
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence2D spatial structure-favored, tandem catalysis-boosted direct transformation of methane to methanol over Cu-FER†
Ning
Liu
 ,
Tingting
Zhang
,
Chengna
Dai
,
Ruinian
Xu
,
Gangqiang
Yu
,
Ning
Wang
and
Biaohua
Chen
*
,
Tingting
Zhang
,
Chengna
Dai
,
Ruinian
Xu
,
Gangqiang
Yu
,
Ning
Wang
and
Biaohua
Chen
*
College of Environmental and Science Engineering, Beijing University of Technology, Beijing, 100124, China. E-mail: chenbh@bjut.edu.cn
First published on 10th June 2025
Abstract
The direct transformation of methane into methanol (DMTM) remains a significant challenge of C1 chemistry. Herein, we investigate continuous N2O-DMTM over Cu-FER zeolite. A two-dimensional (2D) spatial structure-favored tandem catalysis is for the first time elucidated, which leads to boosted (CH3OH + DME) productivity, corresponding to 2736 μmol gcat−1 h−1 or 58![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 368 mmol per molCu per h of CH3OH, and improved reaction stability (passing through a 100 h long-term test). A unique dual Cu single-atom site located at the parallel 6-membered ring (MR) of the 8 MR channel could be generated, which serves as the primary CH3OH production active site exhibiting much higher activity than the traditional monomeric [Cu]+ and Cu dimer sites. The generated CH3OH can subsequently diffuse from the 8 MR channel into a 10 MR main channel and directly react with the radicals of CH3− and OH− to produce DME not only favoring DME production but also efficiently preventing carbon deposition. The present work highlights a tandem catalysis over Cu-FER that would substantially favor the design of other efficient catalysts for N2O-DMTM.
368 mmol per molCu per h of CH3OH, and improved reaction stability (passing through a 100 h long-term test). A unique dual Cu single-atom site located at the parallel 6-membered ring (MR) of the 8 MR channel could be generated, which serves as the primary CH3OH production active site exhibiting much higher activity than the traditional monomeric [Cu]+ and Cu dimer sites. The generated CH3OH can subsequently diffuse from the 8 MR channel into a 10 MR main channel and directly react with the radicals of CH3− and OH− to produce DME not only favoring DME production but also efficiently preventing carbon deposition. The present work highlights a tandem catalysis over Cu-FER that would substantially favor the design of other efficient catalysts for N2O-DMTM.
Introduction
Direct transformation of methane to methanol (DMTM), being recognized as the holy grail of C1 chemistry, has attracted great attention.1–3 In contrast to the traditional syngas route, which needs high temperature and pressure, DMTM, operated under mild conditions, is highly appealing.2,4,5 However, the conflict between the exceptionally high C–H bond energy of CH4 (413 kJ mol−1) and the limited thermostability of CH3OH leads to low CH3OH productivity.6 To address this issue, extensive efforts have been focused on developing highly efficient low-temperature catalysts7 and utilizing alternative oxidants such as N2O8–12 and H2O2 (ref. 13 and 14) to enhance CH3OH production. N2O, known as a greenhouse gas, can be generated in large quantities from adipic acid chemical production, which produces one type of active oxygen species (named αO) through interaction with zeolite catalyst under mild conditions (around 250 °C).11 Thereby, N2O-DMTM, taking advantage of in situ-generated active αO, constitutes a promising approach to achieve high CH3OH production.The Cu-zeolites10,15–20 have been widely investigated in DMTM. Various types of active sites have been reported, including monomeric [Cu]+,17,18 [Cu–O–Cu]2+,10 [Cu3O3]2+ (ref. 19) and the newly reported extraframework [Cu2AlO3]2+.20 Most recently, it is reported that dual single-atom (SA) synergistic catalysis exhibits significantly higher reaction efficiency for DMTM relative to other catalyst systems.13,21–23 For example, the synergistic interaction between the Cu–Ag dual SA active site on ZSM-5 facilitates the formation of highly reactive surface hydroxyl species and promotes the activation of C–H bond in CH4.13 A Cu pair dual SA located at the 8-rings of adjacent gme cavities of MAZ zeolite is reported to be favorable for O2-DMTM.21 A binuclear Fe(II) located at the adjacent six-membered ring (6 MR) of FER zeolite can split O2 even at room temperature for DMTM.22,23 Furthermore, it is also reported that regulation of the distance between the dual Cu SA sites as well as the proximity between Brønsted acid sites (BAS) and Cu active sites are crucial strategies to further enhance CH3OH production,24 due to the close proximity of the dual Cu SA site, which can lead to overoxidation of CH3OH into CO2, while the closely situated Cu site and BAS can facilitate the formation of hydrocarbons (CxHy) through further reaction of CH3OH over the BAS site. Inspired by these literature reports, we can infer that leveraging the unique spatial structure of zeolite catalyst to create a highly efficient dual SA site would constitute a viable route for efficient DMTM catalyst design; meanwhile, selecting large porous topologized zeolites can increase the distance between active site and BAS to mitigate excessive production of CxHy byproducts.24
In the present work, a series of Cu-modified zeolites with different topological structures (FER, BEA, MFI, MOR, Si/Al ≈ 15, Cu = 0.3 wt%) were prepared by wet ion-exchange method and investigated for continuous N2O-DMTM (Fig. 1b and S1a–e†). Boosted productivity can be achieved for the Cu-FER-0.3%, being two orders of magnitude higher than the other counterparts (2736 versus 11–65 μmol gcat−1 h−1, Fig. 1b), and which also notably exceeds most of the recently reported studies (58368 mmol molCu−1 h−1, Fig. 1c and Table S1†). This indicates an exceptionally higher reaction efficiency of the loaded copper species over Cu-FER-0.3%. Additionally, long-term stability is evident (Fig. S2a†), with CH3OH productivity remaining at approximately 1540 μmol gcat−1 h−1 (32853 mmol molCu−1 h−1) even after 100 h of testing. A 2D (two-dimensional) zeolitic spatial structure-favored tandem catalysis is for the first time unravelled over Cu-FER-0.3% (Fig. 1a). Briefly, favored by the 2D spatial structure of FER, a unique dual Cu SA site is formed at the parallel 6 MR site of the 8 MR channel, which possesses much higher CH3OH production efficiency than the traditional [Cu]+ monomer and [Cu]+–[Cu]+ dimer sites. Subsequently, the generated CH3OH could diffuse into the 10 MR channel for tandem catalysis to produce dimethyl ether (DME), wherein the CH3OH, functioning as a “solvent,” can directly react with the radicals of CH3− and OH−, which not only promotes DME production but also efficiently hinders carbon deposition. Generally, the present work presents an efficient, continuous N2O-DMTM system based on Cu-FER zeolite and provides profound mechanistic insights, which would substantially favor other highly efficient catalyst designs.
