
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCefiderocol “under siege”? Understanding the rise of NDM-mediated resistance to novel agents†
Brenda A.
Warecki
 a,
Pablo E.
Tomatis
ab,
María F.
Mojica
cde,
Christopher R.
Bethel
e,
Magdalena
Rodríguez Saravia
f,
Salvador I.
Drusin
bg,
Daisuke
Ono
eh,
Guillermo
Bahr
a,
Krisztina
Papp-Wallace
ehi,
Pranita D.
Tamma
j,
Diego M.
Moreno
bg,
Graciela
Mahler
f,
Robert A.
Bonomo
*cdehikl and
Alejandro J.
Vila
*abd
a,
Pablo E.
Tomatis
ab,
María F.
Mojica
cde,
Christopher R.
Bethel
e,
Magdalena
Rodríguez Saravia
f,
Salvador I.
Drusin
bg,
Daisuke
Ono
eh,
Guillermo
Bahr
a,
Krisztina
Papp-Wallace
ehi,
Pranita D.
Tamma
j,
Diego M.
Moreno
bg,
Graciela
Mahler
f,
Robert A.
Bonomo
*cdehikl and
Alejandro J.
Vila
*abd
aInstituto de Biología Molecular y Celular de Rosario (IBR), CONICET, Universidad Nacional de Rosario, Rosario, Argentina. E-mail: vila@ibr-conicet.gov.ar
bFacultad de Ciencias Bioquímicas y Farmacéuticas, Universidad Nacional de Rosario, Rosario, Argentina
cDepartment of Molecular Biology and Microbiology, Case Western Reserve University School of Medicine, Cleveland, Ohio, USA
dCWRU-Cleveland VAMC Center for Antimicrobial Resistance and Epidemiology (Case VA CARES), Cleveland, Ohio, USA. E-mail: robert.bonomo@va.gov
eResearch Service, Veterans Affairs Northeast Ohio Healthcare System, Cleveland, Ohio, USA
fLaboratorio de Química Farmacéutica, Facultad de Química, Universidad de la República, Montevideo, Uruguay
gInstituto de Química Rosario (IQUIR), CONICET, Universidad Nacional de Rosario, Rosario, Argentina
hDepartment of Medicine, Case Western Reserve University School of Medicine, Cleveland, Ohio, USA
iDepartment Biochemistry, Case Western Reserve University School of Medicine, Cleveland, Ohio, USA
jDepartment of Pediatrics, Division of Infectious Diseases, Johns Hopkins University School of Medicine, Baltimore, Maryland, USA
kDepartments of Pharmacology, Proteomics and Bioinformatics, Case Western Reserve University School of Medicine, Cleveland, Ohio, USA
lClinician Scientist Investigator, Veterans Affairs Northeast Ohio Healthcare System, Cleveland, Ohio, USA
First published on 3rd June 2025
Abstract
The global spread of antimicrobial resistance (AMR) underscores the critical need for the rapid development of new drugs. Particularly alarming is the surge in metallo-β-lactamases (MBLs) – broad spectrum enzymes able to inactivate penicillins, cephalosporins, and carbapenems. Cefiderocol (FDC), a siderophore-containing cephalosporin, was initially reported as resistant to MBL hydrolysis. Indeed, FDC has been designated as the preferred treatment for Gram-negative pathogens producing MBLs – with the most common MBLs of clinical concern belonging to the NDM, VIM, or IMP families. Regrettably, increasing reports of FDC resistance are emerging. Many of these events are linked to overexpression of NDM, sometimes coupled to alterations in iron transporters, challenging the notion of FDC being invulnerable to MBL hydrolysis. Herein, we demonstrate that NDM-1 and -5 are able to inactivate FDC efficiently, while VIM-2 and IMP-1 show impaired catalytic efficiency against this substrate. All these MBL enzymes form a reversible enzyme-product adduct with FDC, whose lifetime varies considerably among MBLs. In IMP-1 and VIM-2, this results in efficient enzyme inhibition. In contrast, NDM variants are poorly inhibited, eliciting efficient turnover rates. We propose a mechanistic explanation for FDC action that aligns with clinical findings suggesting NDMs contributing to FDC resistance. Based on these conclusions, we suggest caution when using this potent cephalosporin against NDM-producers. FDC paired with an NDM-inhibitor may be a strategy to preserve this important antibiotic.
Introduction
Antimicrobial resistance (AMR) is a major public health threat with an international impact, challenging the use of all available antibiotics.1 It is estimated that almost 5 million deaths were associated with AMR in 2021, with 1.14 million deaths directly attributable to this clinical problem. The global burden of AMR is predicted to increase significantly by 2050, with AMR likely to be the primary cause of mortality internationally.2 Arguably, one of the greatest concerns in AMR is the explosion of metallo-β-lactamase (MBL) producing bacteria causing significant disease.3MBLs are zinc-dependent enzymes capable of hydrolyzing most β-lactam antibiotics, including penicillins, cephalosporins and carbapenems.4,5 The ability of MBL genes to almost effortlessly disseminate across bacterial species through plasmids,6 lapses in infection prevention efforts both inside and outside of healthcare settings, and the lack of clinically approved inhibitors with activity against MBL have contributed to bolstering their success.7–11 Two main therapeutic strategies to overcome the challenge of MBL-mediated resistance currently exist: (1) the development of MBL inhibitors and (2) the design of novel β-lactam antibiotics unable to be hydrolyzed by MBLs.12 Successful advancements in both areas are proving to be challenging; however, promising developments are underway. Boronate-based compounds, such as taniborbactam13 and xeruborbactam,14 are efficient MBL inhibitors in advanced phases of development, but available data indicate they are unlikely to inhibit all circulating MBL enzymes currently causing disease.15
Presently, cefiderocol (FDC, formerly S-649266) is the only β-lactam agent with activity against MBLs, in the absence of a β-lactamase inhibitor. FDC is a cephalosporin (Fig. 1A) that features a methylpyrrolidinium group (also found in cefepime) that contributes to penetration through the outer membrane, and a chlorocatechol moiety mimicking a siderophore, which facilitates uptake via iron-transport mechanisms.16 The C7 substituent, instead, is similar to that present in ceftazidime (CAZ), an oxyiminocephalosporin widely used in combination with the serine-β-lactamase inhibitor avibactam against difficult-to-treat infections (Fig. 1A). The carboxypropyl-oxyimino group and the aminothiazole ring at C7 endows CAZ and FDC with antipseudomonal activity, and the ability to penetrate the outer membrane of Gram-negative bacteria, respectively. As a result, FDC is a “dual-mechanism” antibiotic that combines the potency of a cephalosporin core with a siderophore-mimicking moiety which binds extracellular Fe(III) and functions as a “Trojan horse” to enhance drug uptake through iron transporters at the outer membrane.17 These modifications prove advantageous for FDC enabling higher concentrations in the bacterial periplasm compared to other β-lactam antibiotics which enter bacteria through passive transport via non-specific porins.17
 | ||
| Fig. 1 (A) Chemical structures of cefiderocol (left) and ceftazidime (right). The cephalosporin core of FDC and CAZ is substituted on C7 by the same group, while differing in the identity of the C3 moiety (in red). (B) Proposed mechanism of hydrolysis of cefiderocol by NDM-1. The zinc-bound hydroxide group in the active site of NDM-1 performs the nucleophilic attack to the β-lactam ring. This results in ring opening, followed by an electronic rearrangement that leads to elimination of the C3 substituent (P2) and the formation of the exocyclic methylene group (in blue) in the cleaved cephalosporin core (P1). The intermediate species I is indicated between brackets as there was no evidence of its accumulation during turnover. | ||
FDC has previously been reported to be stable against a large panel of clinically relevant MBLs (e.g., NDM-1, VIM-2, and IMP-1).18 Unfortunately, a series of clinical cases reported Enterobacterales isolates developing FDC resistance during treatment with this antibiotic.19–25 In these cases, resistance to FDC most commonly arose from alterations or truncations in genes coding for the iron transporter.19,23 However, several other reports indicated that resistance to FDC was mediated by the production of NDM (New Delhi metallo-β-lactamase),20,21,25–28 challenging the notion that FDC is refractory to hydrolysis by MBLs.
To date, all MBL-mediated resistance reports are limited to NDM variants (namely, NDM-1 and -5).20,21,25 Conflicting evidence exists regarding the catalytic efficiencies of distinct MBLs18,24,29,30 (i.e., NDM-1, VIM-2, and IMP-1 variants). This work demonstrates that NDM can indeed hydrolyze FDC and explains why NDM variants appear to be the only MBL capable of conferring FDC resistance. To these ends, we employ a comprehensive biochemical, spectroscopic, computational and microbiological approach. We demonstrate that FDC binds efficiently to all herein tested MBLs (NDM-1/5, VIM-2, and IMP-1). However, strikingly different catalytic efficiencies exist across these enzymes. While NDM-1 and NDM-5 display a significant turnover rate against FDC, VIM-2, and IMP-1 have a catalytic performance two orders of magnitude smaller. More importantly, we also report the formation of an enzyme-product adduct during FDC hydrolysis by MBLs. This adduct is stable with IMP-1 and VIM-2, leading to enzyme inhibition, but it displays a short half-life with NDM-1 and NDM-5, in line with the higher catalytic efficiencies of these enzymes. We believe the overarching findings of our work challenge the current paradigm that considers all MBLs as equally potent, support caution with the use of cefiderocol treatment for NDM-producing infections and provide insights for future therapeutic strategies using FDC.
Experimental
Protein expression and purification
Truncated blaNDM-1 (blaNDM-1 Δ38) or blaNDM-5 (blaNDM-5 Δ38) were amplified with Phusion DNA polymerase from respective clones using oligomers including Nde1 (forward oligomer) and Xho1 (reverse oligomer) restriction sites which omitted the first 38 amino acids (Δ38) in the case of blaNDM-1 and blaNDM-5.31 This omission removed the periplasmic signal peptide as well as a membrane-binding lipid anchor. This product was ligated into an Nde1/Xho1-digested modified version of the pET-28 b(+) plasmid in which the thrombin cleavage site was replaced by a TEV cleavage site. The construct of blaNDM-1(IMP-1L3) (blaNDM-1(IMP-1L3) Δ38) is described in Palacios et al.32blaVIM-2 (blaVIM-2 Δ25) and blaIMP-1 (blaIMP-1 Δ16) lacking the first 25 and 16 amino acids which accounts for the signal peptide, respectively, were constructed in a pET-28 plasmid containing a His-tag and a TEV cleavage site.
