
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancing the photostability of red fluorescent proteins through FRET with Si-rhodamine for dynamic super-resolution fluorescence imaging†
Xuelian
Zhou
ab,
Lu
Miao
*a,
Wei
Zhou
ab,
Yonghui
Chen
a,
Yiyan
Ruan
a,
Xiang
Wang
a,
Guangying
Wang
ab,
Pengjun
Bao
a,
Qinglong
Qiao
*a and
Zhaochao
Xu
 *ab
*ab
aDalian Institute of Chemical Physics, Chinese Academy of Sciences, 457 Zhongshan Road, Dalian 116023, China. E-mail: miaolu@dicp.ac.cn; qqlqiao@dicp.ac.cn; zcxu@dicp.ac.cn
bSchool of Chemistry, Dalian University of Technology, 2 Linggong Road, Dalian 116024, China
First published on 5th May 2025
Abstract
Red fluorescent proteins (RFPs) are extensively utilized in biological imaging. However, their susceptibility to photobleaching restricts their effectiveness in super-resolution imaging where high photostability is crucial. In this study, we substantially improved the photostability of RFPs by incorporating a hybrid Förster resonance energy transfer (FRET) pair, utilizing RFPs as the energy donor and a photostable fluorophore, tetramethyl-Si-rhodamine (TMSiR), as the acceptor. TMSiR was selectively introduced through fusion with the HaloTag protein linked to the RFPs. We constructed a series of mApple/mCherry–TMSiR pairs with varying FRET efficiencies. Our findings reveal that higher FRET efficiency in the mApple/mCherry–TMSiR complexes correlates with enhanced photostability of RFPs. FRET competes with the singlet-to-triplet state transition of RFPs, while the spatial barrier introduced by the HaloTag protein prevents interaction between sensitized reactive oxygen species near Si-rhodamine and red fluorescent protein, enhancing the photostability of red fluorescent protein. The nearly 6-fold enhancement in mCherry's photostability allows for extended durations of dynamic structured illumination microscopy (SIM) imaging in living cells, facilitating the capture of finer details in organelle interactions. Leveraging the photostable mCherry protein, we tracked various mitochondrial fission processes and their interactions with lysosomes and the endoplasmic reticulum (ER). Interestingly, we observed the involvement of ER in all mitochondrial fission events, whereas lysosomes participated in only 66% of them.
Introduction
Red fluorescent proteins (RFPs) find extensive use in live-cell imaging. Their red-shifted excitation and emission help in reducing auto-fluorescence from tissues or cells, decreasing light scattering absorption, and mitigating phototoxicity.1–5 However, the susceptibility of RFPs to photobleaching restricts imaging duration and achievable image resolution, hindering the comprehensive and prolonged tracking in super-resolution fluorescence imaging.6,7 Photobleaching is primarily attributed to the triplet excited state, leading to the generation of oxidative species that degrade the fluorophore.8–12 Therefore, competing with the transition of triplet states or altering the interaction between reactive oxygen species and fluorophores is a general strategy employed to enhance the photostability of fluorescent proteins.Extensive efforts have been devoted to modifying the chromophore structure to inhibit the generation of triplet states through genetic engineering methods,8,13,14 including directed evolution13,15 and rational mutagenesis.16,17 Given the ongoing challenge of targeted biosynthesis of fluorescent proteins,8,18–20 there is a growing preference for using chemical modification of proteins or physical methods to regulate the interaction between reactive oxygen species and fluorophores. For instance, altering the composition of the cell culture medium to reduce the redox-active components can significantly enhance the photostability of EGFP.21–23 Henrikus et al. enhanced the photostability of GFP at the single-molecule level by covalently linking the triplet quencher azobenzene near the chromophore (A206C/L221C).24 Recently, a photophysical process known as reverse intersystem crossing (RISC) has been developed, employing near-infrared co-illumination to reduce photobleaching of fluorescent proteins both in vitro and in vivo.25,26 However, there is still a lack of effective and direct methods to enhance the photostability of red fluorescent proteins and apply it to super-resolution fluorescence imaging of organelle dynamics.
It has been reported that in certain small molecule dyads, the intersystem crossing (ISC) from the singlet to triplet state of the donor can be competitive with intramolecular donor-to-acceptor Förster resonance energy transfer (FRET).27–29 In the dyads of 2,6-diiodobodipy–styrylbodipy, the 2,6-diiodobodipy moiety was utilized as the singlet state energy donor and the spin converter for triplet state formation, whereas the styrylbodipy was employed as a singlet and triplet state energy acceptor.27 The authors found that the rate of FRET is higher than the rate of ISC, demonstrating the suppression of 2,6-diiodobodipy's ISC. Furthermore, Sreenath et al. constructed an intramolecular FRET conjugate with a zinc(II)-sensitive arylvinylbipyridyl fluoroionophore as donor and BODIPY as acceptor; the rapid intramolecular FRET between the donor and acceptor protects the donor fluorophore from photobleaching.30
The strategy of FRET competing with ISC is also being used to improve the stability of fluorescent proteins. Burrows combined ligand-activated proteins R-phycoerythrin (R-PE) with Alexa Fluor-647 and demonstrated that the photostability of R-PE could be improved through resonance energy transfer.31,32 However, the poor thermal stability and broad excitation spectrum of these ligand-activated proteins limit its applications.33,34 In addition, Basu et al. applied the FRET strategy to improve the photostability of photo-modulatable fluorescent protein in single-molecule tracking. They introduced the dye JF646 into the photoactivatable fluorescent protein mEos3.2 via a Halo tag, achieving single-molecule tracking of chromatin-binding proteins in cells.35 However, it cannot be applied to long-term structured illumination microscopy (SIM) super-resolution imaging to resolve fast subcellular dynamics, and the mechanism for improving photostability has not been demonstrated.
In this paper, we applied the FRET strategy to widely used red fluorescent proteins for SIM super-resolution imaging. We devised a hybrid FRET pair by combining a RFP with the small molecule dye tetramethyl-Si-rhodamine (TMSiR) to enhance the photostability of the RFP (Scheme 1). The RFP was employed as the donor, while the photostable small molecule dye TMSiR served as the acceptor. Specifically, TMSiR was introduced via the HaloTag protein fused to the RFP. By adjusting the connection mode and linker length between RFP and Halo proteins, we generated a series of RFPs–TMSiR pairs with varying FRET effects and examined the interplay between FRET effects and photobleaching dynamics. Linking with TMSiR led to a notable enhancement in the photostability of mCherry and mApple. Moreover, a higher FRET efficiency in the RFP–TMSiR complexes correlated with increased photostability of RFPs, indicative of effective inhibition of RFPs' photobleaching by FRET. Reactive oxygen species (ROS) content detection revealed a significant reduction in ROS production of RFPs post-hybridization, suggesting the triplet state transition of RFP was affected due to the competition between FRET and intersystem hybridization (ISC). Additionally, ROS generated by TMSiR did not induce fluorescence bleaching of RFPs since these ROS were confined within a radius of approximately 5 nm. Remarkably, mCherry, among the most photostable RFPs, demonstrated a nearly 6-fold improvement in photostability, facilitating the capture of finer details and reducing the need for duplicate imaging efforts during long-term dynamic structured illumination microscopy (SIM) imaging in living cells. Leveraging the photostable mCherry, we successfully tracked clear and multiple mitochondrial fission events influenced by the lysosomes/endoplasmic reticulum (ER). A total of 91 mitochondrial fission events were observed through continuous super-resolution tracking of 5 cells for 30 minutes. Intriguingly, the ER was implicated in all mitochondrial fission events, whereas only 66% involved lysosomes. These hybrid photostable RFP complexes hold promise for long-term super-resolution tracking of target proteins in live cells, thereby enabling the resolution of dynamic life processes.