 | ||
| Fig. 1 Schematic diagram of N2O-DMTM over Cu-FER and activity measurement results (T = 330 °C and t = 6 h). (a) A dual Cu SA site can be formed at the opposite 6 MR of the β site of the 8 MR channel, efficiently favoring αO formation, CH4 activation and CH3OH desorption. The CH3OH would diffuse from the 8 MR channel into the 10 MR channel and readily react with CH3− and OH− radicals, generated over the Cu dimer ([Cu]+–[Cu]+), to produce DME. (b) Total productivity (CH3OH + DME) comparison over Cu-zeolite (BEA, MFI, MOR and FER) with Cu loading of 0.3 wt% (average value after 6 h reaction). (c) Productivity comparison with recent literature report of continuous DMTM (see Table S1†). (d) Total (CH3OH + DME) productivity comparison over Cu-FER with diverse Cu loadings (0.11, 0.3, 0.6 and 1.0 wt%) in the absence and presence of H2O (10 vol%). (e) Product selectivities corresponding to activity measurements in panel (d); reaction conditions: N2O/CH4/He/(H2O) = 30:15:55(45):(10), with gas hourly specific velocity (GHSV) = 12000 h−1, mcat = 0.5 g; other activity measurement results are profiled in Fig. S1a–e and 3a–f.† The total productivity, encompassing both CH3OH and DME, was calculated by considering one mole of DME as equivalent to two moles of CH3OH. | ||
Results and discussion
N2O-DMTM over Cu-FER with varying Cu loadings and basic characterizations
The influence of Cu loading (0.11–1 wt%) was also investigated for the N2O-DMTM in the absence and presence of H2O (Fig. 1d, e and S3a–f†), which suggests an optimum Cu loading of 0.3 wt% with the highest CH3OH productivity and (CH3OH + DME) selectivity of 80.3% (in the absence of H2O) and 73.4% (in the presence of H2O), respectively (Table S2†). Comparable CH4 conversions (Fig. S3f†) can be found over the samples of Cu-FER-0.11%, 0.3% and 0.6% (4.6, 4.7 and 4.8% of N2O-DMTM and 3.9, 3.2 and 2.3% of N2O-DMTM-H2O) with relatively low Cu loadings. However, a significant increase in CH4 conversions (50.3% and 49.7%) is evident for the Cu-FER-1% sample during both N2O-DMTM and N2O-DMTM-H2O. This enhancement is closely related to the overoxidation of CH4, resulting in substantial CO2 production, displaying a high CO2 selectivity of 99.1 (N2O-DMTM) and 99.2% (N2O-DMTM-H2O), as shown in Fig. 1e. This finding indicates that there would exist competing pathways (N2O → N2 + O2), leading to the over-oxidation of CH4, which is closely correlated with the coordination environment of Cu species (CuOx over Cu-FER-1%, as unravelled by H2-TPR). Notably, H2O alters the reaction selectivity, with CH3OH being the dominant product when H2O is present, while dimethyl ether (DME) predominates during its absence. This can be closely correlated with the specific reaction mechanism, which will be further discussed later.To characterize the chemical states of the loaded Cu species over Cu-FER samples, X-ray diffraction (XRD), XPS, and H2-TPR were conducted. The XRD patterns (Fig. S4†) primarily display peaks characteristic of the FER framework, with no distinct peaks attributable to CuOx species. The XPS spectra (Fig. 2a) show that the surface Cu species are predominantly in the +2 oxidation state, evidenced by the Cu 2p3/2 binding energy at 933.5 eV.10,25 Shake-up satellite peaks around 945.4 eV for the samples with Cu loading above 0.6% and 944.0 eV for Cu-FER-0.3% were observed, indicating the presence of Cu2+ species.25 No obvious XPS signals related to CuOx species (936 eV10) could be detected, which suggests that the CuOx species, (as detected by H2-TPR of Cu-FER-0.6% and 1.0%, Fig. 2b), are highly dispersed and does not form large, detectable crystalline domains. As noted, no Cu+ species were detected, which commonly display the lower Cu 2p3/2 peak at around 932 eV. H2-TPR results (Fig. 2b) further reveal that Cu2+ cations are the predominant species in samples with lower Cu loadings (0.11 and 0.3 wt%). In contrast, CuOx species gradually form in samples with higher Cu loadings (0.6 and 1.0 wt%). This finding aligns with the decreased product selectivity (Fig. 1e) observed in Cu-FER samples with higher Cu loading (0.6 and 1.0 wt%), due to the formation of CuO species that readily promote CH4 overoxidation.10 Furthermore, quantitative H2-TPR analysis, based on H2 consumption of standard CuO (Table S3†), corroborates the identification of Cu species in the Cu-FER samples. As noted, the loaded Cu species commonly exist in two different states over the Cu-modified zeolites: CuOx oxidation state and Cu2+ cation state (being exchanged with the Brønsted acid site). These two types of Cu species are distinguishable by H2-TPR due to different redox abilities. The CuOx species would be much more easily reduced relative to the Cu2+ cation, which is commonly ion-exchanged inside the zeolite channel and is relatively harder to be reduced. As for the CuOx species, the small-particle CuOx would be much more readily reduced than bulk CuOx, thereby displaying two types of reduction peaks in Fig. 2b over the samples of Cu-FER-0.6% and Cu-FER-1%.
