
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInterrogating the missing links in tandem CO2 hydrogenation: role of intermediate transport, active site proximity, and ion exchange in zeolites
Fatima
Mahnaz
,
Andrew
Iovine
and
Manish
Shetty
 *
*
Artie McFerrin Department of Chemical Engineering, Texas A&M University College Station, TX 77843, USA. E-mail: manish.shetty@tamu.edu
First published on 15th May 2025
Abstract
The tandem hydrogenation of CO2 to fuels and chemicals using bifunctional oxide/zeolite catalysts offers a promising strategy for reducing anthropogenic CO2 emissions while generating sustainable alternatives to fossil fuels. Despite significant advancements in this field, fundamental gaps remain in understanding the inflence of active site-proximity, intermediate transport rates, and the metal oxide migration and their ion-exchange with zeolitic Brønsted acid sites (BAS) on the reaction rates and hydrocarbon (HC) product selectivities. Challenges also include high CO selectivity and understanding the complexities of hydrocarbon pool (HCP) propagation in zeolite pore channels. This perspective integrates insights from analogous bifunctional catalytic systems, such as alkane hydrocracking and isomerization, to refine our understanding of site-proximity and transport artifacts on reaction rates and product selectivities. We examine diffusion-reaction formalisms for elucidating site-proximity effects on rates and HC selectivity, discuss methods to suppress CO selectivity using surface organometallic chemistry (SOMC) approaches, and explore strategies for suppressing ion-exchange and tuning HCP dynamics. By addressing these challenges, we outline a conceptual roadmap for advancing tandem CO2 hydrogenation chemistry, providing potential strategies to enhance catalytic efficiency of bifunctional oxide/zeolite systems.
 Fatima Mahnaz | Fatima Mahnaz is a PhD candidate in chemical engineering at Texas A & M University, specializing in heterogeneous catalysis. She earned her BSc in Chemical Engineering from the Bangladesh University of Engineering and Technology in 2018. Her research focuses on developing bifunctional catalysts for CO2 hydrogenation to sustainable fuels and chemicals, with an emphasis on tandem catalysis and site proximity effects. Passionate about sustainable energy, she aspires to contribute to advancements in carbon-neutral fuel technologies. |
 Andrew Iovine | Andrew Iovine is a senior undergraduate student pursuing a BS in chemical engineering at Texas A & M University. He joined Prof. Manish Shetty's lab in Spring 2025 and is interested in gaining a better understanding of heterogeneous catalysis. After graduation, he plans to pursue a PhD to further study topics in catalysis and reaction engineering field. |
 Manish Shetty | Prof. Manish Shetty is an Assistant Professor in the Artie McFerrin Department of Chemical Engineering at Texas A & M University since Fall 2021. He earned his B.Tech. and M.Tech. in Chemical Engineering from the Indian Institute of Technology Bombay in 2011 and his PhD from the Massachusetts Institute of Technology. He currently leads a research group in heterogeneous catalysis and sustainability, with an emphasis on industrial decarbonization, plastics valorization, hydrogen storage, and improving the efficiency of catalyst materials. |
Introduction
Catalytic tandem reactions have emerged as a powerful tool in chemical synthesis, enabling multi-step transformations within a single system while enhancing energy efficiency1 and breaking or circumventing selectivity-activity tradeoffs.2 This selectivity-activity limitation is described by Sabatier's principle, which states that an ideal catalyst should bind reactants neither too strongly nor too weakly to achieve optimal performance.2–5 However, this balance is often difficult to maintain in single-site catalysts, as strong binding enhances activation but risks poisoning, while weak binding limits reactivity.5,6 Tandem catalysis mitigates this limitation by utilizing multiple catalytic sites with distinct functions, enabling selective activation and intermediate transformation at different stages of the reaction.2 Hence, researchers have increasingly applied tandem catalysis principles to heterogeneous catalytic processes,7–15 particularly in the field of CO2 hydrogenation.16–30Tandem reactions typically involve bifunctional catalytic cascades, with an intermediate forming over one active site and its subsequent conversion over a different active site. Kumar et al. demonstrated that in such cascading systems, the two distinct functionalities (e.g., metal and acid sites, metal and metal-support interface sites, etc.) exhibit different Brønsted–Evans–Polanyi (BEP) scaling relationships for the same intermediates along the reaction pathway and can potentially enhance reaction rates by leveraging separate mechanistic contributions, thereby breaking the selectivity-activity limits.6 While the idea of breaking selectivity-activity limits and converting CO2 to carbon-neutral fuels and chemicals seem appealing, designing efficient catalysts that maximize the utilization of all active sites and enhance selectivity toward desired product is challenging.
During tandem reactions, including CO2 hydrogenation, over bifunctional catalytic systems, the two catalytic functions are linked via the shuttling of reaction intermediates, thus requiring “site-proximity”, which ensures that active sites are spatially arranged to facilitate sequential transformations by minimizing the diffusion distance of intermediates. As such, the transport of intermediates between active sites (e.g., redox and Brønsted acid sites) can influence overall rates and selectivities,31 complicating the interpretation of intrinsic kinetics and reaction mechanism.
In tandem CO2 hydrogenation to hydrocarbons (HCs) in the methanol (CH3OH)-mediated route, CH3OH initially forms over redox sites (or oxygen vacancies) or alloys on metal oxides and then subsequently undergoes dehydration and C–C coupling over Brønsted acid sites (BAS) of zeolites to form HCs.32 This sequential reaction steps make site-proximity particularly crucial for this chemistry as the intermediate CH3OH must efficiently transport from redox sites to BAS for the methanol-to-hydrocarbon (MTH) conversion. Recent studies, including our own, have demonstrated that the efficacy of bifunctional oxide/zeolite systems is largely dictated by the efficiency of CH3OH transport and its consumption over BAS.16,33,34 As such, the catalytic efficiency can be enhanced by improving the site proximity between redox sites and BAS.31 However, at the closest proximity (i.e., nanoscale distance), two key problems exist; (1) the catalytic performance could be hindered by the migration of metal oxides inside zeolite framework and their cation exchange with BAS,17,26,27,35–38 and (2) the improvement in hydrocarbon (HC) yields and formation rates do not necessarily scale with the rate of CH3OH transport.31 As such, the molecular underpinnings of proximity-effects are convoluted and yet not understood for this conversion.
Additional key challenge associated with the CH3OH mediated route is the high CO selectivity caused by the endothermic side reaction of reverse-water–gas-shift (RWGS), which reduces hydrocarbon yield.32 While CO selectivity can be reduced by conducting reactions at low temperatures, the low reaction rates make it difficult to achieve appreciable single-pass CO2 conversions. In addition to that MTH is favored at higher reaction temperatures.32 Therefore, increasing HC yield while keeping CO selectivity low becomes a formidable challenge in this route and requires better CH3OH synthesis catalysts.
It is to be noted that while HC yields can potentially be enhanced by increasing CH3OH yield, achieving precise control over HC selectivity requires a fundamental understanding of how hydrocarbons form within zeolite pore channels.39 The reaction proceeds via a chain carrier mechanism consisting of three distinct phases: initiation, propagation, and termination (Fig. 1).39,40 During the initiation phase, unsaturated hydrocarbons form within the micropores of zeolite. These intermediates then participate in the propagation phase, following the well-established “dual cycle” or hydrocarbon pool (HCP) mechanism, which consists of two interconnected pathways: (1) the olefin cycle, where olefins undergo sequential methylation and cracking, and (2) the aromatic cycle, where aromatic species undergo methylation and demethylation. The interplay between these two cycles occurs through the aromatization of olefins and the dealkylation of aromatics to olefins, ultimately governing hydrocarbon selectivity.39 Although extensive research has focused on modulating the HCP mechanism in MTH conversion through changing reaction conditions, such as co-feeding H2 or CO, or changing zeolite composition, such as adjusting the Si/Al ratio of the zeolite or tuning acid site strength,41 the reaction conditions and catalytic system employed for tandem CO2 hydrogenation is different. A deeper understanding of the factors influencing the HCP mechanism in CO2 hydrogenation is therefore essential for achieving selective conversion to targeted hydrocarbon products.
 | ||
| Fig. 1 Hydrocarbon pool (HCP) mechanism of methanol-to-hydrocarbon (MTH) conversion inside zeolite pore channels including initiation, propagation, and termination phases. The propagation encompasses multiple steps, including olefin methylation, cracking, hydrogen transfer, aromatization, aromatic methylation, and aromatic dealkylation. Adapted with permission from Mahnaz, F. & Shetty, M. et al.38 Copyright J. Catal. 2024. | ||
Overall, despite advances in bifunctional catalyst design, the efficiency of CO2 hydrogenation to selective HC remains constrained by critical knowledge gaps. Key unresolved questions include: (i) What does the “site-proximity” effect entail in tandem CO2 hydrogenation? (ii) What is the role of site-proximity in the transport of reaction intermediates? (iii) Can advection-diffusion-reaction formalisms be leveraged to interpret the proximity effect and its impact on reaction rates and selectivity? (iv) How do metal oxide migration and cation exchange occur at intimate proximities, and what are their effects on reactivity? (v) What strategies can reduce CO selectivity to enhance hydrocarbon yields? and (vi) How can HC selectivity be controlled through modulating HCP?