 | ||
| Scheme 1 FRET strategy employed to enhance the photostability of RFPs. The photostable RFPs were further used for the long-term super-resolution imaging of mitochondrial dynamics. | ||
Results and discussion
Designing FRET pairs to enhance the photostability of RFPs
Here, we anticipate constructing a FRET pair with RFP as the energy donor and a photostable fluorophore as the acceptor, where the FRET from RFP to the photostable fluorophore may compete with the ISC process of RFP, thereby affecting the formation of the RFP triplet state and enhancing its photostability. Small molecular fluorescent dyes, owing to their significantly higher photostability compared to fluorescent proteins and resistance to photobleaching, represent an ideal choice as FRET acceptor fluorophores.36–38 The protein self-labeling methods represented by Halo/SNAP39–41 allow stable and site-specific labeling of RFPs with small molecular fluorophores. mApple and mCherry are widely utilized RFPs with excellent fluorescence properties compared to other RFPs (Table S1†). mCherry stands out as one of the most photostable RFPs, exhibiting a strong half-life (t1/2) of 87.97 ± 0.86 s under 200 μW laser illumination. And mApple possesses a higher quantum yield of 0.49, but its photostability is comparatively lower with a t1/2 of only 28.33 ± 0.12 s. In order to construct a FRET pair with mApple or mCherry as a donor fluorophore to enhance their photostability, the selection of the FRET acceptor and its connection method to the donor are crucial.For efficient FRET, the donor emission and acceptor absorption spectra must overlap, and the donor has high quantum yield, while the acceptor exhibits a favorable extinction coefficient. Additionally, the distance between the donor and acceptor should be less than 10 nm.42 The small molecule fluorescent dye TMSiR exhibits excellent properties, such as membrane permeability, near-infrared emission, high photostability, and signal-to-noise ratio, making it widely applied in super-resolution imaging techniques like STED.43–45 The obvious spectral overlap between the mApple/mCherry emission spectrum and TMSiR absorption spectrum, along with the high molar extinction coefficient of TMSiR (Fig. S1 and Table S1†), led to its selection as the FRET acceptor in this study. Through genetic engineering, we fused the Halo tag to the RFPs, and the TMSiR with a chloroalkane group (Halo–TMSiR) was linked to RFPs via a covalent reaction with the HaloTag. In comparison to direct chemical modifications, a self-labeling approach exhibits higher labeling specificity and better biocompatibility. It has been widely employed for fluorescent labeling of proteins and long-term dynamic imaging in living cells.46–48
Constructing the mApple–TMSiR FRET pairs to enhance the photostability of mApple
We initially fused the Halo protein to both the C-terminal and N-terminal of mApple protein, and obtained AH-L and HA-L, respectively (Fig. 1a). A short linker of five amino acids was added to ensure a distance of less than 10 nm between the donor and acceptor fluorophores. Both fusion proteins were expressed in vitro and labeled with TMSiR, followed by purification. SDS-PAGE proved that the fusion protein can be covalently labeled with Halo-dyes (Fig. 1b). The fluorescence of the HA-L emitted at 660 nm, corresponding to a decrease in donor fluorescence intensity and an increase in acceptor fluorescence, indicating the effectiveness of FRET (Fig. 1c and d). Furthermore, a higher FRET efficiency of 53.3% for AH-L was observed compared to HA-L with a FRET efficiency of 35.4%. Since the N-terminal of the Halo protein is relatively close to its self-labeled fluorophore (Fig. S2†), the fusion strategy of mApple to the N-terminal of Halo (AH-L) resulted in a shorter distance between the mApple fluorophore and TMSiR (Fig. 1a), leading to a higher FRET efficiency. Subsequently, in vitro photostability tests on HA-L and AH-L proteins were performed with continuous illumination at 561 nm for 100 minutes, and the fluorescence intensity of mApple's maximum emission (λem = 590 nm) was collected (Fig. 1e and f). Both fusion proteins exhibited a moderate increase in photostability compared to mApple alone, with the AH-1 construct showing greater improvement due to higher FRET efficiency. This is consistent with our hypothesis that FRET between RFPs and TMSiR may compete with the formation of the RFP triplet state, thereby enhancing photostability (Fig. 1g). Importantly, a higher FRET efficiency from mApple to TMSiR likely competes more effectively with the ISC of donor mApple, resulting in stronger photostability. | ||
| Fig. 1 The design strategy for the initial mApple–TMSiR FRET pairs. (a) Schematic representation of the structure of Halo-tag (PDB 6y7a) fused at the N-terminal or C-terminal of mApple (PDB 2H5Q, a mCherry crystal structure as a substitute). (b) The 12% SDS-PAGE of purified HA-L and AH-L fusion proteins labeled with or without NAP-488 dye.49 CBB: gel stained with Coomassie brilliant blue; FL: fluorescence image of the gel excited with UV. Under SDS-PAGE denaturing conditions, the RFP chromophore undergoes hydrolysis, resulting in lower molecular weight bands. (c) and (e) Fluorescence emission spectra of HA-L (c) and AH-L (e) with and without the addition of TMSiR. (d) and (f) Fluorescence intensity trends of HA-L (d) and AH-L (f) at maximum emission (λem = 590 nm) with or without TMSiR labeling. Continuous illumination was used with an external 561 nm laser (36 mW) over time. Each curve represents the average of three independent experiments ± SD. [Protein]: 2 μM. (g) The Jablonski diagram depicting the competitive processes of ISC and FRET. | ||
Regulating FRET effects could enhance the photostability of mApple and mCherry