 | ||
| Fig. 2 Physicochemical characterizations of Cu-FER samples with various Cu loadings (0.11, 0.3, 0.6, and 1%). (a) XPS, (b) H2-TPR, and (c) CO-probed in situ Fourier transform infrared spectroscopy (FTIR); (d) UV-vis diffuse reflectance spectroscopy (UV-vis DRS); and (e) electron paramagnetic resonance (EPR). He–Cu-FER in panels (c–e) represents the Cu-FER-0.3% being pretreated at 500 °C under a He atmosphere (>99.999%, 40 mL min−1) for 1 h; N2O–Cu-FER represents Cu-FER-0.3% initially pretreated by He (following the above procedure) and then further treated by N2O (30 vol% N2O in He) at 250 °C for 1 h. (f) Cu K-edge X-ray adsorption near-edge spectra (XANES) and (g) Fourier-transformed extended X-ray absorption fine structure (FT-EXAFS) spectra of N2O-pretrated Cu-FER-0.3%, Cu foil and CuO; (h) imaginary and (i) magnitude part of phase-corrected FT-EXAFS spectra for N2O-pretrated Cu-FER-0.3% obtained by Fourier transforming the k3χ(k) curves in Fig. S5† of 2.4–10.8 Å. (j) The wavelet transform of k3-weighted EXAFS spectra; the lobes corresponding to Cu–O and Cu–Cu scattering contributions are pointed out with arrows. (k) The 27Al nuclear magnetic resonance (NMR) spectra of Cu-FER-0.3 and predicted contents of different Al location sites based on peak area integration. | ||
Based on the above basic characterizations, the Cu cations constitute the active species over Cu-FER-0.3%. The Cu cations typically exist as monomeric [Cu]+ or a Cu dimer (adjacent [Cu]+–[Cu]+ that can evolve into [Cu–O–Cu]2+ following oxidation) within the Cu-zeolite.15,16,18,24,26,27 To make a further identification, CO-probed FTIR, UV-vis DRS and EPR were initially employed to characterize Cu-FER-0.3%, wherein the sample was respectively pretreated by He and N2O, denoted as He–Cu-FER and N2O–Cu-FER, for better comparison. A characteristic CO adsorption vibration band over the monomeric [Cu]+ site (vCO = 2154 cm−1) was clearly observed (Fig. 2c),10 and the band intensity of N2O–Cu-FER was decreased to a certain degree relative to He–Cu-FER. This finding indicates the coexistence of both monomeric [Cu]+ and Cu dimer ([Cu]+–[Cu]+). The Cu dimer can be oxidized into [Cu–O–Cu]2+ by N2O, eventually leading to the observed decrease of vCO band at 2154 cm−1. Notably, the Cu+ cations detected by CO-probed in situ FTIR (Fig. 2c) can be closely related to the auto-reduction of Cu2+ cations during high-temperature (T = 500 °C) He pretreatment. Further pretreatment by N2O (T = 250 °C) would lead to the formation of [Cu–O–Cu]2+ by the neighboring monomeric [Cu]+, eventually resulting in the decreasing vibration peak intensity. UV-vis DRS and EPR corroborate this finding, wherein the N2O–Cu-FER shows a characteristic [Cu–O–Cu]2+ UV-vis peak at ∼440 nm (Fig. 2d),28 aligning with its decreased intensity seen in EPR (Fig. 2e) due to the EPR-silent nature of [Cu–O–Cu]2+. Taking advantage of this, the Cu dimer content was further predicted by EPR,10 accounting for 20 wt% of Cu over Cu-FER-0.3%.
To further identify such oxo-Cu dimer motif structure ([Cu–O–Cu]2+), the N2O–Cu-FER was characterized using XAS. The Cu K-edge XANES (Fig. 2f) displayed a distinct pre-edge peak at approximately 8978 eV, which is characteristic of the 1s–3d transition in Cu2+10,17, indicating dominant Cu2+ species in Cu-FER-0.3%. The white line (WL) intensity is greater than that of Cu foil, yet lower than that of CuO, suggesting the Cu species is in a less oxidative environment compared to CuO. Moreover, a distinct FT-EXAFS spectrum was observed for Cu-FER-0.3%, in contrast to CuO and Cu foil (Fig. 2g). The EXAFS fitting (Fig. 2i, S6 and Table S4†), combined with wavelet transform (WT) analysis (Fig. 2j), further confirmed the existence of [Cu–O–Cu]2+, which shows a Cu–Cu distance of 3.38 Å, wherein the EXAFS-WT clearly displays two lobes, at low (2.5–7.5 Å−1) and high (10–15 Å−1) k ranges, being respectively associated with framework O/Si/Al and Cu–Cu contribution in the oxygen-bridged Cu dimer.29 Meanwhile, the EXAFS-WT indicated that the Cu species is majorly located at the ion-exchange site being coordinated by the framework O. Notably, density functional theory (DFT) calculations were conducted to determine the site of such Cu dimer (Fig. S7†), which indicated that the Cu dimer site is energetically favorable to be located at the opposite T3 site of 10 MR, and such structure was utilized for the EXAFS peak fitting.
Next, 27Al NMR was conducted to elucidate the specific location of Al atoms and related occupation ratios over Cu-FER-0.3%. The 27Al NMR spectrum (Fig. S8†) displays a prominent signal around 55 pm, with no significant signals detected between 20–40 ppm indicative of extra-framework Al,8 which indicates that the Al atoms are primarily located within the framework.30 Moreover, the resonance band can be deconvoluted into three distinct bands (Fig. 2k), attributable to framework Al located at T2 (51.5 ppm), T3 (53.3 ppm), and T4 (55.2 ppm).31 The specific ratio was accordingly predicted based on the peak area integral, which revealed that the framework Al is mainly located at the T3 (44.8%, 10 MR channel) and T4 (41.0%, 8 MR channel) sites. This finding indicated that the T3 and T4 sites act as major potential sites for Cu accommodation over Cu-FER-0.3%, which well supports the DFT calculation (Fig. S7†) pinpointing the Cu dimer site at the T3 position of the 10 MR ring.
N2O-DMTM over traditional monomeric [Cu]+ and Cu dimer ([Cu]+–[Cu]+) site
To uncover the reason for the boosted N2O-DMTM performance of Cu-FER, DFT was initially employed to simulate N2O-DMTM over the monomeric [Cu]+ and Cu dimer ([Cu]+–[Cu]+) sites of Cu-FER and with the calculated energy barrier (ΔE) being compared with those of Cu-BEA10 and Cu-ZSM-5 (ref. 32) in our previous works (see Fig. 3a). As noted, the radical mechanism was simulated by DFT, as revealed by in situ FTIR study (Fig. S9a and b†), which is also consistent with our previous reports.10,32 As shown in Fig. 3a, comparable energy barriers can be observed for both scenarios of the monomeric [Cu]+ and Cu dimer sites of Cu-FER relative to other topologized zeolites. This finding indicates that the zeolitic topology structure would exert limited effect on the intrinsic N2O-DMTM efficiency of the traditional monomeric [Cu]+ and Cu dimer sites. Such discrepancy with the boosted CH3OH productivity of Cu-FER (Fig. 1b) implies that there must exist some other super active site over Cu-FER. | ||
| Fig. 3 DFT and microkinetic modeling results of N2O-DMTM. (a) Energy barrier comparisons of Cu-FER with other topologized Cu-zeolites (BEA10 and MFI32) in our previous works; the superscript M indicates the Cu monomeric site, and D represents the Cu dimer. (b) Optimized oxo-Cu active site motif structures. (c) Energy diagram over the monomeric [Cu]+ and Cu dimer sites of Cu-FER and reaction rate comparisons derived by microkinetic modelling (Tables S5 and S6†); the optimized models are depicted in Fig. S10 and S11,† and the monomeric [Cu]+ active site model is inset in panel (c) for a better display. O red, Cu orange, Al pink, Si yellow, C gray, H, white. (d and e) Surface coverage variations along time t derived by microkinetic modeling of (d) monomeric [Cu]+ and (e) the Cu dimer of Cu-FER; as noted, the opposite T3 site (Fig. S7†) and single T3 site (Fig. S12†) in the 10 MR channel were selected as the energetically favorable sites for Cu dimer and monomeric [Cu]+, respectively. | ||
Carefully analyzing the energy diagrams in Fig. 3a and c, one can find that the monomeric [Cu]+ site possesses much lower barriers in CH4 activation (TSII), CH3−, OH− radical rebound (TSIII), and CH3OH desorption steps (0.12, 0.42 and 1.12 eV, Cu-FER) than the Cu dimer (0.49, 0.64 and 1.58 eV, Cu-FER); however, it displays a notably higher barrier in the initial N2O dissociation step (TSI, 1.35 versus 0.89 eV, Cu-FER). Microkinetic modelling (Fig. 3c–e, Tables S5 and S6†) further indicates a much higher reaction efficiency of the monomeric [Cu]+ site, with the reaction rate being two orders of magnitude higher than the Cu dimer (8.7 × 101versus 8.1 × 10−1 s−1, Fig. 3c). Meanwhile, the N2O dissociation step serves as the rate-determining-step (RDS) over the monomeric [Cu]+ site (Fig. 3d), and the CH3OH desorption step (Fig. 3e) constitutes the RDS of the Cu dimer site, with the surface-adsorbed N2O (θN2O) and CH3OH (θCH3OH) respectively constituting the major surface species. In this regard, we speculate that the boosted N2O-DMTM activity could be probably achieved if the N2O dissociation barrier can be extensively reduced over the monomeric [Cu]+ site of Cu-FER.