To address these challenges, this perspective explores insights into proximity effects derived from analogous bifunctional catalytic systems, such as alkane hydrocracking and isomerization chemistry, which share mechanistic parallels with tandem CO2 hydrogenation. We examine seminal work by Weisz42–44 and Iglesia45,46 on n-heptane isomerization, applying diffusion-reaction mathematical formalisms to different proximity length scales to elucidate intermediate transport effects. Additionally, we analyze how catalyst modifications at intimate proximities-via metal oxide migration and cation exchange at redox and acid sites can influence reaction pathways and propose strategies to mitigate these effects. Furthermore, we discuss approaches to overcoming the intrinsic challenge of CO selectivity by employing surface organometallic chemistry (SOMC) to enhance CH3OH selectivity and yield.47,48 Finally, we explore methods to modulate HCP to control HC selectivity. By integrating these perspectives, we aim to provide a roadmap for advancing CO2 hydrogenation through fundamental insights into site-proximity effects, catalyst design, and reaction engineering strategies.
Proximity-effects and role of intermediate transport
Research into the impact of site proximity in bifunctional systems dates back to the 1950s for enhancing the efficiency of alkane isomerization,43,44 which involve similar bifunctional catalytic cascades akin to CO2 hydrogenation with alkanes first forming alkenes on a metal function, alkenes undergoing skeletal isomerization or C–C bond cleavage on an acid function, and their products hydrogenating at the metal function to form their saturated analogs. It was observed that enhancing the proximity between active sites (metal sites on Pt/SiO2 for hydrogenation and dehydrogenation activity and acid-sites on mesoporous SiO2–Al2O3 for isomerization) via modulating their particle size, increased the local concentration of intermediates, driving higher diffusive flux and enabling more efficient conversion (Fig. 2).42 Therefore, the proximity-dependent reactivity was attributed to the internal mass transport limitation of the reaction intermediate. Weisz quantified this proximity requirement as a function of intermediate vapor pressure by applying the condition where reaction rate on the surface would be equal to the net diffusive flux (eqn (1)),44 | (1) |
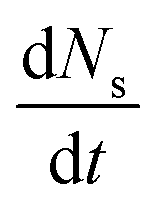 is the surface reaction rate, Deff is the effective internal diffusion, C is the reactant/intermediate concentration and R0 is the catalyst particle size. For the case of negligible inhibition from rate by diffusion, the following condition was assumed,
is the surface reaction rate, Deff is the effective internal diffusion, C is the reactant/intermediate concentration and R0 is the catalyst particle size. For the case of negligible inhibition from rate by diffusion, the following condition was assumed, | (2) |
 | (3) |
 | ||
| Fig. 2 (A) Schematic of n-heptane isomerization over Pt/SiO2 and SiO2–Al2O3 catalyst. (B) Influence of temperature and particle size on the selectivity of iso-heptane in the product. (C) Influence of particle size on iso-heptene selectivity. Adapted with permission from Weisz.42 Copyright Adv. Catal. 1962. | ||
Applying this criterion, Weisz demonstrated that for an intermediate species with a partial pressure of 10−3 atm (as might be observed in olefin production during hydrocarbon reactions), the maximum catalyst particle size should be around 50 μm to avoid diffusion constraints in mesoporous catalysts (Fig. 3). Interestingly, even when an intermediate exists at a partial pressure of 10−10 atm, a stepwise reaction could still proceed without diffusion limitation, provided that the catalytic sites are within ∼100 Å (10 nm) of each other.44 This finding underscores the critical role of site proximity in bifunctional catalysis, where intermediate transport between distinct active sites governs overall catalytic performance.
 | ||
| Fig. 3 Proximity requirement, in terms of component particle size, for typical conditions of reaction rate (10–6 moles s−1 cm−3), as a function of equilibrium vapor pressure of intermediate. Adapted with permission from Weisz.42 Copyright Adv. Catal. 1962. | ||
Akin to hydrocarbon hydroisomerization and hydrocracking reactions, for tandem CO2 hydrogenation, there is a consensus that the HC selectivity could be enhanced by improving CH3OH transport from active sites on metal oxides (e.g., oxygen vacancy sites) to BAS by improving their proximity.18,49,50 However, the improvement in hydrocarbon yields and formation rates do not necessarily scale with the rate of intermediate CH3OH transport (estimated from the ratio of linear velocity and distance between the active sites).31 It is our conjecture that a possible reason could be the diffusional restriction of HC inside microporous zeolites as the acid sites reside within voids of molecular dimensions, restricting their diffusional egress. Therefore, the physical characteristics of the diffusive medium, such as channel size, connectivity, crystal size, and the number of acid sites can become consequential for measured reaction rates.41 We emphasize that using microporous zeolites makes it difficult to apply Weisz's proximity criterion to find a critical length scale with negligible diffusion limitation, as the constituent reactions in MTH involving bulkier hydrocarbons would be inherently mass transport limited. In such systems, where transport artifacts influence selectivity in complex reaction networks, the Thiele modulus (ϕA) emerges as a fundamental non-dimensional parameter for analysis.51,52
 | (4) |
Thiele modulus naturally arises from a mole balance on the reactive domain of a catalyst and is defined as the ratio of the intrinsic reaction rate (in absence of mass transport limitation) to the diffusion rate when the driving force is at its maximum (eqn (4)). A low ϕA value (<1) indicates minimal mass transport limitations, allowing measured reaction rates to reflect intrinsic kinetics.51,52 As reactant concentration gradients become more pronounced, ϕA increases and leads to diffusion constraints that suppress observed reaction rates (rA, meas) relative to their kinetic counterparts (rA, kinetic). This discrepancy can be quantitatively estimated through the effectiveness factor (ηA) (eqn (5)),51,52 which measures the fraction of the intrinsic reaction rate achieved under diffusion-limited conditions.
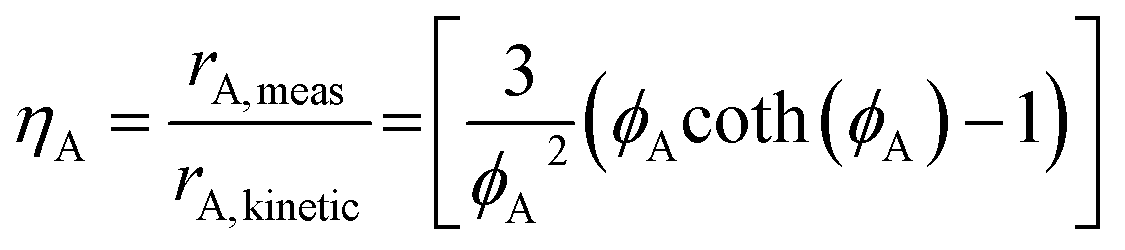 | (5) |
The application of these principles to tandem or cascading catalytic systems, however, presents unique challenges. For example, in such systems, if secondary reaction rates are faster than primary reaction rates and/or if the molecular diffusivities of primary products are low (i.e., ϕA ≫ 1), diffusion limitations can obscure product identity.46 For example, secondary reactions may take place before primary products can be detected in the bulk phase, complicating efforts to elucidate reaction networks and mechanisms.53 Such occurrences necessitate careful diagnostic strategies to ensure accurate interpretation of catalytic performance. A classic example of this phenomenon is n-heptane isomerization over Pt/zeolite catalysts, where reactivity is influenced by the proximity of metal and acid sites.45 The reaction sequence involves the formation of linear heptenes (nH=) as a mixture of equilibrated regioisomers from n-heptane dehydrogenation on Pt sites. These nH= species subsequently undergo isomerization at acid sites to form 2-methylhexenes (2MH=) and 3-methylhexenes (3MH=), which can further isomerize into dimethylpentenes (DMP=) (Fig. 4A). The DMP= isomers act as the precursors to β-scission products.45 Iglesia and co-workers demonstrated that the selectivity toward β-scission products increases with increasing distance between metal and acid sites, highlighting the role of site-proximity on product selectivity.45 Specifically, by dispersing Pt nanoparticles within zeolite crystals thus by increasing the intracrystalline Pt–H+ distance (via decreasing Pt loading), they observed higher n-heptane conversion turnover rates (per H+ site) and shifts in β-scission selectivity. To assess these transport effects on rates and selectivities, Iglesia and coworkers applied mathematical diffusion–reaction formalisms that account for intracrystalline gradients in reactant and product concentrations and for the local equilibration of alkene interconversion steps.45,46 These descriptions were then embedded within a plug-flow convection-reaction formalisms to describe the distribution of products formed as concentration gradients developed within crystallites and along the packed-bed reactor. This rigorous diffusion-convection-reaction analysis (Fig. 4B) linked the product selectivities to Thiele moduli, ultimately revealing that intrinsic selectivities were largely governed by diffusion-enhanced secondary reactions (Fig. 4C). These insights underscore the critical interplay between transport limitations and reaction kinetics in tandem/cascading systems. We emphasize that such analytical frameworks are equally critical for deciphering transport artifacts and the mechanistic intricacies involved in tandem CO2 hydrogenation.