In the subsequent investigation, we aimed to further enhance the photostability of RFPs by regulating the FRET efficiency between RFPs and TMSiR based on the AH-L fusion protein. FRET efficiency can be controlled by modulating the linker length between RFPs and Halo proteins. Therefore, starting with the AH-L fusion protein, we performed varying degrees of truncation at the mApple and Halo protein junction, resulting in three fusion proteins: AH-0, AH-1, and AH-2 (Fig. 2a and Table S2†). Employing the same fusion (Halo fused to the C-terminal of mCherry) and truncation strategy, we also obtained four mCherry–Halo fusion proteins: CH-L, CH-0, CH-1, and CH-2 (Fig. 2a and Table S2†). All fusion proteins were expressed in Escherichia coli and purified with Ni-NTA. SDS-PAGE confirmed that the molecular weights of the purified proteins matched theoretical values, and the fluorescence image demonstrated the covalent connection of the purified proteins with Halo-dye (Fig. 2b). A decrease in donor fluorescence intensity as well as an increase in acceptor fluorescence in the fluorescence emission spectra provided evidence of FRET between mApple/mCherry and TMSiR (Fig. 2c and f). And then the FRET efficiency between each FRET pairs was calculated based on the extent of decrease in donor fluorescence intensity (Table 1). It seemed that all modified proteins exhibited varying FRET efficiencies. The proteins with the longest linker length, AH-L and CH-L, had the lowest FRET efficiency (53.3% and 64.9%, respectively). As the linker was shortened, FRET efficiency gradually increased. However, the proteins with the shortest linker lengths, AH-2 and CH-2, did not achieve the expected maximum FRET efficiency. Instead, proteins with moderate linker lengths, AH-1 and CH-1, exhibited higher FRET efficiencies (80.0% for AH-1 and 83.3% for CH-1). This result may be attributed to the excessively short linker, which causes the RFP and HaloTag to be in close proximity. This proximity can affect the binding efficiency of the small molecule TMSiR to the HaloTag protein.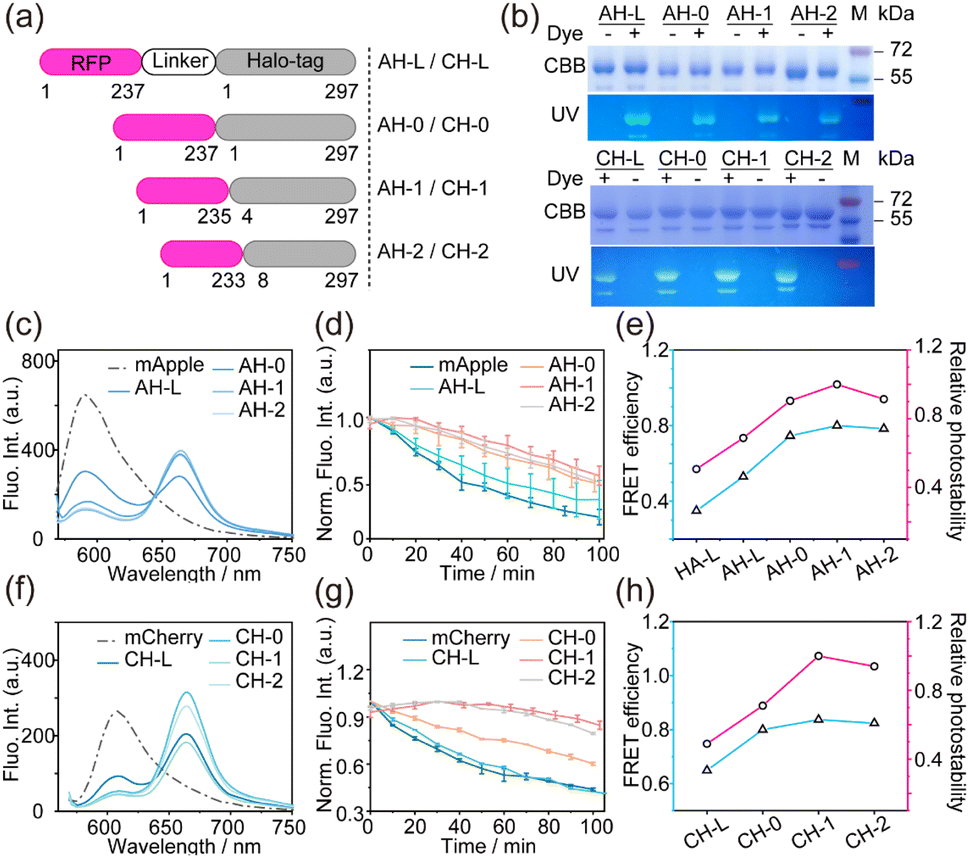 | ||
| Fig. 2 The design strategy for the mApple/mCherry–TMSiR FRET pairs and their photostability in vitro. (a) Schematic representation of the different structure of the mApple/mCherry–Halo fusion protein with different linkers. (b) The 12% SDS-PAGE of the purified fusion proteins labeled with or without NAP-488 dye.49 CBB: gel stained with Coomassie brilliant blue; FL: fluorescence image excited with UV. (c) and (f) Fluorescence emission spectra of mApple–Halo series fusion proteins (c) and mCherry–Halo series fusion proteins (f) with the addition of TMSiR. (d) and (g) Fluorescence intensity trends of the donor at maximum emission (mApple: λem = 590 nm; mCherry: λem = 610 nm) for TMSiR-labeled mApple–Halo proteins (d) and mCherry–Halo proteins (g) under continuous illumination with a 561 nm laser (36 mW). Each curve represents the average of three independent experiments ± SD. [Protein]: 2 μM. (e) and (h) Comparison of FRET efficiency (blue line) and relative photostability (pink line) upon mApple–Halo proteins (e) and mCherry–Halo proteins (h). Relative photostability is represented by the change in fluorescence intensity relative to the maximum value over 100 min. | ||
| Purified protein | Live cell | |||
|---|---|---|---|---|
| Protein name | E FRET (%) | In vitro fluorescence decline (Fmax − F100 min)/Fmax (%) | Confocal half-life t1/2 (s) | SIMa half-life t1/2 (s) |
| a The SIM photobleaching laser power settings for mCherry (0.27 kW cm−2) and mApple (0.16 kW cm−2) are different. | ||||
| mApple | — | 73.9 ± 5.7 | 50.2 ± 4.2 | 426.7 ± 39.7 |
| HA-L | 35.4 | 71.2 ± 5.6 | — | — |
| AH-L | 53.3 | 60.9 ± 1.4 | 64.7 ± 10.9 | 472.0 ± 59.9 |
| AH-0 | 74.2 | 48.6 ± 6.6 | — | — |
| AH-1 | 80.0 | 43.2 ± 7.1 | 125.3 ± 7.4 | 537.8 ± 55.5 |
| AH-2 | 78.6 | 46.2 ± 3.3 | 102.9 ± 3.6 | 512.0 ± 66.4 |
| mCherry | — | 56.1 ± 1.0 | 345.5 ± 47.5 | 168.8 ± 6.6 |
| CH-L | 64.9 | 58.6 ± 0.2 | 405.0 ± 63.7 | 318.0 ± 21.7 |
| CH-0 | 80.1 | 39.8 ± 0.9 | — | — |
| CH-1 | 83.7 | 14.1 ± 1.0 | 1948.8 ± 55.6 | 473.5 ± 51.6 |
| CH-2 | 82.4 | 20.1 ± 0.7 | 1036.2 ± 51.7 | 415.4 ± 36.9 |
In the context of these proteins exhibiting different FRET efficiencies, we evaluated the photostability of all fusion proteins in vitro by continuously irradiating at 561 nm for 100 minutes and measuring the maximum emission fluorescence intensity (mApple: λem = 590 nm; mCherry: λem = 610 nm). The trends in fluorescence intensity showed that mApple without labeling TMSiR decreased by 73.9% of the total intensity after 100 minutes (Fig. 2d). And the photostability of mApple–Halo fusion proteins labeled with TMSiR showed varying degrees of improvement (Fig. 2d), indicating that the introduction of the acceptor TMSiR effectively mitigates photo bleaching of the donor mApple. Significantly, there was a positive correlation between the degree of photostability enhancement and FRET efficiency (Fig. 2e), which were consistent with earlier results (Fig. 1). This observation was further confirmed in the photostability test of mCherry series fusion proteins (Fig. 2g and h). AH-1 and CH-1 with higher FRET efficiencies exhibited superior photostability, where the fluorescence intensity decreased by 43.2% and 14.1% of the total intensity, much more stable than wild mApple and mCherry.
Furthermore, we examined the photostability of all proteins at the acceptor emission wavelength (λem = 660 nm) under 561 nm laser excitation. Interestingly, fusion proteins with different FRET efficiencies exhibited similar photostability at this wavelength (Fig. 3a and b), which was much lower than that of the fluorescence emitted by the donor RFPs (Fig. 3c and d). This is due to the fact that the fluorescence at the wavelength of the acceptor is directly influenced by the stability of the dye TMSiR.
 | ||
| Fig. 3 Mechanistic investigation of FRET-enhanced photobleaching in RFPs. (a) and (b) Trends in the fluorescence intensity of acceptor TMSiR at maximum emission (λem = 660 nm) in TMSiR-labeled mApple–Halo series fusion proteins (a) and mCherry–Halo series fusion proteins (b) under continuous illumination with an external 561 nm laser over time. Each curve represents the average of three independent experiments ± SD. [Protein]: 2 μM. (c) and (d) The photobleaching kinetic curves of AH-1 (c) and CH-1 (d) were compared at the donor and acceptor wavelengths, respectively. (e) and (f) ROS measurements of mApple–Halo fusion proteins (e) and mCherry–Halo fusion proteins (f) with TMSiR labeling under continuous illumination with a 561 nm laser (36 mW) in the presence of 2 μM DCFH. The accumulated changes in fluorescence intensity at 530 nm wavelength were measured (λex = 470 nm; λem = 530 nm). The values represent the average ± SD of three independent experiments. [Protein]: 2 μM. (g) Schematic diagram illustrating the mechanism of ROS generation dependent on distance to inhibit photobleaching of RFPs. | ||
The photobleaching of TMSiR affects all FRET measurements uniformly, resulting in a consistent decline in FRET fluorescence for all proteins. In contrast, the fluorescence at the donor is influenced by the competition between FRET and ISC pathways, leading to variations that correlate with differences in FRET efficiency. These results further prove that FRET assisted strategies can be used to enhance the photostability of donor fluorophores.