N2O-DMTM over dual Cu SA site
According to the pioneering work of Sobalik's group33,34 a distinct Fe cation accommodation site within the FER zeolite has been reported, which allows for the placement of two Fe cations at the parallel 6 MR plane (β site) by the Al pair, with a [Fe]2+–[Fe]2+ distance of 7.4 Å, consequently leading to significantly higher N2O dissociation activity of Fe-FER relative to Fe-ZSM-5 and Fe-BEA, due to the synergistic effect of distant [Fe]2+. Favored by this, Fe-FER has also been extended to O2-DMTM.22,23 Inspired by these literature reports, we supposed that there probably also exists a similar accommodation site for the [Cu]+ cations that can efficiently favor N2O dissociation. However, the large [Cu]+–[Cu]+ distance complicates the site's identification, rendering it “invisible” to extended X-ray absorption fine structure (EXAFS) or diffraction experiments.35 Therefore, to shed more light, ab initio molecular dynamics (AIMD) was employed in the present work to simulate N2O-DMTM over a constructed Cu-FER model featuring two [Cu]+ cations positioned on a parallel 6 MR plane (β site) (Fig. 1a, d[Cu]+–[Cu]+ = 7.49 Å). Notably, each [Cu]+ is coordinated by one AlO4− of the T4 site that has been confirmed as a primary location site for Cu cations based on 27Al NMR (Fig. 2k). We refer to this active site as a “dual Cu SA site” to distinguish it from the Cu dimer site.A remarkably low Gibbs free energy barrier of only 0.26 eV needs to be crossed to generate αO over such dual Cu SA site (Fig. 4a, b, d and Movie S1†), which is substantially lower than the ΔG values of 1.33 and 0.76 eV for the monomeric [Cu]+ and Cu dimer site, respectively. Moreover, due to the extended [Cu]+–[Cu]+ distance, a new motif structure of ([Cu–O]+–[Cu]+, II in Fig. 4a) could be evolved, wherein the subsequent DMTM reaction, including CH4 activation, CH3OH formation and desorption, occurs over the monomeric [Cu–O]+ site by crossing a low Gibbs free energy barrier of 0.33 eV (Fig. 4a, c, e and Movie S2†), and the distant [Cu]+ acts as an observer in these steps. The microkinetic modelling (Fig. 4f) further reveals a significantly promoted reaction rate of such dual Cu SA site (5.64 × 108 s−1), which is respectively six and eight orders of magnitude higher than those of the traditional monomeric [Cu]+ (8.7 × 101 s−1) and Cu dimer (8.1 × 10−1 s−1) site to produce CH3OH. This finding fits well with the boosted activity of Cu-FER, which indicates that such dual Cu SA site would constitute a highly probable candidate active site during N2O-DMTM over Cu-FER. Thereby, in light of the above AIMD simulation and microkinetic modelling, we can infer that favored by the unique topology structure of FER, leading to the formation of such highly active dual Cu SA site, on one hand, it significantly favors N2O dissociation to generate αO through the synergistic effect between distant [Cu]+ sites; and on the other hand, it efficiently prevents the formation of oxo-Cu dimers ([Cu–O–Cu]2+) that possess much lower DMTM reaction efficiency than the evolved monomeric oxo Cu cation site of ([Cu–O]+–[Cu]+).
 | ||
| Fig. 4 AIMD simulation results and microkinetic modeling outcomes of N2O-DMTM over the dual Cu SA of the Cu-FER zeolite. (a) Derived energy diagram along with reaction coordinate. (b–e) 2D and 3D free energy surface of the (b and d) N2O dissociation to generate αO and (c and e) CH4 activation over the [Cu–O]+–[Cu]+ site to generate CH3OH; O red, Cu orange, Al pink, Si yellow, N blue, C gray, H, white. (f) Reaction rate comparisons of monomeric [Cu]+, Cu dimer and dual Cu SA site derived by microkinetic modelling. (g and h) Surface coverage variations along with simulation time t at T = 330 °C (g) and equilibrium surface coverage as a function of reaction temperature T (h) over the dual Cu site of Cu-FER based on microkinetic modelling (Table S7†). As noted, the conversion of structure d to e only involves one transition state (TS), while two types of TS need crossing in Fig. 3. Such diverse reaction routes can be closely related to the different simulation approaches applied (AIMD, Fig. 4, and DFT, Fig. 3). Specifically, metadynamics was employed during the AIMD simulation to screen the Gibbs free energy surface along with two types of collective variables, wherein the reaction temperature was also taken into account by NVT ensemble, while the CI-NEB (climbing image nudged elastic band) approach was employed in searching the TS based on the widely reported radical rebound mechanism, thereby totally involving two TS. | ||
According to the above microkinetic modelling result, we can deduce that the N2O dissociation step is the RDS over the dual Cu SA site during N2O-DMTM, with the surface-adsorbed N2O (θN2O) constituting the major species (Fig. 4g). N2O-DMTM over Cu-FER-0.3% was further conducted at T = 330 °C using N2O/CH4 ratios of 0.5:1 and 1:1. The (CH3OH + DME) production rates were then accordingly calculated and compared with those derived from the scenario of N2O/CH4 = 2:1, attempting to identify such dual Cu SA site by investigating the kinetic effects of reactant pressures, as shown in Fig. S14.† As can be seen, the production rate of (CH3OH + DME) obviously increased along with the increase of N2O/CH4 ratio, from 953, 1532 to 2736 μmol gcat h−1. This finding kinetically verifies the existence of the dual Cu SA site due to the N2O dissociation step constituting the RDS over the dual Cu SA site, and thereby, increasing the CH4 partial pressure would favor an increased reaction rate. As noted, although the monomeric [Cu]+ also displays the RDS step of the N2O dissociation step, it can be ruled out due to its low reaction efficiency (8.7 × 101 s−1).
 | ||
| Fig. 5 AIMD simulation of the DME associative production route over the BAS site. (a and b) 3D and 2D free energy surface as function of the coordination number (CN) of O–C and aO–H, as illustrated in panel (d) of the TS1 model; a value of CN close to 1 represents bond formation, while a value close to 0 represents bond fracture. (c) Variations of CN along the simulation time t; CN is clearly labeled in panel (d) of the IS and TS1 models. (d) The key structural models involved in AIMD simulation. | ||
 | ||
| Fig. 6 DFT and in situ FTIR investigations on tandem catalysis of CH3OH to form DME (CAS-DME route). (a) Derived energy diagram, (b) CH3OH/DME net reaction rate comparison, and (c) surface coverage variations along the reaction time t over the Cu dimer site of Cu-FER; O red, Cu orange, Al pink, Si yellow, C gray, H white. (d and e) In situ FTIR results of cofeeding of CH4 and N2O (2% CH4, 2% N2O in He, 40 mL min−1) over He pretreated Cu-FER-0.3%. (f and g) He sweeping (40 mL min−1) after cofeeding and (h and i) introduction of CH3OH (bubbled by He of 2 mL min−1) after He sweeping at T of 250 °C; the Cu-FER-0.3% was initially pretreated with He for 1 h at 500 °C, then cooled down to 250 °C for the measurement. | ||
Furthermore, it should also be noted that the tandem CAS-DME pathway exhibits a relatively higher energy barrier and lower reaction rate (0.69 eV and 1.39 × 106 s−1 in Fig. 6a and b over the Cu dimer site) compared to the CH3OH production pathway (0.26 and 0.33 eV; 5.68 × 108 s−1 in Fig. 4a and f over dual Cu SA site). This would influence the CH4 oxidation to methanol to some extent, which however does not constitute a determining factor. As revealed by AIMD, the generated CH3OH can be easily desorbed from the dual Cu SA site and would either be released as the final product from Cu-FER or participate in the subsequent tandem catalysis for DME production. In this regard, CH4 oxidation into CH3OH would primarily correlate with the RDS step of N2O dissociation, consequently significantly affecting the generation rate of DME. The in situ FTIR, with He-pretreated Cu-FER-0.3% being initially exposed to (N2O + CH4) followed by He sweeping and interaction with CH3OH, can well support the proposed CAS-DME mechanism. Upon co-feeding N2O and CH4, the radical of CH3− belonging to v[Cu-CH3] can be clearly observed at 2923 cm−1 (Fig. 6d and e) following the radical mechanism.10 It could persist during subsequent He sweeping (Fig. 6f and g), which however nearly disappeared upon the further introduction of CH3OH (Fig. 6h and i), leaving the strong v[CH3] bands of adsorbed CH3OH (2964, 2852 cm−1). This finding confirms the direct reaction of [Cu-CH3] radical with CH3OH. It should be noted that the minor production of DME and the overlap of v[CH3] with CH3OH make it challenging to distinctly identify the generated DME. In this regard, we can deduce that the higher DME productivity relative to that of CH3OH during N2O-DMTM (Fig. 1d) can be majorly related to the tandem catalysis of CH3OH principally over the Cu dimer site of 10 MR channel. Such reaction route would also significantly contribute to long-term reaction stability of Cu-FER-0.3% (Fig. S2a†), wherein the in situ generated CH3OH functioning as a special “solvent” can react with the radicals of CH3− and OH− to produce DME, which thereby would hinder carbon deposition caused by the accumulated radials of CH3−.
The H2O could alter the reaction selectivity (Fig. 1d), resulting in CH3OH as the major product, which can be closely related to two main reasons. The first one lies to that the H2O could directly participate in DMTM reaction, consuming the generated radicals of CH3− and OH− to form CH3OH thereby significantly depressing DME production. As illustrated by TPSR-MS of Fig. 7a, the obvious signal increase of CH3OD (m/e = 33) and HOD (m/e = 19) can be clearly observed along with the decreasing of D2O (m/e = 20), which indicates the direct participation reaction of H2O following the reaction route of (OH− + D-OD + CH3− → HOD + CH3OD). This can be well supported by the DFT simulation that the H2O just needs crossing a low barrier (0.82 eV, Fig. 7b) to produce CH3OH through directly reacting with the radicals of CH3− and OH− over Cu dimer site. The second reason lies to that the H2O could efficiently favor the desorption of CH3OH with the desorption energy being extensively reduced from 1.58 to 0.71 eV (Fig. 7c) and 1.12 to 0.70 eV (Fig. 7d), respectively, over the Cu dimer and monomeric [Cu]+ site. Thereby, the influence of H2O during N2O-DMTM over Cu-FER-0.3% can be well illustrated based on the combined D2O isotopic tracer technique and DFT simulation. Meanwhile, the finding of H2O significantly altering N2O-DMTM selectivity can also well support the proposed CAS-DME route over Cu dimer site. As noted, it is also essential to discuss the influence of H2O on the coordination environment of such dual Cu SA sites. As revealed by the ab initio thermodynamics (AIT) in our previous work,10 H2O complexation with the [Cu]+ site actually cannot thermodynamically affect the methoxy formation to produce [Cu–CH3]+ and [CuOH]+, due to its much lower thermodynamic stability. Moreover, H2O complexations with [Cu]+, [CuOH]+ are reported to commonly occur under low temperature range (T < 300 °C) due to their weak thermodynamic stabilities.55 In this regard, we can speculate that the influence of H2O on the coordination environment of the dual Cu SA site in the present work would also be limited due to its low thermostability under our reaction conditions (T = 330 °C).
 | ||
| Fig. 7 Experimental and theoretical investigation on the effect of H2O during N2O-DMTM. (a) Isotopic transfer study during D2O–N2O-DMTM by TPSR-MS; (b) DFT simulated H2O direct reaction route over the Cu dimer site (10 MR channel) of Cu-FER; energy diagram of H2O-favored CH3OH desorption over the (c) Cu dimer and (d) monomeric [Cu]+ of Cu-FER. | ||
Reaction kinetics of diverse active sites
As for the intrinsic activity of monomeric [Cu+] and Cu dimer sites, the microkinetic modelling results (Fig. 3c) suggest a higher CH3OH production rate of the monomeric [Cu]+ site (8.7 × 101 s−1) relative to the Cu dimer site (8.1 × 10−1 s−1), which can be mainly related to the higher desorption barrier of CH3OH over the Cu dimer site (ΔE = 1.58 eV), although a relatively high N2O dissociation barrier (ΔE = 1.35 eV) can also be observed over the monomeric [Cu+] site. However, the production rate of DME over the Cu dimer site (1.39 × 106 s−1, Fig. 6b) would be much higher than that of monomeric [Cu]+ site (8.91 × 10−1, Fig. S16b†) due to the extensively reduced barrier for the generated CH3OH to produce DME (ΔE = 0.69 eV, Fig. 6a) by following the CAS-DME route.The production rate per Cu atom was further compared for the prepared Cu-FER samples (Fig. S21†). Cu-FER-0.11% exhibits the highest reaction rate per Cu atom of 144233 mmol molCu−1 h−1. The Cu dimer proportion was further predicted based on EPR (Fig. 2e and S20†) and CO-probed in situ FTIR (Fig. 2c and S29†), which however indicates that the Cu-FER 0.11% possesses much lower Cu dimer content than Cu-FER-0.3% and Cu-FER-0.6% (see Table S14 and Fig. S29d†). Such discrepancy strongly implies the higher activity of Cu monomer, which also agrees well with our DFT and AIMD simulation results, showing that the dual Cu SA site, being categorized as Cu monomers as detailed in the ESI† (Identification Category of Dual Cu SA), functioned as the major active site rather than the Cu dimer. As revealed, CH3OH would be primarily produced over the dual Cu SA site, which displays the highest kinetic reaction rate of 5.64 × 108 s−1 (Fig. 4f) relative to the 8.1 × 10−1 and 8.7 × 101 s−1 of Cu dimer and monomeric [Cu]+ site, respectively. Herein, we would also like to note that being of a type of Cu monomer, it is still challenging to quantify the specific content of such dual Cu SA site. However, according to the TOF comparison results (Fig. S21†), we speculate that the Cu-FER with lower Cu loading of 0.11% would probably be much more favorable to form such dual Cu SA site, considering lower Cu loadings would favor Cu cation diffusion inside the FER channel.