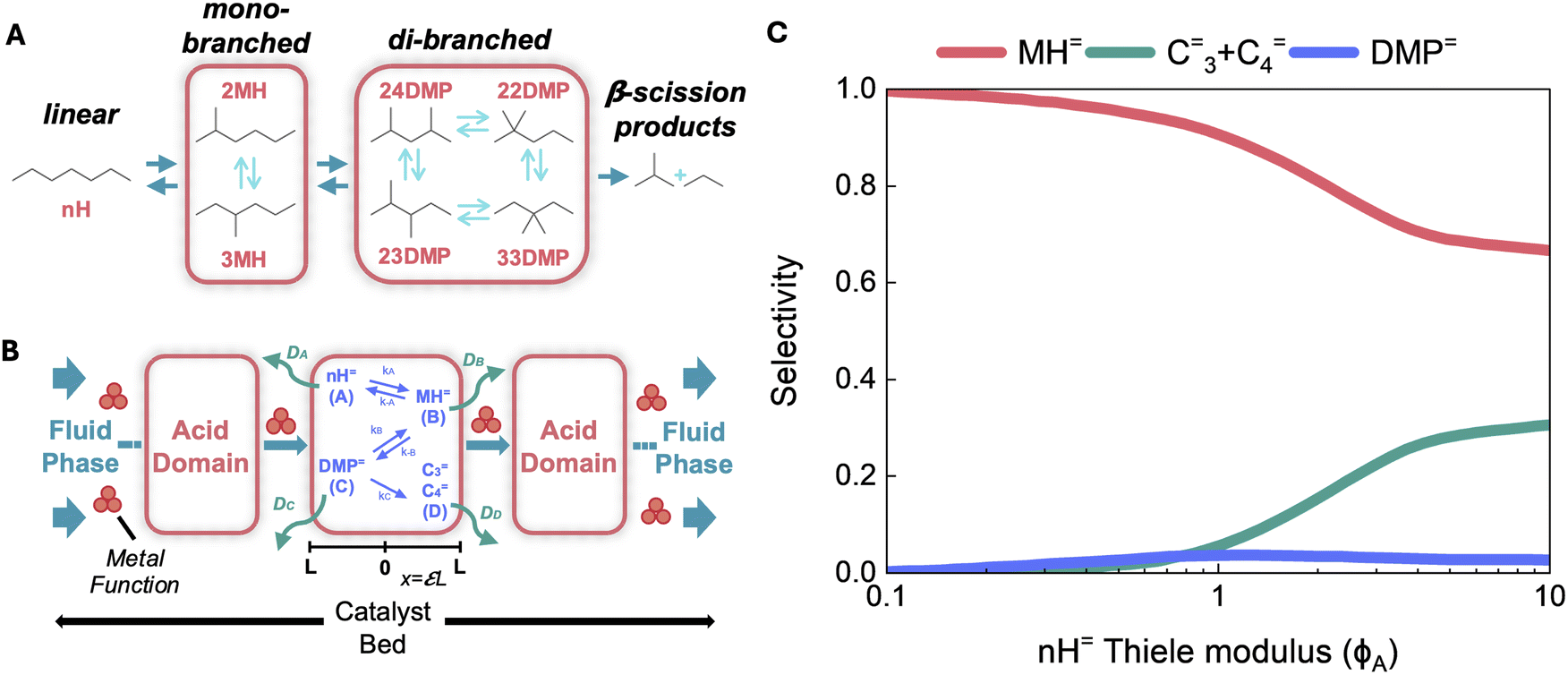 | ||
| Fig. 4 (A) Isomerization and β-scission reaction network for C7 alkenes on bifunctional physical mixtures of Brønsted acid catalyst and Pt/SiO2. Dashed boxes around isomers with the same degree of branching indicate facile interconversion and isomers treated as a kinetic lump. (B) Schematic depiction of the coupled diffusion–convection–reaction system for bifunctional isomerization and -scission cascades. Acid domains are represented by the inset solid rectangles, the catalyst bed is denoted by the surrounding dashed rectangle. Metal functions and acid sites (H+) are also displayed. (C) Selectivities to methyl hexenes (MH=), dimethyl pentenes (DMP=) and -scission products (C3= + C4=) as a function of Thiele modulus, observed by modulating intracrystalline Pt–H+ site distance by confining Pt in MFI. Adapted with permission from Hu, W. & Iglesia, E. et al.45 Copyright J. Catal. 2023. | ||
We note that, while evaluating the applicability of the Thiele modulus (ϕA) to tandem CO2 hydrogenation system, one may question its relevance given the relatively small size of key intermediates, CH3OH, unlike bulky intermediates encountered in alkane isomerization (e.g., n-heptene). Notably, the first step of CH3OH synthesis over metal oxide catalyst can be operated in kinetic regime without mass transport limitation.54 Additionally, for the initiation step of MTH conversion, intermediate CH3OH have smaller kinetic diameter (3.6 Å)55 than typically used zeolite pore size (3.8 Å for CHA, 3.8 Å for AFX, 8.35, 4.8 Å for LEV, 5.6 Å for MFI),56 therefore, CH3OH transport in zeolite pores should not be diffusion limited (assuming negligible external mass transport effects). However, the propagation and termination sequences during MTH are mediated by active chain carriers (e.g., dienes, branched olefins, aromatics etc.), which are transport-limited to varying extents depending on zeolite morphology, pore structure, and crystal size.57,58 The presence of these species and their mass transport limitations impose spatial gradients during MTH on both bed and zeolite crystallite scales, complicating the interpretation of HC selectivity solely based on intrinsic kinetics.57 In such scenarios, the observed volumetric formation rate for bulkier HC (CmHn), robs, CmHn, includes contributions from the intrinsic volumetric rates (rint,p) for a reaction p (i.e., aromatic dealkylation, olefin methylation etc.) in the absence of mass transport limitations and an overall effectiveness factor (ηp) that quantitatively assesses diffusional constraints.57
 | (6) |
 , where R is the zeolite crystallite size and D is the effective diffusivity of component CnHm.59 During MTH on MFI, it was observed that increasing
, where R is the zeolite crystallite size and D is the effective diffusivity of component CnHm.59 During MTH on MFI, it was observed that increasing  (whether by changing zeolite crystal size or acid site density), increased ethylene (C2=) selectivity while other light olefin selectivities were invariant.57 The increase in C2= selectivity and invariance in light olefins selectivity resulted in an increase in ethylene-to-2 methyl-butane (C2=/2MB) ratio (indictive of the relative propagation of olefin cycle relative to aromatic cycle in HCP), as shown in Fig. 5A, suggesting an enhanced propagation of the aromatic cycle. From a mathematical perspective, this phenomenon was rationalized as a consequence of aromatics-based reactions (e.g., aromatic dealkylation to ethylene formation) experiencing more severe transport limitations (i.e., larger ϕA) relative to the other reactions during MTH (e.g., olefins methylation in olefin cycle) such that the rates and effectiveness factor prescribing aromatics cycle propagation depend more sensitively on diffusional constraints, described by,57
(whether by changing zeolite crystal size or acid site density), increased ethylene (C2=) selectivity while other light olefin selectivities were invariant.57 The increase in C2= selectivity and invariance in light olefins selectivity resulted in an increase in ethylene-to-2 methyl-butane (C2=/2MB) ratio (indictive of the relative propagation of olefin cycle relative to aromatic cycle in HCP), as shown in Fig. 5A, suggesting an enhanced propagation of the aromatic cycle. From a mathematical perspective, this phenomenon was rationalized as a consequence of aromatics-based reactions (e.g., aromatic dealkylation to ethylene formation) experiencing more severe transport limitations (i.e., larger ϕA) relative to the other reactions during MTH (e.g., olefins methylation in olefin cycle) such that the rates and effectiveness factor prescribing aromatics cycle propagation depend more sensitively on diffusional constraints, described by,57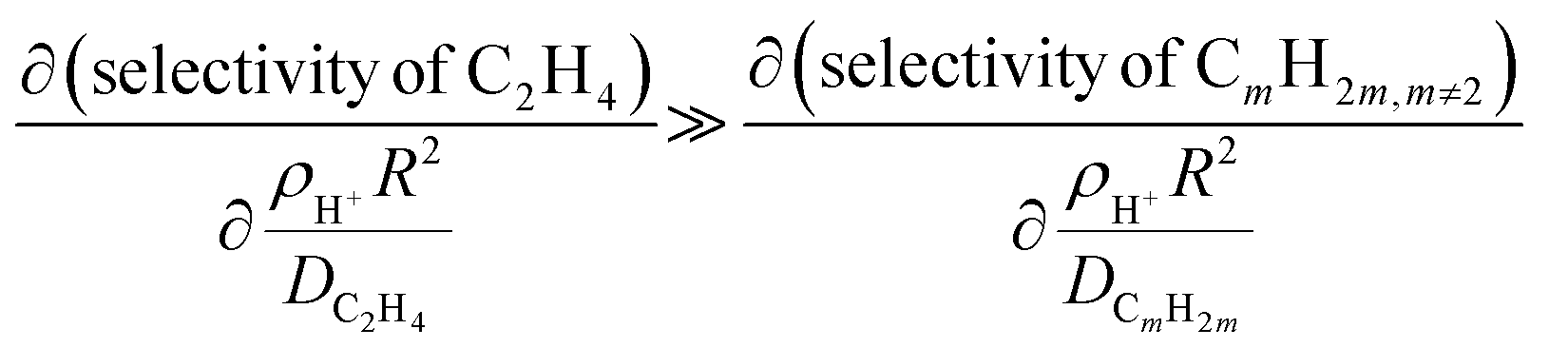 | (7) |
 | ||
Fig. 5 (A) Instantaneous ethylene and 2MB (2-methylbutane, 2-methyl-2-butene) selectivity, ethylene-to-2MB ratio versus for DME conversion over HZSM-5 of varying crystallite size and Si/Al ratio at 623 K. (B) Total turnover number (left axis) and cumulative paraffins selectivity (right axis) for methanol conversion on HSAPO-34 at 673 K, 16 kPa methanol pressure. Adapted with permission from Bhan and coworkers.59 Copyright J. Catal. 2023. for DME conversion over HZSM-5 of varying crystallite size and Si/Al ratio at 623 K. (B) Total turnover number (left axis) and cumulative paraffins selectivity (right axis) for methanol conversion on HSAPO-34 at 673 K, 16 kPa methanol pressure. Adapted with permission from Bhan and coworkers.59 Copyright J. Catal. 2023. | ||
For small cage zeolite with CHA framework, the influence of diffusional constraints was reflected on catalyst lifetime, assessed in terms of cumulative turnover number (TON), where the total turnovers decreased with increasing diffusional constraints regulated by  see (Fig. 5B).41,58 Additionally, reaction-transport analysis of the MTO reaction network over SAPO-34 revealed that the effect of diffusional constraints on total turnovers was caused by dehydrocyclization reactions, which experience stronger diffusional constraints than olefins methylation, methanol transfer hydrogenation, and aromatics dealkylation.58
see (Fig. 5B).41,58 Additionally, reaction-transport analysis of the MTO reaction network over SAPO-34 revealed that the effect of diffusional constraints on total turnovers was caused by dehydrocyclization reactions, which experience stronger diffusional constraints than olefins methylation, methanol transfer hydrogenation, and aromatics dealkylation.58
We note that the diffusional influence on HC product selectivity can further be probed by employing zeolites with varying undulation factor (Ω), which represent the ratio of the maximum diameter HC that can diffuse from zeolite pore to that which can be occluded. Such techniques were utilized to probe diffusional effect on alkene oligomerization and β-scission.53 Iglesia and coworkers revealed that both 1-D 10 membered-ring (MR) TON and mesoporous Al-MCM-41 mitigated β-scission reactions having less diffusional effect (Ω∼1) while zeolites with smaller undulation factor, like MFI and FAU, had broad product distributions, including significant amounts of β-scission products due to diffusion limitation (Fig. 6).60 As such, using zeolites with different undulation factor during CO2 hydrogenation can provide valuable mechanistic insights by distinguishing between primary, secondary and diffusion enhanced reaction pathways.