Molecular mechanism of FRET-enhanced photostability in RFPs
The primary cause of photobleaching in fluorescent proteins is commonly attributed to oxygen dependence, where the electronically excited-state electrons of the fluorophore undergo ISC to the triplet state, interacting with oxygen molecules to generate ROS. The ROS then reacts with the fluorophore, leading to fluorescence quenching.8,20 In this study, we discovered that the photobleaching of RFPs can be inhibited by FRET, and the higher the FRET efficiency, the more effective the inhibition. This observation aligns with our previous speculation that FRET and ISC competition in RFPs affects triplet-state transitions, resulting in a reduction in ROS production and thereby enhancing the photostability of RFPs (Fig. 1g). To further validate this hypothesis, we used 2′,7′-dichlorodihydrofluorescein (DCFH) as a probe to measure the amount of ROS generated by various fusion proteins.50,51 2′,7′-Dichlorodihydrofluorescein (DCFH) reacts with ROS (such as singlet oxygen, superoxide radicals, hydroxyl radicals, etc.) to produce green fluorescent 2′,7′-dichlorofluorescein (DCF) (Fig. S3†). By monitoring the fluorescence intensity change of DCF at 530 nm, the relative content of ROS can be quantitatively analyzed. Fig. 3a and b showed that HA-L and CH-L, with the lowest FRET efficiency, exhibited the most significant fluorescence changes in DCFH, indicating the highest production of ROS. As FRET efficiency increased for different mApple/mCherry–Halo fusion proteins, the production of ROS by mApple/mCherry proteins decreased accordingly. AH-1 and CH-1, with the highest FRET efficiency (and best photostability), displayed the lowest levels of ROS. This result was consistent with previous studies on the competition between FRET and ISC in small molecule diiodobodipy–styrylbodipy dyads.27Additionally, during the ROS detection, we also found that the Halo–TMSiR dye generated more ROS compared to wild RFPs (Fig. 3e and f). However, TMSiR exhibited much higher photostability than RFPs (Fig. 3a and b). This suggests that the photostability of TMSiR was less affected by ROS, but even a small amount of ROS can lead to intense photobleaching of RFPs. However, ROS produced by TMSiR did not affect the photostability of mApple/mCherry in the fusion proteins, because ROS activity was limited to a range with a radius of about 5 nm centered on the TMSiR fluorophore as reported.52 The distance between the fluorophore of the fused RFPs and TMSiR (even in the case of the shortest linkers, AH-1 and CH-1) exceeds the range of ROS activity, effectively isolating the ROS generated by TMSiR from the RFP chromophore (Fig. 3g).
Confocal and super-resolution microscopy were employed to assess the photostability of the fusion proteins within living cells
To validate whether the modified red fluorescent proteins could maintain high photostability in the complex cellular environment, we evaluated the photostability of these hybrid FRET pairs in living HeLa cells. Three groups of proteins with low (AH-L/CH-L), moderate (AH-2/CH-2), and high (AH-1/CH-1) FRET efficiencies were selected, respectively. These proteins were individually cloned into the C-terminal of human histone H2B or mitochondrial membrane protein Tom20, and were transiently expressed in HeLa cells. Confocal fluorescence imaging revealed that these fusion proteins were effectively localized to the cell nucleus or mitochondria performed after the addition of Halo–TMSiR dye, as well as significant FRET effects between RFP and TMSiR upon 561 nm excitation (Fig. 4a and S4†).Subsequently, under the same conditions, the photostability of these proteins was detected by continuous fluorescence imaging with λex = 561 nm. Further single-exponential fitting and normalization yielded Fig. 4b and c. Consistent with the in vitro results, the AH-1 fusion protein with the highest FRET efficiency, exhibited the best photostability of the donor with a half-life (t1/2) of 125.3 s, much higher than that of wild mApple (t1/2 = 50.2 s) (Table 1). Excitingly, the photostability of CH-1 in living cells (t1/2 = 1948.8 s) was approximately 6-fold that of unmodified mCherry (t1/2 = 345.5 s). CH-1 is the most photostable construct based on the overall trend observed throughout the duration of the experiment, including the decay rate. The stabilities of CH-L and CH-2 also showed significant improvements, reaching 405.0 s and 1036.2 s, respectively (Table 1).
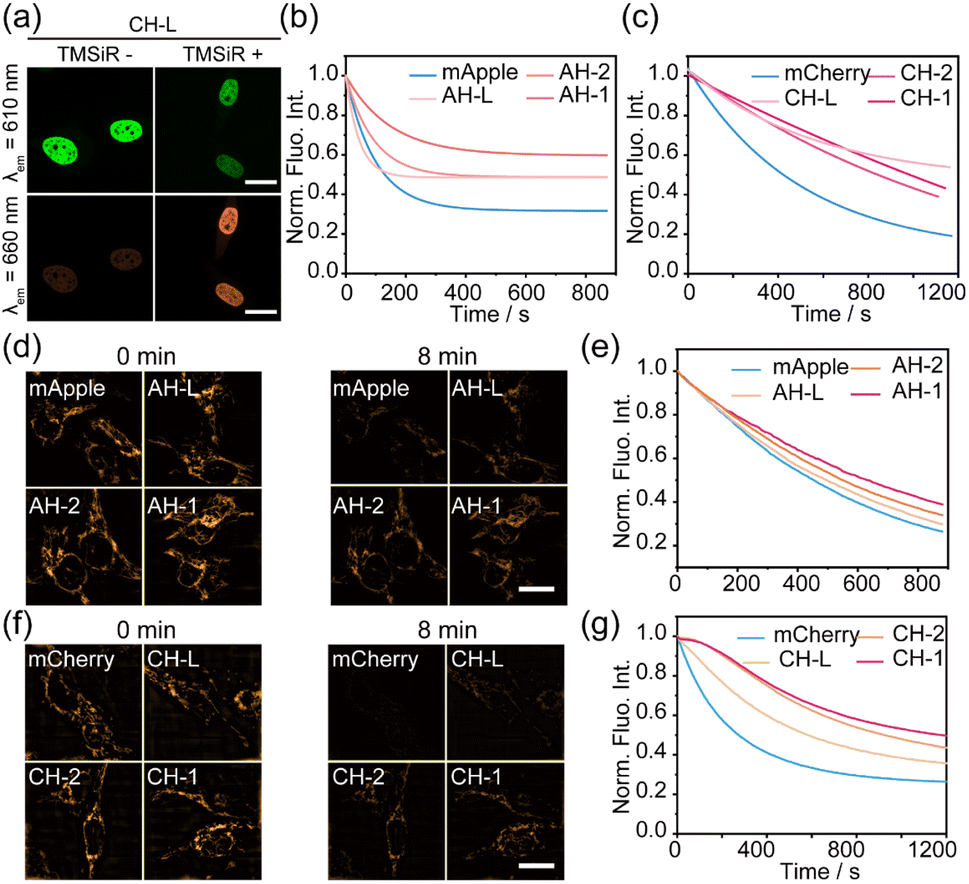 | ||
| Fig. 4 (a) HeLa cells expressing the indicated H2B–CH-L fusion proteins were imaged with confocal microscopy through the donor channel and FRET channel, respectively. Scale bar: 10 μm. (b) and (c) Comparative photobleaching dynamics of mApple series FRET pairs (b) and mCherry series FRET pairs (c) expressed in HeLa cells were captured through confocal fluorescence imaging under continuous illumination with a 561 nm laser (33 mW) over time. Each curve represents the average of three independent experiments. (d) and (f) SIM images of mApple series FRET pairs (d) and Cherry series FRET pairs (f) fused to Tom20, at specified time points. The images show a remarkable set of imaging results. (e) and (g) Photobleaching dynamics comparison of mApple series FRET pairs fused to Tom20 (e) and mCherry series FRET pairs fused to Tom20 (g) in SIM super-resolution imaging under continuous illumination with a 561 nm laser over time. Each curve represents the average of three independent experiments. Laser export power: mApple (0.16 kW cm−2)/mCherry (0.27 kW cm−2). | ||
To demonstrate the universality of this protein system across different microscopy imaging modalities, we further assessed the protein's photostability using SIM. For consistency with mApple, mCherry series proteins were cloned into mitochondrial membrane protein Tom20 for detection. Continuous imaging under SIM mode and single-exponential fitting with normalization (Fig. 4d–g and S5†) revealed that mCherry proteins were nearly completely photobleached within 4 minutes, while high FRET efficiency CH-1 could maintain clear imaging for 8 minutes. AH-1 and CH-1 fusion proteins displayed the strongest photostability and a threefold enhancement of CH-1 to wild mCherry was detected (Fig. 4e and g, Table 1), which was consistent with the results from in vitro and confocal detection. Due to mApple's rapid photobleaching, lower SIM laser power was used to detect mApple photostability. These results suggest that the improvement in the stability of both RFPs assisted by FRET is not affected by the complex cellular environment, and it meets the imaging requirements of different imaging systems. Importantly, mCherry protein, as the most commonly used and relatively stable red fluorescent protein, has nearly tripled its photostability under live-cell conditions using the FRET-assisted strategy. This approach holds potential for long term super-resolution imaging.