As for DME, although it would be mainly generated over the Cu dimer site (located at 10 MR channel), displaying a high kinetic reaction rate of 1.39 × 106 s−1 (Fig. 6b), which is four orders of magnitude higher than that of the monomeric [Cu]+ site (8.9 × 101 s−1, Fig. S16b†), it is actually closely correlated with the in situ generated CH3OH over the dual Cu SA site. In this regard, we can deduce that the dual Cu SA site would play a major role in deciding the reaction efficiency of Cu-FER, the specific content of which would be significantly determined by the distribution of framework Al. To make a further verification, Cu-FER-0.3%, with a higher SiO2/Al2O3 ratio of 70, was further prepared by wet-ion-exchange method and evaluated for N2O-DMTM (Fig. S27†). The derived reaction rate (represented by total productivity per gram of catalyst, 667.8 μmol gcat−1 h−1) is much lower than that of Cu-FER-0.3% (SiO2/Al2O3 = 30), due to the increased SiO2/Al2O3 ratio reducing the formation of such dual Cu SA active site. Therefore, based on the above results, we can verify the significantly important kinetic role the dual Cu SA site plays during N2O-DMTM over Cu-FER.
Finally, we would also like to mention that the H-FER zeolite, with the Fe impurity of 80–90 ppm, was also evaluated for N2O-DMTM, displaying a relatively high and stable (CH3OH + DME) productivity of 843 μmol gcat−1 h−1 (average value of 6 h reaction). As investigated in our previous works, Fe0.6%Cu0.68%-BEA56 and Fe-BEA-1.0%6 display much lower CH3OH productivity (∼150 and 50 μmol gcat−1 h−1) and severe deactivation relative to H-FER during N2O-DMTM in the absence of H2O (under similar reaction conditions). Some other commercial H-zeolites (H-BEA, H-MOR, H-ZSM-5), which may also possess Fe impurities, have also been evaluated for N2O-DMTM (see diagram below, further detailed in Fig. S28†), exhibiting much lower CH3OH productivity (∼10–80 μmol gcat−1 h−1) than H-FER. In this regard, we speculate that the Fe impurity would exert limited effect on N2O-DMTM for the H-FER of the present work. Recently, Xiao et al.8 investigated N2O-DMTM over the transition-metal-free H-FER zeolite catalysts. After calcination at T of 850 °C, H-FER achieved a CH3OH productivity as high as 18300 μmol gcat−1 h−1. The distorted tetracoordinated framework Al and the pentacoordinated Al of the extra-framework were newly proposed to be the potential active centers generated during calcination, activation, and reaction. In this regard, we speculate that the relatively high productivity of 843 μmol gcat−1 h−1 of H-FER can also be probably related to such AlOx species. Due to the specific fundamental reaction mechanism was not present in ref. 8, such exploration based on DFT would be a good research point in future work.
Conclusion
The present work has developed an efficient continuous N2O-DMTM system, achieving boosted productivity (58368 mmol per molCu per h of CH3OH) over Cu-FER zeolite. A 2D spatial structure-favored tandem catalysis is presented for the first time. An unique dual Cu SA, located at the parallel 6 MR site (β site) of the 8 MR channel, was formed. It possesses much higher CH3OH production efficiency (5.64 × 108 s−1) than the traditional monomeric [Cu]+ (8.7 × 101 s−1) and Cu dimer sites (8.1 × 10−1 s−1), which not only significantly reduces the N2O dissociation barrier but also prevents the formation of less active oxo-Cu dimer sites [Cu–O–Cu]2+. Additionally, favored the vertically interconnected 2D channel system of FER, the generated CH3OH could readily diffuse into the 10 MR main channel for tandem catalysis to produce DME. Interestingly, functioning as a special “solvent,” CH3OH could directly react with the radicals of CH3− and OH− to produce DME, which could efficiently hinder carbon deposition and guarantee the long-term stability of Cu-FER-0.3%. Notably, although Cu-FER possesses much higher N2O-DMTM activity than Cu-BEA and Cu-MFI (Cu loading of 0.3%) with the 3D channel system, we still cannot conclude that the 2D channel system would be much more efficient than the 3D channel system for N2O-DMTM. Based on the present study, we would like to emphasize that taking advantage of the zeolite spatial structure to control Cu active site distance would probably constitute a promising route for highly efficient N2O-DMMT catalyst designs; additionally, preferring zeolite with a 2D channel system and suitable pore size can probably limit side reactions to enhance CH3OH (or DME) selectivity.