 | ||
| Fig. 6 Selectivity to oligomers (i.e., not β-scission products) as a function of the diffusion pathway in zeolites. The undulation factor is defined as the ratio of the largest cavity to pore-limiting diameters. Adapted with permission from Sarazen, M. L. et al.60 Copyright ACS Catal. 2016. | ||
Role of site-proximity on thermodynamics constraints
Thermodynamically, CH3OH synthesis from CO2 hydrogenation is not equilibrium-limited at low temperatures (below ∼250 °C),61 although the kinetics become slower resulting in a low single-pass CO2 conversion. However, at higher reaction temperatures, the reaction becomes equilibrium-limited due to the negative entropy of the reaction (ΔG = ΔH – TΔS),62–64 limiting CH3OH yield. Yet high reaction temperatures (350–400 °C) are needed for tandem CO2 hydrogenation as MTH conversion is favorable >350 °C.65,66 Remarkably, despite the unfavorable equilibrium shift for CH3OH synthesis at high temperatures, coupling with the MTH reaction drives the overall tandem reaction forward per Le Chatelier's principle, since CH3OH is simultaneously consumed as an intermediate and converted into thermodynamically more stable olefins and aromatics.64 Jones and coworkers conducted thermodynamic analyses on CH3OH synthesis from CO2 hydrogenation, as well as CO2 conversion to olefins and aromatics.64 Their analysis indicates that the overall Gibbs free energies for the formation of olefins and aromatics are lower (i.e., more negative) than the CH3OH synthesis reaction alone (see Fig. 7),64,67,68 making the tandem reaction more thermodynamically favorable.64,69 This interplay highlights the relevance of site proximity in modulating thermodynamic constraints. We anticipate that when metal oxide and zeolite domains are at intimate proximity, the consumption of CH3OH over adjacent acid sites can lower its effective partial pressure and potentially shift the equilibrium of CH3OH synthesis forward, facilitating higher HC yield. Thus, site proximity can potentially manipulate thermodynamic constraints, favoring CH3OH formation under conditions otherwise deemed equilibrium limited. | ||
| Fig. 7 Gibbs free energy changes at different temperatures for CH3OH synthesis from CO2 hydrogenation, CO2 to lower olefins (ethylene), CO2 to aromatics (toluene), and the reverse water-gas shift reaction. Simulated conditions for equilibrium CO2 conversion: H2/CO2 = 3, total pressure 4 MPa. Adapted with permission from Jones and coworkers.64 Copyright J. CO2. Util. 2021. | ||
Improving hydrocarbon yield
The formation of CO via endothermic RWGS can limit CH3OH yield, as it is favorable under tandem CO2 hydrogenation conditions.70,71 Strategies such as utilizing low reaction temperatures or adding CO as a co-feed to shift RWGS reaction equilibrium have been employed to mitigate the negative impact of CO formation on hydrocarbon yield.26,27,72 However, an efficient way to reduce CO selectivity would be using a better CH3OH synthesis catalyst.The hydrogenation of CO2 to CH3OH is typically performed by catalysts consisting of copper and zinc (Cu/ZnO/Al2O3), and the reaction is commonly reported to proceed via formate and methoxy intermediates.73–75 However, the formate species can also undergo decomposition to produce CO.76,77 Based on these insights, we infer that the stabilization of methoxy species can potentially enhance CH3OH selectivity. A standout report in this context is the recent work by Copéret and coworkers where they incorporated Lewis acidic surface sites at the periphery of Cu to stabilize methoxy intermediates, thereby improving CH3OH selectivity by employing surface organometallic chemistry (SOMC) approach.47 The authors synthesized a series of analogous catalysts containing Cu nanoparticles supported on SiO2 decorated with metal centers of different Lewis acid strength (Cu/M@SiO2, where M = Ti, Zr, Hf, Nb, Ta).47 In this process, first, isolated M sites, free of organic ligands, were generated on SiO2 by grafting a molecular precursor, e.g., M(OSi(OtBu)3)m(OiPr)n, followed by thermal treatment under vacuum to remove organic ligands (Fig. 8A). In the second step (Fig. 8B), Cu nanoparticles were generated on M@SiO2 materials by grafting the copper precursor, [Cu(OtBu)]4 onto these M@SiO2 materials, followed by a reductive thermal treatment. Interestingly, their investigation revealed that while CO formation rates (Fig. 8C) on these materials were nearly identical, CH3OH formation rates varied as a function of the identity of the M atom. The authors demonstrated that the promotion of CH3OH formation rates reflect the increasing acid strength of metal centers in SiO2 support, probed by measuring 13C chemical shift of methoxy surface intermediates from solid-state NMR (Fig. 8D) and pyridine adsorption enthalpies (Fig. 8E). These findings corroborated that the Lewis acid M sites of these catalysts stabilize surface intermediates (formate and methoxy) at the periphery of Cu nanoparticles and influence CH3OH formation rates.47
 | ||
| Fig. 8 Surface organometallic chemistry (SOMC) approach of catalyst synthesis. (A) Isolated M sites, free of organic ligands, were generated on SiO2 by grafting a molecular precursor, e.g., M(OSi(OtBu)3)m(OiPr)n, followed by thermal treatment under vacuum to remove organic ligands. (B) Cu nanoparticles were generated on M@SiO2 materials by grafting the copper precursor, [Cu(OtBu)]4 onto these M@SiO2 materials, followed by a reductive thermal treatment. (C) Formation rate of CH3OH and CO over different Cu/M@SiO2 catalyst. (D) 13C solid-state NMR of formate and methoxy groups over different Cu/M@SiO2 catalyst. (E) 13C chemical shift of methoxy surface intermediates from solid-state NMR plotted against pyridine adsorption enthalpies to probe Lewis acid strength. Adapted with permission from Copéret and coworkers.47 Copyright Angew. Chem. 2021. | ||
In a similar SOMC approach, Copéret and coworkers showed that CuGax alloy formed by grafting Cu on silica containing GaIII sites was highly active and selective for CO2 hydrogenation to CH3OH (∼90% selectivity at a conversion of ∼3%).48 Their investigation reported that under reaction conditions, the silica-supported CuGax de-alloys yielding Cu nanoparticles and GaIII sites, which likely increased interfacial area between Cu0 and GaIIIOx promoting CH3OH formation. Interestingly, only methoxy surface species were observed as intermediates probed via NMR and IR spectroscopy.48 These findings highlight that stabilizing surface intermediates can improve CH3OH yields, offering a potential strategy to increase hydrocarbon yields during tandem CO2 hydrogenation.
