibited in the nanomedicine-treated groups compared with the control group (Fig. 7b). Specifically, the Fe(II)–PW11 group exhibited more efficient tumor inhibition than the MIL-101 groups and the most significant tumor suppression was observed in the A-MIL-101@PW12 group. The lowest tumor weight in the A-MIL-101@PW12 group also confirmed the efficient tumor treatment by in situ synthesized Fe(II)–PW11 (Fig. 7c). In addition, the tumor growth inhibition value (TGI) of the A-MIL-101@PW12 group was determined to be 60%, verifying the efficient therapy by the present strategy (Fig. 7d and e). The satisfactory therapeutic efficacy is not only attributed to NOX-like and LOX-like activities upon the cascade reaction but is also enhanced by GSH depletion. Meanwhile, no apparent change in body weight indicated negligible side effects of these treatments on mice (Fig. 7f).
 | ||
| Fig. 7 In vivo therapeutic efficacy of the in situ synthesized Fe(II)–PW11. (a) The illustration of a HeLa-bearing mice model for cancer therapy. Evaluation of tumor growth (b), tumor weight (c), and tumor growth inhibition ratio (d), and digital photos of excised tumors (e) and body weight (f) for different groups during the period of treatment. (g) The contents of GSH (i), NADPH (ii), and MDA (iii) in tumors after different treatments. (h) H&E, Ki67 and GPX4 images of the tissues from HeLa tumor-bearing mice after different treatments. (I) Saline; (II) MIL-101; (III) Fe(II)–PW11; (IV) MIL-101@PW12; (V) A-MIL-101@PW12. Bars represent mean ± SD (n = 3 independent samples) calculated by the one-way ANOVA multiple comparison test (ns (p > 0.05), *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001). | ||
To evaluate the ferroptosis-induced cancer cell death, in vivo experiments at the tumor sites were conducted. As exhibited (Fig. 7g), the GSH levels in tumors decreased in the A-MIL101@PW12 treated groups, confirming the GSH-induced A-MIL101@PW12 degradation. Moreover, decreased NADPH and increased MDA were recorded in Fe(II)–PW11, MIL-101@PW12 and A-MIL-101@PW12 groups, confirming NADPH depletion and LPO accumulation catalyzed by in situ synthesized Fe(II)–PW11. Besides, the prominent downregulation of GPX4 in the A-MIL-101@PW12 group is in accordance with the ferroptosis-induced anticancer treatments (Fig. 7h). Therefore, the NADPH consumption and LPO accumulation were confirmed to induce ferroptosis for tumor-selective therapy.
Furthermore, the hematoxylin and eosin (H&E) staining assay was carried out to evaluate the antitumor activity, which exhibited significant damage to cancer cells (with a fragmentary cellular structure) in the A-MIL101@PW12 group. This confirmed the severe damage to tumors by the in situ synthesized Fe(II)–PW11 (Fig. 7h). In addition, the severe plasmatorrhexis and weakest cell proliferation were demonstrated by Ki67 staining of tumor slices after being treated with A-MIL-101@PW12, while no obvious normal tissue necrosis was observed in H&E staining of major organs (heart, liver, spleen, lung, and kidney), indicating low toxicity to healthy tissues (Fig. S39†). This suggested that ferroptosis can be selectively initiated at tumor sites by the in situ synthesized Fe(II)–PW11, which is postoperatively degraded into nontoxic debris in normal tissues. Therefore, upon the in situ synthesis of Fe(II)–PW11, A-MIL-101@PW12 exhibited promising tumoricidal efficacy for cancer therapy with low side effects.
Conclusions
In conclusion, the enzymatic NOX-like and LOX-like activities, catalyzed by an in situ synthesized single-site catalyst (Fe(II)–PW11), were reported for tumor-selective therapy via ferroptosis. The in situ synthesis of Fe(II)–PW11 was achieved by the chelation of Fe2+ into PW11 upon TME-activated degradation of iron-based nanocarriers in cancer cells. Upon electron transfer, Fe(II)–PW11 exhibits NOX-like activity for NADPH consumption and O2˙− generation, which promotes the LIP to facilitate ferroptosis. Meanwhile, the formation of the high-valence Fe(IV)O–PW11 intermediate subsequently catalyzes the generation of lipid hydroperoxide radicals through HAT based on LOX-like activity. Consequently, with cascade NOX–LOX activities, Fe(II)–PW11 promotes LPO accumulation for ferroptosis-based cancer therapy in a ROS-independent pathway. Besides, ferroptosis is further enhanced through the suppression of the antioxidant system by depleting GSH and NADPH. After treatment, Fe(II)–PW11 is degraded into low-toxic debris in the neutral environment of normal organs, reducing postoperative off-target toxicity. Furthermore, comprehensive characterization studies have supported the mechanism examination, facilitating the design of ferroptosis therapy upon cascade reactions. This strategy not only achieves selective and efficient ferroptosis therapy through a synergistic multi-enzymatic pathway in a ROS-independent pathway with minimized side effects, but also broadens the applications of single-site catalysts.
Ethical statement
All animal experimental protocols were reviewed and approved by the Animal Care and Use Committee of Institute of Beijing Normal University and complied with all relevant ethical regulations.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
X. Y. Ge and Y. Y. Yin conceived and designed the experiments for the project. X. Y. Ge, X. N. Wang and X. Li performed the characterization of the compounds. J. Ouyang carried out the data analysis. Y. Y. Yin performed the theoretical calculations. X. Y. Ge and N. Na prepared the initial manuscript, with all other authors contributing to revisions. N. Na supervised the project and acquired the funding. All authors discussed the results and provided feedback on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support provided by the National Natural Science Foundation of China (NNSFC, 22474010 and 22274012 to N. N.), the Fundamental Research Funds for the Central Universities (No. 2233300007 to N. N.), the National Key Research and Development Program of China (No. 2024YFA1509600 to N. N.) and the National Natural Science Foundation of China (NNSFC, 21675014 and 21974010 to J. O.).Notes and references
- S. J. Dixon, K. M. Lemberg, M. R. Lamprecht, R. Skouta, E. M. Zaitsev, C. E. Gleason, D. N. Patel, A. J. Bauer, A. M. Cantley, W. S. Yang, B. Morrison and B. R. Stockwell, Cell, 2012, 149, 1060–1072 CrossRef CAS PubMed.
- G. Lei, L. Zhuang and B. Gan, Nat. Rev. Cancer, 2022, 22, 381–396 CrossRef CAS PubMed.
- M. Conrad, S. M. Lorenz and B. Proneth, Trends Mol. Med., 2021, 27, 113–122 CrossRef CAS PubMed.
- Z. Liu, S. Liu, H. Jin, B. Liu, Q. Meng, M. Yuan, X. Ma, J. Wang, M. Wang, K. Li, P. Ma and J. Liu, Angew. Chem., Int. Ed., 2024, e202414879 Search PubMed.
- X. Zhao, X. Wang, W. Zhang, T. Tian, J. Zhang, J. Wang, W. Wei, Z. Guo and X. Wang, Angew. Chem., Int. Ed., 2024, 63, e202400829 CrossRef CAS PubMed.
- R. Hu, X. Chen, Z. Li, G. Zhao, L. Ding, L. Chen, C. Dai, Y. Chen and B. Zhang, Adv. Mater., 2023, 35, 2306469 CrossRef CAS PubMed.
- Q. Yao, J. Fan, S. Long, X. Zhao, H. Li, J. Du, K. Shao and X. Peng, Chem, 2022, 8, 197–209 CAS.
- H. Zhang, Y. Chen, W. Hua, W. Gu, H. Zhuang, H. Li, X. Jiang, Y. Mao, Y. Liu, D. Jin and W. Bu, Angew. Chem., Int. Ed., 2023, 62, e202300356 CrossRef CAS PubMed.
- W. Xu, Y. Yang, L. Liu, B. Hu, Y. Tang, H. Ding, L. Zhang, P. Yang and C. Wang, Adv. Funct. Mater., 2025, 2420540 CrossRef CAS.
- A. Seiler, M. Schneider, H. Förster, S. Roth, E. K. Wirth, C. Culmsee, N. Plesnila, E. Kremmer, O. Rådmark, W. Wurst, G. W. Bornkamm, U. Schweizer and M. Conrad, Cell Metab., 2008, 8, 237–248 CrossRef CAS PubMed.
- Y. Zou, H. Li, E. T. Graham, A. A. Deik, J. K. Eaton, W. Wang, G. Sandoval-Gomez, C. B. Clish, J. G. Doench and S. L. Schreiber, Nat. Chem. Biol., 2020, 16, 302–309 CrossRef CAS PubMed.
- A. Andreou and I. Feussner, Phytochemistry, 2009, 70, 1504–1510 CrossRef CAS PubMed.
- X. Fang, H. Ardehali, J. Min and F. Wang, Nat. Rev. Cardiol., 2022, 20, 7–23 CrossRef PubMed.
- T. M. Seibt, B. Proneth and M. Conrad, Free Radical Biol. Med., 2019, 133, 144–152 CrossRef CAS PubMed.
- K. Bersuker, J. M. Hendricks, Z. Li, L. Magtanong, B. Ford, P. H. Tang, M. A. Roberts, B. Tong, T. J. Maimone, R. Zoncu, M. C. Bassik, D. K. Nomura, S. J. Dixon and J. A. Olzmann, Nature, 2019, 575, 688–692 CrossRef CAS PubMed.
- M. Soula, R. A. Weber, O. Zilka, H. Alwaseem, K. La, F. Yen, H. Molina, J. Garcia-Bermudez, D. A. Pratt and K. Birsoy, Nat. Chem. Biol., 2020, 16, 1351–1360 CrossRef CAS PubMed.
- B. Yang, H. Yao, J. Yang, C. Chen, Y. Guo, H. Fu and J. Shi, J. Am. Chem. Soc., 2021, 144, 314–330 CrossRef PubMed.
- Y. Wang, S. Xu, L. Shi, C. Teh, G. Qi and B. Liu, Angew. Chem., Int. Ed., 2021, 60, 14945–14953 CrossRef CAS PubMed.
- J. Tian, B. Li, F. Zhang, Z. Yao, W. Song, Y. Tang, Y. Ping and B. Liu, Angew. Chem., Int. Ed., 2023, 62, e202307288 CrossRef CAS PubMed.
- Y. Liu, J. Zhang, X. Zhou, Y. Wang, S. Lei, G. Feng, D. Wang, P. Huang and J. Lin, Angew. Chem., Int. Ed., 2024, 63, e202408064 CAS.
- Y. Yin, X. Ge, J. Ouyang and N. Na, Nat. Commun., 2024, 15, 2954 CrossRef CAS PubMed.
- H. Wang, T. Yang, J. Wang, Z. Zhou, Z. Pei and S. Zhao, Chem, 2024, 10, 48–85 CAS.
- J. Han and J. Gun, Coord. Chem. Rev., 2023, 490, 215209 CrossRef CAS.
- L. Zhao, Z. Li, J. Wei, Y. Xiao, Y. She, Q. Su, T. Zhao, J. Li and J. Shao, Chem. Eng. J., 2022, 430, 133057 CrossRef CAS.
- G. Liu, Y. Zhang, L. Xu, B. Xu and F. Li, New J. Chem., 2019, 43, 3469–3475 RSC.
- C. Huang, C. Zhao, Q. Deng, H. Zhang, D. Yu, J. Ren and X. Qu, Nat. Catal., 2023, 6, 729–739 CrossRef CAS.
- C. Wu, D. Xu, M. Ge, J. Luo, L. Chen, Z. Chen, Y. You, Y. Zhu, H. Lin and J. Shi, Nano Today, 2022, 46, 101574 CrossRef CAS.
- Y. Wang, M. Sun, J. Qiao, J. Ouyang and N. Na, Chem. Sci., 2018, 9, 594–599 RSC.
- F. D. Hardcastle, J. Raman Spectrosc., 1995, 26, 397–405 CrossRef CAS.
- B. Viswanadham, N. Nagaraju, C. N. Rohitha, V. Vishwanathan and K. V. R. Chary, Catal. Lett., 2018, 148, 397–406 CrossRef CAS.
- W. Liao, H. Liu, L. Qi, S. Liang, Y. Luo, F. Liu, X. Wang, C. Chang, J. Zhang and L. Jiang, Cell Rep. Phys. Sci., 2021, 2, 100557 CrossRef CAS.
- M. Kamibayashi, S. Oowada, H. Kameda, T. Okada, O. Inanami, S. Ohta, T. Ozawa, K. Makino and Y. Kotake, Free Radical Res., 2009, 40, 1166–1172 CrossRef PubMed.
- K. Teng, L. Ya and Q. Zheng, J. Am. Chem. Soc., 2023, 145, 4081–4087 CrossRef CAS PubMed.
- H. Huang, S. Banerjee, K. Qiu, P. Zhang, O. Blacque, T. Malcomson, M. J. Paterson, G. J. Clarkson, M. Staniforth, V. G. Stavros, G. Gasser, H. Chao and P. J. Sadler, Nat. Chem., 2019, 11, 1041–1048 CrossRef CAS PubMed.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- H. A. Shidy, Dyes Pigm., 2017, 145, 505–513 CrossRef.
- H. Tu, W. Xiong, J. Zhang, X. Zhao and X. Lin, Nat. Commun., 2022, 13, 6603 CrossRef CAS PubMed.
- A. Degterev, Z. Huang, M. Boyce, Y. Li, P. Jagtap, N. Mizushima, G. Cuny, T. Mitchison, M. Moskowitz and J. Yuan, Nat. Chem. Biol., 2005, 1, 112–119 CrossRef CAS PubMed.
- R. Skouta, S. Dixon, J. Wang, D. Dunn, M. Orman, K. Shimada, P. Rosenberg, D. Lo, J. Weinberg, A. Linkermann and B. Stockwell, J. Am. Chem. Soc., 2014, 26, 4551–4556 CrossRef PubMed.
- M. Halasi, M. Wang, T. Chavan, V. Gaponenko, N. Hay and A. Gartel, Biochem. J., 2013, 454, 201–208 CrossRef CAS PubMed.
- J. Zhang, H. Ge, D. Zhou, Q. Yao, S. Long, W. Sun, J. Fan, J. Du and X. Peng, Adv. Mater., 2023, 35, 2308205 CrossRef PubMed.
- J. Cheng, L. Li, D. Jin, Y. Dai, Y. Zhu, J. Zou, M. Liu, W. Yu, J. Yu, Y. Sun, X. Chen and Y. Liu, Adv. Mater., 2023, 35, 2270037 Search PubMed.
- N. Song, R. Tao, H. Li, R. Zhang, Y. Huang, L. Zhang, Y. Liu, D. Yang and C. Yao, Angew. Chem., Int. Ed., 2025, 64, e202500566 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc02512e |
| This journal is © The Royal Society of Chemistry 2025 |