Experimental and theoretical method
Catalyst preparation
The zeolite catalysts H-FER, H-BEA, H-MFI, and H-MOR, each with a Si/Al ratio of approximately 15, were procured from Nankai University Catalyst Co., Ltd, China. The Cu-zeolite samples, comprising Cu loadings of 0.11%, 0.3%, 0.6%, and 1.0% for Cu-FER, and 0.3% for Cu-BEA, Cu-MFI, and Cu-MOR, were synthesized via an ion-exchange method. Specifically, 5 g of H-zeolite and the requisite amount of Cu(NO3)2·6H2O (Sigma-Aldrich) were combined in a round-bottom flask. This mixture was stirred at 400 rpm and maintained at a temperature of 100 °C for 6 h in the presence of 300 mL deionized water. Post-reaction, the slurry was filtered and rinsed multiple times with deionized water, then dried at 100 °C for 24 h. The final product was achieved through calcination at 550 °C for 4 h.Characterizations
 | (1) |
Solid-state 27Al NMR spectra were acquired using a Bruker Avance 400 MHz spectrometer. The 27Al chemical shift was referenced to −0.54 ppm with respect to AlNH4(SO4)2 · 12H2O. Peak fitting was accomplished using MestReNova 14.0 software, with fitting parameters detailed in Table S11.†
Activity measurement
N2O-DMTM activity measurement was carried out in a fixed-bed quartz reactor (outer diameter: 11 mm, inner diameter: 8 mm, length: 485 mm). The catalyst sample was pelletized into particles measuring 0.25–0.425 mm and packed into the constant-temperature zone of the reactor (0.5 g) using quartz wool plugs. Prior to measurement, the sample underwent pretreatment with He (>99.999%, 40 mL min−1) for 1 h at 500 °C, followed by cooling to the reaction temperature of 330 °C. The reactant gases N2O (>99.999%) and CH4 (>99.999%), with He as the balance gas (>99.99%), were introduced into the system at a ratio of 30:15:55. An electronic mass flow meter adjusted the total flow rate to 100 mL min−1, corresponding to a gas hourly space velocity (GHSV) of 12000 h−1. Heating via resistive tape maintained the entire gas line from the point of liquid injection to the gas chromatography (GC) unit at 150 °C to prevent condensation. The GC loop (1 mL) was also heated at 150 °C within a valve box. The details are provided in the ESI.†
Computational method
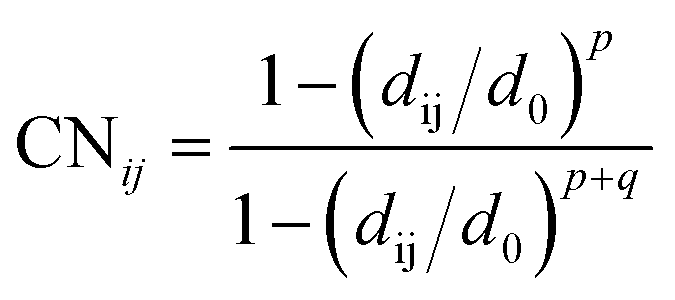 | (2) |
The value of CNij varies from 0 to 1, respectively corresponding to bond breaking and bond formation. Eventually, the free energy surface diagrams in Fig. 4b–e, 5a and b were obtained based on the software PLUMED 2.53
Data availability
The data supporting this study are available within the article and ESI.†Author contributions
Ning Liu: writing – original draft, data curation, formal analysis, software. Tingting Zhang: investigation, methodology, software, visualization. Chengna Dai: data curation, resources, validation. Ruinian Xu:, methodology, conceptualization. Gangqiang Yu: data curation, validation, visualization. Ning Wang: resources, validation, visualization. Biaohua Chen: conceptualization, project administration, funding acquisition, supervision, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the final support from the National Natural Science Foundation of China (No. 22176006).Notes and references
- N. F. Dummer, D. J. Willock, Q. He, M. J. Howard, R. J. Lewis, G. Qi, S. H. Taylor, J. Xu, D. Bethell, C. J. Kiely and G. J. Hutchings, Chem. Rev., 2023, 123, 6359–6411 CrossRef CAS
.
- V. L. Sushkevich, D. Palagin, M. Ranocchiari and J. A. van Bokhoven, Science, 2017, 356, 523–527 CrossRef CAS PubMed
- P. Gupta, B. Rana, R. Maurya, R. Kalita, M. Chauhan and K. Manna, Chem. Sci., 2025, 16, 2785–2795 RSC
- W. Wang, W. Zhou, Y. Tang, W. Cao, S. R. Docherty, F. Wu, K. Cheng, Q. Zhang, C. Copéret and Y. Wang, J. Am. Chem. Soc., 2023, 145, 12928–12934 CrossRef CAS PubMed
- T. Yu, Z. Li, W. Jones, Y. Liu, Q. He, W. Song, P. Du, B. Yang, H. An, D. M. Farmer, C. Qiu, A. Wang, B. M. Weckhuysen, A. M. Beale and W. Luo, Chem. Sci., 2021, 12, 3152–3160 RSC
- N. Liu, Y. Li, C. Dai, R. Xu, G. Yu, N. Wang and B. Chen, J. Catal., 2022, 414, 302–312 CrossRef CAS
- J. Xie, R. Jin, A. Li, Y. Bi, Q. Ruan, Y. Deng, Y. Zhang, S. Yao, G. Sankar, D. Ma and J. Tang, Nat. Catal., 2018, 1, 889–896 CrossRef CAS
- P. Xiao, Y. Wang, Y. Lu, K. Nakamura, N. Ozawa, M. Kubo, H. Gies and T. Yokoi, J. Am. Chem. Soc., 2024, 146, 10014–10022 CrossRef CAS PubMed
- G. Zhao, P. Yan, K. Procter, A. Adesina, Y. Jin, E. Kennedy and M. Stockenhuber, J. Catal., 2023, 417, 140–152 CrossRef CAS
- R. Xu, N. Liu, C. Dai, Y. Li, J. Zhang, B. Wu, G. Yu and B. Chen, Angew. Chem., Int. Ed., 2021, 60, 16634–16640 CrossRef CAS
- G. Zhao, K. Chodyko, E. Benhelal, A. Adesina, E. Kennedy and M. Stockenhuber, J. Catal., 2021, 400, 10–19 CrossRef CAS
- I. Gokce, M. O. Ozbek and B. Ipek, J. Catal., 2023, 427, 115113 CrossRef CAS
- B. Yu, L. Cheng, S. Dai, Y. Jiang, B. Yang, H. Li, Y. Zhao, J. Xu, Y. Zhang, C. Pan, X.-M. Cao, Y. Zhu and Y. Lou, Adv. Sci., 2023, 10, 2302143 CrossRef CAS PubMed
- Á. Szécsényi, G. Li, J. Gascon and E. A. Pidko, Chem. Sci., 2018, 9, 6765–6773 RSC
- Y. Deng, H. Lei, U. Simon, D. Ye and P. Chen, ACS Catal., 2024, 14, 292–298 CrossRef CAS
- L. N. Wilcox, J. Rebolledo-Oyarce, A. D. Mikes, Y. Wang, W. F. Schneider and R. Gounder, ACS Catal., 2024, 14, 3647–3663 CrossRef CAS
- X. Tang, J. Ye, L. Guo, T. Pu, L. Cheng, X.-M. Cao, Y. Guo, L. Wang, Y. Guo, W. Zhan and S. Dai, Adv. Mater., 2023, 35, 2208504 CrossRef CAS PubMed
- H. Zhang, P. Han, D. Wu, C. Du, J. Zhao, K. H. L. Zhang, J. Lin, S. Wan, J. Huang, S. Wang, H. Xiong and Y. Wang, Nat. Commun., 2023, 14, 7705 CrossRef CAS PubMed
- S. Grundner, M. A. C. Markovits, G. Li, M. Tromp, E. A. Pidko, E. J. M. Hensen, A. Jentys, M. Sanchez-Sanchez and J. A. Lercher, Nat. Commun., 2015, 6, 7546 CrossRef PubMed
- I. Lee, M.-S. Lee, L. Tao, T. Ikuno, R. Khare, A. Jentys, T. Huthwelker, C. N. Borca, A. Kalinko, O. Y. Gutiérrez, N. Govind, J. L. Fulton, J. Z. Hu, V.-A. Glezakou, R. Rousseau, M. Sanchez-Sanchez and J. A. Lercher, JACS Au, 2021, 1, 1412–1421 CrossRef CAS PubMed
- A. J. Knorpp, A. B. Pinar, C. Baerlocher, L. B. McCusker, N. Casati, M. A. Newton, S. Checchia, J. Meyet, D. Palagin and J. A. van Bokhoven, Angew. Chem., Int. Ed., 2021, 60, 5854–5858 CrossRef CAS
- K. Mlekodaj, M. Lemishka, A. Kornas, D. K. Wierzbicki, J. E. Olszowka, H. Jirglová, J. Dedecek and E. Tabor, ACS Catal., 2023, 13, 3345–3355 CrossRef CAS
- E. Tabor, J. Dedecek, K. Mlekodaj, Z. Sobalik, P. C. Andrikopoulos and S. Sklenak, Sci. Adv., 2020, 6, eaaz9776 CrossRef CAS PubMed
- P. Xiao, Y. Wang, L. Wang, H. Toyoda, K. Nakamura, S. Bekhti, Y. Lu, J. Huang, H. Gies and T. Yokoi, Nat. Commun., 2024, 15, 2718 CrossRef CAS PubMed
- J. Wang, J. Zhang, C. Xing, T. Jin, J. Liu, M. Ju and X. Tang, Chem. Eng. J., 2023, 455, 140379 CrossRef CAS
- J. Ohyama, Y. Tsuchimura, A. Hirayama, H. Iwai, H. Yoshida, M. Machida, S. Nishimura, K. Kato and K. Takahashi, ACS Catal., 2022, 12, 2454–2462 CrossRef CAS
- M.-L. Tsai, R. G. Hadt, P. Vanelderen, B. F. Sels, R. A. Schoonheydt and E. I. Solomon, J. Am. Chem. Soc., 2014, 136, 3522–3529 CrossRef CAS
- P. Vanelderen, B. E. R. Snyder, M.-L. Tsai, R. G. Hadt, J. Vancauwenbergh, O. Coussens, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, J. Am. Chem. Soc., 2015, 137, 6383–6392 CrossRef CAS PubMed
- C. Negri, T. Selleri, E. Borfecchia, A. Martini, K. A. Lomachenko, T. V. W. Janssens, M. Cutini, S. Bordiga and G. Berlier, J. Am. Chem. Soc., 2020, 142, 15884–15896 CrossRef CAS PubMed
- J. Tuo, J. Wang, X. Gong, C. Zhai, H. Xu, T. Xue, J. Jiang, Y. Guan and P. Wu, Fuel, 2024, 357, 130001 CrossRef CAS
- Z. Xiong, G. Qi, L. Bai, E. Zhan, Y. Chu, J. Xu, N. Ta, A. Hao, F. Deng and W. Shen, Catal. Sci. Technol., 2022, 12, 4993–4997 RSC
- C. Dai, Y. Zhang, N. Liu, G. Yu, N. Wang, R. Xu and B. Chen, Phys. Chem. Phys. Chem., 2023, 25, 24894–24903 RSC
- K. Jíša, J. Nováková, M. Schwarze, A. Vondrová, S. Sklenák and Z. Sobalik, J. Catal., 2009, 262, 27–34 CrossRef
- S. Sklenak, P. C. Andrikopoulos, B. Boekfa, B. Jansang, J. Nováková, L. Benco, T. Bucko, J. Hafner, J. Dědeček and Z. Sobalík, J. Catal., 2010, 272, 262–274 CrossRef CAS
- J. E. Olszówka, P. Kubat, J. Dedecek and E. Tabor, J. Phys. Chem. C, 2023, 127, 7344–7351 CrossRef PubMed
- A. J. Jones and E. Iglesia, Angew. Chem., Int. Ed., 2014, 53, 12177–12181 CrossRef CAS PubMed
- G. Marsden, P. Kostetskyy, R.-S. Sekiya, A. Hoffman, S. Lee, R. Gounder, D. Hibbitts and L. J. Broadbelt, ACS Mater. Au, 2022, 2, 163–175 CrossRef CAS PubMed
- R. Xu, Z. Wang, N. Liu, C. Dai, J. Zhang and B. Chen, ACS Catal., 2020, 10, 6197–6212 CrossRef CAS
- I. D. N. Jugniot, A. Rivot, P. Massot, C. Cardiet, A. Pizzoccaro, M. Jean, N. Vanthuyne, J.-M. Franconi, P. Voisin, G. Devouassoux, E. Parzy, E. Thiaudiere, S. R. A. Marque, A. Bentaher, G. Audran and P. Mellet, Free Radical Biol. Med., 2018, 126, 101–112 CrossRef PubMed
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS
- Y. Wang and J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 13298–13307 CrossRef PubMed
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 6671–6687 CrossRef CAS PubMed
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B, 1976, 13, 5188–5192 CrossRef
- V. Wang, N. Xu, J. Liu, G. Tang and W. Geng, arXiv, 2019, preprint, arXiv:1908.08269, DOI:10.48550/arXiv.1908.08269.
- J. Hutter, M. Iannuzzi, F. Schiffmann and J. VandeVondele, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 15–25 CAS
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS
- S. Goedecker, M. Teter and J. Hutter, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 1703–1710 CrossRef CAS PubMed
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed
- L. Petersen, A. Ardèvol, C. Rovira and P. J. Reilly, J. Am. Chem. Soc., 2010, 132, 8291–8300 CrossRef CAS PubMed
- G. A. Tribello, M. Bonomi, D. Branduardi, C. Camilloni and G. Bussi, Comput. Phys. Commun., 2014, 185, 604–613 CrossRef CAS
- G. Ciccotti, M. Ferrario, J. T. Hynes and R. Kapral, Chem. Phys., 1989, 129, 241–251 CrossRef CAS
- C. Paolucci, A. A. Parekh, I. Khurana, J. R. Di Iorio, H. Li, J. D. Albarracin Caballero, A. J. Shih, T. Anggara, W. N. Delgass, J. T. Miller, F. H. Ribeiro, R. Gounder and W. F. Schneider, J. Am. Chem. Soc., 2016, 138, 6028–6048 CrossRef CAS PubMed
- Y. Li, N. Liu, C. Dai, R. Xu, G. Yu, N. Wang, J. Zhang and B. Chen, Catalysts, 2021, 11, 1444 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Additional descriptions of method as well as experimental and theoretical evidence, including activity measurement results, XRD, XAS, k3χ(k) curve, 27Al NMR, XPS, H2-TPR, optimized DFT models, and microkinetic modelling results. See DOI: https://doi.org/10.1039/d5sc02092a |
| This journal is © The Royal Society of Chemistry 2025 |