Factors affecting hydrocarbon pool (HCP)
The formation of hydrocarbons inside zeolite pores proceeds via a dual cycle mechanism comprised of olefin and aromatic-based methylation/cracking events (Fig. 1).39 The composition of HCP and the relative reactivity of various species in propagation and termination sequences across the catalyst bed govern the HC product selectivity. However, the kinetic and mechanistic elucidation of MTH remains challenging as the concentration and reactivity of olefinic and aromatic chain carriers vary as a function of catalyst properties and reaction conditions, leading to topology, morphology, composition, and time-dependent variations in MTH reaction rates and product selectivity.59 For instance, small pore cage-window type zeolites favor a more aromatic HCP, or increasing BAS density shifts the HCP toward aromatics.41 In the context of CO2 hydrogenation, additional complexities arise, e.g., co-presence of H2 and CO which can further influence HCP. Therefore, a comprehensive understanding of the key factors governing HCP is essential to precisely tune hydrocarbon yield and selectivity in CO2 hydrogenation.Despite advances in bifunctional catalyst design, high CO selectivity remains an intrinsic challenge in CO2 hydrogenation. While CO is often considered an undesired byproduct, recent insights from MTH chemistry suggest that it may play an active role in tuning HCP. Bhan and coworkers demonstrated that CO is mechanistically relevant in increasing ethylene-to-propylene (1.5–3×) and ethylene-to-methylbutenes (1.7×) ratio, both of which indicate a shift in the relative propagation of the aromatic to olefin cycle.78 Additionally, CO can participate in Koch carbonylation reactions with DME, promoting aromatic-cycle propagation and enabling ethylene formation via methyl acetate intermediates.41 Despite these insights, the influence of CO in HCP and HC product selectivity in CO2 hydrogenation remain largely unexplored, warranting the need for targeted investigations into how CO can be leveraged as a potential tuning parameter for HC selectivity.
Regarding the influence of H2 on hydrocarbon selectivity, our recent study has shown that during CO2 hydrogenation, the presence of H2 promotes olefin cycle propagation over the aromatic cycle by facilitating secondary hydrogenation of olefins to paraffins.38 This suppresses olefin aromatization and the formation of deactivation-inducing polycyclic aromatics, thereby mitigating the deactivation of acid function, which is an advantage compared to MTH conversion where zeolites deactivate via coking.79 However, we anticipate that this may also present a stability-selectivity tradeoff, as the suppression of olefin aromatization will likely reduce aromatic selectivity during CO2 hydrogenation to aromatics. Therefore, a detailed understanding of the role of H2 on HCP is essential for optimizing targeted hydrocarbon selectivity.
The composition of zeolites also plays a critical role in shaping HCP and influencing HC selectivity. In this context, our recent study examined the impact BAS strength within the chabazite (CHA) framework on olefin selectivity during CO2 hydrogenation by employing aluminosilicate SSZ-13 and silicoaluminophosphate SAPO-34, both possessing similar acid site densities.38 When integrated as an interpellet admixture with In2O3, SSZ-13 predominantly facilitated secondary hydrogenation of olefins, yielding ∼93% paraffins. In contrast, the In2O3/SAPO-34 admixture produced ∼67% olefins, as the weaker acid strength of SAPO-34 resulted in a lower degree of secondary hydrogenation.38 These findings underscore acid site strength as a crucial parameter for regulating HCP composition and hydrocarbon selectivity. We note that beyond acid strength, acid site density (represented by the Si/Al ratio), also influences HCP dynamics. In this regard, Chen et al. investigated SSZ-13 with varying Si/Al ratios integrated with ZnZrOx and demonstrated that SSZ-13 with only isolated acid sites (i.e., high Si/Al ratio ∼125) effectively mitigated the over-hydrogenation of light olefins to alkanes, thereby enhancing light olefin selectivity (∼89%) compared to lower Si/Al ratio (∼9, yielded ∼93% paraffins) counterpart.28 These findings highlight the tunability of zeolite acidity as a powerful tool for modulating HCP composition and optimizing hydrocarbon selectivity during CO2 hydrogenation.
Solid-state ion-exchange (SSIE) in zeolites
A fundamental challenge in designing efficient bifunctional oxide/zeolite catalysts lies in optimizing the proximity between redox and acid sites. While nanoscale proximity facilitates intermediate transport, it also introduces a caveat of metal oxide migration into zeolite pore channels, leading to solid-state ion exchange (SSIE) with BAS under the harsh reaction conditions (300–450 °C and 20–50 bar pressure).17,35 We note that such SSIE could be avoided by i) conducting reaction at low temperatures and/or ii) employing stable non-migrating metal oxides for CH3OH synthesis (e.g., ZrO2), however, both of these strategies would decrease HC yield as MTH is favorable at higher reaction temperatures and thermally stable metal oxides with fewer oxygen vacancy sites cause less CH3OH yield, respectively.32,80–83 Therefore, elucidating the mechanistic pathways of SSIE and developing strategies to prevent it without compromising catalytic efficiency is a key to designing robust and selective oxide/zeolite catalysts for tandem CO2 hydrogenation.Mechanistic insights on SSIE
A solid-state reaction (Mn+O2n/2 + nH+Z− → Mn+Zn− + nH2O↑) between a zeolite and an oxide, which contains the desired in-going cation, necessitates their intimate contact, e.g., typically achieved through milling or grinding.84–86 This makes SSIE particularly relevant for tandem CO2 hydrogenation, where milling or grinding are often employed to increase the proximity of redox sites and BAS for efficient intermediate transport.22,23,87 Additionally, SSIE requires a reaction temperature of 400–625 K, which falls in the typical operating range for CO2 hydrogenation.35,88,89 As such, careful consideration is required to avoid SSIE and preserve the reactivity of the redox and acid sites. In principle, the transport of in-going cations can proceed through either vapor-phase or surface diffusion mechanisms.90–93 While metals like zinc (Zn) and gallium (Ga) are likely exchanged via intracrystalline gas-phase transport, many metal oxides such as (CuCl2) exhibit low vapor pressures, even at elevated temperatures, making vapor-phase transport less feasible.92 As a result, surface diffusion from the closely contacted oxide to the zeolite particles is considered to be the more dominant mechanism in SSIE.91 However, in either case, the question then remains as to whether ions or molecules of the respective compound diffuse into the zeolite pores after separation or as intact species. Both possibilities are shown schematically in Fig. 9.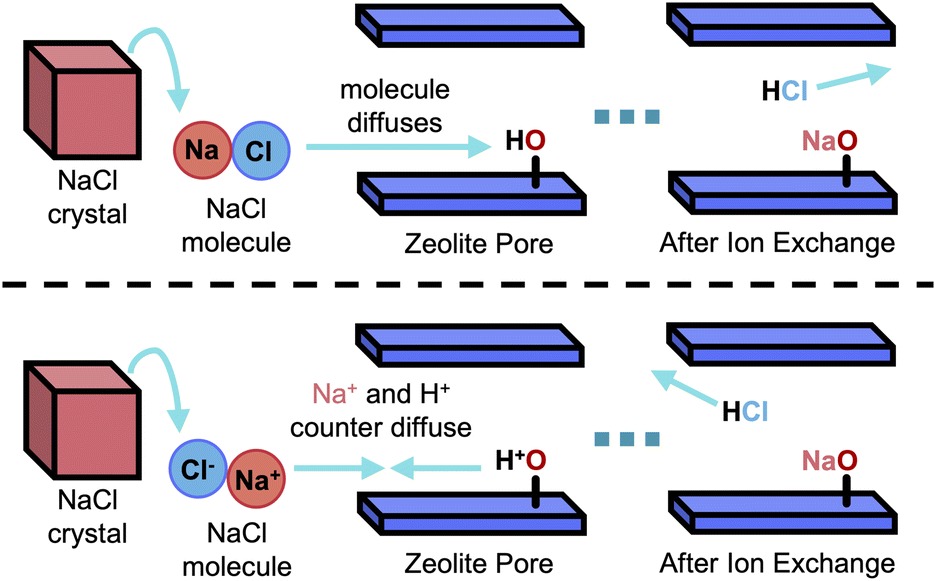 | ||
| Fig. 9 Schematic of two possible mechanism of SSIE. Top scheme: NaCl molecule diffuse, bottom scheme: Na+ and Cl− counter diffuse. Adapted with permission from Karge, H. G.92 Copyright Catal. Today 2008. | ||
In this regard, previous studies suggested that for medium-pore zeolite ferrierite, H-FER, SSIE was not observed with LaCl3 as LaCl3 molecule was too bulky to be able to penetrate the 8 MR pore openings of H-FER.90–93 This indicates that the mechanism of molecular diffusion is more likely to hold. This assumption was further supported by experiments of SSIE of H-ZSM-5 with molecules containing bulky anions (e.g., salts of heteropoly acids such as Cs3[PW12O40]).90–93 Compared to the exchange CsCl, SSIE with Cs3[PW12O40] occurred only to a minor extent, due to partial decomposition of the salt. These studies suggested that the molecules did not dissociate but rather migrated as an intact species for SSIE (top scheme of Fig. 6).
Influence of SSIE on hydrocarbon pool (HCP) mechanisms
Given that SSIE occurs at intimate proximities between redox and acid sites, its catalytic influence on CO2 hydrogenation must be carefully assessed, as it can obscure the intended proximity effects.94,95 Prior studies have demonstrated that SSIE in zeolites can create new active sites, altering HCP mechanism and subsequently affecting HC yield and selectivity. For instance, exchanging the BAS of HZSM-5 with zinc or gallium cations enhanced olefin aromatization,96 while the exchange of BAS in β-zeolite with iron, chromium, or manganese cations promoted oligomerization of lower olefins.97 In the context of tandem CO2 hydrogenation, our recent findings reveal that the nature of the exchanged cation dictates distinct influences to HCP.82 Specifically, SSIE between BAS of SAPO-34 and Inδ+ species shut down HCP propagation inside the channels of SAPO-34 by inhibiting the acidity of SAPO-34, while SSIE with Znδ+ species enhanced hydrogen transfer and secondary hydrogenation likely by creating Lewis acidic sites.82 These SSIE-induced modifications were systematically probed using two key performance metrics: C3 (propane + propylene)/C2 (ethane + ethylene) ratio and paraffin-to-olefin (P/O) ratio during CO2 hydrogenation, which convey the relative propagation of the olefin cycle to the aromatic cycle, and the degree of saturation of olefins in HCP, respectively.59 Using these performance metrics, coupled with the occluded HC analysis and 13C solid-state nuclear magnetic resonance (ssNMR) spectroscopy revealed that ion exchange of BAS with Inδ+ and Znδ+ species influenced the olefin and aromatic pool propagation, consequently altering HC selectivity and yield. These findings underscore the necessity of preventing SSIE of acid sites to mitigate unintended catalytic modifications during CO2 hydrogenation.Mitigating SSIE
Mitigating SSIE in oxide/zeolite systems requires a fundamental understanding of the key factors driving ion exchange, including contact-induced metal oxide migration, volatility-driven gas-phase transport, reduction-assisted SSIE, and moisture-assisted SSIE (Fig. 10).84,92 Identifying the predominant factor is crucial for developing effective mitigation strategies. For instance, volatility-driven SSIE can be assessed by tracking the amount of volatile component in metal oxide, such as Zn in ZnZrOx, after thermal treatment at different temperatures using energy-dispersive X-ray spectroscopy (EDX).98 By correlating the Zn loss with in situ acid site characterization, such as transmission FTIR using pyridine as a probe, the extent of BAS exchange with Zn at different temperatures due to volatility can be estimated.99 We expect such analyses would help determining the optimal operating temperatures to minimize acid site loss. | ||
| Fig. 10 Factors affecting SSIE including volatility-driven gasphase transport (adapted with permission from Redekop, E. A. et al.98), contact-induced migration (adapted with permission from Wang, Y. et al.17 Copyright Angew. Chem. Int. Ed. Engl. 2021), reduction-assisted SSIE (adapted with permission from Mahnaz, F. et al.82 Copyright Chem. Catal. 2024), and moisture-assisted SSIE. | ||
Contact-induced SSIE can be assessed by varying the proximity between redox and acid sites, as evidenced by our recent study showing that despite ZnO being highly volatile, SSIE was less prominent when ZnZrOX was integrated at a microscale distance from BAS of SAPO-34 compared to a nanoscale distance, indicating contact-induced diffusion as the dominant mechanism.82 In such cases, core–shell confinement strategies to create diffusive barriers (e.g., silicalite-1, silica, or alumina) between redox and acid sites can potentially mitigate SSIE.100
Additionally, SSIE can be enhanced under reductive conditions, as observed in our studies where SSIE between BAS of SAPO-34 and Inδ+ was more drastic in the presence of H2, as compared to inert environment.82 Moisture may also facilitate SSIE by stabilizing cations, similar to conventional ion exchange process.92,101,102 In certain cases, moisture aids SSIE by disaggregating oxide species or inhibiting their polymerization. For instance, moisture facilitated the SSIE of acid sites in Y-zeolites with MoO3,84 whereas in dry conditions, the MoO3 species were too bulky to effectively undergo SSIE.103 In such cases, employing hydrophobic coatings on metal oxides or zeolites could be a potential strategy to limit the interactions between BAS and oxide species, limiting excessive SSIE.100 By systematically probing these factors, a more robust framework for suppressing SSIE could be developed, preserving the reactivity of oxide/zeolite bifunctional catalysts.
Future outlook
This perspective highlights the critical challenges in CO2 hydrogenation via the CH3OH-mediated route and potential strategies to circumvent them, focusing on bridging the existing research gaps in the field. A key challenge in using bifunctional oxide/zeolite systems is that the proximity of active sites on metal oxides and BAS plays a crucial role in determining intermediate concentration gradients in acid site domain, therefore mass transport artifacts influence reaction rates and selectivities. Therefore, for elucidating “site-proximity” effect in CO2 hydrogenation, we emphasize on developing diffusion-convection-reaction formulation along with experimentally probing the diffusion artifacts on the selectivity of bulkier hydrocarbons inside zeolite pores. We further propose that, since MTH conversion is inherently mass transport limited, these limitations should be leveraged to control product selectivities by tuning zeolite properties and pore architecture to control diffusion pathways. Probing diffusional effects by using zeolites with different crystallite size and undulation factors may help identifying primary and secondary products and optimizing reactivity and selectivity. When combined with reaction-transport models, these approaches could provide a comprehensive strategy for understanding diffusional influence on observed rates and HC selectivity.Beyond the effects of active-site proximity and intermediate transport, we emphasize on understanding the implications of SSIE of acid sites on rates and HC selectivities and strategies to mitigate it. While conventional acid site characterization methods can detect ion exchange, we propose utilizing useful metrics, such as the propylene-to-ethylene, ethylene-to-2 methyl-butane, paraffin-to-olefin ratios etc., to get better insights into how SSIE influences HCP mechanisms, altering HC selectivities.
A significant challenge in CO2 hydrogenation is the high CO selectivity caused by the RWGS as a side reaction. To tackle this challenge, we emphasize increasing CH3OH yields. An example is innovative synthesis techniques utilizing surface organometallic chemistry (SOMC) techniques to synthesize catalysts that favor high CH3OH selectivity to increase HC yield.
To summarize, a comprehensive understanding of proximity effects, intermediate transport, active site interactions, and their influence on measured reaction rates and selectivities is paramount for advancing CO2 hydrogenation chemistry. Careful interpretation of these phenomena will continue to be strengthened by increasingly advanced experimental and computational techniques. However, without a strong grasp of these concepts, a lack of comprehension could lead to misleading interpretations. Addressing these fundamental challenges is crucial not only for CO2 hydrogenation chemistry but also for similar sustainable process, such as lignocellulosic biomass conversion, or catalytic upcycling of plastics, both of which contain macromolecules where diffusion constraints and catalyst design remain a critical factor in the production of sustainable fuels and chemicals.
Concluding remarks
In our perspective, addressing the challenges of CO2 hydrogenation via the CH3OH-mediated route requires a paradigm shift in how we interpret catalyst performance—moving beyond activity-selectivity studies to a more mechanistic understanding of reaction-transport effects and active site interactions. We believe that the site-proximity effects in bifunctional oxide/zeolite systems and the inherent mass transport artifacts of constituent reactions in MTH step should not only be acknowledged but strategically leveraged through rational modification of zeolites to control product selectivity. Additionally, the use of diagnostic selectivity metrics, such as ethylene-to-propylene, ethylene-to-2-methylbutane, and paraffin-to-olefin ratios, etc., can provide deeper insight into hydrocarbon pool (HCP) dynamics, while advanced synthesis strategies like surface organometallic chemistry (SOMC) can help improving CH3OH yields; together, these approaches can be leveraged to tune hydrocarbon selectivities and yields. As researchers in this field, we see these efforts as essential to bridging current knowledge gaps and guiding the design of next-generation catalytic systems for sustainable CO2 valorization and beyond.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this perspective.Author contributions
F. M. and M. S. – conceptualization. F. M. – writing-original draft. F. M., A. I. and M. S. – figures, writing, review and editing. M. S – funding acquisition and supervision. All authors contributed to the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Artie McFerrin Department of Chemical Engineering at Texas A&M University, the College of Engineering, and the Provost for their financial support. This work was funded by Texas A&M University (TAMU), Texas A&M Engineering Experiment Station (TEES), the Governor's University Research Initiative (GURI), the Oak Ridge Associated Universities through their Ralph E. Powe Junior Faculty Enhancement Award, the National Science Foundation (NSF) CBET grant no 2245474, and the ACS Petroleum Research Fund's Doctoral New Investigator Award. MS acknowledges partial support from Texas A & M University System's National Laboratory Office (NLO) for their Development Fellowship.References
- A. Behr, A. J. Vorholt, K. A. Ostrowski and T. Seidensticker, Green Chem., 2014, 16, 982–1006 RSC.
- K. T. Dinh, M. M. Sullivan, P. Serna, R. J. Meyer and Y. Roman-Leshkov, ACS Catal., 2021, 11, 9262–9270 CrossRef CAS.
- M. Shetty, A. Walton, S. R. Gathmann, M. A. Ardagh, J. Gopeesingh, J. Resasco, T. Birol, Q. Zhang, M. Tsapatsis and D. G. Vlachos, ACS Catal., 2020, 10, 12666–12695 CrossRef CAS.
- J. Gopeesingh, M. A. Ardagh, M. Shetty, S. T. Burke, P. J. Dauenhauer and O. A. Abdelrahman, ACS Catal., 2020, 10, 9932–9942 CrossRef CAS.
- M. A. Ardagh, M. Shetty, A. Kuznetsov, Q. Zhang, P. Christopher, D. G. Vlachos, O. A. Abdelrahman and P. J. Dauenhauer, Chem. Sci., 2020, 11, 3501–3510 RSC.
- G. Kumar, E. Nikolla, S. Linic, J. W. Medlin and M. J. Janik, ACS Catal., 2018, 8, 3202–3208 CrossRef CAS.
- H. Wu, B. Zhang, H. Liang, L. Zhai, G. Wang and Y. Qin, Innovation, 2020, 1, 100029 CAS.
- E. A. Wilson, S. C. Eady, T. Silbaugh, L. T. Thompson and M. A. Barteau, J. Catal., 2021, 404, 977–984 CrossRef CAS.
- J.-C. Wasilke, S. J. Obrey, R. T. Baker and G. C. Bazan, Chem. Rev., 2005, 105, 1001–1020 CrossRef CAS PubMed.
- M. Muzzio, H. Lin, K. Wei, X. Guo, C. Yu, T. Yom, Z. Xi, Z. Yin and S. Sun, ACS Sustain. Chem. Eng., 2020, 8, 2814–2821 CrossRef CAS.
- J. Louie, C. W. Bielawski and R. H. Grubbs, J. Am. Chem. Soc., 2001, 123, 11312–11313 CrossRef CAS PubMed.
- M. C. Haibach, S. Kundu, M. Brookhart and A. S. Goldman, Acc. Chem. Res., 2012, 45, 947–958 CrossRef CAS PubMed.
- D. Gorbunov, M. Nenasheva, E. Naranov, A. Maximov, E. Rosenberg and E. Karakhanov, Appl. Catal., A, 2021, 623, 118266 CrossRef CAS.
- S. D. Drouin, F. Zamanian and D. E. Fogg, Organometallics, 2001, 20, 5495–5497 CrossRef CAS.
- R. Helmer, S. S. Borkar, A. Li, F. Mahnaz, J. Vito, A. Iftakher, M. F. Hasan, S. Rangarajan and M. Shetty, Angew. Chem., Int. Ed., 2025, 64, e202416384 CrossRef CAS PubMed.
- R.-P. Ye, J. Ding, W. Gong, M. D. Argyle, Q. Zhong, Y. Wang, C. K. Russell, Z. Xu, A. G. Russell and Q. Li, Nat. Commun., 2019, 10, 1–15 CrossRef PubMed.
- Y. Wang, G. Wang, L. I. van der Wal, K. Cheng, Q. Zhang, K. P. de Jong and Y. Wang, Angew. Chem., 2021, 133, 17876–17884 CrossRef.
- Y. Wang, L. Tan, M. Tan, P. Zhang, Y. Fang, Y. Yoneyama, G. Yang and N. Tsubaki, ACS Catal., 2018, 9, 895–901 CrossRef.
- J. Wang, R. Li, G. Zhang, C. Dong, Y. Fan, S. Yang, M. Chen, X. Guo, R. Mu and Y. Ning, J. Am. Chem. Soc., 2024, 146, 5523–5531 CrossRef CAS.
- G. Wang, Y. Wang, J. Cao, X. Wang, Y. Yi and F. Liu, Microporous Mesoporous Mater., 2020, 291, 109693 CrossRef.
- P. Tian, G. Zhan, J. Tian, K. B. Tan, M. Guo, Y. Han, T. Fu, J. Huang and Q. Li, Appl. Catal., B, 2022, 315, 121572 CrossRef CAS.
- H. Tian, H. He, J. Jiao, F. Zha, X. Guo, X. Tang and Y. Chang, Fuel, 2022, 314, 123119 CrossRef CAS.
- D. Tian, Y. Men, S. Liu, J. Wang, Z. Li, K. Qin, T. Shi and W. An, Colloids Surf., A, 2022, 653, 129945 CrossRef CAS.
- A. Livescu, R. Navar, J. R. Mangalindan, F. Mahnaz, Y. Ge, M. Shetty and X. Yang, Top. Catal., 2024, 1–22 Search PubMed.
- Z. Liu, Y. Ni, T. Sun, W. Zhu and Z. Liu, J. Energy Chem., 2021, 54, 111–117 CrossRef CAS.
- P. Gao, S. Li, X. Bu, S. Dang, Z. Liu, H. Wang, L. Zhong, M. Qiu, C. Yang and J. Cai, Nat. Chem., 2017, 9, 1019–1024 CrossRef CAS PubMed.
- P. Gao, S. Dang, S. Li, X. Bu, Z. Liu, M. Qiu, C. Yang, H. Wang, L. Zhong and Y. Han, ACS Catal., 2018, 8, 571–578 CrossRef CAS.
- S. Chen, J. Wang, Z. Feng, Y. Jiang, H. Hu, Y. Qu, S. Tang, Z. Li, J. Liu and J. Wang, Angew. Chem., 2024, 136, e202316874 CrossRef.
- J. Chen, X. Wang, D. Wu, J. Zhang, Q. Ma, X. Gao, X. Lai, H. Xia, S. Fan and T.-S. Zhao, Fuel, 2019, 239, 44–52 CrossRef CAS.
- J. R. Mangalindan, F. Mahnaz, J. Vito, N. Suphavilai and M. Shetty, ACS Eng. Au, 2025, e5c00008 CrossRef.
- F. Mahnaz, J. R. Mangalindan, B. C. Dharmalingam, J. Vito, Y.-T. Lin, M. Akbulut, J. J. Varghese and M. Shetty, ACS Sustain. Chem. Eng., 2024, 12, 5197–5210 CrossRef CAS PubMed.
- F. Mahnaz, V. Dunlap, R. Helmer, S. S. Borkar, R. Navar, X. Yang and M. Shetty, ChemCatChem, 2023, 15, e202300402 CrossRef CAS.
- L. Guo, P. Ai, X. Gao, H. Wu, X. Wang, Y. Kugue, J. Liang, W. Gao, X. Guo, J. Sun, EES Catalysis, 2023 Search PubMed.
- P. Sharma, J. Sebastian, S. Ghosh, D. Creaser and L. Olsson, Catal. Sci. Technol., 2021, 11, 1665–1697 RSC.
- K. B. Tan, K. Xu, D. Cai, J. Huang and G. Zhan, Chem. Eng. J., 2023, 463, 142262 CrossRef CAS.
- T. Xie, J. Ding, X. Shang, X. Zhang and Q. Zhong, J. Colloid Interface Sci., 2023, 635, 148–158 CrossRef CAS.
- Y. Li, L. Zeng, G. Pang, X. Wei, M. Wang, K. Cheng, J. Kang, J. M. Serra, Q. Zhang and Y. Wang, Appl. Catal., B, 2023, 324, 122299 CrossRef CAS.
- F. Mahnaz, J. R. Mangalindan, J. Vito, R. Helmer and M. Shetty, J. Catal., 2024, 434, 115518 CrossRef CAS.
- S. Ilias and A. Bhan, ACS Catal., 2013, 3, 18–31 CrossRef CAS.
- S. Ilias and A. Bhan, J. Catal., 2012, 290, 186–192 CrossRef CAS.
- A. Hwang and A. Bhan, Acc. Chem. Res., 2019, 52, 2647–2656 CrossRef CAS.
- P. B. Weisz, in Advances in Catalysis, Elsevier, 1962, vol. 13, pp. 137–190 Search PubMed.
- P. Weisz and E. Swegler, Science, 1957, 126, 31–32 CrossRef CAS.
- P. Weisz, Science, 1956, 123, 887–888 CrossRef CAS PubMed.
- W. Hu, G. Noh and E. Iglesia, J. Catal., 2023, 425, 125–142 CrossRef CAS.
- G. Noh, Z. Shi, S. I. Zones and E. Iglesia, J. Catal., 2018, 368, 389–410 CrossRef CAS.
- G. Noh, E. Lam, D. T. Bregante, J. Meyet, P. Šot, D. W. Flaherty and C. Copéret, Angew. Chem., 2021, 133, 9736–9745 CrossRef.
- E. Lam, G. Noh, K. W. Chan, K. Larmier, D. Lebedev, K. Searles, P. Wolf, O. V. Safonova and C. Copéret, Chem. Sci., 2020, 11, 7593–7598 RSC.
- Z. Li, Y. Qu, J. Wang, H. Liu, M. Li, S. Miao and C. Li, Joule, 2019, 3, 570–583 CrossRef CAS.
- Z. Li, J. Wang, Y. Qu, H. Liu, C. Tang, S. Miao, Z. Feng, H. An and C. Li, ACS Catal., 2017, 7, 8544–8548 CrossRef CAS.
- J. C. Gottifredi, E. E. Gonzo and O. Quiroga, Concept and Design of Chemical Reactors, 1986 Search PubMed.
- A. Krishna, Catal. Rev.: Sci. Eng., 1990, 32, 279–381 CrossRef CAS.
- C. W. Hullfish, J. Z. Tan, H. I. Adawi and M. L. Sarazen, ACS Catal., 2023, 13, 13140–13150 CrossRef CAS.
- F. Arena, G. Mezzatesta, G. Zafarana, G. Trunfio, F. Frusteri and L. Spadaro, J. Catal., 2013, 300, 141–151 CrossRef CAS.
- H. Hu, J. Zhu, F. Yang, Z. Chen, M. Deng, L. Weng, Y. Ling and Y. Zhou, Chem. Commun., 2019, 55, 6495–6498 RSC.
- Y. Bhawe, M. Moliner-Marin, J. D. Lunn, Y. Liu, A. Malek and M. Davis, ACS Catal., 2012, 2, 2490–2495 CrossRef CAS.
- R. Khare, D. Millar and A. Bhan, J. Catal., 2015, 321, 23–31 CrossRef CAS.
- A. Hwang, T. T. Le, Z. Shi, H. Dai, J. D. Rimer and A. Bhan, J. Catal., 2019, 369, 122–132 CrossRef CAS.
- Z. Shi and A. Bhan, J. Catal., 2023, 421, 198–209 CrossRef CAS.
- M. L. Sarazen, E. Doskocil and E. Iglesia, ACS Catal., 2016, 6, 7059–7070 CrossRef CAS.
- Q. Sun, Y.-L. Zhang, H.-Y. Chen, J.-F. Deng, D. Wu and S.-Y. Chen, J. Catal., 1997, 167, 92–105 CrossRef CAS.
- S. S. Iyer, T. Renganathan, S. Pushpavanam, M. V. Kumar and N. Kaisare, J. CO2 Util., 2015, 10, 95–104 CrossRef CAS.
- M. Bowker, ChemCatChem, 2019, 11, 4238–4246 CrossRef CAS PubMed.
- I. Nezam, W. Zhou, G. S. Gusmão, M. J. Realff, Y. Wang, A. J. Medford and C. W. Jones, J. CO2 Util., 2021, 45, 101405 CrossRef CAS.
- I. Yarulina, A. D. Chowdhury, F. Meirer, B. M. Weckhuysen and J. Gascon, Nat. Catal., 2018, 1, 398–411 CrossRef CAS.
- F. J. Keil, Microporous Mesoporous Mater., 1999, 29, 49–66 CrossRef CAS.
- W. Prachumsai, S. Pangtaisong, S. Assabumrungrat, P. Bunruam, C. Nakvachiratrakul, D. Saebea, P. Praserthdam and S. Soisuwan, J. Environ. Chem. Eng., 2021, 9, 104979 CrossRef CAS.
- M. Huš, V. D. Dasireddy, N. S. Štefančič and B. Likozar, Appl. Catal., B, 2017, 207, 267–278 CrossRef.
- Y. Wang, X. Gao, M. Wu and N. Tsubaki, EcoMat, 2021, 3, e12080 CrossRef CAS.
- D. Zhao, S. Han and E. V. Kondratenko, ChemCatChem, 2023, 15, e202300679 CrossRef CAS.
- S. Ghosh, J. Sebastian, L. Olsson and D. Creaser, Chem. Eng. J., 2021, 416, 129120 CrossRef CAS.
- T. Numpilai, C. Wattanakit, M. Chareonpanich, J. Limtrakul and T. Witoon, Energy Convers. Manage., 2019, 180, 511–523 CrossRef CAS.
- A. Beck, M. A. Newton, L. G. van de Water and J. A. van Bokhoven, Chem. Rev., 2024, 124, 4543–4678 CrossRef CAS PubMed.
- G. Pacchioni, ACS Catal., 2024, 14, 2730–2745 CrossRef CAS.
- Y. Yang, C. A. Mims, D. Mei, C. H. Peden and C. T. Campbell, J. Catal., 2013, 298, 10–17 CrossRef CAS.
- S. Kattel, B. Yan, Y. Yang, J. G. Chen and P. Liu, J. Am. Chem. Soc., 2016, 138, 12440–12450 CrossRef CAS.
- S. Kattel, P. Liu and J. G. Chen, J. Am. Chem. Soc., 2017, 139, 9739–9754 CrossRef CAS.
- Z. Shi and A. Bhan, Chem. Eng. J., 2023, 456, 140867 CrossRef CAS.
- S. S. Arora, Z. Shi and A. Bhan, ACS Catal., 2019, 9, 6407–6414 CrossRef CAS.
- M. Stöcker, Microporous Mesoporous Mater., 1999, 29, 3–48 CrossRef.
- R. Singh, K. Tripathi and K. K. Pant, Fuel, 2021, 303, 121289 CrossRef CAS.
- F. Mahnaz, B. C. Dharmalingam, J. R. Mangalindan, J. Vito, J. J. Varghese and M. Shetty, Chem. Catal., 2025, 5(2), 101183 CrossRef CAS.
- H. Chen, H. Cui, Y. Lv, P. Liu, F. Hao and W. Xiong, Fuel, 2022, 314, 123035 CrossRef CAS.
- G. Ertl, H. Knözinger and J. Weitkamp, Handbook of heterogeneous catalysis, VCH Weinheim, 1997, vol. 2 Search PubMed.
- H. G. Karge, Studies in Surface Science and Catalysis, 1997, vol. 105, pp. 1901–1948 Search PubMed.
- H. G. Karge, in Studies in Surface Science and Catalysis, Elsevier, 1994, vol. 83, pp. 135–146 Search PubMed.
- Y. Wang, F. Liu, M. Yao, J. Ma, S. Geng, J. Cao and X. Wang, Sep. Purif. Technol., 2025, 355, 129743 CrossRef CAS.
- H. M. Torres Galvis and K. P. de Jong, ACS Catal., 2013, 3, 2130–2149 CrossRef CAS.
- M. G. Sibi, D. Verma and J. Kim, Catal. Rev., 2022, 1–60 Search PubMed.
- G. Ertl, H. Knözinger and J. Weitkamp, Preparation of solid catalysts, John Wiley & Sons, 2008 Search PubMed.
- H. G. Karge, H. K. Beyer and G. Borbély, Catal. Today, 1988, 3, 41–52 CrossRef CAS.
- H. G. Karge, in Handbook of Heterogeneous Catalysis, Wiley-VCH, 2008, pp. 484–510 Search PubMed.
- D. Yin and D. Yin, Microporous Mesoporous Mater., 1998, 24, 123–126 CrossRef CAS.
- T. Liu, Z. Liu, S. Jiang, P. Peng, Z. Liu, A. D. Chowdhury and G. Liu, Chem. Soc. Rev., 2025, 54, 2726–2761 RSC.
- Y. Yue, J. Tian, J. Ma, S. Yang, W. Li, J. Huang, Q. Li and G. Zhan, Appl. Catal., B, 2024, 355, 124158 CrossRef CAS.
- Y. Ono, H. Adachi and Y. Senoda, J. Chem. Soc., Faraday Trans. 1, 1988, 84, 1091–1099 RSC.
- D. Esquivel, A. J. Cruz-Cabeza, C. Jimenez-Sanchidrian and F. J. Romero-Salguero, Microporous Mesoporous Mater., 2013, 179, 30–39 CrossRef CAS.
- E. A. Redekop, T. Cordero-Lanzac, D. Salusso, A. Pokle, S. Oien-Odegaard, M. F. Sunding, S. Diplas, C. Negri, E. Borfecchia and S. Bordiga, Chem. Mater., 2023, 35, 10434–10445 CrossRef CAS PubMed.
- Y. Yuan, E. Huang, S. Hwang, P. Liu and J. G. Chen, Nat. Commun., 2024, 15, 6529 CrossRef CAS.
- S. Xing, S. Turner, D. Fu, S. van Vreeswijk, Y. Liu, J. Xiao, R. Oord, J. Sann and B. M. Weckhuysen, JACS Au, 2023, 3, 1029–1038 CrossRef CAS.
- X. Ren, S. Yang, R. Xu, M. Guo, W. Huang, M. Ding and J. Zhong, J. Membr. Sci., 2022, 643, 119998 CrossRef CAS.
- R. P. Townsend, E. N. Coker, in Studies in surface science and catalysis, Elsevier, 2001, vol. 137, pp. 467–524 Search PubMed.
- A. V. Kucherov and A. A. Slinkin, J. Mol. Catal., 1994, 90, 323–354 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |