
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA general transition metal binding aptamer following the Irving–Williams series†
Jin Wangab,
Yunus A. Kaiyumc,
Sihan Wangb,
Xiangmei Li a,
Hongtao Leia,
Philip E. Johnsonc and
Juewen Liu*b
a,
Hongtao Leia,
Philip E. Johnsonc and
Juewen Liu*b
aGuangdong Provincial Key Laboratory of Food Quality and Safety, College of Food Science, South China Agricultural University, Guangzhou 510642, China
bDepartment of Chemistry, Waterloo Institute for Nanotechnology, University of Waterloo, Waterloo, Ontario N2L 3G1, Canada. E-mail: liujw@uwaterloo.ca
cDepartment of Chemistry, York University, 4700 Keele Street, Toronto, Ontario M3J 1P3, Canada
First published on 24th June 2025
Abstract
Using both Co2+ and Ni2+ as target metal ions, aptamer selections were carried out by immobilization of a DNA library via hybridization. A set of aptamers were obtained although they all showed binding to both metal ions. The Co-1 aptamer was highly enriched in both selections and it is a general metal binding aptamer with binding affinity following the Irving–Williams series: Mn2+ < Fe2+ < Co2+ < Ni2+ < Cu2+ > Zn2+. Using NMR spectroscopy, Co-1 forms new base pairs with a relatively large structural change upon binding to Zn2+, whereas a previously reported Zn-1 aptamer shows less conformational change and rigidifies upon Zn2+ binding. The induced-fitting of the Co-1 aptamer and the lock-and-key binding of the Zn-1 aptamer may explain the general metal binding of the former. Using isothermal titration calorimetry, Co-1 binding Co2+ was an enthalpy-driven event and the Kd was determined to be 2.7 μM, whereas the Kd from the strand-displacement assay was 76 nM. The Co-1 aptamer is the first reported aptamer to follow the Irving–Williams series, although this series is very prevalent in proteins. This aptamer can thus serve as a model system for understanding metal binding by DNA and can also be a general first-row transition metal sensing element.
Introduction
The first-row transition metal ions are particularly important in biology and many of them are essential cofactors in metalloenzymes.1 In most metalloproteins, the affinity of metal binding follows the Irving–Williams series: Mn2+ < Fe2+ < Co2+ < Ni2+ < Cu2+ > Zn2+.2 This high affinity for copper has led to the so-called “copper tyranny”, and organisms have to employ complex non-equilibrium strategies to keep other metal ions in their binding sites.3Studies in metal binding sites in nucleic acids never seemed to have encountered this “copper tyranny” problem. In the early days, metal binding studies were performed in ribozymes and later in DNAzymes, since they in general require divalent metal ions as cofactors to achieve catalytic activities.4–8 Later, different metal ions were intentionally used to select for DNAzymes that can achieve metal-specific activities, which are useful as biosensors for metal detection.9–12 Cu2+ is probably one of the poorest metal ions in promoting the RNA cleavage reaction,13 although in some modified DNAzymes, it showed a high activity.14 DNAzymes specific for Mn2+,15 Fe2+/Fe3+,11 Ni2+,16 Cu2+,17 and Zn2+ ions18,19 have been reported, although some contained chemical modifications in DNAzymes to achieve the needed specificity and activity. The activity of many ribozymes was found to correlate with affinity to phosphate instead of following the Irving–Williams series.8 These metal ions, however, do not need to stably bind to ribozymes or DNAzymes to exert their catalytic activities. They only need to transiently bind to the transition state and neutralize the negative charges in the scissile phosphate in most cases.7,13,20
To study metal binding sites in DNA, aptamers are more relevant. Aptamers are single-stranded nucleic acids that can selectively bind to target molecules.4,21,22 The isolation of metal-specific aptamers has been carried out for a long time with early examples in the 1990s. Back then, the selection was achieved by the immobilization of metal ions such as Ni2+ and Zn2+ on a chelation bead and the focus was on RNA aptamer selection.23,24 Such aptamers were rarely used for applications due to the poor stability of RNA. Later, aptamer selection by immobilization of a DNA library (so called capture-SELEX) had been developed, which allows the use of free metal ions as targets.25,26 Ellington's group first used this method to isolate an aptamer for Zn2+, although this aptamer contained a fluorophore.27 Soh and coworkers used an interesting strategy to isolate aptamers for Cu2+ and Hg2+.28 Jahan et al. aimed to select aptamers for the PD-L1 protein by immobilizing the PD-L1 protein on nickel nitrilotriacetic acid (Ni–NTA) beads. Interestingly, due to the presence of Ni2+ on the beads, the selected aptamer showed binding to Ni2+ instead of the PD-L1 protein.29 Capture-SELEX has also been used to obtain aptamers for many transition metal ions including Cd2+,30 Pb2+,31 Zn2+,32 and Cu2+.33 In each case, the aptamers were able to bind to their target metal ion with the highest affinity. So far, Irving–Williams series was only seen in a duplex thermal stability assay using DNA containing imidazole-derived nucleobases.34 Thus, it seems that the Irving–Williams series has yet to be observed in unmodified nucleic acids.
Thus, an interesting question is whether nucleic acids are capable of binding metal ions following the Irving–Williams series. The chemical diversity for interacting with metal ligands in nucleic acids is much less compared to that in proteins.35 For example, nucleic acids lack thiol ligands that have strong affinity with copper. Co2+ and Ni2+ are interesting metal ions and they differ from Cu2+ and Zn2+ by having slower ligand exchange rates.36 In this study, we performed two capture-SELEX experiments using Co2+ and Ni2+ as target metal ions. Interestingly, the most abundant aptamers followed the Irving–Williams series, with Cu2+ binding being much stronger than that for Co2+ or Ni2+. Yet, such a sequence was not observed during our previous Cu2+ selection.33 By using NMR and isothermal titration calorimetry (ITC), we attempted to further compare this general metal aptamer to some specific aptamers to gain fundamental insights.
Materials and methods
Chemicals and DNA are described in the ESI.†SELEX
A library-immobilized selection strategy was employed as previously described.33,37 CoCl2·6H2O and NiCl2·6H2O were dissolved in 100 mM Milli-Q water, and diluted with SELEX buffer for selection. For Co2+ selection, the concentrations were set at 10 μM for rounds 1–14 and reduced to 2 μM for rounds 15–17. Similarly, for Ni2+ selection, 10 μM was used for rounds 1–13, followed by 2 μM for rounds 14–17. The last round's PCR products were sequenced at the McMaster University Genome Facility, and the sequencing results were analyzed using Geneious Prime software (Auckland, New Zealand).Thioflavin T (ThT)-based binding assay
The fluorescence spectra of the ThT-based assay were measured using a Varian Eclipse fluorescence spectrophotometer (CA, USA). The assay contained 0.5 μM aptamer and 1 μM ThT in 500 μL of selection buffer. The mixture was transferred to a quartz cuvette. Metal ions were added or titrated until equilibrium was reached. Fluorescence was excited at 440 nm, and the emission spectra were recorded between 460 and 520 nm. Fluorescence at 490 nm served as the basis for calculations. The initial fluorescence of the aptamer/ThT mixture is denoted as F0, and the value after metal ion addition is F. The ratio F/F0 was plotted, and the Kd was determined using the equation y = y0 + aKd/(Kd + [M2+]), where a represents the maximal fluorescence signal change at saturation.Fluorescence strand-displacement assay
Fluorescence strand-displacement assays were performed using a Tecan Spark F200 microplate reader (Switzerland) in a 96-well microplate. The excitation and emission wavelengths were set at 485 nm and 520 nm, respectively. To prepare the displacement sensor, 1 μL of FAM-labeled Co-1 aptamer (100 μM), 2 μL of quencher-cDNA (100 μM), and 97 μL of SELEX buffer were mixed, annealed at 85 °C for 1 min, and gradually cooled to room temperature for 30 min, followed by cooling at 4 °C for 30 min and then at −20 °C for 30 min. For the test, 2 μL of annealed aptamer was added to 98 μL SELEX buffer, and baseline fluorescence was recorded for 5 min. Subsequently, 2 μL of Co2+ or other metal ions at different concentrations were introduced, and fluorescence was monitored for another 10 min. Fluorescence changes were calculated using the formula (F − F0)/F0, where F0 represents the initial fluorescence before metal ion addition and F corresponds to the fluorescence after adding metal ions.NMR
NMR experiments on the DNA aptamer sample were performed using a 600 MHz Bruker Avance spectrometer. All NMR spectra were acquired with an aptamer concentration of 0.5 mM in an aqueous solution with 100 mM NaCl and 2 mM MgCl2 in 10% D2O at 5 °C. Water suppression was achieved using excitation sculpting.38Circular dichroism (CD) spectroscopy
CD spectra were recorded using a Chirascan spectropolarimeter (Applied Photophysics Ltd, Surrey, UK) with procedures reported in previous experiments.39 Aptamers (5 μM) were prepared in 10 mM MES buffer (pH 6.0) containing 100 mM NaCl and 2 mM MgCl2. Measurements were conducted before and after adding 20 μM metal ions. The wavelength range was from 230 to 320 nm with a bandwidth of 4 nm, and a 1 cm path-length cuvette was used.ITC
Isothermal titration calorimetry (ITC) was conducted with a MicroCal iTC200 instrument (Worcestershire, UK) to test binding by the Co-1 aptamer. The aptamer (20 μM) in the SELEX buffer was annealed by heating to 95 °C for 5 min and cooling naturally over 30 min. Subsequently, both the aptamer and ions (333 μM in the SELEX buffer) were degassed for 5 min before loading. Then, 300 μL of the aptamer was loaded into a cell chamber and a syringe was filled with 75 μL of metronidazole. After an initial injection of 0.5 μL, subsequent injections of the target were titrated into the cell at a rate of 2.0 μL per injection over a duration of 5.0 seconds, totaling 20 injections at 25 °C. Each titration was spaced 240 seconds apart. The binding constant was determined by fitting the titration curve to a one-site binding model using Origin software.Results and discussion
Selection of DNA aptamers for Co2+ and Ni2+
The selection of DNA aptamers for Co2+ and Ni2+ was carried out using a library containing a 30-nucleotide (nt) random region as described previously (Table S1†).33 This library was immobilized on streptavidin-coated beads via hybridization to a fixed PCR primer binding region. After adding target metal ions, some aptamer sequences may bind the metal ions and be released from the beads for PCR amplification. Since we aimed to achieve high affinity aptamers for Co2+ and Ni2+, a low target metal ion concentration of 10 μM was used in the initial rounds.40–42 In the final three or four rounds, the metal ion concentration was further decreased to 2 μM (see Table S2† for the selection conditions). The round 17 libraries were deep sequenced and the sequences were highly converged suggesting successful selections.Fig. 1A shows the alignment of the top 10 most abundant Co2+ sequences. The predominant sequence in the Co2+ selection named Co-1 has an abundance of 10.3% and its mFold predicted secondary structure is shown in Fig. 1B. Co-1 is predicted to fold into a three-way junction linked by a few nucleotides, and these linking nucleotides (in red) are highly conserved. Therefore, it is likely that Co2+ binding takes place in the middle pocket formed by the linking nucleotides.
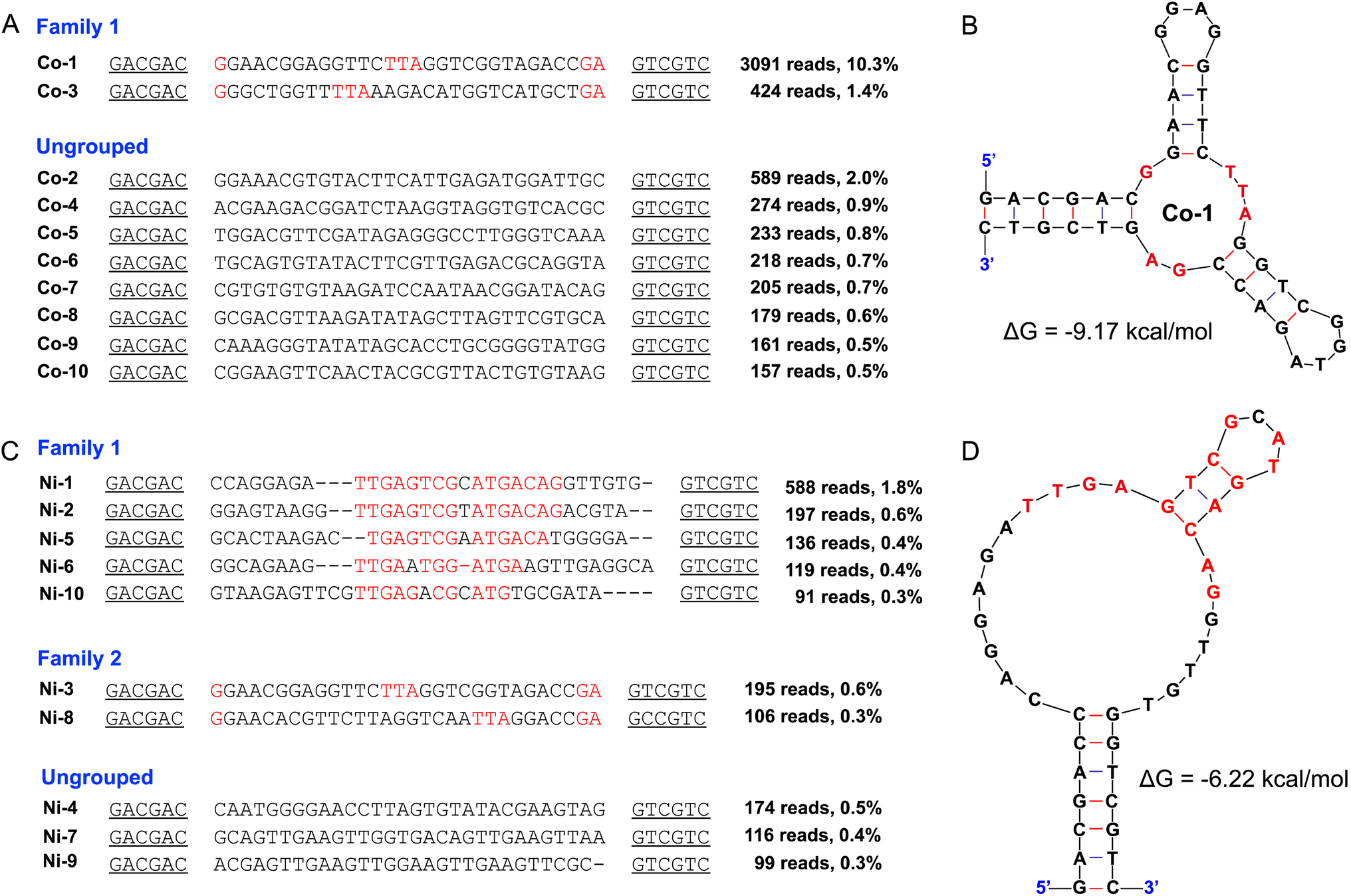 | ||
| Fig. 1 Alignment of the top 10 sequences for (A) the Co2+ selection and (C) the Ni2+ selection. The nucleotides in the primer-binding regions are underlined, and the conserved nucleotides are marked in red or blue. The mFold predicted secondary structure of (B) Co-1 (ΔG = −9.17 kcal mol−1) and (D) Ni-1 (ΔG = −6.22 kcal mol−1) aptamers at 25 °C, 100 mM Na+ and 2 mM Mg2+. | ||
For the Ni2+ sequencing results (Fig. 1C), the Ni-3 sequence is identical to that of Co-1, suggesting that this sequence can bind to both metals. Ni-1 is a representative sequence of another family. We also folded Ni-1 as shown in Fig. 1D. Based on the family assignment, Co-1 (same as Ni-3), Co-2, Ni-1, and Ni-4 were selected for further testing. The alignment of the top 20 sequences from Co2+ and Ni2+ selections is presented in Fig. S1 and S2.†
Aptamer binding to Co2+ and Ni2+ using ThT fluorescence spectroscopy
We first used Thioflavin T (ThT) fluorescence spectroscopy as a rapid assay to evaluate binding of these selected aptamers. ThT, as a free dye, exhibits almost no fluorescence. It can bind to any DNA, including aptamers, leading to fluorescence enhancement. When a DNA aptamer binds to its target, ThT is displaced, resulting in a fluorescence decrease.26,43 The ThT assay has been successfully applied to study aptamer binding to rare earth ions,37 Cu2+,33 and other targets.26,32 We first collected the fluorescence spectra of the Co-1 aptamer/ThT mixture with increasing concentrations of Co2+ and Ni2+. Both metal ions induced over a 50% fluorescence drop at 2 μM metal concentration (Fig. 2A and C). Based on the titration results, the Co-1 aptamer has a Kd of 731 nM for Co2+ and 467 nM for Ni2+ (Fig. 2B and D). Ni-4 exhibited a Kd of 901 nM for Co2+ and 257 nM for Ni2+ in the same titration (Fig. S3†). The Co-2 and Ni-1 aptamers, in contrast, required higher Co2+ and Ni2+ concentrations to show a fluorescence decrease (Fig. S4†). Thus, Co-1 and Ni-4 are higher affinity aptamers. Although the binding affinities of the two were comparable, Co-1 demonstrated the highest enrichment, and it was found in both Co2+ and Ni2+ selections. Therefore, Co-1 was chosen for further studies. As a control, a mutant named Co-1a was designed by swapping the positions of the conserved nucleotides in the loops (Fig. S5†). Titration experiments with Co2+ and Ni2+ revealed no significant fluorescence decrease (Fig. 2B and D, blue lines) for Co-1a, indicating that Co-1 exhibits specific binding for Co2+ and Ni2+.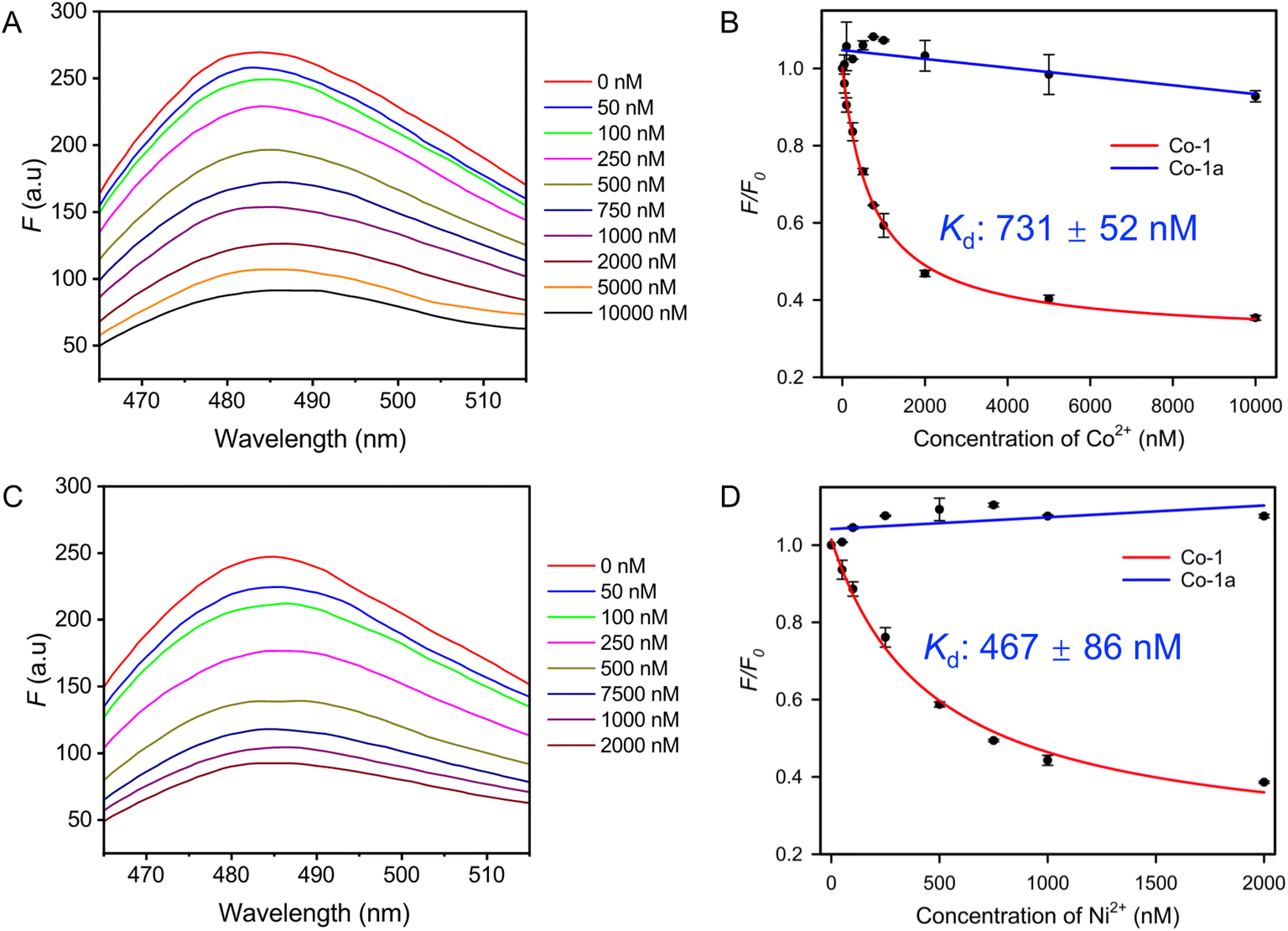 | ||
| Fig. 2 The fluorescence spectra of the ThT/Co-1 mixture in the presence of different concentrations of (A) Co2+ and (C) Ni2+ in the SELEX buffer. Titration curves of (B) Cu2+ and (D) Ni2+ into the Co-1 aptamer and a nonbinding mutant Co-1a (ThT: 0.5 μM, ThT: 1 μM). | ||
DNA strand-displacement binding assays
While ThT fluorescence provides a cost-effective assay for screening a large number of sequences, it is a signal-off assay and suffers from nonspecific fluorescence quenching by metal ions. Thus, we further used the strand-displacement assay (Fig. 3A).44 The Co-1 aptamer was extended on the 5′ end by five nucleotides and it was hybridized to a 12-mer quencher-labeled cDNA (Table S1†). This hybridization complex has quenched fluorescence. In the presence of a target metal ion, the binding of the aptamer can release the quencher-labeled strand, resulting in a fluorescence increase.37,44–46 Using this assay, we tested Co2+ (Fig. 3B) and Ni2+ (Fig. S6C†) by following the kinetics of the sensor response at different metal concentrations. For both metals, a modest fluorescence increase occurred at a metal concentration of 100 nM and a rapid fluorescence increase to the saturated value was achieved with 10 μM of the metals. Indeed, these two metal ions responded very similarly to this sensor. | ||
| Fig. 3 (A) Scheme of the strand displacement assay for the detection of Co2+. (B) Calibration curves of the sensor for Co2+. (C) Kinetics traces of the FAM Co-1 sensor in the presence of various concentrations of Co2+ in SELEX buffer. Inset: the linear response at 1000 nM. (D) The response of Co-1 to various metal ions. After confirming the response range of Co-1 to Co2+ and Ni2+, we further tested the response of Co-1 to other metal ions. Co2+ and Ni2+ produced significant fluorescence enhancement even at 1 μM, whereas Cu2+ induced a noticeable increase at as low as 0.1 μM. In contrast, Fe2+, Zn2+, and Cd2+ showed a pronounced fluorescence increase at 10 μM. At concentrations of 10 μM and even 100 μM, a significant increase in fluorescence was observed with Mn2+, while no noticeable changes were detected with Ca2+, Sc3+, Cr3+, Sr2+, Y3+, Ba2+, Hg2+, and Pb2+. Therefore, this aptamer Co-1 has binding to first-row transition metal ions along with Cd2+. | ||
We then performed careful titrations for these metal ions. The titration curve for Co2+ is shown in Fig. 3C and the rest are shown in Fig. S6 and S7.† The observed Kd values using the strand displacement assay are apparent Kd due to the presence of the quencher-labeled strand acting as a competitor.47–49 The apparent Kd values were determined to be 178 μM for Mn2+, 7.8 μM for Fe2+, 1260 nM for Co2+, 607 nM for Ni2+, 139 nM for Cu2+, 5.9 μM for Zn2+, and 4.9 μM for Cd2+. We then titrated the quencher-labeled strand with the FAM-aptamer strand to calculate the true Kd of the aptamer (see Fig. S8† for details). The corresponding true Kd values were 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 680 nM for Mn2+, 468 nM for Fe2+, 76 nM for Co2+, 37 nM for Ni2+, 8 nM for Cu2+, 352 nM for Zn2+, and 295 nM for Cd2+.
680 nM for Mn2+, 468 nM for Fe2+, 76 nM for Co2+, 37 nM for Ni2+, 8 nM for Cu2+, 352 nM for Zn2+, and 295 nM for Cd2+.
The Co-1 aptamer follows the Irving–Williams series
In 1953, H. Irving and R. J. P. Williams proposed the Irving–Williams series, which describes the binding strength of ligands with divalent 3d transition metal ions. The binding strength increases with the atomic number of the metal, following the trend Mn2+ < Fe2+ < Co2+ < Ni2+ < Cu2+ > Zn2+.2 Fig. 4A plots the true Kd of these metal ions as a function of their positions in the periodic table from Mn2+ to Zn2+, and it matches perfectly with this series. Thus, we have obtained the first DNA aptamer that follows the Irving–Williams series. The Irving–Williams series can be explained as follows: from Mn2+ to Ni2+, the decreasing ionic radius leads to higher charge density, resulting in increased complex stability. Although Cu2+ has a slightly larger radius than Ni2+, the Jahn–Teller effect provides additional stabilization for its octahedral complexes. In contrast, Zn2+ has a similar radius to Cu2+ but possesses a filled d10 configuration, which limits effective orbital overlap with ligands and leads to lower complex stability. | ||
| Fig. 4 (A) The relationship between the Kd of Co-1 for first-row divalent transition metals and their (A) atomic numbers and (B) radii. The predicted secondary structures and sequences of the (B) Cu-133 and (C) Zn-1 aptamers.32 The conserved nucleotides on both sides of the loop region are marked in red or blue. | ||
Metal binding studies using NMR
To investigate the possible conformational changes taking place in the DNA aptamer with a ligand, we then performed 1H-NMR titrations monitoring the imino region (Fig. 5).50 For this purpose, we chose to use Zn2+ as Zn2+ is not paramagnetic and we also have previously reported a Zn2+ aptamer that is specific for Zn2+ (no binding to Co2+ or Ni2+).32 Based on the NMR results, the free Co-1 aptamer has approximately 9 imino proton resonances attributable to base pairs involving G and T nucleotides, approximately 6 of which are from Watson–Crick pairs located between 12 and 14 ppm. Approximately 3 are from non-Watson–Crick pairs located between 10 and 11 ppm (Fig. 5A). By examining the mFold predicted secondary structure of Co-1, 14 Watson–Crick base pairs were predicted to form. Therefore, not all these predicted base pairs were stably formed in the absence of Zn2+. When Zn2+ was titrated, more peaks emerged in both regions, and a total of about 17–18 base pairs were formed. Thus, additional Watson–Crick and non-Watson–Crick pairs form upon binding to Zn2+ indicating that this aptamer undergoes metal-induced folding.51 Assuming that the binding site in Co-1 is at the same location for all metal ions, the binding of Co2+ and Ni2+ should behave similarly and result in metal-induced structure formation. | ||
| Fig. 5 1D 1H-NMR of the imino proton region of the (A) Co-1 and (B) Zn-1 aptamers binding Zn2+ ions. Data were acquired at 5 °C in 100 mM NaCl, 2 mM MgCl2, 10% D2O. | ||
For comparison, we also performed NMR on the Zn-1 aptamer (Fig. 5B). Overall, as the number of peaks observed in the imino region remained similar before and after adding Zn2+ the amount of structural change upon Zn2+ binding is less for the Zn-1 aptamer. Due to the sharpening of the imino signals this aptamer may have a pre-formed binding pocket that is stabilized upon binding to Zn2+. This aptamer might have a higher metal binding selectivity due to this hypothesised pre-formed binding pocket. Without high resolution structures, this hypothesis is speculative.
Metal binding studies using CD spectroscopy
CD spectroscopy reflects the differential absorption of left and right circularly polarized light, and it can provide information regarding the secondary structure of aptamers.52,53 The Co-1 aptamer exhibited a characteristic positive peak at 278 nm and a negative peak at 244 nm (Fig. S9†). The positive peak is attributed to base stacking, while the negative peak indicates the helicity of the polynucleotide chain.54,55 Upon the addition of 20 μM transition metal ions, the intensity of the positive peak decreased significantly, while the negative peak also reduced slightly. Mn2+ did not cause much spectral change attributable to its low affinity to this aptamer. This result suggests that transition metal ions binding induced a substantial conformational rearrangement in the Co-1 aptamer, likely leading to partial disruption or reorganization of the base stacking. This observation is consistent with our NMR results, which showed the appearance of new imino proton resonances upon Zn2+ titration, indicating the formation of additional Watson–Crick and non-canonical base pairs and the overall folding of the aptamer into a more structured conformation. However, such Zn2+-induced folding appears to involve non-B-form structural motifs, resulting in a less regular base stacking pattern and hence a reduced CD signal at 278 nm.In contrast, the Zn-1 aptamer displayed spectral changes in an opposite way upon Zn2+ addition, with a slight increase in the intensity of the positive CD band at 278 nm (Fig. S10†), suggesting that the aptamer adopts a well-folded structure even in the absence of metal ions. The modest enhancement in base stacking upon Zn2+ binding indicates that the Zn-1 aptamer possesses a pre-organized binding pocket that is further stabilized by the metal ion. This interpretation is supported by the NMR results, where no significant changes were observed in the imino proton region after Zn2+ addition, confirming that Zn-1 undergoes only subtle conformational adjustments.
Metal binding studies using ITC
Finally, we characterized metal binding using ITC. Cu2+ and Zn2+ could not give good ITC results likely due to the hydrolysis of these metal ions in buffer. When injected into the sample chamber, the heat associated with diluting hydrolyzed metal species can dominate the signal. Therefore, we could only titrate Co2+ into the Co-1 aptamer (Fig. 6). A typical binding curve was observed with a ΔH of −16.4 kcal mol−1 and a ΔS of −29.6 cal mol−1 K−1 (TΔS = −8.8 kcal mol−1). Therefore, the binding of Co-1 to Co2+ ions was driven by enthalpy, which compensated for the penalty due to entropy loss. The enthalpy gain could be from the formation of new base pairs seen in NMR, along with the coordination of Co2+. | ||
| Fig. 6 Titration of 333 μM Co2+ into the 20 μM Co-1 aptamer. | ||
Selection outcomes
Since this Co-1 aptamer can bind Cu2+ even stronger than Co2+ (Fig. 4A), we then searched the library for previously reported Cu2+ selection.33 For the top 1000 sequences, we did not find the Co-1 aptamer. It is interesting to note that when we used Zn2+ or Cu2+ for the selection, we obtained highly specific aptamers for them. However, when we used Co2+ and Ni2+, we obtained a highly general aptamer. The secondary structures of the Cu-1 aptamer and Zn-1 aptamer are shown in Fig. 4B and C, respectively. They have quite similar secondary structures with conserved metal binding regions formed by the two loops. The Co-1 and Cu-1 aptamers have a similar affinity to Cu2+. The number of conserved nucleotides is much lower in Co-1, and this may explain its lower specificity and its ability to accommodate many divalent metal ions. Co-1 may only be able to capture the information of divalent transition metals (two charges and a certain size range). In contrast, the Cu-1 and Zn-1 aptamers have a larger binding pocket allowing them to form more complex binding structures that can better fit the orbitals of the metal ions. Although metal ions are not in general considered to have shapes, they do have different coordination geometries. Such information may be captured by the more complex aptamers, likely via inner sphere coordination.Based on numerous additional signals appearing in the NMR spectra of the Co-1 aptamer in the presence of Zn2+, new base pairs form in this aptamer upon metal binding. In contrast, for the Zn-1 aptamer, no new imino peaks appeared indicating that a small amount of few new structure forms. Instead, the set of low intensity imino signals in the free Zn-1 aptamer gives rise to a few strong signals indicating the appearance of a more defined structure in the metal-bound Zn-1 aptamer. Therefore, we hypothesize that the Co-1 aptamer has an induced-fit mechanism, and added metal ions induced the formation of the bound structure. This binding site follows the Irving–Williams series and thus the intrinsic properties of the metal ions dominated the stability of the binding. In contrast, the Zn-1 aptamer has a binding mechanism where the binding site rigidifies with metal binding, which may explain its better selectivity.
Conclusions
In this work, an interesting DNA aptamer named Co-1 was isolated using capture-SELEX for both Co2+ and Ni2+ as target metal ions. This is the first aptamer that shows a clear binding affinity trend following the Irving–Williams series. This series is quite common in proteins,56 and we demonstrated here that DNA can also have comparable metal binding sites. This Co-1 aptamer will be a useful model system to study metal binding by DNA aptamers. Having aptamers that are specific for a group of metal ions is also interesting. We previously reported a DNAzyme that can recognize all trivalent lanthanides,57 and all thiophilic metals.20 The 17E DNAzyme can use nearly all divalent metals for catalysis.13 This aptamer may also serve as a general first-row transition metal biosensor.Data availability
The data that support the findings of this study are available from the the Federated Research Data Repository, at https://doi.org/10.20383/103.01328.Author contributions
Jin Wang performed the aptamer selections, collected data, proposed key ideas, and drafted the initial manuscript. Yunus A. Kaiyum and Philip E. Johnson were responsible for the NMR experiments. Sihan Wang conducted the ITC experiments. Xiangmei Li and Hongtao Lei provided valuable suggestions during manuscript preparation and support for some CD experiments. Juewen Liu contributed to conceptualization, data analysis, manuscript revision, and funding acquisition. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding for this work was from the Natural Sciences and Engineering Research Council of Canada (NSERC). Jin Wang received a China Scholarship Council (CSC, 202308440518) scholarship to visit the University of Waterloo.References
- Y. Lu, N. Yeung, N. Sieracki and N. M. Marshall, Nature, 2009, 460, 855–862 CrossRef CAS.
- H. Irving and R. Williams, J. Chem. Soc., 1953, 3192–3210 RSC.
- D. Osman and N. J. Robinson, FEBS Lett., 2023, 597, 141–150 CrossRef CAS PubMed.
- W. Zhou, R. Saran and J. Liu, Chem. Rev., 2017, 117, 8272–8325 CrossRef CAS PubMed.
- R. J. Lake, Z. Yang, J. Zhang and Y. Lu, Acc. Chem. Res., 2019, 52, 3275–3286 CrossRef CAS PubMed.
- R. K. Sigel and A. M. Pyle, Chem. Rev., 2007, 107, 97–113 CrossRef CAS PubMed.
- W. L. Ward, K. Plakos and V. J. DeRose, Chem. Rev., 2014, 114, 4318–4342 CrossRef CAS PubMed.
- J. Schnabl and R. K. Sigel, Curr. Opin. Chem. Biol., 2010, 14, 269–275 CrossRef CAS.
- Y. Lu, Chem.–Eur. J., 2002, 8, 4588–4596 CrossRef CAS.
- J. Liu, A. K. Brown, X. Meng, D. M. Cropek, J. D. Istok, D. B. Watson and Y. Lu, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2056–2061 CrossRef CAS.
- Y. Wu, S.-F. Torabi, R. J. Lake, S. Hong, Z. Yu, P. Wu, Z. Yang, K. Nelson, W. Guo, G. T. Pawel, J. Van Stappen, X. Shao, L. M. Mirica and Y. Lu, Sci. Adv., 2023, 9, eade7622 CrossRef CAS.
- K. Hwang, Q. Mou, R. J. Lake, M. Xiong, B. Holland and Y. Lu, Inorg. Chem., 2019, 58, 13696–13708 CrossRef CAS PubMed.
- W. J. Moon, P.-J. J. Huang and J. Liu, Biochemistry, 2021, 60, 1909–1918 CrossRef CAS PubMed.
- B. Cuenoud and J. W. Szostak, Nature, 1995, 375, 611–614 CrossRef CAS PubMed.
- H. Fan, C. E. McGhee, R. J. Lake, Z. Yang, Z. Guo, X.-B. Zhang and Y. Lu, JACS Au, 2023, 3, 1615–1622 CrossRef CAS PubMed.
- W. Ren, P.-J. J. Huang, D. de Rochambeau, W. J. Moon, J. Zhang, M. Lyu, S. Wang, H. Sleiman and J. Liu, Biosens. Bioelectron., 2020, 165, 112285 CrossRef CAS PubMed.
- J. Liu and Y. Lu, J. Am. Chem. Soc., 2007, 129, 9838–9839 CrossRef CAS PubMed.
- J. Li, W. Zheng, A. H. Kwon and Y. Lu, Nucleic Acids Res., 2000, 28, 481–488 CrossRef CAS PubMed.
- P.-J. J. Huang, D. de Rochambeau, H. F. Sleiman and J. Liu, Angew. Chem., Int. Ed., 2020, 59, 3573–3577 CrossRef CAS PubMed.
- P.-J. J. Huang and J. Liu, Anal. Chem., 2014, 86, 5999–6005 CrossRef CAS PubMed.
- H. Yu, O. Alkhamis, J. Canoura, Y. Liu and Y. Xiao, Angew. Chem., Int. Ed., 2021, 60, 16800–16823 CrossRef CAS PubMed.
- S. Qian, D. Chang, S. He and Y. Li, Anal. Chim. Acta, 2022, 1196, 339511 CrossRef CAS PubMed.
- H.-P. Hofmann, S. Limmer, V. Hornung and M. Sprinzl, RNA, 1997, 3, 1289–1300 CAS.
- J. Ciesiolka, J. Gorski and M. Yarus, RNA, 1995, 1, 538–550 CAS.
- R. Nutiu and Y. Li, Angew. Chem., Int. Ed., 2005, 44, 1061–1065 CrossRef CAS.
- K. Yang, N. M. Mitchell, S. Banerjee, Z. Cheng, S. Taylor, A. M. Kostic, I. Wong, S. Sajjath, Y. Zhang and J. Stevens, Science, 2023, 380, 942–948 CrossRef CAS.
- M. Rajendran and A. D. Ellington, Anal. Bioanal. Chem., 2008, 390, 1067–1075 CrossRef CAS PubMed.
- H. Qu, A. T. Csordas, J. Wang, S. S. Oh, M. S. Eisenstein and H. T. Soh, ACS Nano, 2016, 10, 7558–7565 CrossRef CAS PubMed.
- R. Jahan, A. P. Silwal, S. K. S. Thennakoon, S. P. Arya, R. M. Postema, H. Timilsina, A. M. Reynolds and X. Tan, Chem. Commun., 2023, 59, 12851–12854 RSC.
- H. Wang, H. Cheng, J. Wang, L. Xu, H. Chen and R. Pei, Talanta, 2016, 154, 498–503 CrossRef CAS PubMed.
- Y. Chen, H. Li, T. Gao, T. Zhang, L. Xu, B. Wang, J. Wang and R. Pei, Sens. Actuators, B, 2018, 254, 214–221 CrossRef CAS.
- Y. Liu, X. Wang and J. Liu, Chem. Commun., 2024, 60, 6280–6283 RSC.
- J. Wang, Y. Liu, X. Li, H. Lei and J. Liu, Chem. Commun., 2024, 60, 14272–14275 RSC.
- C. Bardehle and J. Müller, Chem.–Eur. J., 2025, e202404332 CrossRef CAS PubMed.
- R. K. Sigel and H. Sigel, Acc. Chem. Res., 2010, 43, 974–984 CrossRef CAS PubMed.
- J. Reedijk, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3611–3616 CrossRef CAS PubMed.
- J. Wang, Y. A. Kaiyum, X. Li, H. Lei, P. E. Johnson and J. Liu, J. Am. Chem. Soc., 2025, 147, 1831–1839 CrossRef CAS PubMed.
- T.-L. Hwang and A. Shaka, J. Magn. Reson., Ser. A, 1995, 112, 275–279 CrossRef CAS.
- J. Wang, X. Li, H. Lei and J. Liu, Anal. Chem., 2025, 97, 9454–9461 CrossRef CAS PubMed.
- O. Alkhamis and Y. Xiao, J. Am. Chem. Soc., 2022, 145, 194–206 CrossRef PubMed.
- Y. Ding and J. Liu, J. Am. Chem. Soc., 2023, 145, 7540–7547 CrossRef CAS PubMed.
- Y. Ding, Z. Zhang and J. Liu, ChemBioChem, 2024, 25, e202400570 CrossRef CAS.
- F. Y. Khusbu, X. Zhou, H. Chen, C. Ma and K. Wang, Trends Anal. Chem., 2018, 109, 1–18 CrossRef.
- R. Nutiu and Y. Li, J. Am. Chem. Soc., 2003, 125, 4771–4778 CrossRef CAS PubMed.
- N. Nakatsuka, K.-A. Yang, J. M. Abendroth, K. M. Cheung, X. Xu, H. Yang, C. Zhao, B. Zhu, Y. S. Rim and Y. Yang, Science, 2018, 362, 319–324 CrossRef CAS PubMed.
- J. Canoura, T. Nguyen, C. Byrd, R. Hill, Y. Liu and Y. Xiao, Anal. Chem., 2024, 96, 11488–11497 CrossRef CAS.
- J. Hu and C. J. Easley, Analyst, 2011, 136, 3461–3468 RSC.
- N. Nakatsuka, K.-A. Yang, J. M. Abendroth, K. M. Cheung, X. Xu, H. Yang, C. Zhao, B. Zhu, Y. S. Rim, Y. Yang, P. S. Weiss, M. N. Stojanović and A. M. Andrews, Science, 2018, 362, 319–324 CrossRef CAS.
- S. Stangherlin, Y. Ding and J. Liu, Small Methods, 2024, 2401572 Search PubMed.
- Z. R. Churcher and P. E. Johnson, Aptamers, 2020, 4, 3–9 Search PubMed.
- S. R. Eisen, P. Dauphin-Ducharme and P. E. Johnson, Q. Rev. Biophys., 2024, 57, e9 CrossRef CAS PubMed.
- R. del Villar-Guerra, J. O. Trent and J. B. Chaires, Angew. Chem., Int. Ed., 2018, 57, 7171–7175 CrossRef CAS PubMed.
- A. Micsonai, F. Wien, L. Kernya, Y.-H. Lee, Y. Goto, M. Réfrégiers and J. Kardos, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E3095–E3103 CrossRef CAS PubMed.
- C. Wang, K. Zhu, P. Shi, X. Ding and S. Zhang, Chem. Commun., 2022, 58, 4779–4782 RSC.
- N. C. Garbett, P. A. Ragazzon and J. B. Chaires, Nat. Protoc., 2007, 2, 3166–3172 CrossRef CAS.
- T. S. Choi and F. A. Tezcan, Nature, 2022, 603, 522–527 CrossRef CAS.
- P.-J. J. Huang, J. Lin, J. Cao, M. Vazin and J. Liu, Anal. Chem., 2014, 86, 1816–1821 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc02436f |
| This journal is © The Royal Society of Chemistry 2025 |