
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePore engineering in metal–organic frameworks and covalent organic frameworks: strategies and applications
Yanpei
Song
 and
Shengqian
Ma
*
and
Shengqian
Ma
*
Department of Chemistry, University of North Texas, Denton, TX 76201, USA. E-mail: Shengqian.Ma@unt.edu
First published on 14th June 2025
Abstract
Crystalline porous materials, particularly metal–organic frameworks (MOFs) and covalent organic frameworks (COFs), have garnered significant attention for advanced applications due to their tunable pore environments and versatile functionalities. By precisely controlling factors such as size, shape, functional sites, and pore distribution, MOFs and COFs can be tailored to exhibit high selectivity for specific molecules, making them ideal for applications in gas storage and separation, catalysis, and water remediation. This review provides a background overview, beginning with an introduction to pore surface engineering strategies and the design features of MOFs and COFs. It then highlights recent advancements in three key research areas that our group has investigated in-depth over the past decade, discussing the strategies and principles involved. Finally, we outline the remaining challenges and offer our perspectives on future opportunities for pore-engineered MOFs and COFs.
 Yanpei Song | Yanpei Song received his BS from Beijing University of Chemical Technology, China in 2017, and graduated from the University of North Texas with a PhD degree in 2023 under the supervision of Prof. Shengqian Ma. Since 2023, he has been a postdoctoral research associate in the Nanomaterials Chemistry Group, Chemical Science Division at Oak Ridge National Laboratory. His research interests focus on the development of functional covalent organic frameworks, and porous organic polymers for environmental remediation and precious metal recovery. |
 Shengqian Ma | Shengqian Ma obtained his BS degree from Jilin University in 2003 and graduated from Miami University with a PhD Degree in 2008. After finishing a two-year Director's Postdoctoral Fellowship at Argonne National Laboratory, he joined the Department of Chemistry at University of South Florida as an Assistant Professor in 2010 and was promoted to Associate Professor with early tenure in 2015 and to Full Professor in 2018. In August 2020, he joined the University of North Texas as the Robert A. Welch Chair in Chemistry. His current research interest focuses on the task-specific design and functionalization of advanced porous materials for energy-, biological-, and environmental-related applications. |
1. Introduction
The intriguing properties of crystalline porous materials, including zeolites,1–3 metal–organic frameworks (MOFs),4–9 covalent organic frameworks (COFs),10–15 and hydrogen-bonded organic frameworks (HOFs),16–20 have drawn significant interest from diverse research fields over the past several decades. Among these crystalline porous materials, MOFs and COFs have attracted particular attention due to their highly tunable structures.21–23 Unlike traditional porous materials such as activated carbon,24 molecular sieves,25 and mesoporous silica,26 which often exhibit limited structural regularity, poorly defined pore architectures, and minimal chemical tunability, MOFs and COFs offer precise control over pore size, shape, connectivity, and surface functionality. Their modular construction, based on the selection of organic linkers and metal or covalent nodes, enables the formation of frameworks with diverse and well-defined pore structures. This high degree of structural programmability has driven a surge in the development of novel porous materials with broad applicability across various fields.MOFs are a class of hybrid organic–inorganic crystalline materials composed of metal ions or metal clusters (nodes) connected by organic ligands (linkers) through coordination bonds. The vast combinational possibilities between metals and organic ligands result in a wide range of structures with highly tunable properties. In contrast, COFs are fully organic crystalline porous materials composed of organic building blocks connected via strong covalent bonds (e.g., C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, CC, B–O bonds). Unlike MOFs, which have metal nodes, COFs are constructed entirely from organic molecules, resulting in lightweight structures with higher thermal and chemical stability. Recently, a new class of crystalline porous materials, covalent metal–organic frameworks (CMOFs), has emerged, combining the key features of both MOFs and COFs. CMOFs integrate the structural precision and covalent connectivity of COFs with the functional versatility and catalytic potential of metal centers in MOFs.27
N, CC, B–O bonds). Unlike MOFs, which have metal nodes, COFs are constructed entirely from organic molecules, resulting in lightweight structures with higher thermal and chemical stability. Recently, a new class of crystalline porous materials, covalent metal–organic frameworks (CMOFs), has emerged, combining the key features of both MOFs and COFs. CMOFs integrate the structural precision and covalent connectivity of COFs with the functional versatility and catalytic potential of metal centers in MOFs.27
The distinctive morphologies and tunable pore surface chemistries of MOFs and COFs not only provide exceptionally large internal surface areas—up to approximately 7839 m2 g−1 for MOFs28 and 5083 m2 g−1 for COFs29—with uniform pore channels and windows, but also endow them with versatile pore functionalities, making them highly suitable for various industrial applications such as gas storage and separation,30–37 heterogeneous catalysis,38–44 proton conduction,45–49 chemical sensing,50–56 and environmental remediation.57–62 Abundant opportunities for tailoring their pore surfaces through pore engineering can be readily achieved through a wide range of chemical strategies, from de novo design of organic linkers and connecting nodes to post-synthetic modifications (PSMs).63–69De novo synthesis allows precise control over pore size, shape, and connectivity by designing functionalized linkers/monomers and metal nodes, while PSM enables the introduction of specific functional moieties or active sites into pre-formed frameworks. Additionally, sophisticated engineering techniques, such as pore space partitioning and hierarchical pore construction, can further optimize their performance. Pore surface modification, such as grafting functional groups or catalytic sites, enables the alignment of ordered active centers within the pores, facilitating selective molecular interactions. Meanwhile, partitioning or sub-framework assembly can tune the pore environment to accommodate target analytes based on their molecular size, polarity, or electronic properties. These tailored modifications are particularly crucial for meeting industrial requirements, such as selectively storing or separating desired products from complex mixtures, especially small-molecule gases (e.g., H2, CO2, CH4). The synergy between rational framework design and advanced post-synthetic engineering highlights the tremendous potential of MOFs and COFs in addressing critical challenges in energy, environment, and chemical industries.
2. Pore surface engineering
Pore engineering refers to a comprehensive set of processes aimed at modifying the pore structures of MOFs and COFs. These processes include the construction, combination, functionalization, and partitioning of pores at the unit cell level. Additionally, pore engineering can involve the generation of hierarchical pore structures—ranging from micro to meso and even macro pores—within the framework. The primary goal of pore engineering is to optimize the pore characteristics of MOFs and COFs, ensuring that these materials meet the specific requirements of various applications. This optimization typically involves introducing specific functional sites or guest species into targeted pore positions. These species are often immobilized through covalent bonds or coordination interactions, which help tune the chemical environment or morphology of the pore surface. By carefully controlling the pore structure and its chemistry, pore engineering enables the precise customization of the material's properties, making it adaptable to a broad range of applications.Through the pore engineering methodology, MOFs and COFs can be endowed with a variety of versatile pore features, including enhanced surface areas, controlled pore sizes, and specialized functionalities. This level of customization enables the materials to be tailored for specific industrial applications. Additionally, pore engineering acts as a powerful tool for developing crystalline porous materials with targeted, application-specific properties, driving innovation across a wide range of scientific and industrial fields.
2.1 De novo design of linkers and connecting nodes
The fundamental principle of pore construction in MOFs lies in the careful selection of metal clusters and organic ligands, following a bottom-up synthetic approach. The formation of these metal clusters is highly dependent on experimental conditions, including the choice of metal precursors, solvent during the solvothermal process, and reaction temperature. The pioneering MOF structures reported by Yaghi's group, MOF-2 and MOF-5, showcase how differences in coordination modes under varying synthetic conditions can lead to distinct pore architectures.70,71 Both MOFs are constructed from zinc(II) clusters and 1,4-benzenedicarboxylic acid (H2BDC) as the organic ligand. However, MOF-2 features Zn2(COO)4 paddlewheel units, while MOF-5 forms Zn4O(COO)6 secondary building units (SBUs). This difference in coordination geometry results in contrasting pore topologies: MOF-2 adopts a 2D layered structure with lower surface area and less accessible porosity, whereas MOF-5 forms a 3D open framework, offering a higher surface area and greater porosity. These structural distinctions highlight how synthetic control over metal–ligand coordination plays a crucial role in tuning the porous properties and functional potential of MOFs.Incorporating mixed metals is a common strategy to adjust framework flexibility, tune pore chemistry and functionality, and introduce additional active sites. The simplest method for synthesizing mixed-metal MOFs (MM-MOFs) is a direct one-pot solvothermal synthesis using a mixture of different metal precursors. However, this approach often lacks precise control over metal distribution, leading to challenges such as uneven metal arrangement, where one metal species may dominate the connecting nodes, while the other forms (hydroxy)oxide clusters on the pore surfaces instead of integrating into the framework. To address this challenge, selecting metals with similar coulombic charges, ionic radii, and chemical behaviors are essential for achieving a uniform metal distribution within the framework.72 Wang et al. successfully incorporated up to 10 different divalent metals (Mg, Ca, Sr, Ba, Mn, Fe, Co, Ni, Zn, and Cd) into microcrystalline MOF-74 via a one-pot solvothermal reaction, demonstrating the feasibility of controlled metal integration (Fig. 1).73 By optimizing metal–ligand coordination, reaction conditions, and precursor compatibility, the development of mixed-metal MOFs (MM-MOFs) with enhanced pore functionality, diverse active sites, and improved performance in applications such as gas storage, separation, and catalysis becomes achievable.
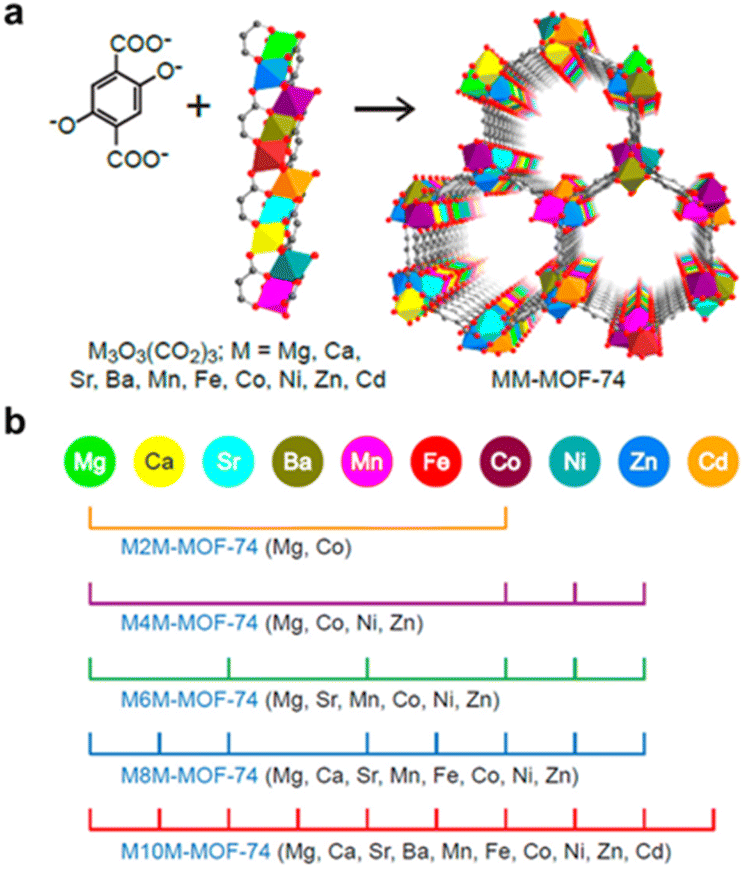 | ||
| Fig. 1 (a) Schematic illustration of synthesizing MM-MOF-74. (b) Combination of metal ions used to synthesize MM-MOF-74. Reproduced with permission.73 Copyright 2014, American Chemical Society. | ||
The use of multiple linkers (mixed linkers) to modify the functionality and pore structure of MOFs is a highly effective strategy for achieving precise pore engineering. This approach is relatively easy to implement because different ligands often share the same coordination mode with metal nodes, differing only slightly in their functional groups. The pioneering work on multivariate (MTV) MOFs was reported by Deng et al., who successfully synthesized MTV-MOF-5 using up to eight different linkers in a one-pot reaction, resulting in a single-phase MTV-MOF-5 crystal.74 In their approach, all selected ligands had identical lengths and coordination sites, ensuring their orderly incorporation into a single MOF crystal without disrupting the framework structure. This multivariate linker strategy was subsequently extended to the development of MTV-COFs, where up to four different monomers with various functionalities were uniformly incorporated into a single-phase COF crystal (Fig. 2).75 The successful distribution of multiple building blocks in both MTV-MOFs and MTV-COFs highlights the versatility of this approach for customizing pore environments and tuning material properties for diverse applications.76,77
 | ||
| Fig. 2 Construction of single-linker COFs and MTV-COFs through pure linkers or mixed linkers and the influence of solvent combinations on their synthesis. Reproduced with permission.75 Copyright 2023, American Chemical Society. | ||
In contrast to MOFs, which have connecting nodes formed through coordination bonds, COFs are typically constructed from reversible covalent bonds via dynamic covalent reactions (DCRs). During COF formation, processes such as bond formation, bond cleavage, and bond exchange occur simultaneously under appropriate conditions. This ongoing dynamic bond breaking and reformation allows the framework to undergo self-healing, effectively repairing defects and resulting in highly ordered pore structures and highly crystalline frameworks. The pore chemistry of COFs can be readily tuned by introducing different linkage units, even when maintaining the same rigid skeleton, enabling the design of COFs with diverse functionalities. The first successful synthesis of COFs via reversible reactions was achieved by Yaghi in 2005, who utilized boronic ester condensation (e.g., boronic ester and boroxine formation).78 This breakthrough not only demonstrated the self-healing ability of purely organic frameworks but also yielded the first porous, crystalline, covalent organic frameworks (COFs). Following this pioneering work, other reversible reactions, particularly Schiff-base condensations (e.g., imine, hydrazone, azine, and Salen linkages),79–86 have become prominent in COF synthesis. Among these, imine-linked COFs have gained notable success due to their high thermal and chemical stability, as well as their broad chemical tunability, derived from the vast selection of functional amines and aldehydes, making them highly suitable for various task-specific applications. Moreover, imine-linked COFs can undergo PSMs to yield specifically evolved functional COFs, further enhancing their properties and expanding their application potential across diverse research fields.
In recent years, irreversible covalent bonds have been increasingly employed to construct COFs with enhanced stability, enabling their application under harsh conditions. Several types of irreversible linkages, including β-ketoenamine,87–89 polyimide,90–92 imidazole,93–95 and benzoxazole,96–98 are primarily derived from imine-based chemistry.99 Among these, β-ketoenamine-linked COFs have emerged as one of the most popular platforms for pore engineering, thanks to their high crystallinity and synthetic feasibility. Beyond imine-derived linkages, vinylenes and fused heterocycle linkages are two additional types of irreversible bonds widely employed for constructing ultrastable COFs.100–104 The Feng group105 and Jiang group106 reported pioneering examples of vinylene-linked COFs in 2016 and 2017, respectively, using the Knoevenagel condensation reaction to form carbon–carbon double bonds. Subsequently, the aldol condensation reaction107 and the Horner–Wadsworth–Emmons reaction,108 both involving aldehyde monomers and active methyl groups, were developed to expand the library of vinylene-linked COFs, further enhancing their synthetic versatility. In another notable advancement, Thomas et al. introduced the concept of covalent triazine frameworks (CTFs),109 synthesizing a series of triazine-linked polymers via highly dynamic condensation reactions. The CN aromatic linkages (triazine units) in CTFs, fused through –CN units, impart these frameworks with high chemical stability, rich nitrogen content, and expanded practical applications, particularly in heterogeneous catalysis and gas storage.110–112 Additionally, other fused heterocycle linkages, such as dioxin,113–115 piperazine,116,117 pyrazine,118 and thiazole,119,120 have been employed to construct ultrastable COFs with excellent acid- and base-resistance. Moreover, these fused-ring structures generally exhibit enhanced electrical conductivity compared to imine-linked COFs, making them highly promising candidates for electrocatalysis and other electrochemical applications. Collectively, the development of irreversible linkages in COFs has significantly broadened their structural diversity, chemical robustness, and functional tunability, driving their deployment in demanding industrial and scientific applications.
Recently, CMOFs have emerged as a promising class of crystalline porous materials that integrate the advantages of MOFs and COFs. Their synthesis typically involves assembling metal complexes or clusters with functional groups (e.g., –CHO, –NH2), which then react with organic linkers via dynamic covalent chemistry to form extended networks. This approach combines the functional versatility of metal centers with the high chemical stability of covalent bonds. The de novo design of linkers and connecting nodes is central to CMOF development. By tailoring the geometry and reactivity of organic linkers and metal-based precursors, it is possible to control the framework structure, pore environment, and overall functionality. Li's group has established a versatile strategy for constructing CMOFs by synthesizing cyclic trinuclear units (CTUs) based on coinage metals (Cu, Ag, Au), which are functionalized with reactive groups.121–128 These CTUs can undergo covalent linkage with a variety of organic monomers, leading to CMOFs with tunable structures and properties, such as redox activity, luminescence, metallophilic interactions, and catalytic functionality. As such, CMOFs offer a robust platform for developing stable and multifunctional porous materials with promising applications in catalysis, sensing, and energy conversion.
2.2 Post-synthetic modifications for tailoring pore chemistry
PSMs offer a powerful and versatile approach to tailoring the pore chemistry of MOFs and COFs, expanding their functionality beyond what is achievable through direct synthesis.129–132 Unlike de novo synthesis, which requires precise control over precursor design and reaction conditions, PSMs enable the site-selective incorporation of functional groups that are otherwise difficult to introduce during initial framework formation. This allows for fine-tuning of host–guest interactions, enhancing selectivity and efficiency in applications such as gas storage, molecular separation, catalysis, proton conduction, and sensing. Beyond functionalization, PSMs play a crucial role in reinforcing structural stability, making MOFs and COFs more resistant to moisture, acids, bases, and high temperatures. By modifying reactive sites or strengthening framework linkages, PSMs prevent framework degradation and improve material robustness under harsh operational conditions. This is particularly beneficial for COFs, where dynamic imine bonds can be post-synthetically converted into stronger covalent linkages such as amide and fused heterocyclic bonds, significantly enhancing chemical and thermal stability.133 Moreover, PSMs facilitate hierarchical pore engineering, allowing precise control over pore size, shape, and surface chemistry to accommodate specific molecular targets. This has profound implications for gas separation, biomolecular recognition, drug delivery, and size-selective catalysis, where optimized pore environments are essential for performance. By leveraging a diverse range of PSM techniques, including oxidation, hydrolysis, acylation, and cross-linking reactions, researchers can systematically enhance both the functionality and durability of MOFs and COFs, broadening their practical applications across multiple scientific and industrial fields.Despite these advantages, PSMs also present several limitations that need to be addressed to fully realize their potential: (1) incomplete or non-uniform functionalization often occurs due to steric hindrance or limited diffusion of modifiers within the framework, leading to heterogeneous surface chemistry and inconsistent performance; (2) excessive or bulky grafting can cause pore blockage and reduced accessible surface area, thereby compromising adsorption and transport properties; (3) certain post-modification reactions—especially those involving harsh reagents or conditions—may induce partial framework degradation, particularly in less robust MOFs; (4) the precise characterization of modification degree and location within confined pore environments remains challenging, hindering the rational design of structure–property relationships. To overcome these issues, several strategies can be adopted: the use of small, highly reactive modifiers and optimized solvent systems can improve the uniformity of functionalization; controlling grafting density and selecting appropriately sized modifiers help preserve porosity; employing mild reaction conditions and orthogonal chemistries can maintain structural integrity; and advanced characterization tools such as solid-state nuclear magnetic resonance, synchrotron-based X-ray techniques, and vibrational spectroscopy can offer deeper insight into the spatial distribution and extent of functionalization.