E. coli BL21(DE3) cells were transformed with the sequence-verified plasmids for protein production. An E. coli BL21 (DE3) pET-28 b(+):blaMBL Δsignal peptide culture was grown at 37 °C in LB media until it reached OD600 = 0.6. Then, MBL production was induced by addition of 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG). At the time of induction of protein expression, the growth medium was supplemented with 0.5 mM ZnSO4. Cells were incubated for 18 hours at 20 °C. All subsequent purification steps were performed at 4 °C. The cells were harvested and resuspended in 50 mM Tris–Cl, pH 8.0, 200 mM NaCl and supplemented with 10 μg per mL DNase, 4 mM MgCl2, 2 mM phenylmethanesulfonyl fluoride (PMSF), and 10 mM β-mercaptoethanol. E. coli cells were disrupted by sonication (five cycles of 30 s with 1 min between), and the insoluble material was removed by centrifugation for 60 min at 15000g. The crude extract was loaded onto a Ni-Sepharose column equilibrated with buffer A (50 mM Tris–Cl, pH 8.0, 200 mM NaCl, 10 mM β-mercaptoethanol), the column was washed with 100 mL of buffer A, and His6x–MBL was eluted with buffer B (50 mM Tris–Cl, pH 8.0, 200 mM NaCl, 10 mM β-mercaptoethanol, 500 mM imidazole) using a linear gradient (0–100% buffer B, in 100 mL). Then, 100 μM His6x–MBL was mixed with the TEV protease (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 TEV:His6x–MBL ratio), and the mixture was incubated for 16 h at 4 °C during dialysis against 100 volumes of 50 mM Tris–Cl, pH 8.0, 200 mM NaCl, 10 mM β-mercaptoethanol. The MBL was then loaded onto a Ni-Sepharose column to separate it from the His6x tag, the uncleaved fusion protein, and the His6x-tagged TEV protease. The MBL was collected in the flow-through of the column with a purity > 95%, as determined by SDS-PAGE. β-Mercaptoethanol was removed from the protein sample by one 12 h dialysis step of 100 μM MBL against 100 volumes of 10 mM HEPES, pH 7.5, 200 mM NaCl, 200 μM ZnSO4, followed by three 4 h dialysis steps against 100 volumes of 10 mM HEPES, pH 7.5, 200 mM NaCl. MBL was concentrated by ultrafiltration. Protein concentrations were determined from the absorbance at 280 nm using a molar absorption coefficient (ε280) of 27960 M−1 cm−1 (NDM-1), 31970 M−1 cm−1 (NDM-1(IMP-1L3)), 28500 M−1 cm−1 (VIM-2), and 44920 M−1 cm−1 (IMP-1).
:50 TEV:His6x–MBL ratio), and the mixture was incubated for 16 h at 4 °C during dialysis against 100 volumes of 50 mM Tris–Cl, pH 8.0, 200 mM NaCl, 10 mM β-mercaptoethanol. The MBL was then loaded onto a Ni-Sepharose column to separate it from the His6x tag, the uncleaved fusion protein, and the His6x-tagged TEV protease. The MBL was collected in the flow-through of the column with a purity > 95%, as determined by SDS-PAGE. β-Mercaptoethanol was removed from the protein sample by one 12 h dialysis step of 100 μM MBL against 100 volumes of 10 mM HEPES, pH 7.5, 200 mM NaCl, 200 μM ZnSO4, followed by three 4 h dialysis steps against 100 volumes of 10 mM HEPES, pH 7.5, 200 mM NaCl. MBL was concentrated by ultrafiltration. Protein concentrations were determined from the absorbance at 280 nm using a molar absorption coefficient (ε280) of 27960 M−1 cm−1 (NDM-1), 31970 M−1 cm−1 (NDM-1(IMP-1L3)), 28500 M−1 cm−1 (VIM-2), and 44920 M−1 cm−1 (IMP-1).
Steady-state kinetic measurements
The kinetic parameters kcat and KM were determined by initial velocity measurements of the hydrolysis rate of CFD by purified NDM-1, NDM-5, VIM-2, IMP-1, and NDM-1(IMP-1L3). These experiments were performed at 30 °C, in 10 mM HEPES, 200 mM NaCl pH 7.5 buffer, supplemented with 20 μM ZnSO4. FDC hydrolysis was monitored by following the time evolution of absorption maximum at 259 nm, using the reported18 value for CFD of Δε259 = −9430 M−1 cm−1 in a 0.1 cm path length cuvette. UV difference spectra (absorption spectra of cefiderocol or ceftazidime reacted with NDM-1, VIM-2, or IMP-1 β-lactamase versus spectra of cefiderocol or ceftazidime in a 200:1 S:E ratio) were measured from λ = 220 nm to 300 nm.
NMR experiments
NMR experiments were recorded in Bruker NMR spectrometers Neo Avance 400 MHz and Avance III 700 MHz. Small molecule NMR spectra were recorded by adding 10% D2O and using standard pulse sequences (1H NMR: zgesgp, zgpr, 13C NMR: zgpg30, HSQC: fhsqcf3gpph, hsqcetgpprsisp2.2, HMBC: hmbcgpl2ndwg) and water suppression conditions.1H NMR experiments were performed by dissolving the FDC powder (provided by Shionogi & Co, Ltd, Osaka, Japan) in HEPES 50 mM pH 7.5 until a concentration of 5 mM in a final volume of 600 μL. In the case of enzymatic hydrolysis, after acquiring a 1H NMR spectrum, the purified enzyme (NDM-1, VIM-2 or IMP-1) was added to a final concentration of 20–25 μM. Alkaline chemical hydrolysis was performed by incubating FDC with excess NaOH (4 equivalents) for 15 min. Spectra were acquired after adding 10% D2O and lowering the pH to 7.5.
FDC was purified by thin layer C18 reverse phase chromatography, using previously activated glass backed plates (20 × 20 cm, 1000 μm, F-254 indicator). A chamber was saturated with formic acid 0.1% acetonitrile 80:20 and the chromatography developed for 40 minutes. After revealing at UV (254 nm), the desired product was extracted from the stationary phase with methanol, filtered off and the solvent was evaporated under vacuum to give pure FDC, assessed by HPLC and 1H NMR (see Fig. S3B†).
Protein NMR experiments were performed out on an Avance III 700 MHz Bruker spectrometer equipped with a triple resonance inverse (TXI) probe at 300 K (NDM-1) and 310 K (VIM-2) using standard HSQC techniques with solvent suppression. Backbone resonance assignments for NDM-1 and VIM-2 were taken from the BMRB bank (NDM-1: BMRB entry 27043; VIM-2: BMRB entry 51165). The interaction of FDC and P1 with NDM-1 and VIM-2 was followed by stepwise titrations on 300 μM 15N-labeled protein samples in 50 mM HEPES buffer at pH 7.5 (NDM-1) and 20 mM phosphate, 100 μM ZnSO4 buffer at pH 7 (VIM-2) and followed by recording HSQC spectra.
Pre-steady state experiments
The time-dependent Trp fluorescence intensity was measured in an Applied Photophysics SX.18-MVR stopped-flow coupled to a fluorometer. Enzyme concentration was 10 μM and 20 μM for substrates in all cases. All determinations were performed in HEPES 10 mM, NaCl 200 mM at pH 7.5 supplemented with 20 μM ZnSO4 at 10 °C using a thermostatic circulator. Enzyme fluorescence measurements were performed by excitation at 280 nm with a slit width of 0.5 mm and detection through a cut-off filter at >305 nm. The photomultiplier voltage was set to obtain an 8 V signal with the free enzyme. Absorbance changes were measured at 260 nm for CAZ and 259 nm for FDC using an absorption photomultiplier. For both absorbance and fluorescence measurements, the excitation/absorption pathlength was 0.2 cm.Electrospray ionization mass spectrometry (ESI-MS)
Mass spectrometry data were measured for purified NDM-1, IMP-1, and VIM-2 incubated with cefiderocol. NDM-1, IMP-1, and VIM-2 were subjected to electrospray ionization-mass spectrometry (ESI-MS) after cefiderocol exposure (1:500 ratio) at t = 0, 10 seconds, 1 minute, 5 minutes, 1 hour, and 24 hours. Reactions were performed in HEPES 10 mM NaCl 200 mM at pH 7.5, supplemented with 50 μM ZnSO4. Reactions were quenched with 1% acetonitrile and 0.1% formic acid. Samples were analyzed using Q-TOF Waters Synapt-G2-Si and a Waters Acquity ultrapressure liquid chromatography (UPLC) BEH C18 column (1.7 μm pore size; 2.1 by 50 mm). MassLynxV4.1 was used to deconvolute protein peaks. The tune settings for each data run were as follows: capillary voltage at 3.5 kV, sampling cone at 35, source offset at 35, source temperature of 100 °C, desolvation temperature of 500 °C, cone gas at 100 L h−1, desolvation gas at 800 L h−1, and nebulizer bar at 6.0. Mobile phase A was 0.1% formic acid–water. Mobile phase B was 0.1% formic acid–acetonitrile. The mass accuracy of this system is ±5 Da.
Minimum inhibitory concentration (MIC) measurements
MICs were measured using broth microdilution method with Iron-Depleted Cation-Adjusted Mueller–Hinton Broth (ID-CAMHB), according to the Clinical Laboratory and Standards Institute (CLSI) protocol.33 Cefiderocol (FDC) was provided by Shionogi. The final concentration of E. coli DH10B cells harboring the pMBLe blaNDM-1 or blaIMP-1 plasmids in each well was adjusted to 5 × 105 CFU mL−1. To induce the expression of blaNDM-1 or blaIMP-1 from the pMBLe plasmid, the bacterial inoculum was prepared in ID-CAMHB with different concentrations of isopropyl β-D-1-thiogalactopyranoside (IPTG), ranging from 0 to 200 μM. Plates were incubated at 37 °C for 18 h. Results shown are the mode of three independent experiments.Immunoblotting
Protein productions levels in E. coli DH10B cells at different IPTG concentrations were measured using the antibiotic-free growth control wells of the 96-well plate used for the MIC determinations. In brief, once the MIC were determined, the content of each FDC-free growth control wells at different IPTG concentrations were recovered (200 μL) and spun down. The cells were lysed to prepare crude extracts by resuspending them in 60 μL of loading buffer (19:1 mixture of Coomassie blue and β-mercaptoethanol) and freezing at −20 °C overnight. Next, after boiling for 10 minutes, 20 μL of the crude extracts were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride membranes. The membranes were blocked in 5% non-fat dry milk in 20 mM Tris–Cl with 150 mM NaCl pH 7.4 (TBS) for 1 h and probed in 5% non-fat dry milk in TBS with 1:1000 dilution of monoclonal anti-strep tag II antibody (Millipore Sigma) and, as a loading control, 1:5000 dilution of monoclonal anti-DNAK antibody (Stressgen) for 18 h at 4 °C. DnaK was used as the loading control. Membranes were washed four times for 15 min with TBS with 0.05% Tween 20 (TBST) and then incubated for 1 h in 1:10000 dilution of goat-anti-mouse-HRP antibodies in 5% non-fat dry milk in TBS. Blots were washed four times for 15 min with TBST, developed using the ECL-Plus™ kit (GE Healthcare Life Sciences) according to the manufacturers' instructions, and imaged on a Fotodyne Luminary/FX. Images were quantified by Fiji image processing package.34 Results shown are the average of two immunoblotting experiments from two different MIC plates.