AH-1 for long-term SIM fluorescence imaging of mitochondrial dynamics
Mitochondria, often referred to as the “powerhouses” of the cell, play a crucial role in various cellular functions such as energy metabolism, redox balance, calcium regulation, and the maintenance of electrochemical gradients.53,54 and it carry out these functions through continuous processes of contact, fusion, and fission, as well as interactions with other cellular organelles.55,56 Mitochondrial fission is particularly significant for cellular stress responses and apoptosis among these.57 Real-time monitoring of mitochondrial fission allows comprehensive insights into the physiological state and response mechanisms within cells, contributing to disease research, drug development, and advancements in cell biology. Fluorescence imaging techniques are the preferred method for tracking mitochondrial dynamics. In conventional imaging tracking of mitochondrial events, limited by the low photostability of fluorescent proteins, mitochondrial dynamics imaging in a short period of time (a few minutes) is usually tracked, and then analyzed by a large number of cell imagings.58–62 Meanwhile, short time imaging can't capture the long, continuous interaction information.63–65 Here, we applied RFPs with high photostability to track mitochondria dynamic using SIM imaging (Fig. 5). | ||
| Fig. 5 Long-term SIM imaging of mitochondrial dynamics in live HeLa cells. (a) SIM images of the mitochondrial outer membrane protein Tom20 labeled with AH-1 in the entire live HeLa cell after TMSiR staining. Scar bar: 5 μm. (b) and (c) Cross-sectional profiles of the mitochondrial outer membrane for mApple and AH-1 and their intensity distributions. (d) Signal-to-noise ratio comparison of the mitochondrial outer membrane for mApple and AH-1. (e) Schematic representation of the mitochondrial fission process. (f) Local magnified image of the solid box area in (a). Pink arrows indicate the site of mitochondrial fission. Scar bar: 1 μm. (g) Line scan analysis of relative fluorescence intensity along the blue lines in the representative images of mitochondrial fission time points (130 s, 250 s) shown in (f). (h) Schematic representation of the mitochondrial contact process. (i) Local magnified image of the solid box area in (a). Pink arrows indicate the site of mitochondrial contact. Scar bar: 1 μm. (j) Line scan analysis of relative fluorescence intensity along the blue lines in the representative images of mitochondrial contact time points (560 s, 610 s) shown in (i). (k) Quantitative statistics of mitochondrial fission events occurring in HeLa cells expressing mApple and AH-1 during the total observation period of 20 minutes. The number of events was counted separately for the intervals of 0–10 minutes and 10–20 minutes. The graph displays the average values ± SD from five cell experiments. (l–m) Quantification of the percentage of mitochondrial fission events for mApple and AH-1 within the intervals of 0–10 minutes and 10–20 minutes. | ||
It is noteworthy that although the FRET-assisted strategy enhances the photostability of RFPs, it inevitably results in a loss of brightness for the donor mApple/mCherry. Nevertheless, under SIM imaging, AH-1 labeled with TMSiR still displayed a high resolution and signal-to-noise ratio to observe mitochondrial outer membrane structure, comparable to mApple before dye labeling (Fig. 5b–d). Mitochondrial fission and contact behaviors were clearly tracked (Fig. 5e and f). Fig. 5f shows that a mitochondrial branch (pink arrows) briefly contacted and separated from another mitochondrion on the right at 220 s, followed by fission at 290 s. Additionally, a mitochondrion (Fig. 5i) underwent a 190 s contact with another mitochondrion below by altering its shape (elongating, shortening, bending) from 560 s to 610 s, separating at 800 s. Changes in fluorescence intensity provided further evidence of these mitochondrial behaviors (Fig. 5g and j).
Furthermore, we demonstrated the imaging advantages of FRET-assisted high photostability RFPs by comparing imaging information collected after mApple and AH-1 labeling of mitochondria. We quantified the total number of mitochondrial contact events in 5 independent cells labeled with mApple and AH-1, respectively (Fig. 5k–m). In comparison with the wild mApple protein, we observed that the total contact number of AH-1 labeled mitochondrial outer membrane increased significantly. We used half of the total duration, 10 minutes as the critical point, separately counting the contact times for 0–10 minutes and 10–20 minutes. In either stage, the number of mitochondrial contact events with AH-1 labeling consistently exceeded those with mApple (Fig. 5k). The statistical difference was mainly evident in the 10–20 minute phase, where the fluorescence of mApple was quenched, resulting in many existing contact events being missed. Consequently, the percentage of contact events observed by mApple in the later stages accounted for only 18.10% of the total contact events, while AH-1 exhibited a proportion of 35.53% (Fig. 5l–m). These results demonstrate that owing to the higher photostability of AH-1, we can observe more details of mitochondrial dynamics than with mApple, making it highly suitable for observing and statistically analyzing long-term dynamic behaviors and events in mitochondria.
CH-1 labeling was used to track the interaction between mitochondria and the endoplasmic reticulum (ER)
Mitochondria typically perform functions through interactions with other subcellular organelles, maintaining cellular physiological balance.58,66–68 Studies have shown that interactions between mitochondria, ER, and lysosomes play a crucial role in guiding mitochondrial fission, influencing processes such as calcium signaling, lipid and protein transport, and the regulation of cell apoptosis.54,55,62 Here, we employed the previously described photo-stable CH-1 to label mitochondria, combined with SIM imaging, to reveal the dynamic interactions between mitochondria and two cellular organelles: ER or lysosomes.Initially, we co-transfected Tom20–CH-1 and ER marker EGFP-Sec61β in HeLa cells. Continuous tracking was performed using SIM over 30 min (Fig. 6a–h). Fig. 6a displays the well-defined morphology of mitochondria and the ER. As the imaging time increased, a significant decrease in EGFP brightness was observed, while CH-1 maintained a constant intensity (Fig. 6b). Cranfill et al. reported that EGFP has a half-life (t1/2) of 50.69 ± 0.56 s under 200 μW laser irradiation, while mCherry has a t1/2 of 87.97 ± 0.86 s, less than double the difference.69 Here, the significantly improved photostability of CH-1 compared to EGFP was demonstrated. In dynamic imaging (Fig. S6†), we observed the elongation of mitochondria and rapid contact with the ER (503 s), followed by rapid repositioning within 15 seconds. These ER–mitochondria contacts occurred rapidly and frequently within seconds to tens of seconds, as observed through statistical analysis revealing a total of 98 ER–mitochondria contact events within 30 minutes, with a 41.84% occurrence rate in the 15–30 minute contact events (Fig. 6h and S6g†). This may be attributed to the diminishing brightness of EGFP in the later stages, resulting in some contact events not being captured. ER–mitochondria contact involves essential cellular activities such as active transport and lipid transfer.62,70
 | ||
| Fig. 6 Dual-color SIM images of the endoplasmic reticulum/lysosomes and mitochondria in live HeLa cells. (a) Dual-color SIM image of the entire live HeLa cell co-transfected with GFP-Sec61β and CH-1–Tom20 (labeled with TMSiR). Delayed dual-color SIM images show the merged channels of the endoplasmic reticulum (green) and mitochondria (orange). Scar bar: 5 μm. (b) Photobleaching dynamics comparison of EGFP and CH-1. (c) Local magnified images of the solid-boxed region in (a). Contact points between the mitochondria and endoplasmic reticulum are highlighted with pink arrows. Scar bar: 1 μm. (d) Model of endoplasmic reticulum-mediated mitochondrial fission. (e and f) Line scan analysis of relative fluorescence intensity along the blue lines in representative endoplasmic reticulum-mediated mitochondrial fission time points (941 s, 1017 s) in (c). (g) Quantification of the percentage of endoplasmic reticulum-mediated mitochondrial fission events occurring in 0–15 min and 15–30 min. The graph shows mean ± SD from seven cell experiments. (h) Quantification of the percentage of endoplasmic reticulum–mitochondria contact events occurring in 0–15 min and 15–30 min stages. (i) SIM image of the entire live HeLa cell expressing CH-1–Tom20, stained with LysoTracker Green and TMSiR. Delayed dual-color SIM images show the merged channels of lysosomes (green) and mitochondria (orange) over time. Scar bar: 5 μm. (j) Photobleaching dynamics comparison of LysoTracker Green and CH-1. (k) Local magnified images of the solid-boxed region in (i). Mitochondrial fission is mediated by lysosomes (pink arrow) and non-lysosomes (blue arrow) at this location. Scar bar: 1 μm. (l) Model of lysosome-mediated mitochondrial fission. (m and n) Line scan analysis of relative fluorescence intensity along the white lines in representative lysosome-mediated mitochondrial fission time points (941 s, 1017 s) in (k). (o) Quantification of the percentage of lysosome-mediated and non-lysosome-mediated mitochondrial fission events. The graph shows mean ± SD from five cell experiments. (p) Comparison of the percentage of endoplasmic reticulum- and lysosome-mediated mitochondrial contact events. (q) Proposed functional model of mitochondrial fission sites, with approximately 65.93% of mitochondrial fission events in cells being simultaneously mediated by the endoplasmic reticulum and lysosomes. | ||
In addition to rapid ER and mitochondrial contact, we also observed the significant role of the ER in mitochondrial fission (Fig. 6c–h). As shown in Fig. 6c, the contact site with the ER–mitochondrial marked by pink arrows exhibited a distinct behavior of contraction followed by fission between 836 and 1017 s. The ER drove a noticeable morphological change of the mitochondria, with the mitochondria breaking and departing at 1017 s. Fluorescence intensity analysis further indicated the continuous presence of the ER at the site of mitochondrial fission (Fig. 6e and f). Subsequently, we counted 58 mitochondrial fission events from seven cells, and all these events involved the participation of the ER. We further quantified the fraction of ER-mediated mitochondrial fission events, and in the total of 58 ER-mediated events, the 15–30 minute fission events accounted for 41.38%, possibly due to EGFP photobleaching results (Fig. 6g). This is consistent with the statistics for ER–mitochondria contacts.
CH-1 labeling was used to track the interaction between mitochondria and lysosomes
The interaction between mitochondria and lysosomes is also part of the complex regulatory network within cells. Here, we further tracked the interaction between mitochondria and lysosomes in live cells. Specifically, we used genetically encoded CH-1 to label mitochondria and commercial dye LysoTracker Green to label lysosomes (Fig. 6i–o). The photostability of CH-1 remains excellent and shows minimal decay within 30 min of SIM imaging (Fig. 6j), indicating that CH-1 can be used for longer fluorescence tracking. We observed multiple mitochondrial fission events involving lysosomes, as shown in Fig. 6k, where lysosomes indicated by pink arrows maintained sustained contact with the mitochondria below for over 400 seconds and interacted with the adjacent mitochondria at 663 s, causing the upper mitochondria to contract and break (1112 s), and then returned to the original contact position with the lower mitochondria after the interaction was completed. Fluorescence intensity analysis further indicated the presence of lysosomes at the site of mitochondrial fission (Fig. 6m and n). Lysosomes, acting as the “garbage disposal” of the cell, have been found to induce mitochondrial fission events through mitochondrial–lysosomal contact sites under the action of the small GTPase Rab7.71 At the same time, the position of mitochondria indicated by the blue arrow also underwent significant morphological changes and fission at 663 s, but this fission did not involve lysosomes. This suggests that not all mitochondrial fission events are mediated by lysosomes. It should be noted that in the mitochondria-ER imaging described above with the addition of TMSiR dye, the mitochondria consistently maintained a well-defined morphology. However, in this section, there was a tendency for the mitochondria to become more rounded in the later stages of SIM imaging, which may be attributed to the introduction of the LysoTracker dye.To quantify this result, we counted a total of 91 mitochondrial fission events from 5 cells, finding that approximately 65.93% of the fission events were mediated by lysosomal contact, while the remaining 34.07% occurred without lysosomal involvement (Fig. 6o). In summary, 100% of the mitochondrial fission events were associated with the endoplasmic reticulum, while only 65.93% of the fission events may be co-mediated by lysosomes and the endoplasmic reticulum (Fig. 6p and q). This statistical result is consistent with the imaging and statistics of ER and lysosome-mediated mitochondrial fission reported by Kim et al., who obtained 73 fission events from tracking the fluorescence of 30 cells within 2 minutes, with the ER participating in all mitochondrial fission events and lysosomes involved in 58.9% of mitochondrial fission events.62 Using the photo-stable CH-1 protein, we achieved 91 fission events and similar statistical data in a 30 minute tracking session, significantly reducing workload. In short, high photostability RFPs obtained by FRET assisted methods allow us to record longer time to monitor more dynamic events and details in living cells.
Conclusions
In summary, we developed a FRET-assisted strategy using RFPs as donors and the photostable small molecule fluorophore TMSiR as a receptor to enhance the photostability of RFPs for long-term dynamic SIM imaging in living cells. Initially, we constructed a series of FRET pairs using the RFPs mCherry and mApple by individually fusing them with the HaloTag and labeling them with TMSiR. In vitro and live cell photostability tests demonstrated a significant enhancement in photostability at the donor RFPs' wavelength (λex = 561 nm; mApple: λem = 590 nm; mCherry: λem = 610 nm) with increased FRET efficiency. To further validate the general applicability of our FRET-assisted strategy, we extended our experiments to include a different FRET pair: sfGFP as the donor and o-rhodamine as the acceptor (Fig. S7,† FRET efficiency is 58.1%). The results showed that sfGFP coupled with O-rhodamine exhibited a significant enhancement in the photostability of SIM imaging under continuous illumination (sfGFP t1/2 = 280.7 s; GH-L t1/2 = 427.1 s). These results validate the effectiveness and versatility of FRET-assisted strategies in enhancing the photostability of donor fluorophores.Furthermore, we have conducted a detailed analysis of the relationship between FRET efficiency and photobleaching dynamics. We confirmed the effect of FRET on triplet state conversion of RFP where the FRET pairs with higher efficiency had lower ROS yields, which was precisely due to the competition between FRET and the photobleaching-related ISC process. We also proposed another possible mechanism involving distance-dependent reactive oxygen species. These findings provide new insights into previous studies through ROS content detection.
Finally, we found CH-1 protein displayed higher FRET efficiency, and exhibited approximately 6-fold enhancement in photostability compared to wild-type mCherry. Leveraging its superior photostability, CH-1 was applied to long-term SIM imaging of mitochondrial dynamics. We employed CH-1 (labeled with mitochondria) in conjunction with EGFP-Sec61β (labeled with ER) and LysoTracker dye (labeled with lysosomes) to examine dynamic processes of mitochondrial fission mediated by the ER and lysosomes in live cells. As anticipated, CH-1 demonstrated exceptionally high photostability compared to EGFP and LysoTracker dyes, facilitating clear tracking of multiple mitochondrial fission events influenced by lysosomes/ER. Furthermore, we observed 91 mitochondrial fission events during 30 minutes of continuous super-resolution tracking. Long-term SIM imaging with these photostable RFPs offers insights into the intricacies and synergies within cells, contributing to a deeper understanding of protein interactions and their impact on cellular physiology and adaptability.
Data availability
All experimental procedures and additional figures are available in the ESI.†Author contributions
X. Z. expressed the proteins, measured the spectrum, did the cell imaging, and wrote the manuscript. L. M. designed experiments, analyzed the data, and wrote the manuscript. W. Z. synthesized the probe. Y. R. cultured the cells. X. W. measured the spectrum. G. W. examined the optical properties. P. B. synthesized the probe. Q. Q. synthesized the probe and examined the optical properties. Z. X. conceived the study and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (22225806, 22078314, 22278394, 22378385) and Dalian Institute of Chemical Physics (DICPI202142, DICPI202436).Notes and references
- A. Miyawaki, D. M. Shcherbakova and V. V. Verkhusha, Curr. Opin. Struct. Biol., 2012, 22, 679–688 CrossRef CAS.
- D. M. Shcherbakova, O. M. Subach and V. V. Verkhusha, Angew. Chem., Int. Ed., 2012, 51, 10724–10738 CrossRef CAS.
- D. Shcherbo, E. M. Merzlyak, T. V. Chepurnykh, A. F. Fradkov, G. V. Ermakova, E. A. Solovieva, K. A. Lukyanov, E. A. Bogdanova, A. G. Zaraisky, S. Lukyanov and D. M. Chudakov, Nat. Methods, 2007, 4, 741–746 CrossRef CAS PubMed.
- F. Montecinos-Franjola, J. Y. Lin and E. A. Rodriguez, Biochem. Soc. Trans., 2020, 48, 2657–2667 CrossRef CAS.
- K. D. Piatkevich, E. N. Efremenko, V. V. Verkhusha and S. D. Varfolomeev, Russ. Chem. Rev., 2010, 79, 243–258 CrossRef CAS.
- A. Cambré and A. Aertsen, Microbiol. Mol. Biol. Rev., 2020, 84, e00008–e00020 CrossRef.
- M. Fernández-Suárez and A. Y. Ting, Nat. Rev. Mol. Cell Biol., 2008, 9, 929–943 CrossRef.
- A. V. Mamontova, A. P. Grigoryev, A. S. Tsarkova, K. A. Lukyanov and A. M. Bogdanov, Russ. J. Bioorg. Chem., 2017, 43, 625–633 CrossRef CAS.
- J. O. Onukwufor, A. J. Trewin, T. M. Baran, A. Almast, T. H. Foster and A. P. Wojtovich, Free Radical Biol. Med., 2020, 147, 1–7 CrossRef CAS PubMed.
- H. Feng, Q. Zhao, B. Zhang, H. Hu, M. Liu, K. Wu, X. Li, X. Zhang, L. Zhang and Y. Liu, Angew. Chem., Int. Ed., 2023, 62, e202215215 CrossRef CAS PubMed.
- R. B. Vegh, K. B. Bravaya, D. A. Bloch, A. S. Bommarius, L. M. Tolbert, M. Verkhovsky, A. I. Krylov and K. M. Solntsev, J. Phys. Chem. B, 2014, 118, 4527–4534 CrossRef CAS.
- A. Roy, P. Carpentier, D. Bourgeois and M. Field, Photochem. Photobiol. Sci., 2010, 9, 1342–1350 CrossRef CAS PubMed.
- N. C. Shaner, M. Z. Lin, M. R. McKeown, P. A. Steinbach, K. L. Hazelwood, M. W. Davidson and R. Y. Tsien, Nat. Methods, 2008, 5, 545–551 CrossRef CAS.
- A. Acharya, A. M. Bogdanov, B. L. Grigorenko, K. B. Bravaya, A. V. Nemukhin, K. A. Lukyanov and A. I. Krylov, Chem. Rev., 2017, 117, 758–795 CrossRef CAS PubMed.
- N. C. Shaner, R. E. Campbell, P. A. Steinbach, B. N. G. Giepmans, A. E. Palmer and R. Y. Tsien, Nat. Biotechnol., 2004, 22, 1567–1572 CrossRef CAS.
- H. Ren, B. Yang, C. Ma, Y. S. Hu, P. G. Wang and L. Wang, ACS Chem. Biol., 2016, 11, 2679–2684 CrossRef CAS.
- M. G. Eason, A. M. Damry and R. A. Chica, Curr. Opin. Struct. Biol., 2017, 45, 91–99 CrossRef CAS.
- V. Marx, Nat. Methods, 2015, 12, 187–190 CrossRef CAS.
- J. Kwon, M. S. Elgawish and S. Shim, Adv. Sci., 2022, 9, 2101817 CrossRef.
- A. P. Demchenko, Methods Appl. Fluoresc., 2020, 8, 022001 CrossRef CAS PubMed.
- A. M. Bogdanov, E. I. Kudryavtseva and K. A. Lukyanov, PLoS One, 2012, 7, e53004 CrossRef CAS PubMed.
- A. M. Bogdanov, E. A. Bogdanova, D. M. Chudakov, T. V. Gorodnicheva, S. Lukyanov and K. A. Lukyanov, Nat. Methods, 2009, 6, 859–860 CrossRef CAS PubMed.
- A. V. Mamontova, A. M. Bogdanov and K. A. Lukyanov, BioTechniques, 2015, 58, 258–261 CrossRef CAS PubMed.
- S. S. Henrikus, K. Tassis, L. Zhang, J. H. M. Van Der Velde, C. Gebhradt, A. Herrmann, G. Jung and T. Cordes, ChemBioChem, 2021, 22, 3283–3291 CrossRef CAS PubMed.
- G. Donnert, C. Eggeling and S. W. Hell, Nat. Methods, 2007, 4, 81–86 CrossRef CAS PubMed.
- L. Ludvikova, E. Simon, M. Deygas, T. Panier, M.-A. Plamont, J. Ollion, A. Tebo, M. Piel, L. Jullien, L. Robert, T. Le Saux and A. Espagne, Nat. Biotechnol., 2023, 1–5 Search PubMed.
- Z. Wang, Y. Xie, K. Xu, J. Zhao and K. D. Glusac, J. Phys. Chem. A, 2015, 119, 6791–6806 CrossRef CAS PubMed.
- J. Zhao, K. Xu, W. Yang, Z. Wang and F. Zhong, Chem. Soc. Rev., 2015, 44, 8904–8939 RSC.
- K. Xu, Y. Xie, X. Cui, J. Zhao and K. D. Glusac, J. Phys. Chem. B, 2015, 119, 4175–4187 CrossRef CAS PubMed.
- K. Sreenath, Z. Yuan, J. R. Allen, M. W. Davidson and L. Zhu, Chemistry, 2015, 21, 867–874 CrossRef.
- S. M. Burrows, P. Patel and D. Pappas, Appl. Spectrosc., 2009, 63, 709–715 CrossRef CAS.
- Y. Tian and D. Pappas, Appl. Spectrosc., 2011, 65, 991–995 CrossRef CAS.
- F. Pagels, A. C. Guedes, H. M. Amaro, A. Kijjoa and V. Vasconcelos, Biotechnol. Adv., 2019, 37, 422–443 CrossRef CAS.
- H. Chen, J. Deng, L. Li, Z. Liu, S. Sun and P. Xiong, Mar. Drugs, 2023, 21, 572 CrossRef CAS PubMed.
- S. Basu, L.-M. Needham, D. Lando, E. J. R. Taylor, K. J. Wohlfahrt, D. Shah, W. Boucher, Y. L. Tan, L. E. Bates, O. Tkachenko, J. Cramard, B. C. Lagerholm, C. Eggeling, B. Hendrich, D. Klenerman, S. F. Lee and E. D. Laue, Nat. Commun., 2018, 9, 2520 CrossRef PubMed.
- Y. Zhang, W. Zhou, N. Xu, G. Wang, J. Li, K. An, W. Jiang, X. Zhou, Q. Qiao, X. Jiang and Z. Xu, Chin. Chem. Lett., 2023, 34, 107472 CrossRef CAS.
- J. Pan, W. Lin, F. Bao, Q. Qiao, G. Zhang, Y. Lu and Z. Xu, Chin. Chem. Lett., 2023, 34, 107519 CrossRef CAS.
- X. Zhang, L. Wang, N. Li and Y. Xiao, Chin. Chem. Lett., 2021, 32, 2395–2399 CrossRef CAS.
- G. V. Los, L. P. Encell, M. G. McDougall, D. D. Hartzell, N. Karassina, C. Zimprich, M. G. Wood, R. Learish, R. F. Ohana, M. Urh, D. Simpson, J. Mendez, K. Zimmerman, P. Otto, G. Vidugiris, J. Zhu, A. Darzins, D. H. Klaubert, R. F. Bulleit and K. V. Wood, ACS Chem. Biol., 2008, 3, 373–382 CrossRef CAS.
- A. Keppler, S. Gendreizig, T. Gronemeyer, H. Pick, H. Vogel and K. Johnsson, Nat. Biotechnol., 2003, 21, 86–89 CrossRef CAS PubMed.
- J. Li, Q. Qiao, Y. Ruan, N. Xu, W. Zhou, G. Zhang, J. Yuan and Z. Xu, Chin. Chem. Lett., 2023, 34, 108266 CrossRef CAS.
- B. Bajar, E. Wang, S. Zhang, M. Lin and J. Chu, Sensors, 2016, 16, 1488 CrossRef.
- D. Si, Q. Li, Y. Bao, J. Zhang and L. Wang, Angew. Chem., Int. Ed., 2023, 62, e202307641 CrossRef CAS PubMed.
- J. B. Grimm, B. P. English, J. Chen, J. P. Slaughter, Z. Zhang, A. Revyakin, R. Patel, J. J. Macklin, D. Normanno, R. H. Singer, T. Lionnet and L. D. Lavis, Nat. Methods, 2015, 12, 244–250 CrossRef CAS.
- Y. Xiao and X. Qian, Coord. Chem. Rev., 2020, 423, 213513 CrossRef CAS.
- C. Jing and V. W. Cornish, Acc. Chem. Res., 2011, 44, 784–792 CrossRef CAS.
- L. Xue, I. A. Karpenko, J. Hiblot and K. Johnsson, Nat. Chem. Biol., 2015, 11, 917–923 CrossRef CAS.
- G. Crivat and J. W. Taraska, Trends Biotechnol., 2012, 30, 8–16 CrossRef CAS PubMed.
- X. Liu, Q. Qiao, W. Tian, W. Liu, J. Chen, M. J. Lang and Z. Xu, J. Am. Chem. Soc., 2016, 138, 6960–6963 CrossRef CAS PubMed.
- B. Miljevic, F. Hedayat, S. Stevanovic, K. E. Fairfull-Smith, S. E. Bottle and Z. D. Ristovski, Aerosol Sci. Technol., 2014, 48, 1276–1284 CrossRef CAS.
- J. Zhou, E. A. Bruns, P. Zotter, G. Stefenelli, A. S. H. Prévôt, U. Baltensperger, I. El-Haddad and J. Dommen, Atmos. Meas. Tech., 2018, 11, 65–80 CrossRef CAS.
- M. Ethirajan, Y. Chen, P. Joshi and R. K. Pandey, Chem. Soc. Rev., 2011, 40, 340–362 RSC.
- H. M. McBride, M. Neuspiel and S. Wasiak, Curr. Biol., 2006, 16, R551–R560 CrossRef CAS.
- N. Pfanner, B. Warscheid and N. Wiedemann, Nat. Rev. Mol. Cell Biol., 2019, 20, 267–284 CrossRef CAS PubMed.
- S. Hoppins, L. Lackner and J. Nunnari, Annu. Rev. Biochem., 2007, 76, 751–780 CrossRef CAS PubMed.
- J. Chen, W. Liu, X. Fang, Q. Qiao and Z. Xu, Chin. Chem. Lett., 2022, 33, 5042–5046 CrossRef CAS.
- D. C. Chan, Annu. Rev. Genet., 2012, 46, 265–287 CrossRef CAS.
- S. C. Lewis, L. F. Uchiyama and J. Nunnari, Science, 2016, 353, aaf5549 CrossRef.
- Y. C. Wong, D. Ysselstein and D. Krainc, Nature, 2018, 554, 382–386 CrossRef CAS.
- Y. C. Wong, W. Peng and D. Krainc, Dev. Cell, 2019, 50, 339–354 CrossRef CAS.
- J. Qin, Y. Guo, B. Xue, P. Shi, Y. Chen, Q. P. Su, H. Hao, S. Zhao, C. Wu, L. Yu, D. Li and Y. Sun, Nat. Commun., 2020, 11, 4471 CrossRef CAS PubMed.
- M. Boutry and P. K. Kim, Nat. Commun., 2021, 12, 5354 CrossRef CAS PubMed.
- X. Li, J. Zheng, W. Liu, Q. Qiao, J. Chen, W. Zhou and Z. Xu, Chin. Chem. Lett., 2020, 31, 2937–2940 CrossRef CAS.
- X. Yang, Z. Yang, Z. Wu, Y. He, C. Shan, P. Chai, C. Ma, M. Tian, J. Teng, D. Jin, W. Yan, P. Das, J. Qu and P. Xi, Nat. Commun., 2020, 11, 3699 CrossRef CAS PubMed.
- W. Liu, J. Chen, Q. Qiao, X. Liu and Z. Xu, Chin. Chem. Lett., 2022, 33, 4943–4947 CrossRef CAS.
- W. Peng, Y. C. Wong and D. Krainc, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 19266–19275 CrossRef CAS.
- C. C. Williams, C. H. Jan and J. S. Weissman, Science, 2014, 346, 748–751 CrossRef CAS.
- S. Nagashima, L.-C. Tábara, L. Tilokani, V. Paupe, H. Anand, J. H. Pogson, R. Zunino, H. M. McBride and J. Prudent, Science, 2020, 367, 1366–1371 CrossRef CAS PubMed.
- P. J. Cranfill, B. R. Sell, M. A. Baird, J. R. Allen, Z. Lavagnino, H. M. de Gruiter, G.-J. Kremers, M. W. Davidson, A. Ustione and D. W. Piston, Nat. Methods, 2016, 13, 557–562 CrossRef CAS PubMed.
- M. J. Phillips and G. K. Voeltz, Nat. Rev. Mol. Cell Biol., 2016, 17, 69–82 CrossRef CAS PubMed.
- F. Guerra and C. Bucci, Cells, 2016, 5, 34 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc02442k |
| This journal is © The Royal Society of Chemistry 2025 |