Ligand exchange, a key PSM strategy, has emerged as a powerful method for incorporating multiple functionalities into MOFs, leading to the formation of MTV-MOFs. Unlike de novo synthesis, which requires precise control over precursor compatibility, ligand exchange allows for the substitution of organic linkers in a pre-formed MOF without disrupting its overall framework integrity. This method provides a versatile route for introducing functional diversity, modifying pore environments, and tuning MOF properties for specific applications. In ligand exchange, pre-assembled MOFs are immersed in a solution containing new ligands, which gradually replace the original linkers through thermodynamic and kinetic exchange processes. The feasibility of this method depends on several factors, including the stability of the framework, the binding affinity of the incoming ligands, and solvent compatibility. Li et al. demonstrated a stepwise ligand exchange strategy to prepare a series of isoreticular bio-MOF-100 analogues (Fig. 3).139 In this approach, the ligand 2,6-naphthalenedicarboxylate (NDC), originally used to construct bio-MOF-101, was replaced by 4,4′-biphenyldicarboxylate (H2BPDC) through a controlled soaking process. Specifically, bio-MOF-101 was immersed in an H2BPDC/DMF/NMP solution at 75 °C for a set duration, leading to its transformation into bio-MOF-100. This method was further employed to expand the pores of bio-MOF-100 by substituting BPDC with longer organic linkers, including azobenzene-4,4′-dicarboxylate (ABDC) and 2′-amino-1,1′:4, 1′′-terphenyl-4,4′′-dicarboxylate (NH2-TPDC), thereby fine-tuning the pore size and functionality. The same post-synthetic ligand exchange strategy, involving the immersion of solid MOFs in a solution containing a secondary ligand, has also been successfully applied to zeolitic imidazolate frameworks (ZIFs). Cohen's group reported that in ZIF-71(Zn), the 4,5-dichloro-imidazole ligand could be partially replaced by 4-bromo-1H-imidazole via solid–liquid PSE.140 Furthermore, Cohen's group extended the ligand exchange approach to solid-state reactions, demonstrating that post-synthetic ligand exchange could occur between two distinct MOFs under relatively mild conditions. This was exemplified with UiO-66(Zr),141 MIL-53(Al), and MIL-68(In),142 where ligand exchange was successfully achieved through solid–solid PSE. For instance, in the case of MIL-53(Al), two functionalized derivatives, one synthesized using BDC-NH2 (aminated) and the other using BDC-Br (brominated), were physically mixed in water and incubated at 85 °C (a significantly lower temperature than the original solvothermal synthesis at 150 °C) for five days. Aerosol time-of-flight mass spectrometry (ATOFMS) analysis confirmed that the chemical composition of the framework had been altered, providing strong evidence that solid–solid PSE had successfully occurred within the MIL-53(Al) derivatives. These studies highlight the versatility and effectiveness of ligand exchange as a PSM technique, allowing for precise control over MOF pore environments, functionality, and framework composition, while maintaining structural integrity.
 | ||
| Fig. 3 Scheme illustrating the pore expansion strategy. (a) As-synthesized bio-MOF-101 was converted to bio-MOF-100 through ligand exchange with BPDC. (b) BPDC in bio-MOF-100 was subsequently replaced with ABDC to form bio-MOF-102, followed by the replacement of ABDC in bio-MOF-102 with NH2-TPDC to yield bio-MOF-103. (c) Light microscope images of the crystalline MOFs, with scale bars representing 0.2 mm. Reproduced with permission.139 Copyright 2013, American Chemical Society. | ||
Similar to ligand exchange in MOFs, monomer exchange in COFs has been explored as a strategy to construct COFs with more complex structures. Qian et al. developed an in situ COF-to-COF transformation approach, where the benzidine linkers in TP-COF-BZ were selectively replaced with 1,4-diaminobenzene linkers under solvothermal conditions (Fig. 4a).143 This method enabled the controlled generation of COFs with three distinct types of pores, demonstrating the potential of monomer exchange for tailoring COF architectures at the solid-state level. Wang and coworkers further advanced this concept by introducing a self-limited dynamic linker exchange strategy for surface functionalization of uniform COF microspheres (Fig. 4b).144 Their approach allowed for precise modification of COF surfaces while preserving the overall morphology. Specifically, they selected six different monomers with varying functional groups and linker lengths, enabling the transformation of a parent COF microsphere (PCOF) into six distinct functionalized COF microspheres, which was originally synthesized via the Schiff-base condensation of 1,3,5-tris(4-aminophenyl)benzene (TPB) and 2,5-dimethoxyterephthalaldehyde (DMTP). This strategy significantly enriched the library of COF microspheres, offering a versatile approach to tune COF properties for diverse applications.
 | ||
| Fig. 4 (a) Precise construction of dual-pore COFs via a multiple-linking-site strategy and fabrication of triple-pore COFs through the integration of vertex-truncation design with the multiple-linking-site strategy. Reproduced with permission.143 Copyright 2017, American Chemical Society. (b) Structures of six functional linkers and a schematic illustration of the in situ linker exchange process for functionalizing ECOF stationary phases. Reproduced with permission.144 Copyright 2024, John Wiley and Sons. | ||
A number of MOF structures possess the ability to release auxiliary ligands from their metal ion nodes, often leading to the formation of coordinatively unsaturated metal centers. These OMSs can subsequently coordinate with other molecules, thereby introducing new functionalities or modulating pore geometries. One of the earliest demonstrations of this phenomenon was reported in 1999 by Williams and co-workers, who investigated the well-known HKUST-1 compound.145 Their study revealed that a partial building block ligand, 1,3,5-benzenetricarboxylate (BTC), on the paddle-wheel SBUs of HKUST-1, could be replaced by pyridine upon treatment with dry pyridine. This replacement occurred due to the lability of the axial aqua ligands, yet the 3D lattice of the MOF remained intact. This PSM not only altered the pore structure and chemistry of HKUST-1 but also introduced tunable functionalization at the metal nodes. Interestingly, the study demonstrated that pyridine-decorated HKUST-1 could not be obtained through direct solvothermal synthesis from Cu(II) salts, BTC, and pyridine, but could be successfully prepared via post-synthetic modification, thereby highlighting the unique advantage of MOFs as structurally robust and tunable platforms. This ability to precisely tailor the local microenvironment through ligand exchange greatly enhances the versatility and functional adaptability of these materials.
Both the ligands in MOFs and the monomers in COFs can be readily functionalized with additional groups through coordinated or covalent interactions, significantly expanding their structural diversity and functional tunability. Among these, 2,2′-bipyridyl (bpy) is a widely used chelating ligand for transition metal (TM) complexes due to its strong coordination ability and electronic properties. Carboxylate-functionalized bpy derivatives have been extensively explored as either pre-integrated or post-modified ligands for MOF synthesis with various metal salts, enabling the precise incorporation of catalytically active metal centers. Yaghi's group pioneered the incorporation of metal sites into MOF-253, a framework featuring open 2,2′-bpy coordination sites.146 MOF-253 was synthesized from 2,2′-bipyridine-5,5′-dicarboxylic acid (bpydc) and AlCl3 in DMF, followed by post-synthetic metalation with PdCl2 and Cu(BF4)2 in MeCN solution to yield bimetallic MOFs. This strategy allowed for the controlled introduction of catalytically active metal centers, significantly enhancing the material's functional versatility. Building on this concept, Lin's group reported the synthesis of chemically stable and recyclable bpy-UiO-Pd and bpy-UiO-Ir by treating a UiO-type MOF containing bipyridyl moieties (bpy-UiO) with Pd and Ir precursors.147 Notably, bpy-UiO-Ir exhibited exceptional catalytic activity in the dehydrogenative borylation of aromatic C–H bonds using B2(pin)2 (pin = pinacolate) as the borylating agent, highlighting the potential of MOFs containing nitrogen donor ligands as highly stable and efficient catalysts for key organic transformations. These studies underscore the effectiveness of post-synthetic metalation in tailoring MOFs for specific catalytic applications.
Beyond MOFs, the bpy moiety has also been widely employed in COF frameworks to immobilize metal sites, enabling the design of highly recyclable and structurally robust catalysts. Our group developed an effective strategy for incorporating metal-active sites into COFs by synthesizing COF-TpBpy through the condensation of 1,3,5-triformylphloroglucinol (Tp) and 5,5′-diamino-2,2′-bipyridine (Bpy), followed by coordination with Cu sites (Fig. 5).148 Additionally, flexible polymeric phosphonium salts (PPS) were confined within the COF channels via in situ polymerization. The resulting material, PPS ⊂ COF-TpBpy-Cu, leveraged the mobility of catalytically active sites on the highly flexible PPS, along with synergistic interactions between the polymeric moieties and Lewis acid sites (Cu species) anchored on the COF walls. This unique system exhibited superior catalytic performance in the cycloaddition of epoxides with CO2 to form cyclic carbonates, demonstrating enhanced reactivity and recyclability. This work not only unveils a novel strategy for designing bifunctional catalysts with dual activation behavior but also opens new avenues for developing multifunctional systems that mimic biocatalysis. To date, numerous functionalized bipyridine-based COFs have been reported, incorporating a range of metal sites including Co, Ni, Cu, Pd, Ru, Rh, Re, and Ir, among others.149,150 These metallized bpy-based COFs have exhibited significant potential for various catalytic applications, such as asymmetric catalysis, electrocatalysis, photocatalysis, and small-molecule activation. The tunability of the bpy-metal coordination environment, coupled with the high stability and porosity of COFs, continues to drive innovations in designing next-generation porous catalysts for sustainable chemical transformations.
 | ||
| Fig. 5 (a) The concept of heterogeneous concerted catalysis involving active sites on porous materials and highly flexible linear polymers. (b) Schematic representation of PPS ⊂ COF-TpBpy-Cu synthesis, along with the structures of COF-TpBpy and PPS ⊂ COF-TpBpy-Cu. Reproduced with permission.148 Copyright 2016, American Chemical Society. | ||
The Salen unit is one of the most significant ligands in coordination chemistry, widely utilized for the design of metal–organic frameworks (MOFs) and covalent organic frameworks (COFs) due to its strong coordination ability and structural versatility.151 Wang and coworkers were the first to report the construction of a Salen-based COF, achieving both the formation of the COF structure and the functionalization with Salen moieties in a single step (Fig. 6).152 This work paved the way for the development of a series of metallo-Salen-based COFs through post-synthetic metalation, broadening their applicability in catalysis and molecular adsorption. They synthesized the unique Salen-COF via the solvothermolysis of 1,3,5-tris[(5-tert-butyl-3-formyl-4-hydroxyphenyl)ethynyl]benzene and ethylenediamine, resulting in a highly crystalline material with a high Brunauer–Emmett–Teller (BET) surface area of 1366 m2 g−1. Post-synthetic metalation was performed using various metal ions, including Cu, Ni, Zn, Co, and Mn, yielding a family of M/Salen-COFs. Powder X-ray diffraction (PXRD) patterns confirmed that the crystalline structure of Salen-COF remained intact after metalation, demonstrating its structural robustness. These metallo-Salen-COFs have since been extensively investigated for catalytic applications, capitalizing on the diverse catalytic properties of the incorporated metal centers. Prior to this, in 2011, Nguyen and coworkers had reported a series of metallosalen-based MOFs containing Co, Zn, Cr, Cu, and Ni.153 They synthesized these frameworks by treating MnIIISO-MOF, a Mn3+(Salen)-based MOF, with H2O2 to remove the Mn3+ ions from the Salen struts. This process generated a vacant coordination environment within the framework, allowing for the subsequent introduction of various metal precursors to form isostructural MSO-MOF materials. This re-metalation strategy significantly expanded the functionality and tunability of Salen-based MOFs, offering a platform for diverse catalytic transformations and selective adsorption processes. These pioneering studies on both COFs and MOFs underscore the versatility of Salen linkers in framework materials, highlighting their potential in designing multifunctional porous materials for advanced applications in catalysis, separation, and sensing.
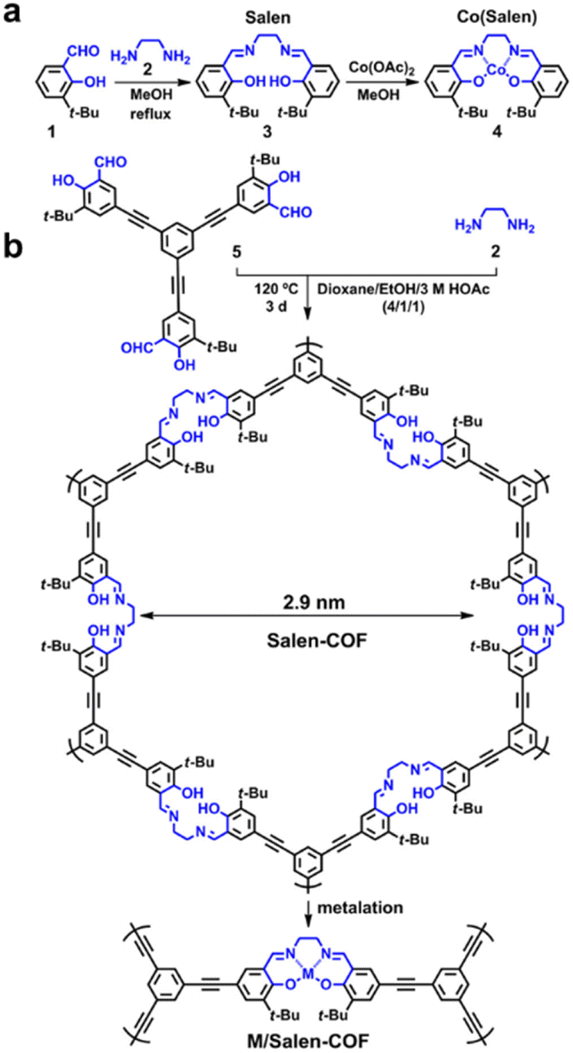 | ||
| Fig. 6 (a) Classical synthesis of salen units and (b) one-step construction of salen-COFa. Reproduced with permission.152 Copyright 2016, American Chemical Society. | ||
While functionality can be introduced into COFs or MOFs through coordination-based PSM, covalent PSM offers an alternative and widely adopted strategy for precisely modifying organic active sites on pore surfaces. This method is particularly valuable for fine-tuning the chemical environment within the frameworks, enabling enhanced control over their properties and functionalities. The concept of PSM in MOFs was first introduced by Cohen's group in 2007, marking a significant advancement in the field.154 They demonstrated the covalent modification of the IRMOF-3 using acetic anhydride. IRMOF-3, featuring a cubic topology and amino-functionalized pore walls, was synthesized from Zn(NO3)2·4H2O and 2-amino-1,4-benzenedicarboxylic acid (R3-BDC). The free amino groups in IRMOF-3 provided a versatile platform for post-synthetic covalent modification. In a typical PSM reaction, crystals of IRMOF-3 were suspended in CH2Cl2 (or CHCl3) and treated with pure acetic anhydride at room temperature, resulting in IRMOF-3-AM1, where the amino groups were converted to acetanilide groups. This pioneering study opened new possibilities for designing functionalized crystalline porous materials through post-synthetic modification strategies, allowing the precise tuning of chemical properties without disrupting the overall framework integrity. In the realm of COFs, Jiang's group successfully utilized PSM to functionalize COFs through click chemistry, employing azide-appended building units that reacted with various alkynes to yield triazole-functionalized COFs with tunable pore surface chemistry.155 They synthesized X% N3-COF-5 by combining 2,5-bis(azidomethyl)benzene-1,4-diboronic acid (N3-BDBA), benzene-1,4-diboronic acid (BDBA), and hexahydroxytriphenylene (HHTP). The azide groups in the framework enabled further functionalization via 1,2,3-triazole rings through click reactions with alkynes, yielding X% RTrz-COF-5. By adjusting the proportion of azide units (X = 5, 25, 50, 75, and 100), they precisely controlled the density of functional groups, demonstrating a scalable strategy for surface engineering of COFs. Furthermore, the pore chemistry of these COFs was finely tuned by modifying the triazole moieties present on the pore walls.
Different triazole substitutions enabled precise control over the framework's binding affinity toward various gas molecules, enhancing their potential for efficient gas storage and selective gas separation. These modifications allowed COFs to be tailored for specific applications, such as the selective sorption of CO2 over N2. The advancements in PSM strategies for both MOFs and COFs underscore their versatility in functional material design. By leveraging covalent modifications, researchers can impart new properties to these frameworks, expanding their applications in catalysis, gas storage, molecular sieving, and sensing technologies. The ability to introduce and fine-tune functional groups post-synthetically provides a powerful tool for optimizing porous materials for targeted applications while preserving their inherent structural stability.
 | ||
| Fig. 7 Illustration of pore space partitioning via symmetry-matching regulated ligand insertion. (a) View along the c-axis and (b) side view of the channels, depicting the cylindrical channel before and after partitioning (green: Ni, red: O, blue: N, gray: C). Reproduced with permission.160 Copyright 2015, American Chemical Society. | ||
The PSP strategy has also been applied to 2D COFs to subdivide their mesopores (2–50 nm) and micropores (1–2 nm) into ultramicropores (<1 nm). Two recent studies on pore partitioning in 2D COFs utilized aldehyde groups remaining in the channels of preformed COFs as anchoring sites, enabling amine-based insertions to partition the pore space into multiple smaller domains through post-synthetic Schiff-base reactions, achieving precise pore segmentation. Xu et al. employed a hexagonal boronic ester-linked COF, DBAAn-BTBA-COF, constructed from 1,3,5-benzenetriboronic acid (BTBA) and 4,4′-(2,3,6,7-tetrahydroxyanthracene-9,10-diyl)dibenzaldehyde (DBAAn), as the parent framework (Fig. 8).161 This mesoporous COF features well-defined hexagonal pores with a diameter of 2.9 nm. The six aldehyde groups on each hexagonal unit, oriented toward the center of the 1D channels, were further reacted with a planar hexagonal amine-based building block possessing C6 symmetry. This reaction formed imine bonds, partitioning each pore into six equal domains. Hexaminophenyl benzene (HAPB) was identified as the optimal partitioning agent, yielding a new crystalline material, DBAAn-BTBA-HAPB-COF, where the original 2.9 nm pores were divided into six uniform ultramicropores of 6.5 Å. However, DBAAn-BTBA-HAPB-COF could not be synthesized via a one-pot procedure, underscoring the advantages of the post-synthetic PSP strategy in achieving precise pore size control. The resulting wedge-shaped ultramicroporous 1D channels endowed the COF with exceptional efficiency in separating five hexane isomers based on molecular sieving effects. Hao et al. extended this strategy to imine-linked 2D COFs, designing and synthesizing a series of tetragonal and hexagonal MTV-COFs with aldehyde groups anchored within their channels.162 Additional linear or triangular linkers were then inserted into the center of the pore spaces, acting as partitioning agents to transform the micropores and mesopores into ultramicropores. Notably, the introduction of multi-N components into the partitioned COF 3-2P resulted in high I2 and CH3I uptake capacities of 0.42 and 0.24 g g−1 at 150 °C, respectively. These findings demonstrate that pore partitioning enables precise control over the pore environment and functionality of COFs, highlighting the significant potential of COF-based adsorbents for diverse applications.
 | ||
| Fig. 8 (a) Graphical representation of pore partitioning in hexagonal channels. (b) Conversion of DBAAn-BTBA-COF into DBAAn-BTBA-HAPB-COF via symmetry-matching knot insertion. Reproduced with permission.161 Copyright 2023, Springer Nature. | ||
Very recently, Zhang et al. reported a powerful and generalizable strategy for constructing metal-halide porous framework superlattices with spatially modulated chemical compositions within the pores of various MOFs.163 By employing a one-pot synthesis approach, the authors achieved the confined growth of selected metal halides (PbI2, PbBr2, CdI2, and NiBr2) within a series of 3D Zr(IV)-based MOFs, resulting in the formation of highly ordered, single-crystalline porous superlattices. Critically, the local pore environments of the host MOFs—specifically, the cavity sizes, geometries, and spatial orientation of anchoring sites—played a key role in directing the dimensionality (0D, 1D, or 2D) and structural arrangement of the metal-halide building units. This control arises from the influence of inter-node distances and spatial confinement on the nucleation and growth pathways of the halide crystals. Moreover, further treatment of these superlattices with selected amine molecules led to the formation of perovskite-like superlattices exhibiting highly tunable photoluminescence and chiroptical properties. This work highlights an innovative and versatile platform for engineering high-order, multi-dimensional single-crystalline superlattices with precisely tailored chemical functionality and emergent physical properties. The integration of inorganic components into MOF scaffolds in this manner not only provides a new avenue for pore regulation, but also opens up exciting possibilities for designing multifunctional hybrid materials for optoelectronic, chiral, and quantum applications.