Binding experiments and IC50
Binding experiments and IC50 values were determined using the hydrolysis product of FDC. To obtain the hydrolyzed product (P1), intact FDC was incubated with NDM-1 for 5 min. The reaction progress was monitored by measuring absorbance at 259 nm using a Jasco V-650 spectrophotometer, ensuring complete hydrolysis of FDC. P1 and P2 were then separated from the enzyme using an Amicon® Ultra Centrifugal Filter with a 10 kDa molecular weight cut-off. Notably, the catechol moiety exhibited no interaction with the enzymes.Dissociation constants for the interaction of P1 with NDM-1, VIM-2, and IMP-1 were estimated using a Cary Eclipse Varian spectrofluorometer. Measurements were performed with 2 μM of MBLs in a buffer containing 10 mM HEPES, 200 mM NaCl (pH 7.5), and 20 μM ZnSO4, using a 0.5 cm path length cuvette. Tryptophan fluorescence emission spectra were recorded by titrating the MBLs with increasing equivalents of P1 until no further changes were observed. Kd values were determined by fitting the titration curves to a one-site specific binding model using nonlinear regression in GraphPad Prism 10.3.0. All determinations were performed in triplicate.
The inhibitory activity of FDC and P1 against NDM-1, VIM-2, and IMP-1 were tested using the chromogenic substrate nitrocefin. The assay buffer consisted of 10 mM HEPES, 200 mM NaCl (pH 7.5), and 20 μM ZnSO4. Briefly, NDM-1, VIM-2, or IMP-1 (2 nM in all cases) was incubated with various concentrations of FDC or P1, or without inhibitors, for 5 minutes at 30 °C. The incubation mixture was then added to a 0.1 cm path length cuvette containing nitrocefin at a final concentration of 50 μM, and absorption at 495 nm was immediately monitored on a Jasco V-670 spectrophotometer for 300 seconds to record the initial velocities of nitrocefin hydrolysis. Data were used to fit IC50 curves using GraphPad Prism 10.3.0. All determinations were performed in triplicate.
Docking experiments
Docking experiments were performed using AutoDock-GPU.35,36 The structures for the NDM-1, VIM-2 and IMP-1 proteins were obtained from the Protein Data Bank (codes 5ZGX,375YD7 [crystal structure of VIM-2 metallo-beta-lactamase, Wachino, J. (to be published)] and 7XHW,38 respectively).QM/MM modeling of the nucleophilic attack
The productive docking pose with the lowest binding energy of FDC with each protein was used as a starting point to simulate the hydrolysis reaction. All simulations were performed using Amber20 (ref. 39) software as follows. Each complex was first submerged in a periodic octahedral box of water molecules. Then an initial water MM optimization phase was done, with a harmonic restraint potential of 1000 kcal mol−1 on all protein and ligand atoms, so that only water molecules can move. In this phase, the ff14SB forcefield40 was used for the protein, while the TIP3P model41 was used for the water atoms. Finally, a QM/MM optimization was done with a harmonic potential of 40 kcal mol−1 on a distance of 1.32 Å between the oxygen atom of the nucleophile and the carbon atom of the carbonyl group of the β-lactam, to force the reaction. The QM region was treated at DFTB level and consisted of the ligand (cefiderocol), the Zn ions, the hydroxyl ion, and the sidechains of the residues His116, His118, Asp120, His196, Cys221 and His263 which are coordinated to the Zn ions. The QM/MM boundary in the sidechains are placed in the Cβ–Cγ bond in the case of histidine residues and the Cα–Cβ bond in the aspartate and cysteine residues, capping with hydrogen link atoms. The electrostatic embedding method is used for electrostatic QM/MM interactions, using the PME approach for long-range interactions, with a cutoff of 8 Å.QM/MM simulations of the EP complexes
The EP complexes were built in silico starting from the crystallographic structures of each protein bound to the hydrolysis product of a ligand (5O2E42 for NDM-1 and 7A5Z43 for VIM-2). In both cases the ligand scaffold was used and the R groups were substituted with those of the product P1. The EP complex of IMP-1 was created by aligning the apo protein (7XHW) with the previous models. As in the hydrolysis modeling, the simulations were done with AMBER 20, submerging the protein in water molecules, which were then optimized with a restraint on the rest of the system. The final QM/MM optimization was done with no restraints. Parameters and QM subsystems were treated as described above.Results & discussion
MBLs hydrolyze FDC with different catalytic efficiencies
FDC and CAZ differ only by the C3 substituent. The bulky substituent at C3 may thus be regarded as the main limitation for hydrolysis of FDC by various β-lactamases. However, as a class, cephalosporins remain favored MBL substrates, including nitrocefin, which possesses a long and bulky substituent at C3. We explored binding of FDC by docking this compound into the active sites of three clinically relevant MBLs (NDM-1, VIM-2, and IMP-1). These simulations suggest that FDC is able to bind productively to all these proteins, i.e., with the carbonyl group of the β-lactam ring appropriately positioned with the attacking nucleophile, a zinc-bound hydroxide. The C3 substituent is a long, flexible side chain that can adopt different conformations, fitting into the active site of these MBLs (Fig. S1†).We then performed UV difference spectroscopy to monitor the hydrolysis of FDC and CAZ (as a reference substrate) by a panel of clinically relevant B1 MBLs: NDM-1, VIM-2, and IMP-1.44 These experiments revealed that NDM-1 efficiently hydrolyzes FDC, whereas IMP-1 and VIM-2 exhibit a negligible activity. In contrast, all three enzymes can turnover CAZ (Fig. S2†). To assess the differential ability of these MBLs to hydrolyze FDC, we determined the steady-state kinetic parameters for FDC hydrolysis. NDM-1 and NDM-5 are efficient at hydrolyzing FDC with catalytic efficiencies in the order of 105 M−1 s−1 (Table 1). In comparison, VIM-2 and IMP-1 show poor activity against FDC, with efficiencies two orders of magnitude less than the NDM variants, mostly due to low kcat values. KM values are relatively high but fall within the same range for all tested enzymes, revealing that the turnover rate is the main determinant of the differing FDC hydrolysis efficiency by these MBLs. The catalytic efficiencies for CAZ hydrolysis were larger for all MBLs, with the following trend: NDM-1 > IMP-1 > VIM-2 (Table 1). KM values for CAZ were one order of magnitude lower than those for FDC, a fact that can be attributed to the bulkier substituent at the C3 position in FDC. Instead, a similar pattern was observed for kcat values, NDM-1 exhibiting the highest turnover rate, followed by IMP-1 and then VIM-2. These data show a better performance of NDM variants compared to VIM-2 and IMP-1 toward FDC that is due to a large difference in the kcat values.
| Enzyme/substrate | K M (μM) | k cat (s−1) | k cat /K M (M−1 s−1) |
|---|---|---|---|
| a Standard deviations were <10%, as reported by the authors. | |||
| NDM-1 | |||
| FDC | 443 ± 59 | 84 ± 11 | 1.91 × 105 |
| CAZ | 45 ± 12 | 187 ± 13 | 4.16 × 106 |
|
|||
| NDM-5 | |||
| FDC | 405 ± 40 | 75 ± 3 | 1.85 × 105 |
| CAZ45 | 86 ± 11 | 15 | 1.8 × 105 |
|
|||
| VIM-2 | |||
| FDC | 250 ± 80 | 1.10 ± 0.14 | 4.4 × 103 |
| CAZ46a | 72 | 3.6 | 5 × 104 |
|
|||
| IMP-1 | |||
| FDC | 300 ± 120 | 1.25 ± 0.23 | 4.1 × 103 |
| CAZ47 | 44 ± 3 | 8 ± 1 | 1.8 × 105 |
Cephalosporin hydrolysis can lead to elimination or reorganization of the substituent at C3.48 We therefore studied NDM-mediated hydrolysis of FDC by NMR spectroscopy to identify the hydrolysis products. The 1H NMR spectrum of the reaction showed changes in most chemical shifts, particularly from protons bound to C6 and C7, as well as the appearance of two signals at 5.53 and 5.61 ppm that can be attributed to vinylic protons (Fig. S3C†). Heteronuclear 1H–13C HSQC spectra revealed that these protons are bound to a single carbon with a signal at 124 ppm, confirming that they correspond to an exocyclic methylene group (Fig. S3F†). The 1H–13C HMBC spectrum shows the interaction between the methylene signals and carbons C2 and C4 from the dihydrothiazine ring, (Fig. S3G†) revealing that, after β-lactam cleavage, FDC experiences double bond isomerization, with a concomitant elimination of the C3 substituent leading to products P1 and P2 (Fig. 1B).
FDC hydrolysis by VIM-2 or IMP-1 also proceeded with elimination of the C3 substituent (Fig. S3D†). Despite the slower rate, NMR did not reveal accumulation of the intermediate I (Fig. 1B). The catalytic mechanism of cephalosporins by MBLs and the differences with carbapenem hydrolysis have been exhaustively studied by computational approaches,49–51 highlighting differences in the protonation step. Less is known about the elimination of the C3 substituent. To further understand this step, we modeled the initial nucleophilic attack by performing QM/MM simulations. These calculations show that, when the oxygen from the nucleophilic hydroxide is within bonding distance of the carbonyl group of the β-lactam ring, the C–N bond is simultaneously cleaved and there is a lengthening of the C–C bond between C3 and the bound leaving group (Fig. S4†). These calculations support our experimental results, enabling us to conclude that the elimination of the C3 substituent occurs immediately after β-lactam cleavage in the enzyme active site.
Cephalosporins are prone to undergo epimerization at C6,52via opening and closure of the dihydrothiazine ring (Fig. S5†). This process results in the formation of diastereomeric degradation products, which can interfere in the interpretation of hydrolysis kinetics and the identity of degradation products. To assess the structure of P1, we studied the alkaline hydrolysis of FDC and CAZ at pH 12. In both cases, 1H and 1H–13C NMR revealed the presence of an epimeric mixture of P1 (Fig. S6†). When comparing these results to the enzyme-catalyzed reaction, the finding of only one product allows to discard C6 epimerization in the enzyme active site.
Pre-steady state kinetics reveals the accumulation of enzyme-bound adducts that account for distinct catalytic efficiencies
We used pre-steady state kinetics to investigate the mechanistic basis of the different catalytic behavior of the three MBLs toward FDC. The kinetic traces of the absorbance at 259 nm (reporting on cephalosporin hydrolysis) under pre-steady state conditions are in line with the kinetic parameters under steady state conditions: NDM-1 exhibited the fastest substrate hydrolysis, followed by IMP-1 and then VIM-2 (Fig. S7†). All hydrolysis traces displayed a monophasic decay under different assayed conditions, consistent with the lack of a significant accumulation of the intermediate species I.The intrinsic Trp fluorescence of enzymes can be exploited to probe substrate binding. FDC binding to NDM-1, VIM-2, and IMP-1 was studied by following Trp fluorescence during turnover under pre-steady state conditions with a stopped flow equipment to gain mechanistic insights on the binding event.53,54 These MBLs have four (NDM-1), three (VIM-2) and six (IMP-1) Trp residues. Inspection of the crystal structures reveals that some of these residues are close to the active site and can serve as sensitive binding probes, such as Trp93 in NDM-1 (BBL 87), Trp87 (BBL 87) and Trp 219 (BBL 242) in VIM-2, and Trp28 in IMP-1 (BBL 64). Binding and hydrolysis of CAZ as a reference substrate for all three MBLs was also examined.
Binding of CAZ to the three MBLs is reflected in a large quenching of the Trp fluorescence after mixing (Fig. 2), which in the case of NDM-1 is fast and occurs during the dead time of the experiment (Fig. S8†). When CAZ hydrolysis is completed, the fluorescence of the MBLs is fully recovered, indicating total product release from the active site. The different time frames for fluorescence recovery (VIM-2 > IMP-1 > NDM-1) in the three enzymes are consistent with the steady-state kinetic parameters reported in Table 1 (Fig. 2).
 | ||
| Fig. 2 Trp fluorescence intensity followed upon substrate (FDC or CAZ) addition. Fluorescence quenching occurs until complete substrate turnover in the case of CAZ hydrolysis by NDM-1 (A), VIM-2 (B) and IMP-1 (C) where the fluorescence intensity is fully recovered after substrate depletion, leaving an unbound enzyme. In the case of FDC, Trp fluorescence in the three enzymes show an intense and fast quenching process after addition of the substrate, with a slight recovery for NDM-1 (A). This suggests a partial adduct formation. In contrast, Trp fluorescence intensity in the case of VIM-2 (B) and IMP-1 (C) maintains quenched, suggesting a strong adduct formation that lasts in time. Exposure at longer times results in a fluorescence decay due to photobleaching, which is more evident in panel 2A. | ||
The interaction of MBLs with FDC was completely different to CAZ, and even among MBLs. FDC binds rapidly to both IMP-1 and VIM-2, as revealed by the fluorescence quenching, but there is no fluorescence recovery in the interaction of these two MBLs with FDC (Fig. 2). This suggests that both IMP-1 and VIM-2 form an adduct upon FDC binding, without restoring the resting-state conformation of the enzymes. We interpret that IMP-1 and VIM-2 are inhibited by formation of this adduct, accounting for the significantly smaller kcat values measured for these enzymes toward FDC (Table 1). Instead, in the case of FDC, there is a partial recovery of the fluorescence after the initial quenching event (Fig. 2A), suggesting that a fraction of NDM-1 molecules forms this adduct, while the bound species is released in a population of NDM-1 molecules. In other words, this suggests a more labile adduct in the case of NDM-1.
Overall, the pre-steady state binding studies agree with the steady state parameters for CAZ and FDC. These experiments show that all three MBLs are able to bind and hydrolyze FDC, but leading to the accumulation of a bound species, whose population differs among the distinct MBLs. We interpret that the high concentration of the bound form in IMP-1 and VIM-2 inhibits these enzymes, impairing turnover. In contrast, NDM-1 is able to release the bound species during turnover, resulting in a concentration of active NDM-1 molecules that can turnover FDC.
MS reveals that the adducts are formed by a species that resembles a product
In order to identify the bound species, electrospray ionization-mass spectrometry (ESI-MS) was performed by quenching the reaction between FDC and the MBLs at different times by addition of 1% acetonitrile. After 1 min of incubation of NDM-1 with FDC, a signal with an additional mass of +485.4 Da was identified (Fig. 3), which is the mass expected for the hydrolysis product P1 of FDC (Fig. 1B), forming the EP1 adduct. Different time points of incubation revealed an increasing abundance of the EP1 adduct, with a subsequent decrease after 5 min (Fig. S9†). After 24 h of incubation, only the unbound enzyme could be identified. This reveals that EP1 is a time-dependent, reversible, adduct. A similar reaction with apo-NDM-1 and FDC after different incubation times did not disclose the presence of any adduct, revealing that the adduct requires FDC hydrolysis, and that the intact compound does not remain bound to the active site (Fig. S10†). | ||
| Fig. 3 ESI-MS spectra of NDM-1, VIM-2 and IMP-1 enzymes (A) and after incubation for 1 minute with a 1:500 molar ratio of enzyme-cefiderocol (B). | ||
When VIM-2 and IMP-1 were incubated with FDC, an additional peak with +488.2 Da and +485.3 Da was identified, respectively, and an adduct-free enzyme ratio > 1 could be detected even after 1 hour of incubation (Fig. S11†). These results suggest that the adduct formed with IMP-1 and VIM-2 exists longer and more abundant than with NDM-1, in agreement with the fluorescence traces observed in pre-steady state kinetics.
There is sound evidence for the trapping of similar covalent adducts in denaturing conditions during the hydrolysis of cephalosporins by MBLs, including cephaclor with NDM-1,55 cefoxitin with the B2 MBLs CphA56 and cefuroxime with the B3 MBL FEZ.57 This observation is explained by the following: after β-lactam cleavage, the dihydrothiazine ring of cephalosporins can epimerize at C6 under denaturing conditions, generating a thiolate intermediate that can form disulfide bonds with free Cys residues. To validate the identity of this adduct, we repeated the MS experiments in the presence of β-mercaptoethanol (BME) and (tris(2-carboxyethyl)phosphine) (TCEP), which maintains the thiols in a reduced state. Under these conditions, the adduct was not detected (Fig. S12†), confirming that its formation results from a covalent binding during sample processing that enables trapping of the bound species.
Taken together, these different approaches reflect the same phenomenon, and therefore, we posit that the three MBLs form an adduct with the hydrolysis product P1. Thus, IMP-1 and VIM-2 are able to hydrolyze FDC but undergo a strong product inhibition, which may account for the low kcat values (Table 1).
The hydrolyzed product is bound to the active site of NDM-1 and VIM-2
NMR was used to map the binding site of P1 to NDM-1 and VIM-2. We recorded 1H–15N HSQC spectra of 15N-labeled NDM-1 against increasing concentrations of FDC, until a final ratio of 1:10. Addition of FDC led to the appearance of a new set of peaks, with the concomitant disappearance of several resonances corresponding to the unbound form of NDM-1, as well as the shifting and intensity decrease of other signals in the HSQC spectrum (Fig. S13†), revealing differing time exchange, depending on resonances. These results enabled the identification of protein regions perturbed upon binding to FDC, either by chemical shift perturbation or intensity loss. Fig. 4A shows that some metal ligands, residues belonging to the active site loops L3 and L10 (which surround the active site and define the active site walls), and some second shell residues are perturbed by the formation of this adduct. To explore this further, we performed a titration of 15N-labeled NDM-1 with the hydrolysis product P1, which gave rise to a perturbation pattern similar to that elicited by FDC binding to NDM-1 (Fig. S14†). From this observation we concluded that P1 (with a mass of 485 Da) is bound to the active site of NDM-1, giving rise to the adduct observed in the stopped-flow experiments.
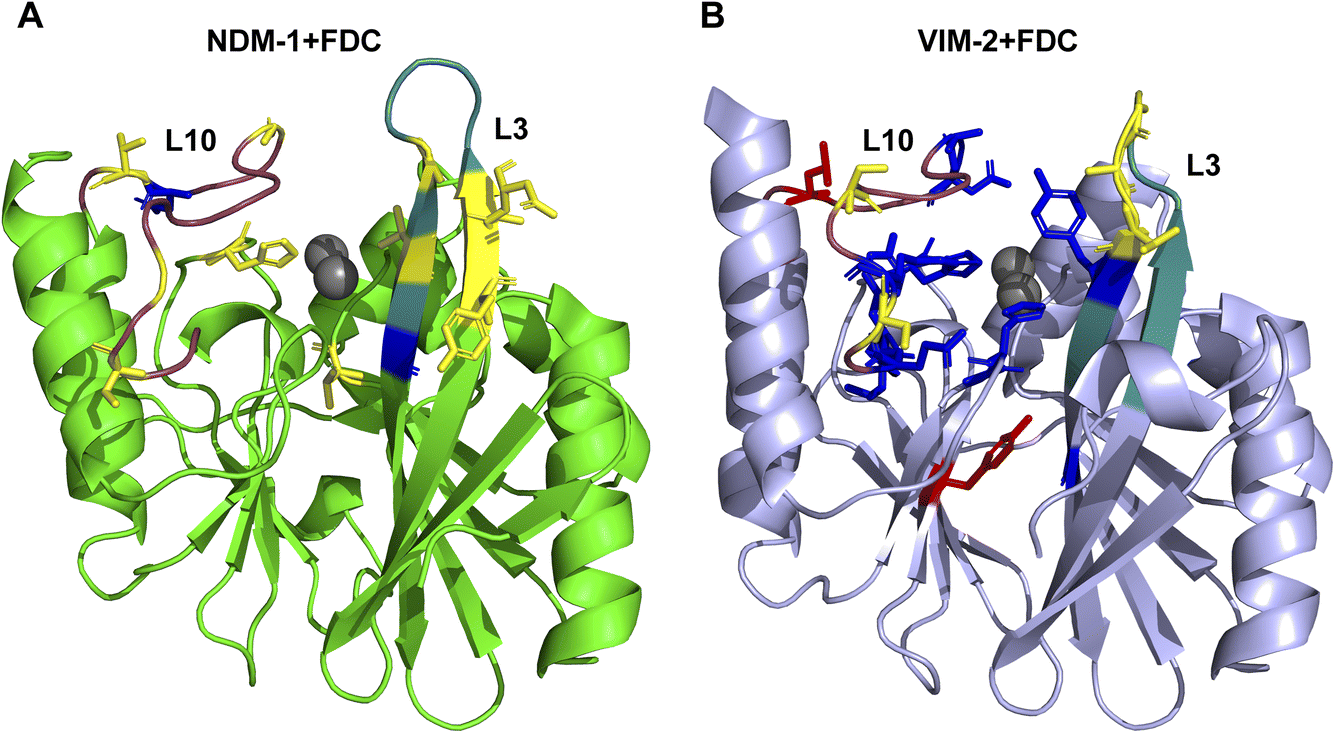 | ||
| Fig. 4 Mapping of the spectral perturbations into the three-dimensional structure of NDM-1 (A) and VIM-2 (B) based on the 1H–15N HSQC spectra in the presence of FDC. Loop L3 is highlighted in cyan, while loop L10 is shown in magenta. Residues exhibiting chemical shift changes upon FDC addition are colored yellow, those that disappear are marked in blue, and residues with reduced intensity are shown in red. | ||
Titration of 15N-labeled VIM-2 with FDC and the hydrolysis product P1 also gave rise to a set of perturbed signals in the protein NMR spectrum, which correspond to active site residues (Fig. S15 and S16†). Interestingly, a larger number of residues from loop L10 were altered in comparison with NDM-1, suggesting a greater interaction of the adduct with this active site wall (Fig. 4B). Since the spectrum of VIM-2 in the presence of the hydrolysis product P1 resembled the perturbation pattern of the species formed upon exposure to FDC, it was concluded that the arrangement of P1 in the active site is slightly different in NDM-1 and VIM-2.
Overall, these data reveal that the bound species to NDM-1 and VIM-2 correspond to the hydrolysis product P1. However, the accommodation of the substrate in the active site differs, in the case of NDM-1, loop L3 residues show the largest perturbation, while in VIM-2, loop L10 displays a stronger interaction with the bound species.
The hydrolysis product of FDC is an MBL inhibitor
We tested the inhibitory potency of FDC and the reaction product P1 against NDM-1, VIM-2, and IMP-1 using nitrocefin as a substrate reporter. Nitrocefin is a chromophoric cephalosporin, and its hydrolysis can be monitored in the visible range, beyond the absorbance at 260 nm of all cephalosporins. As shown in Fig. 5A, the nitrocefin hydrolysis activities of VIM-2 and IMP-1 are inhibited by addition of FDC, while the activity of NDM-1 is largely unperturbed at high concentrations of FDC. Incubation of the hydrolysis product P1 with the different MBLs results in a significant inhibition, disclosing a large difference between the impact in NDM-1 versus VIM-2 and IMP-1 and further confirming the identity of the inhibitory molecule. However, the high instability of P1 in the assay conditions precluded the measurement of a bona fide inhibition parameter. Instead, we measured the affinity of P1 against the resting state enzymes by a titration followed by Trp fluorescence, as shown in Fig. 5B. Surprisingly, binding of P1 to the three enzymes was within the same range (Kd values around 30–50 μM). | ||
| Fig. 5 Inhibition of the nitrocefinase activity of NDM-1, VIM-2 and IMP-1 by addition of FDC (A). Binding of the hydrolysis product P1 to NDM-1, VIM-2 and IMP-1 followed by Trp fluorescence. The calculated dissociation constants are 41.55 μM (NDM-1), 47.27 μM (VIM-2) and 38.63 μM (IMP-1) (B). | ||
These experiments indicate that, although the affinity of the hydrolysis product P1 to the three enzymes is similar, it results in different inhibitory profiles. We postulate the following schematic reaction mechanism for the three enzymes:

Given that the affinity of the bound product P1 is similar for all three MBLs, we speculate that different kinetic dissociation constants may give rise to distinct inhibitory effects. Consequently, we modeled product binding to these three enzymes based on available structures of cephalosporin hydrolysis products, and performed a geometry optimization of the structures of the resulting adducts by QM/MM calculations (Fig. 6).
 | ||
| Fig. 6 Structures obtained from QM/MM optimizations of (A) NDM-1, (B) VIM-2 and (C) IMP-1 with the hydrolysis product P1. Carbon atoms of P1 are colored in orange, while in the protein the L3 loop is shown in cyan, the L10 loop in pink, and the rest of the protein in green. Detailed view of the interactions between the bound product P1 and residues from the active site cavity of (D) NDM-1 (E) VIM-2 and (F) IMP-1. Carbon atoms of the ligand are shown in orange, while those of the residues of the MBLs are shown in cyan. Zn atoms are shown as grey spheres, hydrogen bonds are shown as dotted lines and hydrogen atoms are omitted for clarity. | ||
These simulations show that the active site of NDM-1 in the EP1 adduct presents a more open conformation of loops L3 and L10 (Fig. 6A) that may favor a fast dissociation of the EP1 adduct. Instead, VIM-2 and IMP-1 present a more closed active site due to the conformations of loop L3 and L10 (Fig. 6B and C). These findings led us to assert that the differential inhibitory effect of FDC of the product-bound form in VIM-2 and IMP-1 is due to a slower dissociation rate compared to NDM-1, which is indeed reflected in the difference of turnover numbers (Table 1). A more detailed analysis of the interactions between P1 and the different enzymes reveals differences due to the distinct active site cavities. In NDM-1, the product interacts via hydrogen bonds and salt bridges with Glu149, Lys224 and Asn233, and has hydrophobic interactions with Trp87 and Met61 (Fig. 6D). In VIM-2, the product interacts via hydrogen bonds with Trp87, Glu149 and Asn233, but the conformation of the L3 loop creates a more closed space with hydrophobic interactions with Phe61 and Tyr67 (Fig. 6E). Similarly, IMP-1 has hydrogen bond interactions with Asp149, Lys224, Asn233 and Ser119, and is surrounded with hydrophobic interactions with Glu59, Val61, Trp64, Val67 and Phe87 (Fig. 6F).
FDC resistance depends on the FDC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :NDM ratio in the periplasm
:NDM ratio in the periplasm
The current biochemical results are in line with the clinical reports informing NDM variants as the only MBLs able to confer resistance to FDC. However, NDM-mediated resistance is associated with the finding of high-number copy plasmids, suggesting that a high concentration of NDM in the periplasm may be necessary to effectively thwart the antibiotic action of FDC. To explore if the overproduction of MBLs increases FDC resistance – regardless of the specific MBL, we used E. coli DH10B cells to express blaNDM-1 and blaIMP-1 from the pMBLe plasmid, which is optimized for controlled production of MBLs with their native signal peptides from an IPTG-inducible promoter.31 This strain provides an isogenic background to compare protein production and resistance to FDC devoid of mutations in the iron transporter gene and of other resistance mechanisms present in clinical isolates.
We determined the minimum inhibitory concentration (MIC) values of FDC in E. coli producing NDM-1 and IMP-1 in Iron-Depleted Cation-Adjusted Mueller–Hinton Broth (ID-CAMHB) as well as the production levels of both enzymes. Comparable protein levels of IMP-1 and NDM-1 resulted in differing resistance profiles. Higher levels of NDM-1 led to an increase of FDC MIC values of four two-fold dilutions (from 0.03 to 0.5 mg L−1), while higher levels of IMP-1 only led to two two-fold dilution increase in the FDC MICs (0.015 to 0.06 mg L−1) (Fig. S17†). These results indicate that the overproduction of NDM-1 may lead to higher increases in MIC values, in contrast to IMP-1, and support the herein proposed inhibition model.
Conclusion
Clinical reports challenging the efficacy of FDC have raised significant concern especially in certain circumstances. These resistance events involve and evolve mutations in the specific iron transporter that FDC utilizes to enter bacterial cells, as well as the hydrolytic activity of specific NDM variants.19–24 Unfortunately, this is not unprecedented: the evolutionary pressure on bacteria has led to a diverse ensemble of resistance determinants, giving rise to the emergence of resistance to newly developed drugs upon their approval for clinical use.58 This scenario highlights the need of a mechanistic understanding of these resistance events to guide informed therapeutic strategies that can effectively leverage and preserve the potential of these new drugs. Deeper understandings also inform likely cross-resistance between β-lactam agents to assist clinical microbiology laboratories with prioritizing susceptibility testing and clinicians with empiric antibiotic selection.Here, we demonstrate that four clinically relevant metallo-β-lactamases (NDM-1, NDM-5, VIM-2, and IMP-1) bind FDC, but only NDM variants display high catalytic efficiencies against this cephalosporin. Stopped-flow binding, ESI-MS and NMR experiments provide evidence for the formation of an enzyme-product adduct (EP1) that we identify as the hydrolyzed FDC molecule that has undergone elimination of the C3 substituent, leading to MBL inhibition. Heteronuclear 1H–15N HSQC NMR shows that the product remains bound to the active site, and the observed perturbations do not result from allosteric binding. The inhibitory effect is potent for VIM-2 and IMP-1, accounting for the low kcat values of these enzymes. In contrast, NDM-1 exhibits weaker inhibition, resulting in a partial population of inhibited enzyme-product (EP1) complexes alongside unbound, active NDM-1 molecules, which is reflected in the high kcat values for NDM-1. NDM-5 exhibits a similar catalytic performance. Since NDM variants accumulate substitutions outside the active site, we anticipate that this situation can be extended to most members of the NDM family. Indeed, the catalytic efficiency of NDM-9 (also resistant to the inhibitory action of the β-lactamase inhibitor taniborbactam) falls within the same range as the NDM variants studied here (Table S1†).24
The modeled structures of the enzyme-product adducts suggest that the potent inhibitory effects observed with VIM-2 and IMP-1 may be attributed to a more extensive interaction network of the product within the active site (Fig. 6). This favors a more closed conformation of the active site loops L3 and L10, resulting in slower dissociation rates. Product-inhibition of MBLs by cephalosporins which underwent C3 elimination has been recently reported,59,60 including clinically used drugs and a cephalosporin prochelator described by Franz and coworkers. Furthermore, a series of novel cephalosporin conjugates synthesized by Martin and coworkers have disclosed a MBL-dependent inhibition,61 with product inhibition potencies ranked as follows: IMP-1 ≫ VIM-2 > NDM-1. In the case of these compounds, the strong inhibition of IMP-1 was attributed to a more closed conformation of loop L3 and the interaction of a Trp residue in this loop with the bound product, in line with our finding for FDC. Furthermore, MBL inhibition also correlated to lower kcat values for these cephalosporin conjugates.
The bound product species (P1) is similar for FDC and CAZ once these antibiotics have undergone C3 elimination (Fig. 1). However, substrate binding experiments (Fig. 2) show a clearly distinct behavior of CAZ, since the fluorescence of the resting state enzyme recovers in all cases, even for VIM-2, which shows the slowest turnover rate. This can be explained based on the KM values of CAZ, which are one order of magnitude smaller than those of FDC (Table 1) and fall within the same range of the dissociation constants of the enzyme-product complex, which is identical for both substrates. Mechanistically, an incoming molecule of CAZ can displace the bound product from the active site of any of the three enzymes based on its higher affinity, while this is not the case for FDC.
The proposed mechanism of inhibition accounts for the fact that MBL-mediated resistance events have been linked only to NDM-1 and NDM-5. One case reported by Simner et al. described the development of FDC resistance in E. coli after two weeks of treatment. Genomic analysis revealed a ten-fold increase in the blaNDM-5 copy number, which, in turn, boosted the FDC MIC from 2 to 32 μg mL−1, i.e., four two-fold dilutions.21 The herein reported in vitro data in E. coli laboratory strains show an equivalent 16-fold increase in MIC values starting from lower basal levels (consistent with the performance of a laboratory strain) and supporting the extrapolations of our study to the resistance phenotypes reported in the clinic.
These results help us propose the following model that accounts for the action of FDC and the resistant events (Fig. 7). As already described, FDC enters the cells through the iron transporters (CirA, in the case of E. coli), and in the presence of low levels of NDM production, most NDM molecules are inhibited and the high periplasmic concentration of FDC makes it possible to inhibit its target, PBP3 (Fig. 7A). Deletions on the CirA transporter impair uptake of FDC that enters through non-specific porins, and the lower levels of FDC in the periplasm enable efficient turnover by NDM in a synergistic mechanism,19 so that FDC cannot inhibit PBP3 (Fig. 7B). Overproduction of NDM variants results in a mixed population of product-bound and unbound MBLs in the periplasm, the latter being able to hydrolyze FDC despite its high concentration, therefore leading to resistance27 (Fig. 7C). In contrast, production of either IMP-1 or VIM-2 results in inhibition of most MBLs molecules in the periplasm, enabling FDC to inhibit PBP3 (Fig. 7D).
 | ||
| Fig. 7 Model of action of FDC under different conditions. (A) FDC is efficient at low expression levels of NDM variants and intact CirA; (B) the activity of FDC is challenged when the cirA gene is mutated impairing FDC uptake combined with the catalytic activity of NDM, that can turnover FDC; (C) high expression levels of NDM variants can elicit resistance to FDC even with an active CirA transporter, and (D) IMP-1 (and also VIM-2) are inhibited by the product of hydrolysis of FDC, being unable to confer resistance. | ||
Current ESCMID Guidelines and IDSA Guidance for treating multidrug resistant bacteria recommend the use of FDC to treat carbapenem-resistant Enterobacterales expressing MBLs.62 Although this remains an important clinical consideration, our findings suggest that caution should be used when using FDC as monotherapy to treat infections caused by NDM-producers given the potential for FDC “MIC creep” and even frank resistance. FDC is a novel therapy that should be preserved as a “precision” infectious disease treatment.58 In the case of NDM, a combination therapy strategy of FDC paired with β-lactamase inhibitors (e.g., xeruborbactam), maybe a critical and necessary strategy to preserve this potent cephalosporin. Bismuth-based drugs are also able to resensitize FDC-resistant strains bearing NDM, and may offer an alternative strategy to prolong the life-span of FDC.63 Instead, our data suggest that FDC can be safely used for VIM and IMP producers.
Our findings also underscore the importance of understanding the specific MBL produced by an infecting organism even to the allelic level (“precision microbiological diagnostics”). The dynamic nature of antimicrobial resistance necessitates anticipation of mechanisms directed to overcome even our best treatments.58 Further research to comprehensively understand, anticipate and address resistance mechanisms to ensure the continued efficacy of new drugs such as FDC in combating challenging infections is mandated.
Data availability
The data supporting this article have been included as part of the ESI.† This study was carried out using NMR assignments of VIM-2 and NDM-1 publicly available data from the Biological Magnetic Resonance Data Bank at https://bmrb.io with accession number BMR51165 and BMR27043, respectively.Author contributions
BAW performed protein expression and purification, enzymatic measurements, stopped-flow experiments and protein NMR experiments, conceptualized experiments, analyzed and interpreted data and wrote the first draft, PET and GB performed protein expression and purification, and enzymatic measurements, MFM conceptualized and performed microbiological experiments, analyzed and interpreted data and wrote the paper, CRB performed protein expression and purification and MS experiments, MRS and GM conceptualized and performed NMR experiments on FDC, analyzed and interpreted data and wrote the paper, SID performed docking and QM/MM calculations, analyzed and interpreted data and wrote the paper, DO performed microbiological experiments, KPW performed MS experiments, PDT reviewed, commented and edited the paper, DMM conceptualized docking and QM/MM calculations, analyzed and interpreted data and wrote the paper, RAB and AJV conceptualized research and wrote the paper. All authors discussed the results and commented on the manuscript.Conflicts of interest
This work was partially supported by Shionogi & Co., Ltd.Acknowledgements
This work was supported by a research grant from Shionogi and the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (NIH) to RAB, GM and AJV under award number R01AI100560. PDT was supported by the NIH (R21-AI173475 and K24-AI185302). This work was supported by grants PICT-2016-1657 and PICT 2020-03769 from Agencia I+D+i to AJV and DMM, a grant from ASaCTeI (PEICID 2023-191) to AJV, and the REPARA network grant (Redes Federales de Alto Impacto, Subsecretaría de Ciencia y Tecnología de la Nación) to AJV. This study was also supported in part by funds and/or facilities provided by the Cleveland Department of Veterans Affairs, award number 1I01BX001974 to RAB from the Biomedical Laboratory Research & Development Service of the VA Office of Research and Development, and the Geriatric Research Education and Clinical Center VISN 10. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH or the Department of Veterans Affairs. BAW, PET, GB and AJV thank Marina Avecilla (IBR-CONICET) for her valuable technical support and dedication. GM and MR would like to thank Gonzalo Hernández Dossi for collaborating designing and performing NMR experiments at Laboratorio de Resonancia Magnética Nuclear, Facultad de Química, Universidad de la República. GM is a researcher at the Programa de Desarrollo de Ciencias Basicas-UY. AJV, PET, SID and DDM are staff members from CONICET. BAW is recipient of a fellowship from Agencia I+D+I, and MR is recipient of a fellowship from Comisión Académica de Posgrado (CAP-Udelar).References
- S. Kariuki, Lancet, 2024, 404, 1172–1173 CrossRef CAS PubMed.
- M. Naghavi, S. Emil Vollset, K. S. Ikuta, L. R. Swetschinski, A. P. Gray, E. E. Wool, G. Robles Aguilar, T. Mestrovic, G. Smith, C. Han, R. L. Hsu, J. Chalek, D. T. Araki, E. Chung, C. Raggi, A. Gershberg Hayoon, N. Davis Weaver, P. A. Lindstedt, A. E. Smith, U. Altay, N. V Bhattacharjee, K. Giannakis, F. Fell, B. McManigal, N. Ekapirat, J. Andretta Mendes, T. Runghien, O. Srimokla, A. Abdelkader, S. Abd-Elsalam, R. Gyan Aboagye, H. Abolhassani, H. Abualruz, U. Abubakar, H. J. Abukhadijah, S. Aburuz, A. Abu-Zaid, S. Achalapong, I. Yeboah Addo, V. Adekanmbi, T. Esther Adeyeoluwa, Q. Estiningtyas Sakilah Adnani, L. Akua Adzigbli, M. Sohail Afzal, S. Afzal, A. Agodi, A. J. Ahlstrom, A. Ahmad, S. Ahmad, T. Ahmad, A. Ahmadi, A. Ahmed, H. Ahmed, I. Ahmed, M. Ahmed, S. Ahmed, S. Anees Ahmed, M. Ahmed Akkaif, S. Al Awaidy, Y. Al Thaher, S. O. Alalalmeh, M. T. AlBataineh, W. A. Aldhaleei, A. Ali Saeed Al-Gheethi, N. Bida Alhaji, A. Ali, L. Ali, S. Shujait Ali, W. Ali, K. Allel, S. Al-Marwani, A. Alrawashdeh, A. Altaf, A. B. Al-Tammemi, J. A. Al-Tawfiq, K. H. Alzoubi, W. Adnan Al-Zyoud, B. Amos, J. H. Amuasi, R. Ancuceanu, J. R. Andrews, A. Anil, I. Adesola Anuoluwa, S. Anvari, A. Edward Anyasodor, G. Louis Carace Apostol, J. Arabloo, M. Arafat, A. Y. Aravkin, D. Areda, A. Aremu, A. A. Artamonov, E. A. Ashley, M. O. Asika, S. Shamsadin Athari, M. Moh, W. Atout, T. Awoke, S. Azadnajafabad, J. Mba Azam, S. Aziz, A. Y. Azzam, M. Babaei, F.-X. Babin, M. Badar, A. Amin Baig, M. Bajcetic, S. Baker, M. Bardhan, H. Jawdat Barqawi, Z. Basharat, A. Basiru, M. Bastard, S. Basu, N. Simegnew Bayleyegn, M. Ashagrie Belete, O. Omolaja Bello, A. Beloukas, J. A. Berkley, A. Srikanth Bhagavathula, S. Bhaskar, S. S. Bhuyan, J. A. Bielicki, N. Ivanovich Briko, C. Stewart Brown, A. J. Browne, D. Buonsenso, Y. Bustanji, C. G. Carvalheiro, C. A. Castañeda-Orjuela, M. Cenderadewi, J. Chadwick, S. Chakraborty, R. Mohan Chandika, S. Chandy, V. Chansamouth, V. Kumar Chattu, A. Ahmad Chaudhary, P. R. Ching, H. Chopra, F. Rabbi Chowdhury, D.-T. Chu, M. Chutiyami, N. Cruz-Martins, A. Gomes da Silva, O. Dadras, X. Dai, S. D. Darcho, S. Das, F. Pio De la Hoz, D. Myriam Dekker, K. Dhama, D. Diaz, B. Felix Rothschild Dickson, S. Ghislain Djorie, M. Dodangeh, S. Dohare, K. Georgieva Dokova, O. Prakashbhai Doshi, R. Kokou Dowou, H. Larson Dsouza, S. J. Dunachie, A. Marian Dziedzic, T. Eckmanns, A. Ed-Dra, A. Eftekharimehrabad, T. Cyrus Ekundayo, I. El Sayed, M. Elhadi, W. El-Huneidi, C. Elias, S. J. Ellis, R. Elsheikh, I. Elsohaby, C. Eltaha, B. Eshrati, M. Eslami, D. William Eyre, A. Oluwaseun Fadaka, A. Francis Fagbamigbe, A. Fahim, A. Fakhri-Demeshghieh, F. Oludayo Fasina, M. Margaret Fasina, A. Fatehizadeh, N. A. Feasey, A. Feizkhah, G. Fekadu, F. Fischer, I. Fitriana, K. M. Forrest, C. Fortuna Rodrigues, J. E. Fuller, M. A. Gadanya, M. Gajdács, A. P. Gandhi, E. E. Garcia-Gallo, D. O. Garrett, R. K. Gautam, M. Welay Gebregergis, M. Gebrehiwot, T. Gebru Gebremeskel, C. Geffers, L. Georgalis, R. Mohamed Ghazy, M. Golechha, D. Golinelli, M. Gordon, S. Gulati, R. Das Gupta, S. Gupta, V. Kumar Gupta, A. Derbie Habteyohannes, S. Haller, H. Harapan, M. L. Harrison, A. I. Hasaballah, I. Hasan, R. Syeda Hasan, H. Hasani, A. Haekyung Haselbeck, M. Saquib Hasnain, I. Ibrahim Hassan, S. Hassan, M. Sadat Hassan Zadeh Tabatabaei, K. Hayat, J. He, O. E. Hegazi, M. Heidari, K. Hezam, R. Holla, M. Holm, H. Hopkins, M. Mahbub Hossain, M. Hosseinzadeh, S. Hostiuc, N. R. Hussein, L. Duc Huy, E. D. Ibáñez-Prada, A. Ikiroma, I. M. Ilic, S. Mohammed Shariful Islam, F. Ismail, N. Elkudssiah Ismail, C. Declan Iwu, C. Juliana Iwu-Jaja, A. Jafarzadeh, F. Jaiteh, R. Jalilzadeh Yengejeh, R. G. Dominic Jamora, J. Javidnia, T. Jawaid, A. W. J Jenney, H. Jin Jeon, M. Jokar, N. Jomehzadeh, T. Joo, N. Joseph, Z. Kamal, K. Kazeem Kanmodi, R. S. Kantar, J. Apollo Kapisi, I. M. Karaye, Y. Saleh Khader, H. Khajuria, N. Khalid, F. Khamesipour, A. Khan, M. Jobair Khan, M. Tariq Khan, V. Khanal, F. Fatima Khidri, J. Khubchandani, S. Khusuwan, M. Seo Kim, A. Kisa, V. Andreevich Korshunov, F. Krapp, R. Krumkamp, M. Kuddus, M. Kulimbet, D. Kumar, E. A. P Kumaran, A. Kuttikkattu, H. Hmwe Kyu, I. Landires, B. Kankia Lawal, T. Thi Thu Le, I. Maria Lederer, M. Lee, S. Won Lee, A. Lepape, T. Leka Lerango, V. S. Ligade, C. Lim, S. S. Lim, L. Workie Limenh, C. Liu, X. Liu, X. Liu, M. J. Loftus, H. M. Ibrahim Amin, K. Lynn Maass, S. B. Maharaj, M. Adam Mahmoud, P. Maikanti-Charalampous, O. M. Makram, K. Malhotra, A. Azam Malik, G. D. Mandilara, F. Marks, B. Alfonso Martinez-Guerra, M. Martorell, H. Masoumi-Asl, A. G. Mathioudakis, J. May, T. A. McHugh, J. Meiring, H. Negash Meles, A. Melese, E. Belayneh Melese, G. Minervini, N. Saad Mohamed, S. Mohammed, S. Mohan, A. H. Mokdad, L. Monasta, A. Moodi Ghalibaf, C. E. Moore, Y. Moradi, E. Mossialos, V. Mougin, G. Duke Mukoro, F. Mulita, B. Muller-Pebody, E. Murillo-Zamora, S. Musa, P. Musicha, L. A. Musila, S. Muthupandian, A. Jayaraman Nagarajan, P. Naghavi, F. Nainu, T. Sadasivan Nair, H. Hama Rashid Najmuldeen, Z. S. Natto, J. Nauman, B. Prakash Nayak, G. Takop Nchanji, P. Ndishimye, I. Negoi, R. Irina Negoi, S. Aria Nejadghaderi, Q. P. Nguyen, E. Ali Noman, D. C. Nwakanma, T. J. Ochoa, I. A. Odetokun, O. Adeolu Ogundijo, T. R. Ojo-Akosile, S. Reuben Okeke, O. Christabel Okonji, A. T. Olagunju, A. Olivas-Martinez, A. Abayomi Olorukooba, P. Olwoch, K. Ikenna Onyedibe, E. Ortiz-Brizuela, O. Osuolale, P. Ounchanum, O. T. Oyeyemi, M. P. Padukudru A, J. L. Paredes, R. R. Parikh, J. Patel, S. Patil, S. Pawar, A. Y. Peleg, P. Peprah, J. Perdigão, C. Perrone, I.-R. Petcu, K. Phommasone, Z. Zahid Piracha, D. Poddighe, A. J. Pollard, R. Poluru, A. Ponce-De-Leon, J. Puvvula, F. Naz Qamar, N. Hashim Qasim, C. Donatien Rafai, P. Raghav, L. Rahbarnia, F. Rahim, V. Rahimi-Movaghar, M. Rahman, M. Aziz Rahman, H. Ramadan, S. Kumaran Ramasamy, P. Sinduvadi Ramesh, P. W. Ramteke, R. Kumar Rana, U. Rani, M.-M. Rashidi, D. Rathish, S. Rattanavong, S. Rawaf, E. Moustafa Mohamed Redwan, L. Felipe Reyes, T. Roberts, J. V Robotham, V. Daniel Rosenthal, A. Guy Ross, N. Roy, K. E. Rudd, C. John Sabet, B. Ahmad Saddik, M. Reza Saeb, U. Saeed, S. Saeedi Moghaddam, W. Saengchan, M. Safaei, A. Saghazadeh, N. Saheb Sharif-Askari, A. Sahebkar, S. Swaroop Sahoo, M. Sahu, M. Saki, N. Salam, Z. Saleem, M. A. Saleh, Y. Leonardo Samodra, A. M. Samy, A. Saravanan, M. Satpathy, A. E. Schumacher, M. Sedighi, S. Seekaew, M. Shafie, P. A. Shah, S. Shahid, M. Jamal Shahwan, S. Shakoor, N. Shalev, M. Aaqib Shamim, M. Ali Shamshirgaran, A. Shamsi, A. Sharifan, R. P. Shastry, M. Shetty, A. Shittu, S. Shrestha, E. Edwar Siddig, T. Sideroglou, J. Sifuentes-Osornio, L. Manuel Lopes Rodrigues Silva, E. A. F Simões, A. J. H Simpson, A. Singh, S. Singh, R. Sinto, S. S. M Soliman, S. Soraneh, N. Stoesser, T. Zhekova Stoeva, C. Kumar Swain, L. Szarpak, S. T. Sudha Y, S. Tabatabai, C. Tabche, Z. Mohammed-Ameen Taha, K.-K. Tan, N. Tasak, N. Y. Tat, A. Thaiprakong, P. Thangaraju, C. Chepngeno Tigoi, K. Tiwari, M. Roberto Tovani-Palone, T. Huu Tran, M. Tumurkhuu, P. Turner, A. John Udoakang, A. Udoh, N. Ullah, S. Ullah, A. Govindaraj Vaithinathan, M. Valenti, T. Vos, H. T. L Vu, Y. Waheed, A. Sarah Walker, J. L. Walson, T. Wangrangsimakul, K. Gayan Weerakoon, H. F. L Wertheim, P. C. M Williams, A. Arja Wolde, T. M. Wozniak, F. Wu, Z. Wu, M. Kumar Kumar Yadav, S. Yaghoubi, Z. Sule Yahaya, A. Yarahmadi, S. Yezli, Y. Engida Yismaw, D. Keon Yon, C.-W. Yuan, H. Yusuf, F. Zakham, G. Zamagni, H. Zhang, Z.-J. Zhang, M. Zieli, A. Zumla, H. H. Zyoud, S. H. Zyoud, S. I. Hay, A. Stergachis, B. Sartorius, B. S. Cooper, C. Dolecek, C. J. L Murray and A. Resistance Collaborators, Lancet, 2024, 404, 1199–1226 CrossRef PubMed.
- M. F. Mojica, M. A. Rossi, A. J. Vila and R. A. Bonomo, Lancet Infect. Dis., 2022, 22, e28–e34 CrossRef CAS PubMed.
- T. Palzkill, Ann. N. Y. Acad. Sci., 2013, 1277, 91–104 CrossRef CAS PubMed.
- G. Bahr, L. J. González and A. J. Vila, Chem. Rev., 2021, 121, 7957–8094 Search PubMed.
- R. A. Bonomo, E. M. Burd, J. Conly, B. M. Limbago, L. Poirel, J. A. Segre and L. F. Westblade, Clin. Infect. Dis., 2018, 66, 1290–1297 CrossRef CAS PubMed.
- K. Bush and P. A. Bradford, Cold Spring Harbor Perspect. Med., 2016, 6, a025247 Search PubMed.
- J. F. Fisher, S. O. Meroueh and S. Mobashery, Chem. Rev., 2005, 105, 395–424 CrossRef CAS PubMed.
- C. L. Tooke, P. Hinchliffe, E. C. Bragginton, C. K. Colenso, V. H. A. Hirvonen, Y. Takebayashi and J. Spencer, J. Mol. Biol., 2019, 431, 3471–3500 CrossRef.
- C. M. Rotondo and G. D. Wright, Curr. Opin. Microbiol., 2017, 39, 96–105 CrossRef CAS.
- L. C. Ju, Z. Cheng, W. Fast, R. A. Bonomo and M. W. Crowder, Trends Pharmacol. Sci., 2018, 39, 635–647 CrossRef CAS PubMed.
- K. Bush and P. A. Bradford, Nat. Rev. Microbiol., 2019, 17, 295–306 Search PubMed.
- B. Liu, R. E. L. Trout, G. H. Chu, D. Mcgarry, R. W. Jackson, J. C. Hamrick, D. M. Daigle, S. M. Cusick, C. Pozzi, F. De Luca, M. Benvenuti, S. Mangani, J. D. Docquier, W. J. Weiss, D. C. Pevear, L. Xerri and C. J. Burns, J. Med. Chem., 2020, 63, 2789–2801 CrossRef CAS.
- S. J. Hecker, K. R. Reddy, O. Lomovskaya, D. C. Griffith, D. Rubio-Aparicio, K. Nelson, R. Tsivkovski, D. Sun, M. Sabet, Z. Tarazi, J. Parkinson, M. Totrov, S. H. Boyer, T. W. Glinka, O. A. Pemberton, Y. Chen and M. N. Dudley, J. Med. Chem., 2020, 63, 7491–7507 CrossRef CAS PubMed.
- S. I. Drusin, C. Le Terrier, L. Poirel, R. A. Bonomo, A. J. Vila and D. M. Moreno, Antimicrob. Agents Chemother., 2024, 68, e01168 Search PubMed.
- N. Kohira, J. West, A. Ito, T. Ito-Horiyama, R. Nakamura, T. Sato, S. Rittenhouse, M. Tsuji and Y. Yamano, Antimicrob. Agents Chemother., 2016, 60, 729–734 Search PubMed.
- A. Ito, T. Nishikawa, S. Matsumoto, H. Yoshizawa, T. Sato, R. Nakamura, M. Tsuji and Y. Yamano, Antimicrob. Agents Chemother., 2016, 60, 7396–7401 CrossRef CAS PubMed.
- T. Ito-Horiyama, Y. Ishii, A. Ito, T. Sato, R. Nakamura, N. Fukuhara, M. Tsuji, Y. Yamano, K. Yamaguchi and K. Tateda, Antimicrob. Agents Chemother., 2016, 60, 4384–4386 Search PubMed.
- S. Klein, S. Boutin, K. Kocer, M. O. Fiedler, D. Störzinger, M. A. Weigand, B. Tan, D. Richter, C. Rupp, M. Mieth, A. Mehrabi, T. Hackert, S. Zimmermann, K. Heeg and D. Nurjadi, Clin. Infect. Dis., 2022, 74, 905–908 Search PubMed.
- D. Nurjadi, K. Kocer, Q. Chanthalangsy, S. Klein, K. Heeg and S. Boutin, Antimicrob. Agents Chemother., 2022, 66, e0201121 Search PubMed.
- P. J. Simner, H. H. Mostafa, Y. Bergman, M. Ante, T. Tekle, A. Adebayo, S. Beisken, K. Dzintars and P. D. Tamma, Clin. Infect. Dis., 2022, 75, 47–54 Search PubMed.
- P. J. Simner, Y. Bergman, R. Conzemius, E. Jacobs, T. Tekle, S. Beisken and P. D. Tamma, Open Forum Infect. Dis., 2023, 10, ofad276 Search PubMed.
- N. B. Fiona Senchyna, K. Murugesan, W. Rotunno, S. S. Nadimpalli and S. Deresinski, Clin. Infect. Dis., 2024, 78, 1425–1428 CrossRef PubMed.
- S. Gaillot, S. Oueslati, J. B. Vuillemenot, M. Bour, B. I. Iorga, P. Triponney, P. Plésiat, R. A. Bonnin, T. Naas, K. Jeannot and A. Potron, Front. Microbiol., 2023, 14, 1253160 CrossRef.
- G. Haidar, E. G. Kline, G. D. Kitsios, X. Wang, E. J. Kwak, A. Newbrough, K. Friday, K. H. Kramer and R. K. Shields, JAC Antimicrob. Resist., 2024, 6, dlae141 Search PubMed.
- C. Tascini, M. Coppi, A. Antonelli, C. Niccolai, A. Bartolini, D. Pecori, A. Sartor, T. Giani and G. M. Rossolini, Clin. Microbiol. Infect., 2024, 30, 398–400 CrossRef CAS.
- S. Mushtaq, Z. Sadouki, A. Vickers, D. M. Livermore and N. Woodford, Antimicrob. Agents Chemother., 2020, 64, e01582 CrossRef CAS PubMed.
- Q. Wang, L. Jin, S. Sun, Y. Yin, R. Wang, F. Chen, X. Wang, Y. Zhang, J. Hou, Y. Zhang, Z. Zhang, L. Luo, Z. Guo, Z. Li, X. Lin, L. Bi and H. Wang, Microbiol. Spectrum, 2022, 10, e0267021 CrossRef.
- L. Poirel, J. M. O. de la Rosa, Z. Sakaoglu, A. Kusaksizoglu, M. Sadek and P. Nordmann, Antimicrob. Agents Chemother., 2022, 66, 1–5 Search PubMed.
- C. Lasarte-Monterrubio, P. Guijarro-Sánchez, J. C. Vázquez-Ucha, I. Alonso-Garcia, L. Alvarez-Fraga, M. Outeda, M. Martinez-Guitian, A. Peña-Escolano, R. Maceiras, E. Lence, C. González-Bello, J. Arca-Suárez, G. Bou, A. Beceiro, I. Merino, E. Cercenado, R. Gómez, T. Soler, I. Gracia-Ahufinger, L. Martín, F. Galán, N. Tormo, J. C. Rodríguez, S. Capilla, F. Marco, M. D. Quesada, E. Padilla, F. Tubau, J. González, A. I. López-Calleja, J. L. del Pozo, M. I. García, M. Martinez, J. Calvo, X. Mulet, F. Peña, A. I. Rodríguez, M. J. Gude, A. Fernández, J. Fernández, F. Fernandez-Cuenca and A. Pascual, Antimicrob. Agents Chemother., 2023, 67, e0150522 CrossRef PubMed.
- L. J. González, G. Bahr, T. G. Nakashige, E. M. Nolan, R. A. Bonomo and A. J. Vila, Nat. Chem. Biol., 2016, 12, 516–522 Search PubMed.
- A. R. Palacios, M. F. Mojica, E. Giannini, M. A. Taracila, C. R. Bethel, P. M. Alzari, L. H. Otero, S. Klinke, L. I. Llarrull, R. A. Bonomo and A. J. Vila, Antimicrob. Agents Chemother., 2019, 63, e01754 CrossRef CAS.
- Clinical and Laboratory Standards Institute (CLSI), Performance Standards for Antimicrobial Susceptibility Testing, CLSI supplement M100, Clinical and Laboratory Standards Institute, Wayne, PA, 34th edn, 2024 Search PubMed.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J. Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS.
- G. M. Morris, H. Ruth, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 Search PubMed.
- D. Santos-Martins, L. Solis-Vasquez, A. F. Tillack, M. F. Sanner, A. Koch and S. Forli, J. Chem. Theory Comput., 2021, 17, 1060–1073 Search PubMed.
- H. Zhang, G. Ma, Y. Zhu, L. Zeng, A. Ahmad, C. Wang, B. Pang, H. Fang, L. Zhao and Q. Hao, Antimicrob. Agents Chemother., 2018, 62, e01579 Search PubMed.
- K. Yamamoto, H. Tanaka, G. Kurisu, R. Nakano, H. Yano and H. Sakai, J. Biochem., 2022, 173, 21–30 CrossRef.
- D. A. Case, K. Belfon, I. Y. Ben-Shalom, S. R. Brozell, D. S. Cerutti, T. E. III, V. W. D. Cruzeiro, T. A. Darden, R. E. Duke, G. Giambasu, M. K. Gilson, H. Gohlke, A. W. Goetz, R. Harris, S. Izadi, S. A. Iz mailov, K. Kasavajhala, A. Kovalenko, R. Krasny, T. Kurtzman, T. S. Lee, S. LeGrand, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, V. Man, K. M. Merz, Y. Miao, O. Mikhailovskii, G. Monard, H. Nguyen, A. Onufriev, F. Pan, S. Pantano, R. Qi, D. R. Roe, A. Roitberg, C. Sagui, S. Schott-Verdugo, J. Shen, C. L. Simmerling, N. R. Skrynnikov, J. Smith, J. Swails, R. C. Walker, J. Wang, L. Wilson, R. M. Wolf, X. Wu, Y. Xiong, Y. Xue, D. M. York and P. A. Kollman, Amber20, University of California, San Francisco, 2020 Search PubMed.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, J. Chem. Theory Comput., 2015, 11, 3696–3713 Search PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- J. E. Raczynska, I. G. Shabalin, W. Minor, A. Wlodawer and M. Jaskolski, Drug Resistance Updates, 2018, 40, 1–12 CrossRef PubMed.
- A. Lucic, P. Hinchliffe, T. R. Malla, C. L. Tooke, J. Brem, K. Calvopiña, C. T. Lohans, P. Rabe, M. A. McDonough, T. Armistead, A. M. Orville, J. Spencer and C. J. Schofield, Eur. J. Med. Chem., 2021, 215, 113257 CrossRef CAS.
- D. Sulton, D. Pagan-Rodriguez, X. Zhou, Y. Liu, A. M. Hujer, C. R. Bethel, M. S. Helfand, J. M. Thomson, V. E. Anderson, J. D. Buynak, L. M. Ng and R. A. Bonomo, J. Biol. Chem., 2005, 280, 35528–35536 Search PubMed.
- A. Makena, J. Brem, I. Pfeffer, R. E. J. Geffen, S. E. Wilkins, H. Tarhonskaya, E. Flashman, L. M. Phee, D. W. Wareham and C. J. Schofield, J. Antimicrob. Chemother., 2015, 70, 463–469 CrossRef CAS.
- J. D. Docquier, J. Lamotte-Brasseur, M. Galleni, G. Amicosante, J. M. Frère and G. M. Rossolini, J. Antimicrob. Chemother., 2003, 51, 257–266 CrossRef CAS PubMed.
- N. Laraki, N. Franceschini, G. M. Rossolini, P. Santucci, C. Meunier, E. De Pauw, G. Amicosante, J. M. Frère and M. Galleni, Antimicrob. Agents Chemother., 1999, 43, 902–906 CrossRef CAS PubMed.
- H. Feng, J. Ding, D. Zhu, X. Liu, X. Xu, Y. Zhang, S. Zang, D. C. Wang and W. Liu, J. Am. Chem. Soc., 2014, 136, 14694–14697 CrossRef CAS PubMed.
- R. Tripathi and N. N. Nair, ACS Catal., 2015, 5, 2577–2586 CrossRef CAS.
- C. K. Das and N. N. Nair, Phys. Chem. Chem. Phys., 2017, 19, 13111–13121 Search PubMed.
- E. O. Levina, M. G. Khrenova, A. A. Astakhov and V. G. Tsirelson, RSC Adv., 2020, 10, 8664–8676 RSC.
- A. Badarau, A. Llińa, A. P. Laws, C. Damblon and M. I. Page, Biochemistry, 2005, 44, 8578–8589 CrossRef CAS PubMed.
- J. Spencer, A. R. Clarke and T. R. Walsh, J. Biol. Chem., 2001, 276, 33638–33644 CrossRef CAS PubMed.
- R. M. Rasia and A. J. Vila, J. Biol. Chem., 2004, 279, 26046–26051 CrossRef CAS PubMed.
- P. W. Thomas, M. Cammarata, J. S. Brodbelt and W. Fast, ChemBioChem, 2014, 15, 2541–2548 CrossRef CAS.
- A. Zervosen, M. H. Valladares, B. Devreese, C. Prosperi-Meys, H. W. Adolph, P. S. Mercuri, M. Vanhove, G. Amicosante, J. Van Beeumen, J. M. Frère and M. Galleni, Eur. J. Biochem., 2001, 268, 3840–3850 CrossRef CAS PubMed.
- P. S. Mercuri, I. García-Sáez, K. De Vriendt, I. Thamm, B. Devreese, J. Van Beeumen, O. Dideberg, G. M. Rossolini, J. M. Frère and M. Galleni, J. Biol. Chem., 2004, 279, 33630–33638 Search PubMed.
- R. A. Bonomo, F. Perez, A. M. Hujer, K. M. Hujer and A. J. Vila, Clin. Infect. Dis., 2024, 78, 1429–1433 CrossRef CAS PubMed.
- N. J. Torelli, A. Akhtar, K. Defrees, P. Jaishankar, O. A. Pemberton, X. Zhang, C. Johnson, A. R. Renslo and Y. Chen, ACS Infect. Dis., 2019, 5, 1013–1021 Search PubMed.
- A. C. Jackson, J. M. Zaengle-Barone, E. A. Puccio and K. J. Franz, ACS Infect. Dis., 2020, 6, 1264–1272 CrossRef CAS PubMed.
- K. H. M. E. Tehrani, N. Wade, V. Mashayekhi, N. C. Brüchle, W. Jespers, K. Voskuil, D. Pesce, M. J. Van Haren, G. J. P. Van Westen and N. I. Martin, J. Med. Chem., 2021, 64, 9141–9151 CrossRef CAS PubMed.
- P. D. Tamma, E. L. Heil, J. A. Justo, A. J. Mathers, M. J. Satlin and R. A. Bonomo, Clin. Infect. Dis., 2024, ciae403 CrossRef.
- C. Wang, Y. Xia, R. Wang, J. Li, C. L. Chan, R. Y. T. Kao, P. H. Toy, P. L. Ho, H. Li and H. Sun, Nat. Commun., 2023, 14, 5311 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc02122g |
| This journal is © The Royal Society of Chemistry 2025 |