3. Functional design and applications
With the versatile chemical tools available for tuning pore structures and chemistry in MOFs and COFs, their practical applications have been extensively explored over the past two decades. To obtain target materials with precise functionalities, the de novo synthetic strategy is an ideal approach, as it ensures a controlled degree of functional group incorporation at the ligand or monomer level. However, many desired framework materials with specific active sites cannot be directly synthesized through this method due to structural and synthetic limitations. As an alternative, PSM has emerged as a powerful strategy, allowing for the functionalization of organic backbones or metal clusters while preserving the integrity of the pristine framework and maintaining porosity. The ease of functionalizing pore surfaces in MOFs and COFs through sophisticated pore engineering has enabled the development of highly tailored materials for a wide range of practical applications. These materials have exhibited outstanding performance in gas storage and separation, heterogeneous catalysis, and environmental remediation, three key research areas in which we have conducted in-depth studies over the past decade. Through these efforts, we have proposed a series of innovative design and synthesis strategies for functionalizing MOFs and COFs, enabling the development of highly tailored materials with enhanced performance and expanded application potential. In the following discussion, we will highlight significant advancements in these fields, emphasizing how structural tuning and surface modifications in MOFs and COFs contribute to their superior functionalities and practical applications.3.1 Gas storage/separation
MOFs and COFs, featuring uniform pore structures, large surface areas, tunable pore sizes, and customizable pore surface chemistry, have been extensively explored for gas storage and separation applications. Among these, MOFs have gained particular attention due to the efficiency of pore engineering in controlling pore structure and functionality, which significantly enhances their capability for differential molecular recognition. By precisely tailoring pore sizes, channel geometries, surface areas, and functional sites, novel MOF materials can be designed for highly selective.164–168 Given the growing importance of clean energy,169 considerable efforts have been dedicated to identifying materials for hydrogen storage,170,171 methane storage,172–174 and carbon dioxide capture.175,176 The potential of MOFs in hydrogen storage was first demonstrated in 2003 when Yaghi's group reported MOF-5 as an H2 adsorbent.177 Since then, extensive research has led to the evaluation of hundreds of MOFs as physi-sorbents for hydrogen storage applications. It has been established that the gravimetric hydrogen storage capacities at 77 K and high pressures are strongly correlated with the pore volume and/or surface area of MOFs. For instance, an isoreticular series of MOFs with a rht topology, NOTT-112, NU-111, and NU-100 (also known as PCN-610), exhibits systematically increasing BET surface areas of 3800 m2 g−1, ∼5000 m2 g−1, and 6143 m2 g−1, respectively. Correspondingly, their total gravimetric hydrogen uptake at 77 K and 70 bar follows the same trend, with capacities of 10.0, 13.6, and 16.4 wt%, respectively.178–181 This demonstrates the critical role of surface area and pore volume in optimizing hydrogen adsorption performance.COFs have also been extensively investigated for hydrogen storage. Yaghi's group designed and synthesized a series of COFs tailored for H2 adsorption.182 Among them, COF-102 and COF-103 exhibit high BET surface areas of 3472 m2 g−1 and 4210 m2 g−1, respectively, while COF-108 stands out for its exceptionally low crystalline density of 0.17 g cm−3. Theoretical predictions suggest that 3D COFs, such as COF-102, COF-103, COF-105, and COF-108, demonstrate 2.5 to 3 times higher hydrogen storage capacities compared to 2D COFs (e.g., COF-1 and COF-5), primarily due to their greater surface areas and free volumes. Further computational studies on prototypical COFs predict that COF-105 could achieve an excess H2 storage capacity of up to 10 wt% at 77 K and 80 bar, while COF-108 could reach similar capacities at 77 K and 100 bar. These values significantly surpass some well-known MOFs, such as MOF-177 (7.0 wt%)183 and MOF-5 (7.1 wt%)184 under similar conditions. In terms of volumetric storage, COF-102 demonstrated a maximum excess H2 uptake of 40.4 g L−1 at 100 bar, highlighting its potential as a highly efficient hydrogen storage material. To further enhance hydrogen adsorption in COFs, Pramudya et al. proposed a strategy involving the chelation of transition metals (TMs) inside 3D COFs, utilizing various linker sites to host metal ions and improve H2 binding energies (Fig. 9).185 Computational studies revealed that first-row transition metals (Sc to Cu) could achieve interaction strengths comparable to, or even surpassing, those of precious late transition metals (Pd and Pt). This finding suggests that it is unnecessary to rely on expensive and heavy transition metals to achieve strong H2 binding. Beyond COFs, lightweight non-transition metal ions, such as Li+, incorporated via ion exchange into anionic coordination MOFs, have also been explored to enhance hydrogen adsorption performance. A series of Li+-exchanged anionic In-diisophthalate frameworks (NOTT-200, NOTT-206, and NOTT-208) demonstrated simultaneous increases in both hydrogen uptake capacity and adsorption enthalpy, offering a promising approach for optimizing H2 storage in porous materials.186
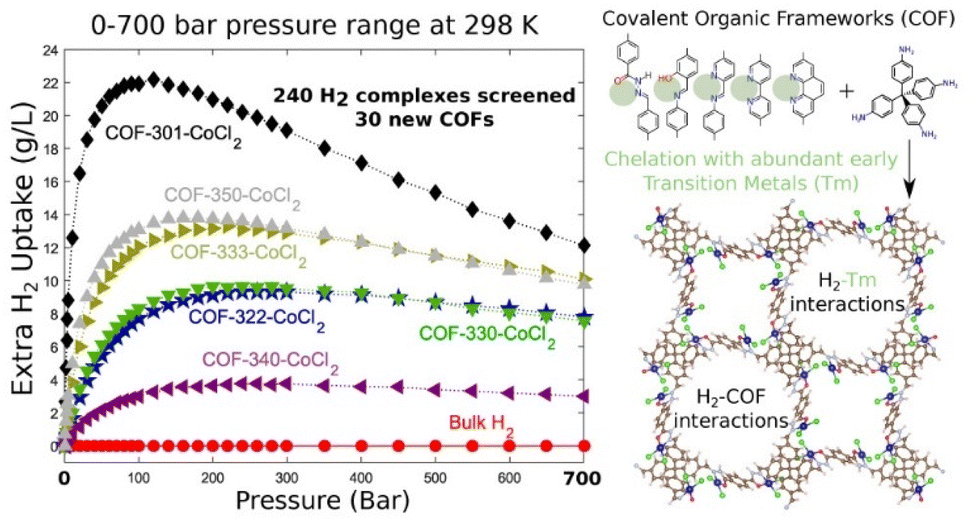 | ||
| Fig. 9 Design principles for high H2 storage through chelation of abundant transition metals in covalent organic frameworks under 0–700 bar at 298 K. Reproduced with permission.185 Copyright 2016, American Chemical Society. | ||
Beyond hydrogen storage, MOFs and COFs have also demonstrated remarkable efficiency in methane storage, offering a promising alternative for natural gas storage and transportation. Their tunable pore structures enable strong methane adsorption at moderate pressures, addressing key challenges in gas delivery systems. Additionally, both MOFs and COFs have been widely explored for CO2 capture, benefiting from their highly selective adsorption sites, which can be chemically functionalized to enhance CO2 affinity while minimizing energy-intensive regeneration processes. In the case of methane storage, the incorporation of specific functional sites in MOFs can significantly enhance CH4 uptake by increasing interactions with methane molecules. One effective strategy involves introducing OMSs into the framework to improve CH4 affinity. For instance, HKUST-1, featuring well-suited pore cavities and abundant accessible Cu sites, exhibits an exceptional CH4 volumetric uptake of 267 cm3 (STP) cm−3 at room temperature and 65 bar, where STP is standard temperature and pressure.187 This makes HKUST-1 the first MOF material with the potential to meet the U.S. Department of Energy (DOE) targets for volumetric CH4 uptake. Further advancements in MOF design have led to even greater methane storage capacities. Chen and coworkers developed a novel NbO-type porous MOF, UTSA-76a, incorporating pyrimidine groups and OMSs, which demonstrated an outstanding methane storage capacity of 257 cm3 (STP) cm−3 at 298 K and 65 bar.188 Moreover, UTSA-76a surpassed HKUST-1 in methane storage performance, exhibiting a higher working capacity of 197 cm3 (STP) cm−3 and a storage density of 0.263 g g−1. This superior performance is attributed to the extensive rotational freedom of its central “dynamic” pyrimidine cluster, which optimizes CH4 packing under high-pressure conditions.
Recently, Yin et al. developed two isostructural 3D COFs featuring a rare self-catenated alb-3,6-Ccc2 topology with a pore size of 1.1 nm.189 Utilizing a [6 + 3] topology design strategy, the authors constructed highly porous 3D COFs by selecting 1,3,5-trimethyl-2,4,6-tri[3,5-di(4-aminophenyl-1-yl)phenyl-1-yl]benzene (TAPB-Me) and its analog 1,3,5-triethyl-2,4,6-tri[3,5-di(4-aminophenyl-1-yl)phenyl-1-yl]benzene (TAPB-Et) as 6-connected polyhedral nodes, while 1,3,5-triformylbenzene (TFB) served as the 3-connected building block, yielding 3D-TFB-COF-Me and 3D-TFB-COF-Et, respectively. Both COFs exhibited exceptionally high BET surface areas exceeding 4000 m2 g−1, ranking among the highest reported for imine-linked COFs and placing them in the top tier of all known microporous materials. Given their high surface areas and microporous architectures, their high-pressure CH4 adsorption performance was evaluated up to 100 bar at 298 K. The gravimetric methane uptake values for 3D-TFB-COF-Me and 3D-TFB-COF-Et reached 423 mg g−1 and 429 mg g−1, respectively. Based on their crystal densities, their volumetric methane uptakes were measured at 249 cm3 (STP) cm−3 and 264 cm3 (STP) cm−3, with 3D-TFB-COF-Et surpassing the latest DOE CH4 storage target of 263 cm3 (STP) cm−3 at 298 K and 100 bar. These findings further underscore the significant potential of MOFs and COFs in methane storage applications, with ongoing research directed toward enhancing adsorption capacities through pore engineering and functional site modifications.
Beyond gas storage, MOFs have demonstrated exceptional potential for gas separations, leveraging their precisely tunable pore structures and surface chemistry. These materials have been widely investigated for key industrial separations, including ethylene/ethane (C2H4/C2H6), propylene/propane (C3H6/C3H8), acetylene/ethylene (C2H2/C2H4), acetylene/carbon dioxide (C2H2/CO2), propyne/propylene (C3H4/C3H6), C4 hydrocarbon separations, C6/C8 isomer separation, CO2 capture and separation.190–192 Chen's group pioneered the application of microporous MOFs in industrial gas separation, demonstrating their separation performance and efficiency through fixed-bed adsorption and breakthrough experiments.193–195 As early as 2005, Chen and Dai first reported the use of microporous MOFs for alkane isomer separation.196 MOF-508, synthesized from H2BDC, 4,4′-bipyridine (Bipy), and Zn(NO3)2·6H2O, was employed as a gas-chromatographic (GC) filler, successfully separating linear and branched isomers of pentane and hexane. MOF-508, a double-interpenetrated MOF, consists of 6-connected paddle-wheel zinc clusters bridged by bdc2− and Bipy linkers, forming a three-dimensional framework with pcu topology. Its one-dimensional channels (∼4.0 Å in diameter) selectively accommodate linear alkanes while excluding branched isomers, demonstrating the size-exclusion effect. Furthermore, MOF pore sizes can be rationally tuned by adjusting their degree of interpenetration, as exemplified by the non-interpenetrated microporous MOF [Zn2(bdc)2(dabco)] (dabco = 1,4-diazabicyclo[2,2,2]octane), which exhibits a similar pcu topology but with optimized pore accessibility.197 In 2016, Chen and co-workers conducted a comprehensive study on acetylene (C2H2) and ethylene (C2H4) adsorption using a series of SIFSIX materials, including SIFSIX-1-Cu, SIFSIX-2-Cu, SIFSIX-2-Cu-i, SIFSIX-3-Cu, SIFSIX-3-Zn, and SIFSIX-3-Ni.198 By finely tuning the pore chemistry and size, all of these materials exhibited higher C2H2 uptake than C2H4, demonstrating selective acetylene adsorption. Notably, SIFSIX-2-Cu-i displayed an exceptionally high C2H2 adsorption capacity (2.1 mmol g−1 at 298 K and 0.025 bar), with a C2H2/C2H4 selectivity of 39.7–44.8, effectively overcoming the typical trade-off between C2H2 selectivity and uptake capacity. Chen and co-workers further extended their research into C2H2/CO2 separation by designing MOFs with enhanced selectivity. A notable example is UTSA-74, an isomeric Zn-MOF-74, with the formula Zn2(H2O)(dobdc)·0.5(H2O).199 UTSA-74 exhibits comparable C2H2 adsorption capacity (145 cm3 cm−3) to Zn-MOF-74 but significantly lower CO2 uptake (90 cm3 cm−3) due to differences in Zn2 coordination environments. Unlike Zn-MOF-74, where Zn2 sites are terminally bound to CO2, in UTSA-74, the Zn2 sites are bridged by CO2 molecules, reducing CO2 binding strength and resulting in a high C2H2/CO2 selectivity of 9 at 100 kPa and 298 K. These findings highlight the remarkable adaptability of MOFs for gas separations, with ongoing research focusing on pore size control, functionalization, and metal-site engineering to optimize selectivity and adsorption capacity for industrial applications.
Our group has recently developed a series of microporous MOFs featuring gas-specific nanotraps for the efficient capture and separation of key industrial gases. In early 2021, we introduced an ultra-strong C2H2 nanotrap designed to selectively capture acetylene and separate C2H2/CO2 mixtures (Fig. 10a).200 This was achieved using the alkyl-functionalized MOF ATC-Cu, which possesses uniquely positioned, oppositely adjacent Cu paddlewheel open metal sites, significantly enhancing its affinity for acetylene molecules. ATC-Cu was synthesized via a hydrothermal reaction between 1,3,5,7-adamantane tetracarboxylic acid (H2ATC) and copper nitrate hydrate, forming a robust 4,4-coordinated network where four Cu paddlewheels are linked by ATC ligands. The exceptionally short Cu–Cu distance of 4.43 Å (after subtracting van der Waals radii) creates a dual potential coordination site, enabling strong C2H2 binding. Additionally, the aliphatic hydrocarbon cavities in ATC-Cu contain twelve inward-facing hydrogen atoms positioned at an average distance of 3.5 Å from the cavity center, providing additional adsorption sites for C2H2. The synergistic effect of these structural features results in an unprecedented acetylene binding affinity, as demonstrated by a record-high isosteric heat of adsorption (QST = 79.1 kJ mol−1). ATC-Cu exhibited the highest reported pure C2H2 uptake at 1 × 10−2 bar and remarkable selectivity in a binary equimolar C2H2/CO2 mixture at 298 K and 1 bar, setting a new benchmark for C2H2 capture and separation. Following this, we developed a MOF-based hydrogen-bonding nanotrap strategy to further enhance acetylene storage and separation (Fig. 10b).201 By introducing hydrogen-bonding acceptor sites on the pore surface, we optimized the separation of C2H2/CO2 mixtures across three isostructural MOFs: MIL-160, CAU-10H, and CAU-23. Among them, MIL-160, synthesized from 2,5-furandicarboxylic acid and Al(OH)(CH3COO)2, demonstrated exceptional C2H2 storage capacity (191 cm3 g−1, or 213 cm3 cm−3) while maintaining a significantly lower CO2 uptake (90 cm3 g−1) under ambient conditions, highlighting its superior C2H2 selectivity.
 | ||
| Fig. 10 (a) The channel structure of ATC-Cu and the two primary C2H2 binding sites within the framework. Reproduced with permission.200 Copyright 2021, American Chemical Society. (b) 1D chains [Al(μ2-OH)(COO)2]n and V-shaped ligands (H2FDC, m-H2BDC, and H2TDC) assemble into three isostructural 3D frameworks of MIL-160, CAU-10H, and CAU-23, respectively. Hydrogen atoms are omitted for clarity. Color code, Al: pale blue; O, red and rose; C, 50% gray. Reproduced with permission.201 Copyright 2021, American Chemical Society. | ||
To further refine gas separation performance, we implemented a dynamic spacer installation (DSI) strategy to construct a series of multifunctional MOFs (LIFM-61/31/62/63) with optimized pore spaces and environments for ethane/ethylene separation.202 These MOFs were derived from the prototype LIFM-28, with pore volumes expanded (from 0.41 to 0.82 cm3 g−1) and pore sizes reduced (from 11.1 × 11.1 Å2 to 5.6 × 5.6 Å2) by incorporating linear dicarboxylic acids into the replaceable coordination sites. Among them, LIFM-63, post-synthetically modified with biphenyl-4,4′-dicarboxylic acid (H2BPDC) and 2′-methyl-[1,1′:4′,1′-terphenyl]-4,4′′-dicarboxylic acid (H2MTPDC), exhibited exceptional C2H6 uptake and C2H6/C2H4 selectivity. This was attributed to enhanced C–H⋯F and C–H⋯π interactions enabled by its optimized pore volume, small pore size, and tailored pore surface chemistry. LIFM-63 demonstrated a remarkable ethane adsorption capacity (4.8 mmol g−1), nearly three times higher than its prototypical counterpart (1.7 mmol g−1) at 273 K and 1 bar. Additionally, we explored the impact of pore functionalization on gas separation by synthesizing two isostructural Ni-based MOFs, [Ni(btz)Cl]·Cl (Ni-MOF-1) and [Ni(bdp)] (Ni-MOF-2), using organic linkers with rich aromatic moieties: btz(1,4′-bis(4H-1,2,4-triazol-4-yl)benzene) and bdp(1,4-benzenedipyrazole), respectively.203 In Ni-MOF-1, the presence of Cl− ions partially obstructed the pore spaces and weakened C–H⋯π interactions, leading to negligible C2H6/C2H4 separation capability. In contrast, Ni-MOF-2, with open pore spaces and fully accessible aromatic rings, exhibited multiple and stronger C–H⋯π interactions favoring C2H6 adsorption over C2H4. As a result, Ni-MOF-2 demonstrated exceptional ethane/ethylene separation performance, ranking among the best porous materials for this critical industrial gas separation process. These studies collectively underscore the power of rational MOF design and pore engineering in achieving precise gas capture and separation, paving the way for advanced materials tailored for industrial applications.
Due to their typically larger pore sizes (>1 nm) than MOFs, most unmodified 2D COFs are not well-suited for gas separation, as the lack of tight molecular sieving limits their selectivity. However, certain 3D COFs have demonstrated promising potential in hydrocarbon separation. For instance, McGrier and co-workers synthesized and metalated a 3D COF containing π-electron conjugated dehydrobenzoannulene (DBA), yielding Ni-DBA-3D-COF via metalation of the pristine DBA-3D-COF.29 The metalation process resulted in only a slight reduction in BET surface area, from 5083 m2 g−1 to 4763 m2 g−1, preserving the material's high porosity. The ethane/ethylene separation performance of Ni-DBA-3D-COF was assessed through gas adsorption isotherms at 273 K and 295 K over a pressure range of 0 to 1.2 bar, revealing a moderate enhancement in adsorption capacity, with an increase of 0.07 mmol for ethane and 0.13 mmol for ethylene at 295 K. Although the enhancement was modest, this proof-of-concept study highlights the feasibility of utilizing metalated COFs for hydrocarbon separations and paves the way for further optimization. Beyond hydrocarbon separations, COFs have also been explored for precise gas separation at the molecular level. Fan et al. introduced an ultramicropore-in-nanopore concept, employing a matryoshka-like pore-channel engineering strategy to achieve angstrom-level control over COF channel sizes.204 This was accomplished via in situ encapsulation of α-cyclodextrin (α-CD) within the 1D nanochannels of a COF membrane during interfacial polymerization. The resulting LA-α-CD-in-TpPa-1 membrane demonstrated exceptional H2 separation performance, exhibiting a high H2 permeance (∼3000 GPU) along with significantly enhanced selectivity (>30) for H2 over CO2 and CH4. This enhancement was attributed to the formation of fast and selective H2 transport pathways, showcasing a novel approach for precisely tuning COF pore structures for advanced gas separation applications. These studies underscore the growing versatility of COFs in gas separation, particularly through pore chemistry and pore size engineering, which open up new possibilities for designing highly selective, high-performance separation membranes or adsorbents.
3.2 Catalysis
COFs and MOFs have emerged as highly promising materials for heterogeneous catalysis due to their high surface area, tunable pore structures, and versatile active sites. Through pore engineering, the size, shape, and surface chemistry of their pores can be precisely tailored to enhance substrate diffusion, reactant accessibility, and selectivity, making them highly efficient catalytic platforms. MOFs, with metal nodes, provide Lewis acid sites that facilitate oxidation, hydrogenation, and C–C coupling reactions. In contrast, COFs, composed of fully organic linkages, can be designed with catalytically active sites through the incorporation of specific functional groups or by coordinating with metal species, enabling diverse catalytic applications. Both materials allow PSM, including metalation, ligand engineering, and biomimetic site incorporation, further enhancing catalytic efficiency. The π-conjugated backbones of COFs make them ideal for photocatalysis and electrocatalysis, enabling applications in CO2 reduction and water splitting. Additionally, their excellent chemical and thermal stability ensures long-term catalytic performance with easy recovery and recyclability, making them superior to many homogeneous catalysts. By leveraging precise pore engineering, researchers have designed MOFs and COFs with hierarchical porosity, OMSs, and functionalized channels, optimizing catalytic activity for oxidation, hydrogenation, cross-coupling, photocatalysis, and electrocatalysis.During the synthesis of MOF materials, solvents often participate in coordinating with central metal nodes. These solvents can be completely or partially removed by subjecting the MOFs to high temperatures under vacuum or other controlled conditions, exposing unsaturated metal sites that serve as Lewis acid sites for multiphase catalytic applications. To further enhance catalytic performance, our group developed a feasible strategy to incorporate linear ionic polymers within MOF pores via in situ free radical polymerization of monomer salts, creating synergistic catalytic sites within a single material.205 A well-known MOF, MIL-101,206 synthesized from Cr(NO3)3·9H2O and terephthalic acid under solvothermal conditions, was treated with ethanol to remove the trapped DMF molecules within its pores. Subsequently, MIL-101 was mixed with a monomer salt, 3-ethyl-1-vinyl-1H-imidazol-3-ium bromide, in DMF and stirred at room temperature, allowing the monomer to settle inside the MOF pores, ensuring successful in situ polymerization of the ionic polymer. MIL-101, featuring mesoporous cages, high stability, and open chromium(III) metal sites, provides abundant accessible Lewis acid sites ideal for hosting flexible catalytic sites and acting as a co-catalyst. The resulting material, MIL-101-IP, was evaluated for CO2 fixation and the transformation of various epoxides into cyclic carbonates. By integrating OMSs inside rigid MOF frameworks and introducing halide ions via immobilized ionic polymers, the unification of two catalytically active components was achieved, demonstrating that pore chemistry can be readily tailored through a host–guest strategy. This highlights the potential of engineering synergistic catalytic systems within porous materials for a wide range of applications.
Heterometallic MOFs have also demonstrated superior catalytic performance compared to their single-metallic counterparts due to their enhanced chemical stability and tunable pore chemistry, which can be precisely controlled by adjusting the metal ratios within the framework. Fu et al. developed a double-metallic MOF-74 (Cu/Co) with various Cu/Co ratios via a facile one-pot synthesis method, ensuring a uniform distribution of Cu2+ and Co2+ throughout the structure.207 By carefully tuning the Cu/Co ratio, not only was the oxidation activity modulated, but the selectivity of oxidation products was also finely controlled. Furthermore, the incorporation of secondary metal ions or clusters in heterometallic MOFs can regulate the electronic properties of active sites, significantly enhancing electrocatalysis and photocatalysis performance. Using a similar method, Tang and co-workers synthesized ultrathin bimetal-organic framework nanosheets (NiCo-UMOFNs) using a mixed solution of Ni2+, Co2+, and benzenedicarboxylic acid (BDC).208 The resulting UMOFNs exhibited nanometer-scale thicknesses, enabling rapid mass transport and superior electron transfer, along with a high percentage of exposed catalytically active surfaces containing coordinatively unsaturated metal sites, ensuring exceptional catalytic activity. Additionally, the distinct surface atomic structures and bonding arrangements of NiCo-UMOFNs could be readily identified and tuned. Electron transfer from Ni to Co clusters resulted in optimized electronic states, significantly improving the electrochemical oxygen evolution reaction (OER) activity compared to monometallic Co-UMOFNs, Ni-UMOFNs, and bulk NiCo-MOFs. The metal ions/clusters can also be manipulated by PSM, Cohen and co-workers prepared UiO-66-NH2(Zr/Ti) catalysts through post-synthetic method, whose electron accepting levels were lowered by the Ti doping, which can be readily achieved by post-synthetic ion exchange of UiO-66-NH2(Zr) with TiCl4(THF)2 in DMF solution (Fig. 11).209 The newly modified UiO-66-NH2(Zr/Ti) showed higher CO2 photoreduction activity and HCOOH selectivity (5.8 mmol mol−1), 1.7 times higher than the monometallic UiO-66-NH2(Zr).
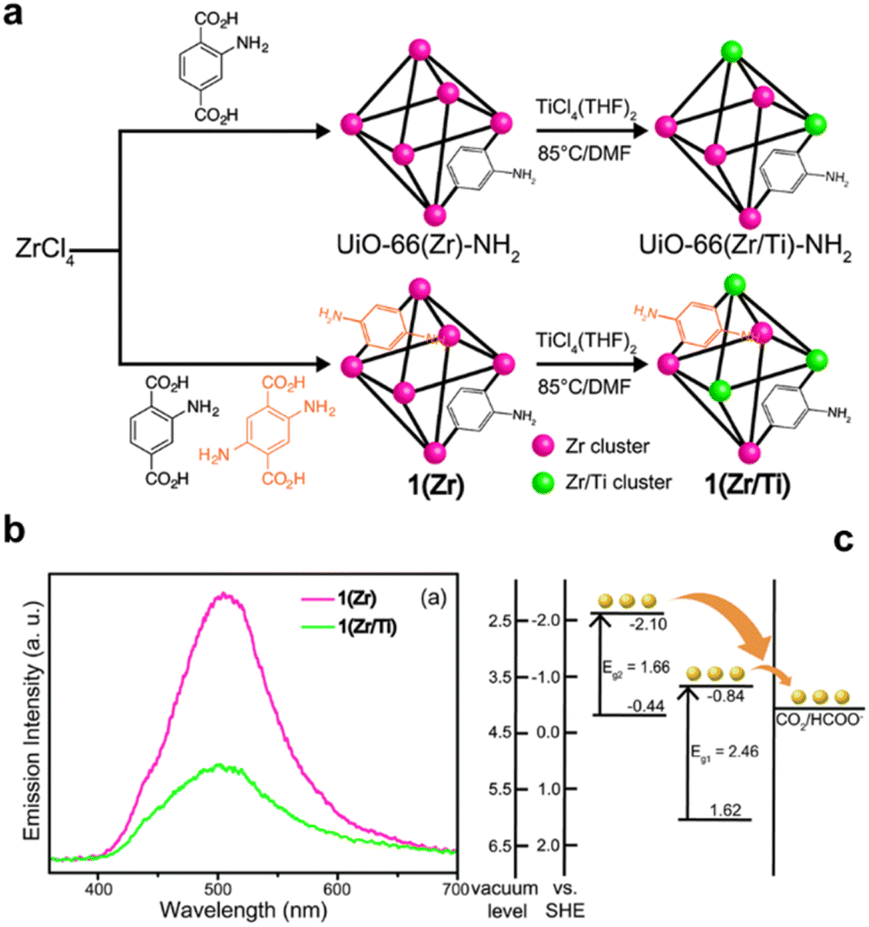 | ||
| Fig. 11 (a) Synthesis of mixed-ligand MOF 1(Zr) via PSE to obtain mixed-metal MOFs 1(Zr/Ti) and UiO-66(Zr/Ti)-NH2. (b) Photoluminescence spectra of 1(Zr) and 1(Zr/Ti). (c) Energy band structure of 1(Zr/Ti) derived from UPS and F(R) results, showing that heterogeneous ligands create two energy levels within the MOF, potentially facilitating CO2 catalysis. Reproduced with permission.209 Copyright 2015, Royal Society of Chemistry. | ||
Jiang's group has been at the forefront of developing MOFs with precise pore engineering to enhance catalytic performance across multiple reactions.210–212 Recently, they introduced a simple yet effective strategy to regulate the spin states of catalytic Co centers by altering their coordination environments. Through post-synthetic metal exchange, Co species were incorporated into a stable Zn-based MOF, yielding three distinct active sites, Co–OAc, Co–Br, and Co–CN, within the MOF pores for CO2 photoreduction (Fig. 12).213 By changing the Co precursor, Co centers coordinated with –CH3COO, –Br, or –CN, precisely tuning their coordination environment and spin states. Experiments and theory showed that this spin-state modulation—without changing Co's oxidation state—greatly enhanced charge separation, CO2 adsorption, and activation. Among the three variants, Co–OAc, which exhibited the highest spin state, demonstrated superior charge separation and optimized energy barriers for CO2 adsorption and activation, resulting in an outstanding photocatalytic CO2 reduction rate of 2325.7 μmol g−1 h−1 with 99.1% selectivity toward CO. To further explore MOFs as hosts for catalytically active species, Jiang's group employed UiO-66 as a template to encapsulate metal nanoparticles (NPs) and nanoclusters (NCs) within its pore cages. While NCs with atomically precise structures hold great promise for catalysis, their tendency to aggregate and the spatial resistance of surface ligands pose significant challenges. The spatial confinement effect of MOFs provides an ideal protective environment to address these issues. In their studies, Au25 NCs214 and a series of MAg24 (M = Ag, Pd, Pt, and Au) NCs215 were successfully encapsulated into UiO-66-X, forming Au25@UiO-66-X (X = H, NH2, OH, and NO2) and MAg24@UiO-66-NH2. These composites allowed for precise tuning of electron density and substrate adsorption properties through variations in both the encapsulated NCs and the MOF ligands. Additionally, various NPs, including Pt NPs and PdCu NPs, were incorporated into MOFs for enhanced catalytic applications. For example, Jiang's group synthesized UiO-66-SO3H (Zr6O4(OH)4(BDC-SO3H)6, denoted as UiO-S) to host PdCu NPs via PSM.216 To further optimize its catalytic properties, a hydrophobic polydimethylsiloxane (PDMS) coating was introduced, yielding a composite catalyst denoted as PdCu@UiO-S@PDMS. The electron transfer within PdCu NPs facilitated N2 activation, while the protonated sulfonic acid groups provided an essential proton source for nitrogen reduction reaction (NRR). Meanwhile, the PDMS coating selectively suppressed water contact with PdCu while still allowing the sulfonate (–SO3−) groups to extract protons from water. This unique microenvironment efficiently supplied protons while suppressing the competitive hydrogen evolution reaction (HER), resulting in an optimized system for electrocatalytic NRR with enhanced performance. Expanding on this work, Jiang's group further engineered MOF-based core–shell structures to fine-tune catalytic activity.217 By varying the functional groups of MOF ligands, they designed a set of UiO-66-X (X = –H, –Br, –NA (naphthalene), –OCH3, –Cl, –NO2) shells coated onto UiO-66-NH2@Pt, forming UiO-66-NH2@Pt@UiO-66-X. These core–shell MOF composites enabled simultaneous modulation of both the microenvironment around the Pt active sites and the photosensitive UiO-66-NH2 core. This precise structural control significantly influenced the photocatalytic H2 production performance. Among the various functionalized composites, UiO-66-NH2@Pt@UiO-66-H exhibited the highest H2 production rate of 2708.2 μmol g−1 h−1, approximately 222 times higher than that of UiO-66-NH2@Pt@ UiO-66-NO2. Their pioneering work has opened new avenues for designing supported catalysts with precisely tailored pore chemistry, demonstrating the immense potential of MOF-based materials for advanced catalytic applications.
 | ||
| Fig. 12 Synthesis of Co–OAc, Co–Br, and Co–CN with different spin states of CoII and CoIIIvia a postsynthetic exchange strategy. Reproduced with permission.213 Copyright 2024, American Chemical Society. | ||
Our group was the first to report the grafting of frustrated Lewis pairs (FLPs) into the pore space of MOFs by tailoring the pore environment.218 MIL-101 (Cr) was selected as the platform to host FLPs due to its exposed CrIII open sites in the secondary building unit (Fig. 13a).219 One site serves as an anchor for the FLP, while the other, along with the hydroxyl group residing at the third CrIII site, preferentially interacts with the targeted double bond in the substrate to facilitate its activation. In this work, the FLP, B(C6F5)2(Mes)/DABCO, was successfully anchored into MIL-101(Cr), yielding MIL-101(Cr)-FLP. This catalyst demonstrated the selective hydrogenation of imine bonds in α,β-unsaturated imine compounds to produce unsaturated amine compounds using H2 gas at room temperature, while this transformation is not achievable with FLPs in a homogeneous system. This represents the first example of an FLP-based heterogeneous catalyst capable of selectively hydrogenating α,β-unsaturated organic compounds, thereby paving the way for the development of precious-metal-free chemoselective heterogeneous catalysts. Building on this breakthrough, our group and co-workers explored various combinations of FLPs and MOFs to investigate their catalytic performance. A novel P/B-type FLP containing a phosphorus-based Lewis base was synthesized within the MOF NU-1000, demonstrating high efficiency in the selective hydrogenation of quinoline and indole to yield tetrahydroquinoline and indoline-type drug compounds with excellent yield and recyclability (Fig. 13b).220 Additionally, chiral FLPs (CFLPs) derived from RB(C6F5)2 (R = C6F5 or C9H11) and various chiral Lewis bases were synthesized and anchored into MOFs, resulting in CFLP@MIL-101(Cr)221 and EMR@MOF,222 respectively. These MOFs exhibited efficient heterogeneous asymmetric hydrogenation with excellent recyclability and regenerability, establishing CFLP@MOF as a promising platform for asymmetric catalysis. This approach bridges different fields of chemistry to address challenges in asymmetric catalysis and beyond.
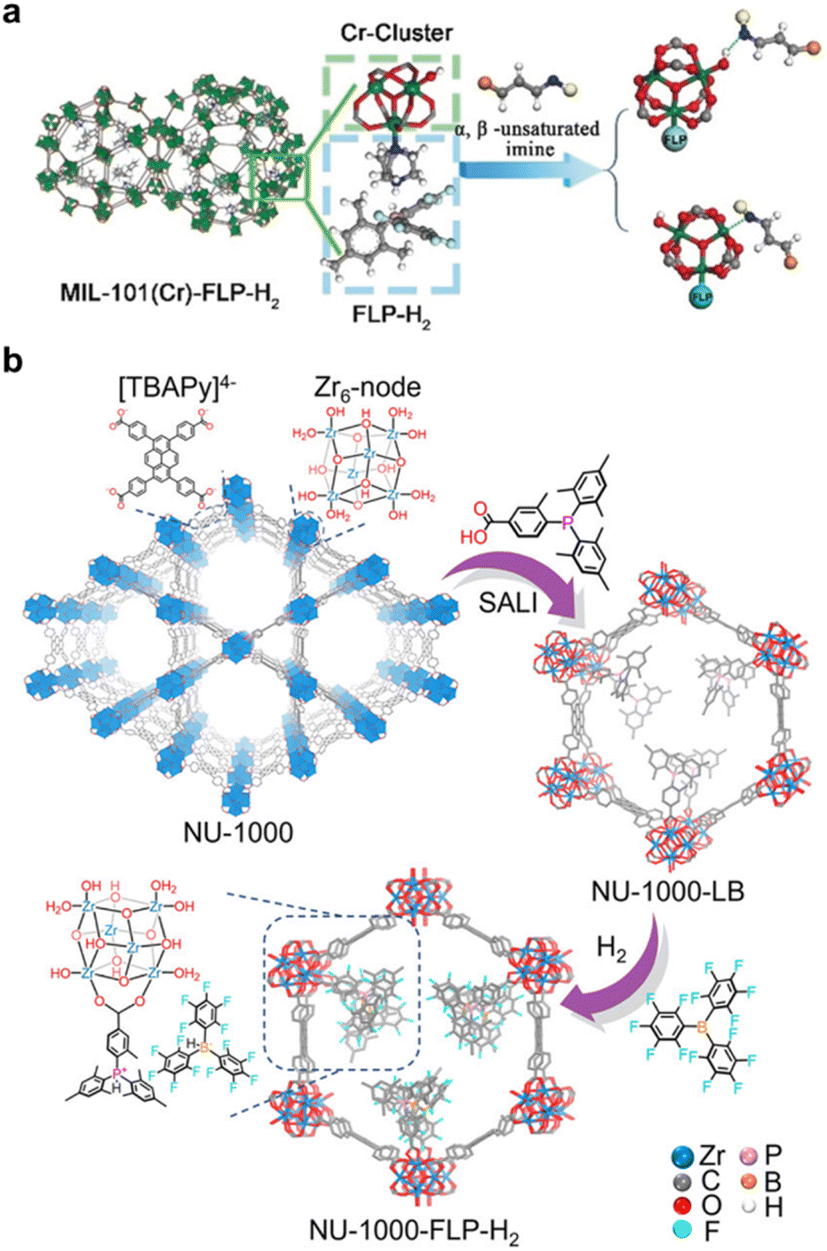 | ||
| Fig. 13 (a) The “activation” process involving a Cr3(μ3-O) (COO)6(OH)(H2O)2 cluster in MIL-101(Cr)-FLP-H2. Reproduced with permission.219 Copyright 2019, John Wiley and Sons. (b) Construction pathway of NU-1000-FLP-H2. Reproduced with permission.220 Copyright 2023, American Chemical Society. | ||
Following these advancements, our group realized a set of metal-free chiral frustrated Lewis pair frameworks within COFs as well. We proposed a general strategy for integrating rationally designed crystalline COFs with newly developed CFLPs to create chiral frustrated Lewis pair frameworks (CFLPFs).223 COF-TAPB-3P-COOH was first synthesized using 1,3,5-tris(4-aminophenyl)benzene (TPB) and p-terphenyl-2′,5′-dicarboxylic acid-4,4′′-dicarboxaldehyde (3P-COOH). Through covalent post-synthetic modification, chiral N1,N1-di-tert-butyl-3,3-dimethylbutane-1,2-diamine (R1NH2) and chiral 4,5-dihydro-4-phenyl-2-oxazolemethanol (R2OH) were introduced as chiral Lewis bases, yielding COF-TAPB-3P-COHNR1* and COF-TAPB-3P-COOR2*, respectively, which were further associated with B(C6F5)3 to form CFLPF1 and CFLPF2. Both metal-free CFLPFs exhibited superior activity and enantioselectivity for asymmetric olefin hydrogenation, along with outstanding stability and recyclability.
Our group has also developed various strategies to tune pore geometry and active sites in COFs to optimize inner pore chemistry and catalytic performance.224–226 Due to their purely organic skeletons, large pore sizes, high chemical stability, and easy recyclability, COFs have emerged as a promising platform for hosting biocatalysts, such as enzymes, to overcome challenges in cell-free enzyme catalysis. COFs provide robust protection against enzyme deactivation, enhance recyclability, and improve operational stability. In a study published in 2017, our group selected TPB-DMTP-COF (COF-OMe), synthesized by the condensation of dimethoxyterephthaldehyde (DMTP) and TPB, as a host material due to its exceptional stability, high surface area, and ordered one-dimensional (1D) channel-like pores (3.3 nm) (Fig. 14).225 Lipase PS (3.0 nm × 3.2 nm × 6.0 nm) was immobilized into COF-OMe in a phosphate buffer solution, yielding lipase@COF-OMe with a high enzyme uptake capacity of 0.95 mg mg−1. Recognizing that protein migration into porous materials is driven by surface interactions, we investigated the impact of pore environments on enzyme loading. Three isoreticular COFs (COF-V, COF-OH, and COF-ONa) with varying degrees of hydrophilicity were synthesized for comparison. COF-V was obtained from 2,5-divinylterephthalaldehyde and TPB, COF-OH from 2,5-dihydroxyterephthalaldehyde and TPB, and COF-ONa by treating COF-OH with NaOH. Under identical conditions, COF-Vexhibited a slightly lower lipase PS uptake (0.78 mg mg−1) than COF-OMe, likely due to its reduced surface area. COF-OH and COF-ONa displayed even lower uptake capacities (0.75 and 0.59 mg mg−1, respectively), supporting the hypothesis that a hydrophobic pore environment favors enzyme infiltration. Beyond surface properties, pore structure also plays a crucial role in enzyme immobilization. To verify this, we synthesized an amorphous analogue (POP-OMe) of COF-OMe using the same monomers but in DMSO as the solvent. POP-OMe exhibited a significantly lower BET surface area (1740 m2 g−1vs. 1056 m2 g−1 for COF-OMe) and a reduced enzyme uptake (0.58 mg mg−1), achieving only 65% of COF-OMe's performance. Subsequent studies focused on tuning pore heterogeneity in COFs to enhance enzyme accessibility and resistance to denaturants.226 COFs with dual pores were designed and synthesized to improve enzyme immobilization. The dual-pore structure of COF-ETTA-EDDA was confirmed by DFT pore size distribution analysis, revealing micropores (∼13.9 Å) and mesopores (∼38.5 Å). The larger mesopores facilitated enzyme immobilization, while the smaller micropores remained unoccupied, serving as transport channels for substrates and products. This structural optimization effectively reduced diffusion path lengths and significantly enhanced flux rates, thereby improving mass transport within the material. As a result, it established clear structure–property design rules for creating hierarchical pore architectures, which are crucial for optimizing enzyme-encapsulation applications and improving catalytic efficiency.
 | ||
| Fig. 14 (a) Schematic representation of lipase PS and porous materials used for enzyme immobilization (blue: N, gray: C, red: O, white: H, yellow: Si, purple: Na). (b) Enzyme uptake capacity of various porous materials after 6 hours of incubation in a 30 mg per mL lipase PS solution. Reproduced with permission.225 Copyright 2018, American Chemical Society. | ||
3.3 Environmental remediation
Given their high surface area, well-defined pore structures, and versatile functionalization potential, MOFs and COFs have gained significant attention as promising materials for environmental remediation. They demonstrate great potential for the binding and extraction of various pollutants, offering innovative solutions to pressing environmental challenges, including water purification, air pollution control, and toxic waste treatment. Task-specific binding sites can be incorporated into MOFs and COFs through de novo synthesis or post-synthetic modification, enabling the introduction of complexing functionalities to effectively capture and neutralize diverse contaminants.Among the various environmental concerns, the adsorption of organic dyes and heavy metals from water has long been a major focus, and MOFs and COFs have been extensively studied for their effectiveness in removing these pollutants from water supplies. One notable example of an environmentally friendly MOF is MOF-235(Fe), a structural analog of the well-known MIL-101, both synthesized from FeCl3·6H2O and BDC, but differing in crystal structures.227 Despite being classified as a nonporous or low-porosity MOF, MOF-235(Fe) has demonstrated strong adsorption capabilities for both anionic (e.g., methyl orange, MO) and cationic (e.g., methylene blue, MB) dyes in aqueous solutions.228 Additionally, the presence of Fe2+ and Fe3+ sites within MOF-235(Fe) enables it to function as a solid-state analog of Fenton reagents in advanced oxidation processes (AOPs), facilitating the degradation of MB in solution through H2O2-derived reactive species.229
Beyond dye adsorption, MOFs have also been developed as highly efficient platforms for heavy metal ion removal. Zhong and co-workers reported a versatile MOF-based broad-spectrum heavy metal ion trap, capable of capturing a total of 22 different heavy metal ions, encompassing hard, soft, and borderline Lewis metal ions (Fig. 15).230 This MOF material, derived from MOF-808(Zr) (constructed from BTC and ZrOCl2·8H2O),231,232 was post-synthetically modified through a solvent-assisted linker exchange method, where the formate groups on the Zr6 clusters were replaced with EDTA, yielding MOF-808-EDTA (BS-HMT). This modification introduced highly efficient chelating sites within the MOF's pores, significantly enhancing its heavy metal binding capabilities. The modified MOF demonstrated ultrahigh removal efficiencies (>99%) for all 22 tested metal ions, irrespective of their classification as soft, hard, or borderline acids. The exceptional performance of BS-HMT highlights how pore chemistry in MOFs can be effectively tuned through simple PSM strategies, broadening their application in selective pollutant separation and environmental remediation. This research underscores the vast potential of MOFs and COFs as tunable, high-performance materials for environmental applications, offering sustainable and efficient solutions for pollutant removal and resource recovery.
 | ||
| Fig. 15 (a) Schematic illustration of the BS-HMT concept. Ordered HCOOH molecules in MOF-808 can be replaced by EDTA, leading to MOF-808 with an ordered EDTA arrangement, which functions as a BS-HMT for metal ion capture. (b) The removal efficiency of hard Lewis metal ions, soft Lewis metal ions, and borderline Lewis metal ions for MOF-808-EDTA. Reproduced with permission.230 Copyright 2018, Springer Nature. | ||
Our group developed a series of post-synthetically modified COFs functionalized with various thiol groups and demonstrated their efficacy in mercury removal. We designed a vinyl-functionalized monomer, 2,5-divinylterephthalaldehyde, in which the vinyl groups remain intact during COF synthesis, allowing for further chemical modifications.233 The resulting vinyl-functionalized COF (COF-V) was synthesized via the condensation of 2,5-divinylterephthalaldehyde and TPB. Given the strong affinity of sulfur-containing groups for soft heavy metal species like mercury and the high efficiency of thiol-ene click chemistry, a library of thiol compounds with varying flexibility and sulfur densities was selected to react with the vinyl groups on the COF pore surface. The modified sulfur-functionalized COFs retained their crystallinity and porosity, enabling effective mercury removal from both aqueous solutions and the gas phase. These COFs exhibited outstanding uptake capacities of up to 1350 mg g−1 (at an equilibrium concentration of 110 ppm) for Hg2+ and 863 mg g−1 for Hg0. Additionally, they could rapidly reduce Hg2+ concentrations to as low as 0.1 ppb, even in the presence of high concentrations of competing ions. Notably, the flexibility of the chelating groups influenced mercury accessibility, demonstrating that the capture and adsorption performance of COF-based adsorbents can be precisely tailored through pore engineering.
Building on the versatile chemical modification potential of COF-V, we further integrated superwettability into COFs by treating COF-V with 1H,1H,2H,2H-perfluorodecanethiol via thiol-ene click chemistry to yield COF-VF (Fig. 16).234 The successful incorporation of perfluoroalkyl groups onto the COF pore surface was confirmed by water contact angle (CA) measurements. COF-VF exhibited a static water CA of approximately 167°, indicating a superhydrophobic surface, whereas COF-V and its alkyl-modified derivative showed CAs of only 113° and 122°, respectively. In contrast, oil droplets were instantly absorbed upon contact with COF-VF, confirming its superhydrophobic and superoleophilic properties. To explore its practical applications, COF-VF was integrated into commercial melamine foams, resulting in a cost-effective superhydrophobic foam (COF-VF@foam). This was achieved by immersing monolithic melamine foam in a precursor solution, followed by solvothermal synthesis to in situ coat COF-V onto the foam structure. Subsequent grafting of perfluoroalkyl groups onto the COF-V coating produced COF-VF@foam. The uptake performance of COF-VF@foam was evaluated using various organic liquids, demonstrating sorption capacities ranging from 67 to 142 times its own weight, highlighting its potential for oil-spill cleanup. Furthermore, COF-VF@foam exhibited excellent recyclability. The foam-based adsorbent could be easily regenerated by squeezing out the absorbed oil, maintaining its performance over at least ten adsorption cycles without significant efficiency loss. These results demonstrate that superhydrophobicity can be effectively incorporated into COFs through pore-surface engineering, offering valuable insights into structure–property relationships between functional groups and practical applications.
 | ||
| Fig. 16 Schematic illustration of imparting superhydrophobicity on COF-V. (a) Synthetic scheme of COF-V and COF-VF. (b and c) Schematic representation of the eclipsed AA stacking structure of COF-V. (d) Schematic representation of COF-VF. Reproduced with permission.234 Copyright 2018, Elsevier. | ||
MOFs and COFs have demonstrated superior radionuclide sequestration performance compared to traditional porous materials, offering enhanced uptake capacity and selectivity. Carboni et al. pioneered the application of MOFs as novel sorbents for actinide extraction from aqueous media.235 Specifically, they synthesized three MOFs with UiO-68 network topology using amino-TPDC or TPDC bridging ligands functionalized with orthogonal phosphorylurea groups (TPDC = p,p′-terphenyldicarboxylic acid). Among them, the UiO-MOF with diethoxyphosphorylurea functionalization [UiO-68-P(O)(OEt)2, MOF 2] was synthesized from ZrCl4 and a diethoxyphosphorylurea-derived TPDC ligand (H2L2), obtained through the condensation of 2′-amino-1,1′:4′,1′′-terphenyl-4,4′′-dicarboxylic acid (H2tpdc-NH2) with diethoxyphosphinyl isocyanate. MOF 2 exhibited near-quantitative uranium removal from both water and artificial seawater, achieving a remarkable saturation sorption capacity of 217 mg U per g, corresponding to the binding of one uranyl ion per two phosphorylurea groups. Building upon this progress, our group designed and synthesized a novel, highly stable carboxylated MOF, BUT-12-3COOH, featuring an ultra-high density of free carboxyl groups, exceptional chemical stability, rapid adsorption kinetics, and a high U(VI) sorption capacity.236 Through precise pore engineering, we optimized the pore environment of BUT-12-3COOH to maximize uranium capture efficiency, achieving an impressive maximum sorption capacity of 235 mg U per g, demonstrating its outstanding extraction ability from aqueous solutions.
Beyond MOFs, our group proposed a pore-environment engineering strategy to optimize COFs for radionuclide sequestration.237–240 Given that amidoxime is the state-of-the-art functional moiety for uranium capture, we incorporated amidoxime groups into COFs via a two-step synthesis (Fig. 17).241 First, a nitrile-functionalized COF (COF-TpDb) was synthesized by condensing 2,5-diaminobenzonitrile (Db) with Tp under solvothermal conditions, aligning cyano groups within the 1D pore channels. These cyano groups were subsequently converted into amidoxime moieties through hydroxylamine treatment, yielding an amidoxime-functionalized COF adsorbent (COF-TpDb-AO). Through this pore-surface functionalization approach, the eclipsed stacking arrangement of 2D extended polygons in COF-TpDb-AO positions the amidoxime chelating groups in adjacent layers parallel to each other, facilitating cooperative interactions for enhanced uranium binding. As a result, COF-TpDb-AO exhibited far superior adsorption performance compared to its amorphous analog (POP-TpDb-AO), with saturation adsorption capacities of 408 mg U per g and 355 mg U per g, respectively. Notably, COF-TpDb-AO reduced uranium concentrations in contaminated water samples from 1 ppm to below 0.1 ppb within minutes, surpassing the stringent drinking water limit (30 ppb). Moreover, in spiked seawater, it achieved an exceptionally high uranium uptake of 127 mg U per g, illustrating its potential for uranium mining applications. These findings highlight the critical role of pore engineering in MOFs and COFs, demonstrating how precise functionalization and tailored pore environments can significantly enhance their performance in environmental remediation, particularly for radionuclide sequestration.
 | ||
| Fig. 17 (a) Synthetic scheme of COF-TpDb, formed through the condensation of Tp (black) and Db (blue), followed by a chemical transformation of cyano groups into amidoxime groups, yielding COF-TpDb-AO. (b and c) Schematic representation of the eclipsed AA stacking structure of COF-TpDb (blue: N; gray: C; red: O; hydrogen omitted for clarity). (d) Schematic representation of COF-TpDb-AO (blue: N; gray: C; red: O; hydrogen omitted for clarity). Reproduced with permission.241 Copyright 2018, John Wiley and Sons. | ||
4. Outlooks and conclusion
In this review, we provide a comprehensive and timely overview of the advancements in pore engineering of MOFs and COFs, highlighting how precise modifications in pore structures and surface chemistry can significantly enhance their functionality and expand their applicability across various domains. We discuss in detail the strategies employed to functionalize and tailor these crystalline porous frameworks, including de novo synthesis and post-synthetic modifications. Furthermore, drawing from our extensive research experience in gas storage and separation, catalysis, and environmental remediation, we discuss the design principles and strategic approaches for optimizing MOFs and COFs in these applications. We highlight how rational pore engineering, through precise control over pore size, topology, and surface chemistry, enhances adsorption capacity, selectivity, and stability. Building on our group's advancements, we illustrate how these pore-engineered frameworks surpass conventional porous materials by exhibiting superior efficiency, accelerated kinetics, and enhanced recyclability, thereby broadening their practical applicability. Through precise control over composition, pore geometry, and surface functionality, MOFs and COFs can be strategically tailored for specific applications, paving the way for next-generation materials with optimized performance and expanded functionality. This review not only provides insight into the current state-of-the-art in pore engineering but also sheds light on future research directions and challenges that need to be addressed for broader industrial implementation.Despite significant advancements, the practical application of MOFs and COFs remains challenging, particularly in scalable synthesis and processing.242–245 High production costs and low yields often hinder large-scale manufacturing. Addressing these issues requires the development of cost-effective organic ligands and monomers alongside more efficient, sustainable synthesis strategies that improve yield while reducing energy consumption and waste. In addition to synthetic challenges, the chemical and mechanical stability, as well as the recyclability of MOFs and COFs under harsh industrial conditions—such as exposure to moisture, high temperature, or chemical contaminants—pose significant barriers to their long-term deployment. Many MOFs and COFs exhibit degradation or loss of structural integrity upon repeated use, which limits their feasibility in continuous operation environments such as adsorption-based separation or catalytic cycles. Therefore, improving the robustness and reusability of these materials is essential for real-world applications. Additionally, the complex and often multistep synthesis required for designing task-specific MOFs and COFs presents another hurdle to their large-scale adoption. To address this, future research should prioritize the development of more streamlined and scalable synthetic strategies that retain key structural features such as crystallinity, porosity, and stability. Furthermore, progress in material processing—such as shaping MOFs and COFs into membranes,246,247 monoliths,248–250 or composites structures251–254—will be vital to bridge the gap between laboratory research and practical applications.
Artificial intelligence (AI) and machine learning (ML) offer transformative solutions by enabling data-driven crystalline porous material design.255–262 ML algorithms can analyze vast chemical datasets, predict structure–property relationships, and optimize synthetic pathways, significantly reducing reliance on trial-and-error experimentation. AI-driven models facilitate high-throughput screening of candidate frameworks, accelerating the discovery of MOFs and COFs with tailored adsorption, separation, and catalytic properties. Deep learning further refines this process by predicting material stability, porosity, and functional performance before synthesis, streamlining development. In pore engineering, AI enables precise modulation of pore environments to enhance material selectivity and efficiency. Advanced ML models uncover correlations between pore structure and host–guest interactions, guiding the rational design of highly selective materials. Generative algorithms, including deep neural networks and reinforcement learning, can autonomously propose novel MOF and COF architectures with optimized pore chemistry and spatial configurations. AI-driven robotic synthesis platforms further enhance reproducibility and accelerate the experimental validation of computationally designed structures, bridging the gap between theoretical predictions and practical applications.
In the future, integrating AI with MOF and COF research will revolutionize material innovation, making it more predictive, scalable, and application-driven. By merging AI-guided discovery with advanced synthesis techniques, next-generation crystalline porous materials can be precisely engineered, unlocking breakthroughs in a variety of practical applications.
Data availability
The data that support the findings of this study are available from the corresponding author, S. Ma, upon reasonable request.Author contributions
Yanpei Song: conceptualization, methodology, writing – original draft, visualization; Shengqian Ma: investigation, funding acquisition, writing – review & editing, validation, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the Robert A. Welch Foundation (B-0027) for this work.References
- Y. Li, L. Li and J. Yu, Applications of Zeolites in Sustainable Chemistry, Chem, 2017, 3, 928–949 CAS.
- X. Meng and F. S. Xiao, Green routes for synthesis of zeolites, Chem. Rev., 2014, 114, 1521–1543 CrossRef CAS PubMed.
- M. Dusselier and M. E. Davis, Small-Pore Zeolites: Synthesis and Catalysis, Chem. Rev., 2018, 118, 5265–5329 CrossRef CAS PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, The chemistry and applications of metal-organic frameworks, Science, 2013, 341, 1230444 CrossRef.
- H. C. Zhou, J. R. Long and O. M. Yaghi, Introduction to metal-organic frameworks, Chem. Rev., 2012, 112, 673–674 CrossRef CAS PubMed.
- N. Stock and S. Biswas, Synthesis of metal-organic frameworks (MOFs): routes to various MOF topologies, morphologies, and composites, Chem. Rev., 2012, 112, 933–969 CrossRef CAS.
- A. J. Howarth, Y. Liu, P. Li, Z. Li, T. C. Wang, J. T. Hupp and O. K. Farha, Chemical, thermal and mechanical stabilities of metal–organic frameworks, Nat. Rev. Mater., 2016, 1, 15018 CrossRef CAS.
- H. C. Zhou and S. Kitagawa, Metal-organic frameworks (MOFs), Chem. Soc. Rev., 2014, 43, 5415–5418 RSC.
- M. Ding, X. Cai and H. L. Jiang, Improving MOF stability: approaches and applications, Chem. Sci., 2019, 10, 10209–10230 RSC.
- Y. Song, Q. Sun, B. Aguila and S. Ma, Opportunities of Covalent Organic Frameworks for Advanced Applications, Adv. Sci., 2019, 6, 1801410 CrossRef PubMed.
- C. S. Diercks and O. M. Yaghi, The atom, the molecule, and the covalent organic framework, Science, 2017, 355, aal1585 CrossRef PubMed.
- S. Kandambeth, K. Dey and R. Banerjee, Covalent Organic Frameworks: Chemistry beyond the Structure, J. Am. Chem. Soc., 2019, 141, 1807–1822 CrossRef CAS PubMed.
- K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Covalent Organic Frameworks: Design, Synthesis, and Functions, Chem. Rev., 2020, 120, 8814–8933 CrossRef CAS PubMed.
- S. Y. Ding and W. Wang, Covalent organic frameworks (COFs): from design to applications, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- X. Feng, X. Ding and D. Jiang, Covalent organic frameworks, Chem. Soc. Rev., 2012, 41, 6010–6022 RSC.
- R.-B. Lin and B. Chen, Hydrogen-bonded organic frameworks: Chemistry and functions, Chem, 2022, 8, 2114–2135 CAS.
- R. B. Lin, Y. He, P. Li, H. Wang, W. Zhou and B. Chen, Multifunctional porous hydrogen-bonded organic framework materials, Chem. Soc. Rev., 2019, 48, 1362–1389 RSC.
- B. Wang, R. B. Lin, Z. Zhang, S. Xiang and B. Chen, Hydrogen-Bonded Organic Frameworks as a Tunable Platform for Functional Materials, J. Am. Chem. Soc., 2020, 142, 14399–14416 CrossRef CAS PubMed.
- D. Yu, H. Zhang, J. Ren and X. Qu, Hydrogen-bonded organic frameworks: new horizons in biomedical applications, Chem. Soc. Rev., 2023, 52, 7504–7523 RSC.
- Z. Zhang, Y. Ye, S. Xiang and B. Chen, Exploring Multifunctional Hydrogen-Bonded Organic Framework Materials, Acc. Chem. Res., 2022, 55, 3752–3766 CrossRef CAS.
- R. Freund, O. Zaremba, G. Arnauts, R. Ameloot, G. Skorupskii, M. Dinca, A. Bavykina, J. Gascon, A. Ejsmont, J. Goscianska, M. Kalmutzki, U. Lachelt, E. Ploetz, C. S. Diercks and S. Wuttke, The Current Status of MOF and COF Applications, Angew. Chem., Int. Ed., 2021, 60, 23975–24001 CrossRef CAS.
- E. Ploetz, H. Engelke, U. Lächelt and S. Wuttke, The Chemistry of Reticular Framework Nanoparticles: MOF, ZIF, and COF Materials, Adv. Funct. Mater., 2020, 30, 1909062 CrossRef CAS.
- Y. Li, M. Karimi, Y.-N. Gong, N. Dai, V. Safarifard and H.-L. Jiang, Integration of metal-organic frameworks and covalent organic frameworks: Design, synthesis, and applications, Matter, 2021, 4, 2230–2265 CrossRef CAS.
- Z. Heidarinejad, M. H. Dehghani, M. Heidari, G. Javedan, I. Ali and M. Sillanpää, Methods for preparation and activation of activated carbon: a review, Environ. Chem. Lett., 2020, 18, 393–415 CrossRef CAS.
- C. Martínez and A. Corma, Inorganic molecular sieves: Preparation, modification and industrial application in catalytic processes, Coord. Chem. Rev., 2011, 255, 1558–1580 CrossRef.
- S.-H. Wu, C.-Y. Mou and H.-P. Lin, Synthesis of mesoporous silica nanoparticles, Chem. Soc. Rev., 2013, 42, 3862–3875 RSC.
- R.-J. Wei, X. Luo and D. Li, Covalent Metal–Organic Frameworks: Fusion of Covalent Organic Frameworks and Metal–Organic Frameworks, Acc. Chem. Res., 2025, 58, 746–761 CrossRef CAS PubMed.
- I. M. Honicke, I. Senkovska, V. Bon, I. A. Baburin, N. Bonisch, S. Raschke, J. D. Evans and S. Kaskel, Balancing Mechanical Stability and Ultrahigh Porosity in Crystalline Framework Materials, Angew. Chem., Int. Ed., 2018, 57, 13780–13783 CrossRef.
- L. A. Baldwin, J. W. Crowe, D. A. Pyles and P. L. McGrier, Metalation of a Mesoporous Three-Dimensional Covalent Organic Framework, J. Am. Chem. Soc., 2016, 138, 15134–15137 CrossRef CAS.
- W. Fan, X. Zhang, Z. Kang, X. Liu and D. Sun, Isoreticular chemistry within metal–organic frameworks for gas storage and separation, Coord. Chem. Rev., 2021, 443, 213968 CrossRef CAS.
- X. Zhao, Y. Wang, D. S. Li, X. Bu and P. Feng, Metal-Organic Frameworks for Separation, Adv. Mater., 2018, 30, e1705189 CrossRef.
- J. R. Li, R. J. Kuppler and H. C. Zhou, Selective gas adsorption and separation in metal-organic frameworks, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- Q. Yang, D. Liu, C. Zhong and J. R. Li, Development of computational methodologies for metal-organic frameworks and their application in gas separations, Chem. Rev., 2013, 113, 8261–8323 CrossRef CAS.
- A. Knebel and J. Caro, Metal-organic frameworks and covalent organic frameworks as disruptive membrane materials for energy-efficient gas separation, Nat. Nanotechnol., 2022, 17, 911–923 CrossRef CAS.
- Z. Wang, S. Zhang, Y. Chen, Z. Zhang and S. Ma, Covalent organic frameworks for separation applications, Chem. Soc. Rev., 2020, 49, 708–735 RSC.
- B. Hosseini Monjezi, K. Kutonova, M. Tsotsalas, S. Henke and A. Knebel, Current Trends in Metal-Organic and Covalent Organic Framework Membrane Materials, Angew. Chem., Int. Ed., 2021, 60, 15153–15164 CrossRef CAS.
- S. Yuan, X. Li, J. Zhu, G. Zhang, P. Van Puyvelde and B. Van der Bruggen, Covalent organic frameworks for membrane separation, Chem. Soc. Rev., 2019, 48, 2665–2681 RSC.
- S. M. J. Rogge, A. Bavykina, J. Hajek, H. Garcia, A. I. Olivos-Suarez, A. Sepulveda-Escribano, A. Vimont, G. Clet, P. Bazin, F. Kapteijn, M. Daturi, E. V. Ramos-Fernandez, I. X. F. X. Llabres, V. Van Speybroeck and J. Gascon, Metal-organic and covalent organic frameworks as single-site catalysts, Chem. Soc. Rev., 2017, 46, 3134–3184 RSC.
- A. Dhakshinamoorthy, Z. Li and H. Garcia, Catalysis and photocatalysis by metal organic frameworks, Chem. Soc. Rev., 2018, 47, 8134–8172 RSC.
- Y. B. Huang, J. Liang, X. S. Wang and R. Cao, Multifunctional metal-organic framework catalysts: synergistic catalysis and tandem reactions, Chem. Soc. Rev., 2017, 46, 126–157 RSC.
- L. Zhu, X. Q. Liu, H. L. Jiang and L. B. Sun, Metal-Organic Frameworks for Heterogeneous Basic Catalysis, Chem. Rev., 2017, 117, 8129–8176 CrossRef CAS PubMed.
- W. Zhao, Q. Zhu, X. Wu and D. Zhao, The development of catalysts and auxiliaries for the synthesis of covalent organic frameworks, Chem. Soc. Rev., 2024, 53, 7531–7565 RSC.
- A. Bavykina, N. Kolobov, I. S. Khan, J. A. Bau, A. Ramirez and J. Gascon, Metal-Organic Frameworks in Heterogeneous Catalysis: Recent Progress, New Trends, and Future Perspectives, Chem. Rev., 2020, 120, 8468–8535 CrossRef CAS PubMed.
- D. Li, H.-Q. Xu, L. Jiao and H.-L. Jiang, Metal-organic frameworks for catalysis: State of the art, challenges, and opportunities, EnergyChem, 2019, 1, 100005 CrossRef.
- X. Meng, H. N. Wang, S. Y. Song and H. J. Zhang, Proton-conducting crystalline porous materials, Chem. Soc. Rev., 2017, 46, 464–480 RSC.
- D. W. Lim and H. Kitagawa, Rational strategies for proton-conductive metal-organic frameworks, Chem. Soc. Rev., 2021, 50, 6349–6368 RSC.
- Y. Ye, L. Gong, S. Xiang, Z. Zhang and B. Chen, Metal-Organic Frameworks as a Versatile Platform for Proton Conductors, Adv. Mater., 2020, 32, e1907090 CrossRef PubMed.
- D. W. Lim and H. Kitagawa, Proton Transport in Metal-Organic Frameworks, Chem. Rev., 2020, 120, 8416–8467 CrossRef CAS PubMed.
- R. Sahoo, S. Mondal, S. C. Pal, D. Mukherjee and M. C. Das, Covalent-Organic Frameworks (COFs) as Proton Conductors, Adv. Energy Mater., 2021, 11, 2102300 CrossRef CAS.
- H. Xu, S. Tao and D. Jiang, Proton conduction in crystalline and porous covalent organic frameworks, Nat. Mater., 2016, 15, 722–726 CrossRef CAS PubMed.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Metal-organic framework materials as chemical sensors, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed.
- Z. Meng and K. A. Mirica, Covalent organic frameworks as multifunctional materials for chemical detection, Chem. Soc. Rev., 2021, 50, 13498–13558 RSC.
- Z. Hu, B. J. Deibert and J. Li, Luminescent metal-organic frameworks for chemical sensing and explosive detection, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC.
- X. Liu, D. Huang, C. Lai, G. Zeng, L. Qin, H. Wang, H. Yi, B. Li, S. Liu, M. Zhang, R. Deng, Y. Fu, L. Li, W. Xue and S. Chen, Recent advances in covalent organic frameworks (COFs) as a smart sensing material, Chem. Soc. Rev., 2019, 48, 5266–5302 RSC.
- L. Guo, L. Yang, M. Li, L. Kuang, Y. Song and L. Wang, Covalent organic frameworks for fluorescent sensing: Recent developments and future challenges, Coord. Chem. Rev., 2021, 440, 213957 CrossRef CAS.
- W.-T. Koo, J.-S. Jang and I.-D. Kim, Metal-Organic Frameworks for Chemiresistive Sensors, Chem, 2019, 5, 1938–1963 CAS.
- I. Ahmed and S. H. Jhung, Covalent organic framework-based materials: Synthesis, modification, and application in environmental remediation, Coord. Chem. Rev., 2021, 441, 213989 CrossRef CAS.
- S. Daliran, A. R. Oveisi, C. W. Kung, U. Sen, A. Dhakshinamoorthy, C. H. Chuang, M. Khajeh, M. Erkartal and J. T. Hupp, Defect-enabling zirconium-based metal-organic frameworks for energy and environmental remediation applications, Chem. Soc. Rev., 2024, 53, 6244–6294 RSC.
- S. Ge, K. Wei, W. Peng, R. Huang, E. Akinlabi, H. Xia, M. W. Shahzad, X. Zhang, B. B. Xu and J. Jiang, A comprehensive review of covalent organic frameworks (COFs) and their derivatives in environmental pollution control, Chem. Soc. Rev., 2024, 53, 11259–11302 RSC.
- J. Wang and S. Zhuang, Covalent organic frameworks (COFs) for environmental applications, Coord. Chem. Rev., 2019, 400, 213046 CrossRef CAS.
- S. Dhaka, R. Kumar, A. Deep, M. B. Kurade, S.-W. Ji and B.-H. Jeon, Metal–organic frameworks (MOFs) for the removal of emerging contaminants from aquatic environments, Coord. Chem. Rev., 2019, 380, 330–352 CrossRef CAS.
- S. Rojas and P. Horcajada, Metal-Organic Frameworks for the Removal of Emerging Organic Contaminants in Water, Chem. Rev., 2020, 120, 8378–8415 CrossRef CAS.
- O. K. Farha and J. T. Hupp, Acc. Chem. Res., 2010, 43, 1166–1175 CrossRef CAS PubMed.
- F. A. Paz, J. Klinowski, S. M. Vilela, J. P. Tome, J. A. Cavaleiro and J. Rocha, Ligand design for functional metal-organic frameworks, Chem. Soc. Rev., 2012, 41, 1088–1110 RSC.
- W. Lu, Z. Wei, Z.-Y. Gu, T.-F. Liu, J. Park, J. Park, J. Tian, M. Zhang, Q. Zhang and T. Gentle Iii, Tuning the structure and function of metal–organic frameworks via linker design, Chem. Soc. Rev., 2014, 43, 5561–5593 RSC.
- K. Wang, K. N. Hui, K. San Hui, S. Peng and Y. Xu, Recent progress in metal-organic framework/graphene-derived materials for energy storage and conversion: design, preparation, and application, Chem. Sci., 2021, 12, 5737–5766 RSC.
- X. Guan, F. Chen, Q. Fang and S. Qiu, Design and applications of three dimensional covalent organic frameworks, Chem. Soc. Rev., 2020, 49, 1357–1384 RSC.
- B. Gui, G. Lin, H. Ding, C. Gao, A. Mal and C. Wang, Three-Dimensional Covalent Organic Frameworks: From Topology Design to Applications, Acc. Chem. Res., 2020, 53, 2225–2234 CrossRef CAS.
- X. Han, C. Yuan, B. Hou, L. Liu, H. Li, Y. Liu and Y. Cui, Chiral covalent organic frameworks: design, synthesis and property, Chem. Soc. Rev., 2020, 49, 6248–6272 RSC.
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Design and synthesis of an exceptionally stable and highly porous metal-organic framework, Nature, 1999, 402, 276–279 CrossRef CAS.
- H. Li, M. Eddaoudi, T. L. Groy and O. M. Yaghi, Establishing microporosity in open metal–organic frameworks: gas sorption isotherms for Zn (BDC)(BDC= 1, 4-benzenedicarboxylate), J. Am. Chem. Soc., 1998, 120, 8571–8572 CrossRef CAS.
- M. Y. Masoomi, A. Morsali, A. Dhakshinamoorthy and H. Garcia, Mixed-Metal MOFs: Unique Opportunities in Metal-Organic Framework (MOF) Functionality and Design, Angew. Chem., Int. Ed., 2019, 58, 15188–15205 CrossRef CAS PubMed.
- L. J. Wang, H. Deng, H. Furukawa, F. Gandara, K. E. Cordova, D. Peri and O. M. Yaghi, Synthesis and characterization of metal-organic framework-74 containing 2, 4, 6, 8, and 10 different metals, Inorg. Chem., 2014, 53, 5881–5883 CrossRef CAS PubMed.
- H. Deng, C. Doonan, H. Furukawa, R. Ferreira, J. Towne, C. Knobler, B. Wang and O. Yaghi, Multiple functional groups of varying ratios in metal-organic frameworks, Science, 2010, 327, 846–850 CrossRef CAS PubMed.
- M. Wang, T. Zeng, Y. Yu, X. Wang, Y. Zhao, H. Xi and Y. B. Zhang, Flexibility On-Demand: Multivariate 3D Covalent Organic Frameworks, J. Am. Chem. Soc., 2024, 146, 1035–1041 CrossRef CAS PubMed.
- S. K. Sobczak, J. Drweska, W. Gromelska, K. Roztocki and A. M. Janiak, Multivariate Flexible Metal-Organic Frameworks and Covalent Organic Frameworks, Small, 2024, 20, e2402486 CrossRef PubMed.
- X. Yin, A. Alsuwaidi and X. Zhang, Hierarchical metal-organic framework (MOF) pore engineering, Microporous Mesoporous Mater., 2022, 330, 111633 CrossRef CAS.
- A. P. Cote, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Porous, crystalline, covalent organic frameworks, Science, 2005, 310, 1166–1170 CrossRef CAS PubMed.
- J. L. Segura, M. J. Mancheno and F. Zamora, Covalent organic frameworks based on Schiff-base chemistry: synthesis, properties and potential applications, Chem. Soc. Rev., 2016, 45, 5635–5671 RSC.
- F. J. Uribe-Romo, J. R. Hunt, H. Furukawa, C. Klöck, M. O'Keeffe and O. M. Yaghi, A crystalline imine-linked 3-D porous covalent organic framework, J. Am. Chem. Soc., 2009, 131, 4570–4571 CrossRef CAS PubMed.
- L. Stegbauer, K. Schwinghammer and B. V. Lotsch, A hydrazone-based covalent organic framework for photocatalytic hydrogen production, Chem. Sci., 2014, 5, 2789–2793 RSC.
- F. J. Uribe-Romo, C. J. Doonan, H. Furukawa, K. Oisaki and O. M. Yaghi, Crystalline covalent organic frameworks with hydrazone linkages, J. Am. Chem. Soc., 2011, 133, 11478–11481 CrossRef CAS PubMed.
- S. Dalapati, S. Jin, J. Gao, Y. Xu, A. Nagai and D. Jiang, An azine-linked covalent organic framework, J. Am. Chem. Soc., 2013, 135, 17310–17313 CrossRef CAS PubMed.
- V. S. Vyas, F. Haase, L. Stegbauer, G. Savasci, F. Podjaski, C. Ochsenfeld and B. V. Lotsch, A tunable azine covalent organic framework platform for visible light-induced hydrogen generation, Nat. Commun., 2015, 6, 8508 CrossRef CAS PubMed.
- W. Zhou, Q. W. Deng, H. J. He, L. Yang, T. Y. Liu, X. Wang, D. Y. Zheng, Z. B. Dai, L. Sun, C. Liu, H. Wu, Z. Li and W. Q. Deng, Heterogenization of Salen Metal Molecular Catalysts in Covalent Organic Frameworks for Photocatalytic Hydrogen Evolution, Angew. Chem., Int. Ed., 2023, 62, e202214143 CrossRef CAS.
- H. Li, X. Feng, P. Shao, J. Chen, C. Li, S. Jayakumar and Q. Yang, Synthesis of covalent organic frameworks via in situ salen skeleton formation for catalytic applications, J. Mater. Chem. A, 2019, 7, 5482–5492 RSC.
- S. Kandambeth, A. Mallick, B. Lukose, M. V. Mane, T. Heine and R. Banerjee, Construction of crystalline 2D covalent organic frameworks with remarkable chemical (acid/base) stability via a combined reversible and irreversible route, J. Am. Chem. Soc., 2012, 134, 19524–19527 CrossRef CAS PubMed.
- C. R. DeBlase, K. E. Silberstein, T. T. Truong, H. D. Abruna and W. R. Dichtel, beta-Ketoenamine-linked covalent organic frameworks capable of pseudocapacitive energy storage, J. Am. Chem. Soc., 2013, 135, 16821–16824 CrossRef CAS PubMed.
- M. A. Khayum, M. Ghosh, V. Vijayakumar, A. Halder, M. Nurhuda, S. Kumar, M. Addicoat, S. Kurungot and R. Banerjee, Zinc ion interactions in a two-dimensional covalent organic framework based aqueous zinc ion battery, Chem. Sci., 2019, 10, 8889–8894 RSC.
- Q. Fang, J. Wang, S. Gu, R. B. Kaspar, Z. Zhuang, J. Zheng, H. Guo, S. Qiu and Y. Yan, 3D Porous Crystalline Polyimide Covalent Organic Frameworks for Drug Delivery, J. Am. Chem. Soc., 2015, 137, 8352–8355 CrossRef CAS PubMed.
- Q. Fang, Z. Zhuang, S. Gu, R. B. Kaspar, J. Zheng, J. Wang, S. Qiu and Y. Yan, Designed synthesis of large-pore crystalline polyimide covalent organic frameworks, Nat. Commun., 2014, 5, 4503 CrossRef PubMed.
- X. Chen, Q. Dang, R. Sa, L. Li, L. Li, J. Bi, Z. Zhang, J. Long, Y. Yu and Z. Zou, Integrating single Ni sites into biomimetic networks of covalent organic frameworks for selective photoreduction of CO2, Chem. Sci., 2020, 11, 6915–6922 RSC.
- T. X. Luan, L. Du, J. R. Wang, K. Li, Q. Zhang, P. Z. Li and Y. Zhao, Highly Effective Generation of Singlet Oxygen by an Imidazole-Linked Robust Photosensitizing Covalent Organic Framework, ACS Nano, 2022, 16, 21565–21575 CrossRef CAS PubMed.
- Y. Zhang, Z. Qiao, R. Zhang, Z. Wang, H. J. Wang, J. Zhao, D. Cao and S. Wang, Multicomponent Synthesis of Imidazole-Linked Fully Conjugated 3D Covalent Organic Framework for Efficient Electrochemical Hydrogen Peroxide Production, Angew. Chem., Int. Ed., 2023, 62, e202314539 CrossRef CAS PubMed.
- M. D. Zhang, J. R. Huang, W. Shi, P. Q. Liao and X. M. Chen, Self-Accelerating Effect in a Covalent-Organic Framework with Imidazole Groups Boosts Electroreduction of CO2 to CO, Angew. Chem., Int. Ed., 2023, 62, e202308195 CrossRef CAS PubMed.
- P. F. Wei, M. Z. Qi, Z. P. Wang, S. Y. Ding, W. Yu, Q. Liu, L. K. Wang, H. Z. Wang, W. K. An and W. Wang, Benzoxazole-Linked Ultrastable Covalent Organic Frameworks for Photocatalysis, J. Am. Chem. Soc., 2018, 140, 4623–4631 CrossRef CAS PubMed.
- C. J. Wu, X. Y. Li, T. R. Li, M. Z. Shao, L. J. Niu, X. F. Lu, J. L. Kan, Y. Geng and Y. B. Dong, Natural Sunlight Photocatalytic Synthesis of Benzoxazole-Bridged Covalent Organic Framework for Photocatalysis, J. Am. Chem. Soc., 2022, 144, 18750–18755 CrossRef CAS PubMed.
- D. A. Pyles, W. H. Coldren, G. M. Eder, C. M. Hadad and P. L. McGrier, Mechanistic investigations into the cyclization and crystallization of benzobisoxazole-linked two-dimensional covalent organic frameworks, Chem. Sci., 2018, 9, 6417–6423 RSC.
- C. Qian, L. Feng, W. L. Teo, J. Liu, W. Zhou, D. Wang and Y. Zhao, Imine and imine-derived linkages in two-dimensional covalent organic frameworks, Nat. Rev. Chem., 2022, 6, 881–898 CrossRef CAS PubMed.
- S. Xu, M. Richter and X. Feng, Vinylene-Linked Two-Dimensional Covalent Organic Frameworks: Synthesis and Functions, Acc. Mater. Res., 2021, 2, 252–265 CrossRef CAS.
- T. He, K. Geng and D. Jiang, All sp2 carbon covalent organic frameworks, Trends Chem., 2021, 3, 431–444 CrossRef CAS.
- Q. Guan, L. L. Zhou and Y. B. Dong, Construction of Covalent Organic Frameworks via Multicomponent Reactions, J. Am. Chem. Soc., 2023, 145, 1475–1496 CrossRef CAS PubMed.
- Z. Meng, A. Aykanat and K. A. Mirica, Proton Conduction in 2D Aza-Fused Covalent Organic Frameworks, Chem. Mater., 2018, 31, 819–825 CrossRef.
- J. Feng, Y. J. Zhang, S. H. Ma, C. Yang, Z. P. Wang, S. Y. Ding, Y. Li and W. Wang, Fused-Ring-Linked Covalent Organic Frameworks, J. Am. Chem. Soc., 2022, 144, 6594–6603 CrossRef CAS PubMed.
- X. Zhuang, W. Zhao, F. Zhang, Y. Cao, F. Liu, S. Bi and X. Feng, A two-dimensional conjugated polymer framework with fully sp2-bonded carbon skeleton, Polym. Chem., 2016, 7, 4176–4181 RSC.
- E. Jin, M. Asada, Q. Xu, S. Dalapati, M. A. Addicoat, M. A. Brady, H. Xu, T. Nakamura, T. Heine, Q. Chen and D. Jiang, Two-dimensional sp2 carbon–conjugated covalent organic frameworks, Science, 2017, 357, 673–676 CrossRef CAS PubMed.
- T. Jadhav, Y. Fang, W. Patterson, C. H. Liu, E. Hamzehpoor and D. F. Perepichka, 2D Poly(arylene vinylene) Covalent Organic Frameworks via Aldol Condensation of Trimethyltriazine, Angew. Chem., Int. Ed., 2019, 58, 13753–13757 CrossRef CAS PubMed.
- D. L. Pastoetter, S. Xu, M. Borrelli, M. Addicoat, B. P. Biswal, S. Paasch, A. Dianat, H. Thomas, R. Berger, S. Reineke, E. Brunner, G. Cuniberti, M. Richter and X. Feng, Synthesis of Vinylene-Linked Two-Dimensional Conjugated Polymers via the Horner-Wadsworth-Emmons Reaction, Angew. Chem., Int. Ed., 2020, 59, 23620–23625 CrossRef CAS PubMed.
- P. Katekomol, J. Roeser, M. Bojdys, J. Weber and A. Thomas, Covalent Triazine Frameworks Prepared from 1,3,5-Tricyanobenzene, Chem. Mater., 2013, 25, 1542–1548 CrossRef CAS.
- K. Kamiya, Selective single-atom electrocatalysts: a review with a focus on metal-doped covalent triazine frameworks, Chem. Sci., 2020, 11, 8339–8349 RSC.
- P. Xiong, S. Zhang, R. Wang, L. Zhang, Q. Ma, X. Ren, Y. Gao, Z. Wang, Z. Guo and C. Zhang, Covalent triazine frameworks for advanced energy storage: challenges and new opportunities, Energy Environ. Sci., 2023, 16, 3181–3213 RSC.
- M. Liu, L. Guo, S. Jin and B. Tan, Covalent triazine frameworks: synthesis and applications, J. Mater. Chem. A, 2019, 7, 5153–5172 RSC.
- X. Guan, H. Li, Y. Ma, M. Xue, Q. Fang, Y. Yan, V. Valtchev and S. Qiu, Chemically stable polyarylether-based covalent organic frameworks, Nat. Chem., 2019, 11, 587–594 CrossRef CAS PubMed.
- B. Zhang, M. Wei, H. Mao, X. Pei, S. A. Alshmimri, J. A. Reimer and O. M. Yaghi, Crystalline Dioxin-Linked Covalent Organic Frameworks from Irreversible Reactions, J. Am. Chem. Soc., 2018, 140, 12715–12719 CrossRef CAS PubMed.
- M. Lu, M. Zhang, C. G. Liu, J. Liu, L. J. Shang, M. Wang, J. N. Chang, S. L. Li and Y. Q. Lan, Stable Dioxin-Linked Metallophthalocyanine Covalent Organic Frameworks (COFs) as Photo-Coupled Electrocatalysts for CO2 Reduction, Angew. Chem., Int. Ed., 2021, 60, 4864–4871 CrossRef CAS PubMed.
- Q. Zhi, W. Liu, R. Jiang, X. Zhan, Y. Jin, X. Chen, X. Yang, K. Wang, W. Cao, D. Qi and J. Jiang, Piperazine-Linked Metalphthalocyanine Frameworks for Highly Efficient Visible-Light-Driven H2O2 Photosynthesis, J. Am. Chem. Soc., 2022, 144, 21328–21336 CrossRef CAS PubMed.
- Y. Yue, H. Li, H. Chen and N. Huang, Piperazine-Linked Covalent Organic Frameworks with High Electrical Conductivity, J. Am. Chem. Soc., 2022, 144, 2873–2878 CrossRef CAS PubMed.
- M. Wang, M. Ballabio, M. Wang, H. H. Lin, B. P. Biswal, X. Han, S. Paasch, E. Brunner, P. Liu, M. Chen, M. Bonn, T. Heine, S. Zhou, E. Canovas, R. Dong and X. Feng, Unveiling Electronic Properties in Metal-Phthalocyanine-Based Pyrazine-Linked Conjugated Two-Dimensional Covalent Organic Frameworks, J. Am. Chem. Soc., 2019, 141, 16810–16816 CrossRef CAS PubMed.
- F. Haase, E. Troschke, G. Savasci, T. Banerjee, V. Duppel, S. Dorfler, M. M. J. Grundei, A. M. Burow, C. Ochsenfeld, S. Kaskel and B. V. Lotsch, Topochemical conversion of an imine- into a thiazole-linked covalent organic framework enabling real structure analysis, Nat. Commun., 2018, 9, 2600 CrossRef PubMed.
- V. Singh, J. Kim, B. Kang, J. Moon, S. Kim, W. Y. Kim and H. R. Byon, Thiazole-Linked Covalent Organic Framework Promoting Fast Two-Electron Transfer for Lithium-Organic Batteries, Adv. Energy Mater., 2021, 11, 2003735 CrossRef CAS.
- R.-J. Wei, H.-G. Zhou, Z.-Y. Zhang, G.-H. Ning and D. Li, Copper (I)–Organic Frameworks for Catalysis: Networking Metal Clusters with Dynamic Covalent Chemistry, CCS Chem., 2020, 2, 2045–2053 Search PubMed.
- R.-J. Wei, M. Xie, R.-Q. Xia, J. Chen, H. -J Hu, G.-H. Ning and D. Li, Gold(I)-Organic Frameworks as Catalysts for Carboxylation of Alkynes with CO2, J. Am. Chem. Soc., 2023, 145, 22720–22727 CrossRef CAS PubMed.
- P.-Y. You, K.-M. Mo, Y.-M. Wang, Q. Gao, X.-C. Lin, J.-T. Lin, M. Xie, G.-H. Ning and D. Li, Reversible modulation of interlayer stacking in 2D copper-organic frameworks for tailoring porosity and photocatalytic activity, Nat. Commun., 2023, 15, 194 CrossRef PubMed.
- X.-C. Lin, Y.-M. Wang, X. Chen, P.-Y. You, K.-M. Mo, G.-H. Ning and D. Li, A photosensitizing metal–organic framework as a tandem reaction catalyst for primary alcohols from terminal alkenes and alkynes, Angew. Chem., Int. Ed., 2023, 62, e202306497 CrossRef CAS PubMed.
- H. Duan, X. Chen, Y.-N. Yang, J. Zhao, X.-C. Lin, W.-J. Tang, Q. Gao, G.-N. Ning and D. Li, Tailoring stability, catalytic activity and selectivity of covalent metal–organic frameworks via steric modification of metal nodes, J. Mater. Chem. A, 2023, 11, 12777–12783 RSC.
- H. G. Zhou, R.-Q. Xia, J. Zheng, D. Yuan, G.-N. Ning and D. Li, Acid-triggered interlayer sliding of two dimensional copper(I)–organic frameworks: more metal sites for catalysis, Chem. Sci., 2021, 12, 6280–6286 RSC.
- X. Chen, J.-Y. Song, J. Zheng, Y.-M. Wang, J. Luo, P. Weng, B.-C. Cai, X.-C. Lin, G.-H. Ning and D. Li, Metal Variance in Multivariate Metal–Organic Frameworks for Boosting Catalytic Conversion of CO2, J. Am. Chem. Soc., 2024, 146, 19271–19278 CrossRef CAS PubMed.
- J. Luo, X. Luo, M. Xie, J.-T. Lin, J. Pang, N. Yin, Y.-Y. Li, G.-H. Ning and D. Li, Molecular regulation of metal-organic frameworks for rapid and efficient extraction of uranium from seawater, Sci. China: Chem., 2025, 68, 1906–1915 CrossRef CAS.
- Z. Wang and S. M. Cohen, Postsynthetic modification of metal-organic frameworks, Chem. Soc. Rev., 2009, 38, 1315–1329 RSC.
- S. Mandal, S. Natarajan, P. Mani and A. Pankajakshan, Post-Synthetic Modification of Metal–Organic Frameworks Toward Applications, Adv. Funct. Mater., 2020, 31, 2006291 CrossRef.
- J. L. Segura, S. Royuela and M. Mar Ramos, Post-synthetic modification of covalent organic frameworks, Chem. Soc. Rev., 2019, 48, 3903–3945 RSC.
- N. A. Rejali, M. Dinari and Y. Wang, Post-synthetic modifications of covalent organic frameworks (COFs) for diverse applications, Chem. Commun., 2023, 59, 11631–11647 RSC.
- L. Cusin, H. Peng, A. Ciesielski and P. Samori, Chemical Conversion and Locking of the Imine Linkage: Enhancing the Functionality of Covalent Organic Frameworks, Angew. Chem., Int. Ed., 2021, 60, 14236–14250 CrossRef CAS PubMed.
- S. Abednatanzi, P. Gohari Derakhshandeh, H. Depauw, F. X. Coudert, H. Vrielinck, P. Van Der Voort and K. Leus, Mixed-metal metal-organic frameworks, Chem. Soc. Rev., 2019, 48, 2535–2565 RSC.
- L. Chen, H. F. Wang, C. Li and Q. Xu, Bimetallic metal-organic frameworks and their derivatives, Chem. Sci., 2020, 11, 5369–5403 RSC.
- S. Das, H. Kim and K. Kim, Metathesis in single crystal: Complete and reversible exchange of metal ions constituting the frameworks of metal–organic frameworks, J. Am. Chem. Soc., 2009, 131, 3814–3815 CrossRef CAS PubMed.
- X. Song, S. Jeong, D. Kim and M. S. Lah, Transmetalations in two metal–organic frameworks with different framework flexibilities: Kinetics and core–shell heterostructure, CrystEngComm, 2012, 14, 5753–5756 RSC.
- C. K. Brozek and M. Dincă, Ti(3+)-, V(2+/3+)-, Cr(2+/3+)-, Mn(2+)-, and Fe(2+)-substituted MOF-5 and redox reactivity in Cr- and Fe-MOF-5, J. Am. Chem. Soc., 2013, 135, 12886–12891 CrossRef CAS PubMed.
- T. Li, M. T. Kozlowski, E. A. Doud, M. N. Blakely and N. L. Rosi, Stepwise ligand exchange for the preparation of a family of mesoporous MOFs, J. Am. Chem. Soc., 2013, 135, 11688–11691 CrossRef CAS PubMed.
- H. Fei, J. F. Cahill, K. A. Prather and S. M. Cohen, Tandem postsynthetic metal ion and ligand exchange in zeolitic imidazolate frameworks, Inorg. Chem., 2013, 52, 4011–4016 CrossRef CAS PubMed.
- M. Kim, J. F. Cahill, Y. Su, K. A. Prather and S. M. Cohen, Postsynthetic ligand exchange as a route to functionalization of 'inert' metal–organic frameworks, Chem. Sci., 2012, 3, 126–130 RSC.
- M. Kim, J. F. Cahill, H. Fei, K. A. Prather and S. M. Cohen, Postsynthetic ligand and cation exchange in robust metal-organic frameworks, J. Am. Chem. Soc., 2012, 134, 18082–18088 CrossRef CAS PubMed.
- C. Qian, Q. Y. Qi, G. F. Jiang, F. Z. Cui, Y. Tian and X. Zhao, Toward Covalent Organic Frameworks Bearing Three Different Kinds of Pores: The Strategy for Construction and COF-to-COF Transformation via Heterogeneous Linker Exchange, J. Am. Chem. Soc., 2017, 139, 6736–6743 CrossRef CAS PubMed.
- J. Xu, G. Feng, D. Ao, X. Li, M. Li, S. Lei and Y. Wang, Functional Covalent Organic Frameworks' Microspheres Synthesized by Self-Limited Dynamic Linker Exchange for Stationary Phases, Adv. Mater., 2024, 36, e2406256 CrossRef PubMed.
- S. S. Y. Chui, S. M. F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, A Chemically Functionalizable Nanoporous Material [Cu3(TMA)2(H2O)3]n, Science, 1999, 283, 1148–1150 CrossRef CAS PubMed.
- E. D. Bloch, D. Britt, C. Lee, C. J. Doonan, F. J. Uribe-Romo, H. Furukawa, J. R. Long and O. M. Yaghi, Metal insertion in a microporous metal–organic framework lined with 2, 2'-bipyridine, J. Am. Chem. Soc., 2010, 132, 14382–14384 CrossRef CAS PubMed.
- K. Manna, T. Zhang and W. Lin, Postsynthetic metalation of bipyridyl-containing metal-organic frameworks for highly efficient catalytic organic transformations, J. Am. Chem. Soc., 2014, 136, 6566–6569 CrossRef CAS PubMed.
- Q. Sun, B. Aguila, J. Perman, N. Nguyen and S. Ma, Flexibility Matters: Cooperative Active Sites in Covalent Organic Framework and Threaded Ionic Polymer, J. Am. Chem. Soc., 2016, 138, 15790–15796 CrossRef CAS PubMed.
- W. Liu, Y. Zhang, J. Wang, X. Shang, C. Zhang and Q. Wang, Advances of functionalized bipyridine-based covalent-organic frameworks for boosting photocatalysis, Coord. Chem. Rev., 2024, 516, 215997 CrossRef CAS.
- A. Jati, K. Dey, M. Nurhuda, M. A. Addicoat, R. Banerjee and B. Maji, Dual Metalation in a Two-Dimensional Covalent Organic Framework for Photocatalytic C-N Cross-Coupling Reactions, J. Am. Chem. Soc., 2022, 144, 7822–7833 CrossRef CAS PubMed.
- W. Tu, Y. Xu, S. Yin and R. Xu, Rational Design of Catalytic Centers in Crystalline Frameworks, Adv. Mater., 2018, 30, 1707582 CrossRef PubMed.
- L. H. Li, X. L. Feng, X. H. Cui, Y. X. Ma, S. Y. Ding and W. Wang, Salen-Based Covalent Organic Framework, J. Am. Chem. Soc., 2017, 139, 6042–6045 CrossRef CAS PubMed.
- A. M. Shultz, A. A. Sarjeant, O. K. Farha, J. T. Hupp and S. T. Nguyen, Post-synthesis modification of a metal-organic framework to form metallosalen-containing MOF materials, J. Am. Chem. Soc., 2011, 133, 13252–13255 CrossRef CAS PubMed.
- Z. Wang and S. M. Cohen, Postsynthetic Covalent Modification of a Neutral Metal–Organic Framework, J. Am. Chem. Soc., 2007, 129, 12368–12369 CrossRef CAS PubMed.
- A. Nagai, Z. Guo, X. Feng, S. Jin, X. Chen, X. Ding and D. Jiang, Pore surface engineering in covalent organic frameworks, Nat. Commun., 2011, 2, 536 CrossRef PubMed.
- A. N. Hong, H. Yang, X. Bu and P. Feng, Pore space partition of metal-organic frameworks for gas storage and separation, EnergyChem, 2022, 4, 100080 CrossRef CAS.
- Q. G. Zhai, X. Bu, X. Zhao, D. S. Li and P. Feng, Pore Space Partition in Metal-Organic Frameworks, Acc. Chem. Res., 2017, 50, 407–417 CrossRef CAS PubMed.
- Q. G. Zhai, X. Bu, C. Mao, X. Zhao, L. Daemen, Y. Cheng, A. J. Ramirez-Cuesta and P. Feng, An ultra-tunable platform for molecular engineering of high-performance crystalline porous materials, Nat. Commun., 2016, 7, 13645 CrossRef CAS PubMed.
- S. T. Zheng, X. Zhao, S. Lau, A. Fuhr, P. Feng and X. Bu, Entrapment of metal clusters in metal-organic framework channels by extended hooks anchored at open metal sites, J. Am. Chem. Soc., 2013, 135, 10270–10273 CrossRef CAS PubMed.
- X. Zhao, X. Bu, Q. G. Zhai, H. Tran and P. Feng, Pore space partition by symmetry-matching regulated ligand insertion and dramatic tuning on carbon dioxide uptake, J. Am. Chem. Soc., 2015, 137, 1396–1399 CrossRef CAS PubMed.
- X. Xu, X. Wu, K. Xu, H. Xu, H. Chen and N. Huang, Pore partition in two-dimensional covalent organic frameworks, Nat. Commun., 2023, 14, 3360 CrossRef CAS PubMed.
- M. Hao, Y. Xie, M. Lei, X. Liu, Z. Chen, H. Yang, G. I. N. Waterhouse, S. Ma and X. Wang, Pore Space Partition Synthetic Strategy in Imine-linked Multivariate Covalent Organic Frameworks, J. Am. Chem. Soc., 2024, 146, 1904–1913 CrossRef CAS PubMed.
- W. Zhang, H. Jiang, Y. Liu, Y. Hu, A. S. Palakkal, Y. Zhou, M. Sun, E. Du, W. Gong, Q. Zhang, J. Jiang, J. Dong, Y. Liu, Y. Zhu, Y. Cui and X. Duan, Metal-halide porous framework superlattices, Nature, 2025, 638, 418–424 CrossRef CAS PubMed.
- R.-B. Lin, S. Xiang, W. Zhou and B. Chen, Microporous Metal-Organic Framework Materials for Gas Separation, Chem, 2020, 6, 337–363 CAS.
- S. J. Geier, J. A. Mason, E. D. Bloch, W. L. Queen, M. R. Hudson, C. M. Brown and J. R. Long, Selective adsorption of ethylene over ethane and propylene over propane in the metal–organic frameworks M2(dobdc) (M = Mg, Mn, Fe, Co, Ni, Zn), Chem. Sci., 2013, 4, 2054–2061 RSC.
- S. Qiu, M. Xue and G. Zhu, Metal-organic framework membranes: from synthesis to separation application, Chem. Soc. Rev., 2014, 43, 6116–6140 RSC.
- A. U. Czaja, N. Trukhan and U. Muller, Industrial applications of metal-organic frameworks, Chem. Soc. Rev., 2009, 38, 1284–1293 RSC.
- K. Adil, Y. Belmabkhout, R. S. Pillai, A. Cadiau, P. M. Bhatt, A. H. Assen, G. Maurin and M. Eddaoudi, Gas/vapour separation using ultra-microporous metal-organic frameworks: insights into the structure/separation relationship, Chem. Soc. Rev., 2017, 46, 3402–3430 RSC.
- S. Ma and H. C. Zhou, Gas storage in porous metal-organic frameworks for clean energy applications, Chem. Commun., 2010, 46, 44–53 RSC.
- L. J. Murray, M. Dinca and J. R. Long, Hydrogen storage in metal-organic frameworks, Chem. Soc. Rev., 2009, 38, 1294–1314 RSC.
- M. P. Suh, H. J. Park, T. K. Prasad and D. W. Lim, Hydrogen storage in metal-organic frameworks, Chem. Rev., 2012, 112, 782–835 CrossRef CAS PubMed.
- J. A. Mason, M. Veenstra and J. R. Long, Evaluating metal–organic frameworks for natural gas storage, Chem. Sci., 2014, 5, 32–51 RSC.
- B. Li, H.-M. Wen, W. Zhou, J. Q. Xu and B. Chen, Porous Metal-Organic Frameworks: Promising Materials for Methane Storage, Chem, 2016, 1, 557–580 CAS.
- Y. He, W. Zhou, G. Qian and B. Chen, Methane storage in metal-organic frameworks, Chem. Soc. Rev., 2014, 43, 5657–5678 RSC.
- A. R. Millward and O. M. Yaghi, Metal–Organic Frameworks with Exceptionally High Capacity for Storage of Carbon Dioxide at Room Temperature, J. Am. Chem. Soc., 2005, 127, 17998–17999 CrossRef CAS PubMed.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T. H. Bae and J. R. Long, Carbon dioxide capture in metal-organic frameworks, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- N. L. Rosi, J. Eckert, M. Eddaoudi, D. T. Vodak, J. Kim, M. O'Keeffe and O. M. Yaghi, Hydrogen Storage in Microporous Metal-Organic Frameworks, Science, 2003, 300, 1127–1129 CrossRef CAS PubMed.
- D. Yuan, D. Zhao, D. Sun and H. C. Zhou, An isoreticular series of metal-organic frameworks with dendritic hexacarboxylate ligands and exceptionally high gas-uptake capacity, Angew. Chem., Int. Ed., 2010, 49, 5357–5361 CrossRef CAS PubMed.
- Y. Yan, X. Lin, S. Yang, A. J. Blake, A. Dailly, N. R. Champness, P. Hubberstey and M. Schroder, Exceptionally high H2 storage by a metal-organic polyhedral framework, Chem. Commun., 2009, 1025–1027 RSC.
- O. K. Farha, C. E. Wilmer, I. Eryazici, B. G. Hauser, P. A. Parilla, K. O'Neill, A. A. Sarjeant, S. T. Nguyen, R. Q. Snurr and J. T. Hupp, Designing higher surface area metal-organic frameworks: are triple bonds better than phenyls?, J. Am. Chem. Soc., 2012, 134, 9860–9863 CrossRef CAS PubMed.
- O. K. Farha, A. O. Yazaydin, I. Eryazici, C. D. Malliakas, B. G. Hauser, M. G. Kanatzidis, S. T. Nguyen, R. Q. Snurr and J. T. Hupp, De novo synthesis of a metal-organic framework material featuring ultrahigh surface area and gas storage capacities, Nat. Chem., 2010, 2, 944–948 CrossRef CAS PubMed.
- S. S. Han, H. Furukawa, O. M. Yaghi and W. A. Goddard, Covalent organic frameworks as exceptional hydrogen storage materials, J. Am. Chem. Soc., 2008, 130, 11580–11581 CrossRef CAS PubMed.
- R. Zacharia, D. Cossement, L. Lafi and R. Chahine, Volumetric hydrogen sorption capacity of monoliths prepared by mechanical densification of MOF-177, J. Mater. Chem., 2010, 20, 2145–2451 RSC.
- S. S. Kaye, A. Dailly, O. M. Yaghi and J. R. Long, Impact of Preparation and Handling on the Hydrogen Storage Properties of Zn4O(1,4-benzenedicarboxylate)3 (MOF-5), J. Am. Chem. Soc., 2007, 129, 14176–14177 CrossRef CAS PubMed.
- Y. Pramudya and J. L. Mendoza-Cortes, Design Principles for High H2 Storage Using Chelation of Abundant Transition Metals in Covalent Organic Frameworks for 0-700 bar at 298 K, J. Am. Chem. Soc., 2016, 138, 15204–15213 CrossRef CAS PubMed.
- S. Yang, G. S. Martin, J. J. Titman, A. J. Blake, D. R. Allan, N. R. Champness and M. Schroder, Pore with gate: enhancement of the isosteric heat of adsorption of dihydrogen via postsynthetic cation exchange in metal-organic frameworks, Inorg. Chem., 2011, 50, 9374–9384 CrossRef CAS PubMed.
- Y. Peng, V. Krungleviciute, I. Eryazici, J. T. Hupp, O. K. Farha and T. Yildirim, Methane storage in metal-organic frameworks: current records, surprise findings, and challenges, J. Am. Chem. Soc., 2013, 135, 11887–11894 CrossRef CAS PubMed.
- B. Li, H. M. Wen, H. Wang, H. Wu, M. Tyagi, T. Yildirim, W. Zhou and B. Chen, A porous metal-organic framework with dynamic pyrimidine groups exhibiting record high methane storage working capacity, J. Am. Chem. Soc., 2014, 136, 6207–6210 CrossRef CAS PubMed.
- Y. Yin, Y. Zhang, X. Zhou, B. Gui, W. Q. Wang, W. T. Jiang, Y. B. Zhang, J. L. Sun and C. Wang, Ultrahigh–surface area covalent organic frameworks for methane adsorption, Science, 2024, 386, 693–696 CrossRef CAS PubMed.
- H. Li, L. Li, R. B. Lin, W. Zhou, Z. Zhang, S. Xiang and B. Chen, Porous metal-organic frameworks for gas storage and separation: Status and challenges, EnergyChem, 2019, 1, 100006 CrossRef PubMed.
- A. Ebadi Amooghin, H. Sanaeepur, R. Luque, H. Garcia and B. Chen, Fluorinated metal-organic frameworks for gas separation, Chem. Soc. Rev., 2022, 51, 7427–7508 RSC.
- L. Yang, S. Qian, X. Wang, X. Cui, B. Chen and H. Xing, Energy-efficient separation alternatives: metal-organic frameworks and membranes for hydrocarbon separation, Chem. Soc. Rev., 2020, 49, 5359–5406 RSC.
- Y. Yang, L. Li, R. B. Lin, Y. Ye, Z. Yao, L. Yang, F. Xiang, S. Chen, Z. Zhang, S. Xiang and B. Chen, Ethylene/ethane separation in a stable hydrogen-bonded organic framework through a gating mechanism, Nat. Chem., 2021, 13, 933–939 CrossRef CAS PubMed.
- L. Li, R.-B. Lin, R. Krishna, H. Li, S. Xiang, H. Wu, J. Li, W. Zhou and B. Chen, Ethane/ethylene separation in a metal-organic framework with iron-peroxo sites, Science, 2018, 362, 443–446 CrossRef CAS PubMed.
- R. B. Lin, L. Li, H. L. Zhou, H. Wu, C. He, S. Li, R. Krishna, J. Li, W. Zhou and B. Chen, Molecular sieving of ethylene from ethane using a rigid metal-organic framework, Nat. Mater., 2018, 17, 1128–1133 CrossRef CAS PubMed.
- B. Chen, C. Liang, J. Yang, D. S. Contreras, Y. L. Clancy, E. B. Lobkovsky, O. M. Yaghi and S. Dai, A microporous metal-organic framework for gas-chromatographic separation of alkanes, Angew. Chem., Int. Ed., 2006, 45, 1390–1393 CrossRef CAS PubMed.
- P. S. Barcia, F. Zapata, J. A. C. Silva, A. E. Rodrigues and B. Chen, Kinetic Separation of Hexane Isomers by Fixed-Bed Adsorption with a Microporous Metal–Organic Framework, J. Phys. Chem. B, 2007, 111, 6101–6103 CrossRef CAS PubMed.
- X. Cui, K. Chen, H. Xing, Q. Yang, R. Krishna, Z. Bao, H. Wu, W. Zhou, X. Dong, Y. Han, B. Li, Q. Ren, M. J. Zaworotko and B. Chen, Pore chemistry and size control in hybrid porous materials for acetylene capture from ethylene, Science, 2016, 353, 141–144 CrossRef CAS PubMed.
- F. Luo, C. Yan, L. Dang, R. Krishna, W. Zhou, H. Wu, X. Dong, Y. Han, T. L. Hu, M. O'Keeffe, L. Wang, M. Luo, R. B. Lin and B. Chen, UTSA-74: A MOF-74 Isomer with Two Accessible Binding Sites per Metal Center for Highly Selective Gas Separation, J. Am. Chem. Soc., 2016, 138, 5678–5684 CrossRef CAS PubMed.
- Z. Niu, X. Cui, T. Pham, G. Verma, P. C. Lan, C. Shan, H. Xing, K. A. Forrest, S. Suepaul, B. Space, A. Nafady, A. M. Al-Enizi and S. Ma, A MOF-based Ultra-Strong Acetylene Nano-trap for Highly Efficient C2H2/CO2 Separation, Angew. Chem., Int. Ed., 2021, 60, 5283–5288 CrossRef CAS PubMed.
- Y. Ye, S. Xian, H. Cui, K. Tan, L. Gong, B. Liang, T. Pham, H. Pandey, R. Krishna, P. C. Lan, K. A. Forrest, B. Space, T. Thonhauser, J. Li and S. Ma, Metal-Organic Framework Based Hydrogen-Bonding Nanotrap for Efficient Acetylene Storage and Separation, J. Am. Chem. Soc., 2022, 144, 1681–1689 CrossRef CAS PubMed.
- C. X. Chen, Z. W. Wei, T. Pham, P. C. Lan, L. Zhang, K. A. Forrest, S. Chen, A. M. Al-Enizi, A. Nafady, C. Y. Su and S. Ma, Nanospace Engineering of Metal-Organic Frameworks through Dynamic Spacer Installation of Multifunctionalities for Efficient Separation of Ethane from Ethane/Ethylene Mixtures, Angew. Chem., Int. Ed., 2021, 60, 9680–9685 CrossRef CAS PubMed.
- Y. Ye, Y. Xie, Y. Shi, L. Gong, J. Phipps, A. M. Al-Enizi, A. Nafady, B. Chen and S. Ma, A Microporous Metal-Organic Framework with Unique Aromatic Pore Surfaces for High Performance C2H6/C2H4 Separation, Angew. Chem., Int. Ed., 2023, 62, e202302564 CrossRef CAS PubMed.
- H. Fan, H. Wang, M. Peng, H. Meng, A. Mundstock, A. Knebel and J. Caro, Pore-in-Pore Engineering in a Covalent Organic Framework Membrane for Gas Separation, ACS Nano, 2023, 17, 7584–7594 CrossRef CAS PubMed.
- B. Aguila, Q. Sun, X. Wang, E. O'Rourke, A. M. Al-Enizi, A. Nafady and S. Ma, Lower Activation Energy for Catalytic Reactions through Host-Guest Cooperation within Metal-Organic Frameworks, Angew. Chem., Int. Ed., 2018, 57, 10107–10111 CrossRef CAS PubMed.
- G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surblé and I. Margiolaki, A Chromium Terephthalate-Based Solid with Unusually Large Pore Volumes and Surface Area, Science, 2005, 309, 2040–2042 CrossRef PubMed.
- Y. Fu, L. Xu, H. Shen, H. Yang, F. Zhang, W. Zhu and M. Fan, Tunable catalytic properties of multi-metal–organic frameworks for aerobic styrene oxidation, Chem. Eng. J., 2016, 299, 135–141 CrossRef CAS.
- S. Zhao, Y. Wang, J. Dong, C.-T. He, H. Yin, P. An, K. Zhao, X. Zhang, C. Gao, L. Zhang, J. Lv, J. Wang, J. Zhang, A. M. Khattak, N. A. Khan, Z. Wei, J. Zhang, S. Liu, H. Zhao and Z. Tang, Ultrathin metal–organic framework nanosheets for electrocatalytic oxygen evolution, Nat. Energy, 2016, 1, 16184 CrossRef CAS.
- Y. Lee, S. Kim, J. K. Kang and S. M. Cohen, Photocatalytic CO2 reduction by a mixed metal (Zr/Ti), mixed ligand metal-organic framework under visible light irradiation, Chem. Commun., 2015, 51, 5735–5738 RSC.
- C. Zhang, C. Xie, Y. Gao, X. Tao, C. Ding, F. Fan and H. L. Jiang, Charge Separation by Creating Band Bending in Metal-Organic Frameworks for Improved Photocatalytic Hydrogen Evolution, Angew. Chem., Int. Ed., 2022, 61, e202204108 CrossRef CAS PubMed.
- K. Sun, Y. Huang, F. Sun, Q. Wang, Y. Zhou, J. Wang, Q. Zhang, X. Zheng, F. Fan, Y. Luo, J. Jiang and H. L. Jiang, Dynamic structural twist in metal-organic frameworks enhances solar overall water splitting, Nat. Chem., 2024, 16, 1638–1646 CrossRef CAS PubMed.
- G. Yang, D. Wang, Y. Wang, W. Hu, S. Hu, J. Jiang, J. Huang and H. L. Jiang, Modulating the Primary and Secondary Coordination Spheres of Single Ni(II) Sites in Metal-Organic Frameworks for Boosting Photocatalysis, J. Am. Chem. Soc., 2024, 146, 10798–10805 CrossRef CAS PubMed.
- K. Sun, Y. Huang, Q. Wang, W. Zhao, X. Zheng, J. Jiang and H. L. Jiang, Manipulating the Spin State of Co Sites in Metal-Organic Frameworks for Boosting CO2 Photoreduction, J. Am. Chem. Soc., 2024, 146, 3241–3249 CrossRef CAS PubMed.
- H. Wang, X. Liu, W. Yang, G. Mao, Z. Meng, Z. Wu and H. L. Jiang, Surface-Clean Au25 Nanoclusters in Modulated Microenvironment Enabled by Metal-Organic Frameworks for Enhanced Catalysis, J. Am. Chem. Soc., 2022, 144, 22008–22017 CrossRef CAS PubMed.
- H. Wang, X. Zhang, W. Zhang, M. Zhou and H. L. Jiang, Heteroatom-Doped Ag25 Nanoclusters Encapsulated in Metal-Organic Frameworks for Photocatalytic Hydrogen Production, Angew. Chem., Int. Ed., 2024, 63, e202401443 CrossRef CAS PubMed.
- L. Wen, K. Sun, X. Liu, W. Yang, L. Li and H. L. Jiang, Electronic State and Microenvironment Modulation of Metal Nanoparticles Stabilized by MOFs for Boosting Electrocatalytic Nitrogen Reduction, Adv. Mater., 2023, 35, e2210669 CrossRef PubMed.
- S. Wang, Z. Ai, X. Niu, W. Yang, R. Kang, Z. Lin, A. Waseem, L. Jiao and H. L. Jiang, Linker Engineering of Sandwich-Structured Metal-Organic Framework Composites for Optimized Photocatalytic H2 Production, Adv. Mater., 2023, 35, e2302512 CrossRef PubMed.
- Z. Niu, W. D. C. Bhagya Gunatilleke, Q. Sun, P. C. Lan, J. Perman, J.-G. Ma, Y. Cheng, B. Aguila and S. Ma, Metal-Organic Framework Anchored with a Lewis Pair as a New Paradigm for Catalysis, Chem, 2018, 4, 2587–2599 CAS.
- Z. Niu, W. Zhang, P. C. Lan, B. Aguila and S. Ma, Promoting Frustrated Lewis Pairs for Heterogeneous Chemoselective Hydrogenation via the Tailored Pore Environment within Metal-Organic Frameworks, Angew. Chem., Int. Ed., 2019, 58, 7420–7424 CrossRef CAS PubMed.
- Z. M. Xu, Z. Hu, Y. Huang, S. J. Bao, Z. Niu, J. P. Lang, A. M. Al-Enizi, A. Nafady and S. Ma, Introducing Frustrated Lewis Pairs to Metal-Organic Framework for Selective Hydrogenation of N-Heterocycles, J. Am. Chem. Soc., 2023, 145, 14994–15000 CrossRef CAS PubMed.
- Y. Zhang, S. Chen, A. M. Al-Enizi, A. Nafady, Z. Tang and S. Ma, Chiral Frustrated Lewis Pair@Metal-Organic Framework as a New Platform for Heterogeneous Asymmetric Hydrogenation, Angew. Chem., Int. Ed., 2023, 62, e202213399 CrossRef CAS PubMed.
- Y. Zhang, Y. Jiang, A. Nafady, Z. Tang, A. M. Al-Enizi, K. Tan and S. Ma, Incorporation of Chiral Frustrated Lewis Pair into Metal-Organic Framework with Tailored Microenvironment for Heterogeneous Enantio- and Chemoselective Hydrogenation, ACS Cent. Sci., 2023, 9, 1692–1701 CrossRef CAS PubMed.
- Y. Zhang, J. Guo, P. VanNatta, Y. Jiang, J. Phipps, R. Roknuzzaman, H. Rabaa, K. Tan, T. AlShahrani and S. Ma, Metal-Free Heterogeneous Asymmetric Hydrogenation of Olefins Promoted by Chiral Frustrated Lewis Pair Framework, J. Am. Chem. Soc., 2024, 146, 979–987 CrossRef CAS PubMed.
- Q. Sun, Y. Tang, B. Aguila, S. Wang, F. S. Xiao, P. K. Thallapally, A. M. Al-Enizi, A. Nafady and S. Ma, Reaction Environment Modification in Covalent Organic Frameworks for Catalytic Performance Enhancement, Angew. Chem., Int. Ed., 2019, 58, 8670–8675 CrossRef CAS PubMed.
- Q. Sun, C. W. Fu, B. Aguila, J. Perman, S. Wang, H. Y. Huang, F. S. Xiao and S. Ma, Pore Environment Control and Enhanced Performance of Enzymes Infiltrated in Covalent Organic Frameworks, J. Am. Chem. Soc., 2018, 140, 984–992 CrossRef CAS PubMed.
- Q. Sun, B. Aguila, P. C. Lan and S. Ma, Tuning Pore Heterogeneity in Covalent Organic Frameworks for Enhanced Enzyme Accessibility and Resistance against Denaturants, Adv. Mater., 2019, 31, e1900008 CrossRef PubMed.
- I. Simonsson, P. Gardhagen, M. Andren, P. L. Tam and Z. Abbas, Experimental investigations into the irregular synthesis of iron(iii) terephthalate metal-organic frameworks MOF-235 and MIL-101, Dalton Trans., 2021, 50, 4976–4985 RSC.
- E. Haque, J. W. Jun and S. H. Jhung, Adsorptive removal of methyl orange and methylene blue from aqueous solution with a metal-organic framework material, iron terephthalate (MOF-235), J. Hazard. Mater., 2011, 185, 507–511 CrossRef CAS PubMed.
- S. C. Moore and M. L. Sarazen, Stability and kinetics of iron-terephthalate MOFs with diverse structures in aqueous pollutant degradation, AIChE J., 2023, 69, e18205 CrossRef CAS.
- Y. Peng, H. Huang, Y. Zhang, C. Kang, S. Chen, L. Song, D. Liu and C. Zhong, A versatile MOF-based trap for heavy metal ion capture and dispersion, Nat. Commun., 2018, 9, 187 CrossRef PubMed.
- H. Furukawa, F. Gandara, Y. B. Zhang, J. Jiang, W. L. Queen, M. R. Hudson and O. M. Yaghi, Water adsorption in porous metal-organic frameworks and related materials, J. Am. Chem. Soc., 2014, 136, 4369–4381 CrossRef CAS PubMed.
- J. Jiang, F. Gandara, Y. B. Zhang, K. Na, O. M. Yaghi and W. G. Klemperer, Superacidity in sulfated metal-organic framework-808, J. Am. Chem. Soc., 2014, 136, 12844–12847 CrossRef CAS PubMed.
- Q. Sun, B. Aguila, J. Perman, L. D. Earl, C. W. Abney, Y. Cheng, H. Wei, N. Nguyen, L. Wojtas and S. Ma, Postsynthetically Modified Covalent Organic Frameworks for Efficient and Effective Mercury Removal, J. Am. Chem. Soc., 2017, 139, 2786–2793 CrossRef CAS PubMed.
- Q. Sun, B. Aguila, J. A. Perman, T. Butts, F.-S. Xiao and S. Ma, Integrating Superwettability within Covalent Organic Frameworks for Functional Coating, Chem, 2018, 4, 1726–1739 CAS.
- M. Carboni, C. W. Abney, S. Liu and W. Lin, Highly porous and stable metal–organic frameworks for uranium extraction, Chem. Sci., 2013, 4, 2396–2402 RSC.
- C. H. Hendon, A. J. Rieth, M. D. Korzynski and M. Dinca, Grand Challenges and Future Opportunities for Metal-Organic Frameworks, ACS Cent. Sci., 2017, 3, 554–563 CrossRef CAS PubMed.
- H. Yang, M. Hao, Y. Xie, X. Liu, Y. Liu, Z. Chen, X. Wang, G. I. N. Waterhouse and S. Ma, Tuning Local Charge Distribution in Multicomponent Covalent Organic Frameworks for Dramatically Enhanced Photocatalytic Uranium Extraction, Angew. Chem., Int. Ed., 2023, 62, e202303129 CrossRef CAS PubMed.
- Y. Xie, Q. Rong, F. Mao, S. Wang, Y. Wu, X. Liu, M. Hao, Z. Chen, H. Yang, G. I. N. Waterhouse, S. Ma and X. Wang, Engineering the pore environment of antiparallel stacked covalent organic frameworks for capture of iodine pollutants, Nat. Commun., 2024, 15, 2671 CrossRef CAS PubMed.
- M. Hao, Z. Chen, H. Yang, G. I. N. Waterhouse, S. Ma and X. Wang, Pyridinium salt-based covalent organic framework with well-defined nanochannels for efficient and selective capture of aqueous 99TcO4−, Sci. Bull., 2022, 67, 924–932 CrossRef CAS PubMed.
- Q. Sun, L. Zhu, B. Aguila, P. K. Thallapally, C. Xu, J. Chen, S. Wang, D. Rogers and S. Ma, Optimizing radionuclide sequestration in anion nanotraps with record pertechnetate sorption, Nat. Commun., 2019, 10, 1646 CrossRef PubMed.
- Q. Sun, B. Aguila, L. D. Earl, C. W. Abney, L. Wojtas, P. K. Thallapally and S. Ma, Covalent Organic Frameworks as a Decorating Platform for Utilization and Affinity Enhancement of Chelating Sites for Radionuclide Sequestration, Adv. Mater., 2018, 30, e1705479 CrossRef PubMed.
- X. Yan, Y. Zhao, G. Cao, X. Li, C. Gao, L. Liu, S. Ahmed, F. Altaf, H. Tan, X. Ma, Z. Xie and H. Zhang, 2D Organic Materials: Status and Challenges, Adv. Sci., 2023, 10, e2203889 CrossRef PubMed.
- J. Guo and D. Jiang, Covalent Organic Frameworks for Heterogeneous Catalysis: Principle, Current Status, and Challenges, ACS Cent. Sci., 2020, 6, 869–879 CrossRef CAS PubMed.
- P. Kumar, B. Anand, Y. F. Tsang, K. H. Kim, S. Khullar and B. Wang, Regeneration, degradation, and toxicity effect of MOFs: Opportunities and challenges, Environ. Res., 2019, 176, 108488 CrossRef CAS PubMed.
- X. Kang, E. R. Stephens, B. M. Spector-Watts, Z. Li, Y. Liu, L. Liu and Y. Cui, Challenges and opportunities for chiral covalent organic frameworks, Chem. Sci., 2022, 13, 9811–9832 RSC.
- Q. Qian, P. A. Asinger, M. J. Lee, G. Han, K. Mizrahi Rodriguez, S. Lin, F. M. Benedetti, A. X. Wu, W. S. Chi and Z. P. Smith, MOF-Based Membranes for Gas Separations, Chem. Rev., 2020, 120, 8161–8266 CrossRef CAS PubMed.
- C. Zhang, B. H. Wu, M. Q. Ma, Z. Wang and Z. K. Xu, Ultrathin metal/covalent-organic framework membranes towards ultimate separation, Chem. Soc. Rev., 2019, 48, 3811–3841 RSC.
- L. Huang, J. Yang, Y. Zhao, H. Miyata, M. Han, Q. Shuai and Y. Yamauchi, Monolithic Covalent Organic Frameworks with Hierarchical Architecture: Attractive Platform for Contaminant Remediation, Chem. Mater., 2023, 35, 2661–2682 CrossRef CAS.
- P. Albacete, M. Asgari, Y. Yang, A. N. Al-Shanks and D. Fairen-Jimenez, Self-Shaping Monolithic Reticular Materials: Ingredients for Success, Adv. Funct. Mater., 2023, 34, 2305979 CrossRef.
- X. Qi, K. Liu and Z. Chang, Beyond powders: Monoliths on the basis of metal-organic frameworks (MOFs), Chem. Eng. J., 2022, 441, 135953 CrossRef CAS.
- J. Yu, C. Mu, B. Yan, X. Qin, C. Shen, H. Xue and H. Pang, Nanoparticle/MOF composites: preparations and applications, Mater. Horiz., 2017, 4, 557–569 RSC.
- M. Kalaj, K. C. Bentz, S. Ayala Jr, J. M. Palomba, K. S. Barcus, Y. Katayama and S. M. Cohen, MOF-Polymer Hybrid Materials: From Simple Composites to Tailored Architectures, Chem. Rev., 2020, 120, 8267–8302 CrossRef CAS PubMed.
- Y. Liu, W. Zhou, W. L. Teo, K. Wang, L. Zhang, Y. Zeng and Y. Zhao, Covalent-Organic-Framework-Based Composite Materials, Chem, 2020, 6, 3172–3202 CAS.
- B. Sun, Z. Sun, Y. Yang, X. L. Huang, S. C. Jun, C. Zhao, J. Xue, S. Liu, H. K. Liu and S. X. Dou, Covalent Organic Frameworks: Their Composites and Derivatives for Rechargeable Metal-Ion Batteries, ACS Nano, 2024, 18, 28–66 CrossRef CAS PubMed.
- K. Neikha and A. Puzari, Metal-Organic Frameworks through the Lens of Artificial Intelligence: A Comprehensive Review, Langmuir, 2024, 40, 21957–21975 CrossRef CAS PubMed.
- Z. Zheng, O. Zhang, H. L. Nguyen, N. Rampal, A. H. Alawadhi, Z. Rong, T. Head-Gordon, C. Borgs, J. T. Chayes and O. M. Yaghi, ChatGPT Research Group for Optimizing the Crystallinity of MOFs and COFs, ACS Cent. Sci., 2023, 9, 2161–2170 CrossRef CAS.
- Z. Zheng, O. Zhang, C. Borgs, J. T. Chayes and O. M. Yaghi, ChatGPT Chemistry Assistant for Text Mining and the Prediction of MOF Synthesis, J. Am. Chem. Soc., 2023, 145, 18048–18062 CrossRef CAS PubMed.
- Y. Kang, W. Lee, T. Bae, S. Han, H. Jang and J. Kim, Harnessing Large Language Models to Collect and Analyze Metal-Organic Framework Property Data Set, J. Am. Chem. Soc., 2025, 147, 3943–3958 CrossRef CAS PubMed.
- L. Gagliardi and O. M. Yaghi, Three Future Directions for Metal–Organic Frameworks, Chem. Mater., 2023, 35, 5711–5712 CrossRef CAS.
- Z. Zheng, N. Rampal, T. J. Inizan, C. Borgs, J. T. Chayes and O. M. Yaghi, Large language models for reticular chemistry, Nat. Rev. Mater., 2025, 10, 369–381 CrossRef.
- Z. Zheng, A. H. Alawadhi, S. Chheda, S. E. Neumann, N. Rampal, S. Liu, H. L. Nguyen, Y. H. Lin, Z. Rong, J. I. Siepmann, L. Gagliardi, A. Anandkumar, C. Borgs, J. T. Chayes and O. M. Yaghi, Shaping the Water-Harvesting Behavior of Metal-Organic Frameworks Aided by Fine-Tuned GPT Models, J. Am. Chem. Soc., 2023, 145, 28284–28295 CrossRef CAS PubMed.
- Q. Ouyang, Y. Rong, G. Xia, Q. Chen, Y. Ma and Z. Liu, Integrating Humidity-Resistant and Colorimetric COF-on-MOF Sensors with Artificial Intelligence Assisted Data Analysis for Visualization of Volatile Organic Compounds Sensing, Adv. Sci., 2025, 12, 2411621 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |