DOI:
10.1039/D5SC01616A
(Edge Article)
Chem. Sci., 2025,
16, 15066-15074
Photoactive monofunctional Pt(II)–cyanine complex for nucleus and mitochondria dual-targeted antitumor therapy†
Received
28th February 2025
, Accepted 17th June 2025
First published on 26th June 2025
Abstract
Monofunctional platinum (Pt) complexes, which have single reactive site for binding to DNA, exhibit distinct mechanisms of action compared to those of currently approved Pt-based chemotherapeutic drugs. Although monofunctional Pt complexes offer a promising strategy for overcoming chemotherapeutic resistance and reducing systemic toxicity, their antitumor effect is limited. Herein, we developed a photoactive monofunctional Pt(II)–cyanine dye complex (denoted as Pt–CDPEN) for cellular nucleus and mitochondria dual-targeted antitumor therapy. Due to the introduction of heavy atom Pt, Pt–CDPEN showed a 2.1-fold higher singlet oxygen quantum yield than that of cyanine dye. The half-maximal inhibitory concentration (IC50) of photoactivated Pt–CDPEN is over 16-fold lower than that of cisplatin. Pt–CDPEN exhibited the lysosomal escape property, enabling dual targeting of the cellular nucleus and mitochondria, thus helping to mitigate the chemotherapeutic resistance of Pt drugs. After systemic administration, PEGylated Pt–CDPEN (named as LET-9) exhibited high tumor accumulation, efficient antitumor therapy, and good biocompatibility. This Pt(II)–cyanine monofunctional complex provides a new platform for dual-targeted antitumor therapy, through simultaneously improving the efficacy and safety of Pt-based therapies.
Introduction
Platinum (Pt)-based chemotherapeutics, prominently represented by cisplatin (Cis-Pt), have been widely used clinically for over 50% of cancer patients. However, there are some potential drawbacks, such as systemic toxicity and drug resistance.1–6 Interestingly, monofunctional Pt complexes offer an intriguing alternative for Pt-based chemotherapeutics.7,8 Their unique single reactive site capable of binding to DNA can reduce toxicity by minimizing DNA damage to healthy cells. Moreover, it was demonstrated that tolyl terpyridine–Pt complex, a type of monofunctional Pt complex, effectively interacts with mitochondrial DNA (mtDNA) to form G-quadruplex structures, leading to mitochondrial dysfunction.9 Mitochondria serve as the energy-producing organelles in eukaryotic cells and play an essential role in tumor cell proliferation.10 Unlike nuclear DNA, mtDNA operates as an independent genetic system with a weaker repair mechanism.7,11 The different mechanism to that of traditional bifunctional Pt drugs promises a potential breakthrough in overcoming drug resistance.12,13 As we know, the non-specific mitochondrial dysfunction of Pt is associated with substantial toxic side effects,14–16 thus targeting mtDNA could be a potential therapeutic strategy for overcoming resistance to Pt-based chemotherapeutics.17,18 Monofunctional Pt complexes have the potential to overcome chemotherapeutic resistance and mitigate systemic toxicity. However, the antitumor effect of these monofunctional Pt complexes is limited due to their relatively lower potency compared to conventional Pt-based drugs.19 Therefore, there is an urgent need for the development of monofunctional Pt complexes with high therapeutic efficiency.
Photodynamic therapy (PDT) has emerged as a promising strategy due to its unique spatiotemporal selectivity, minimal invasiveness, and low systemic toxicity.20,21 The combination of PDT and monofunctional Pt chemotherapy offers a promising solution to address this issue,22–24 through the design of a hybrid system integrating a photosensitizer (PS) with a Pt complex unit.25–28 Guo et al. prepared a monofunctional Pt complex incorporating a BODIPY chromophore.13 The complex generated reactive oxygen species (ROS) to promote lysosomal escape. Apart from this benefit, introducing heavy atoms could promote intersystem crossing (ISC), thus increasing the singlet oxygen (1O2) quantum yield (QY) of PSs.29–33 For instance, introducing halogen,33–35 chalcogen,36–38 or other heavy atoms39,40 into PS molecules can effectively improve their QY of 1O2. Among various PSs, cyanine-based PSs, with good biocompatibility, strong absorption, and a large Stokes shift in the near-infrared (NIR) region, offer potential for both fluorescence (FL)/photoacoustic (PA) imaging and PDT of tumors.33,41 Indocyanine green (ICG) is currently the only approved NIR FL cyanine.42,43 However, ICG has a low QY of 1O2 (Φ = 0.077), which limits its PDT application.44,45
Considering the aforementioned information, a monofunctional Pt complex-modified cyanine dye is a good candidate, which has the following advantages: (i) the integration of Pt and cyanine could escalate the 1O2 QY of cyanine, (ii) the combination of chemotherapeutic drugs and cyanine can improve the therapeutic effect of monofunctional Pt complexes. Therefore, the development of monofunctional Pt(II)–cyanine complexes holds promise for achieving photodynamic-chemo synergistic therapy, thus simultaneously improving the efficiency of cyanine-based PDT and overcoming the chemotherapeutic resistance of Pt drugs. Here, we developed a monofunctional Pt(II) complex-modified cyanine dye (denoted as Pt–CDPEN) with enhanced ISC for FL/PA duplex imaging-guided nucleus/mitochondria dual-targeted antitumor therapy (Scheme 1). The versatile Pt–CDPEN serves as a FL/PA duplex imaging contrast agent, a PS and a monofunctional Pt-based chemotherapeutic agent. The introduction of Pt in Pt–CDPEN enhanced the ISC process, so the as-prepared Pt–CDPEN exhibited enhanced 1O2 generation compared to that of CDPEN. Such abundant 1O2 generation would induce the lysosomal escape of Pt–CDPEN, which effectively targets the cellular nucleus and mitochondria, thus causing abundant DNA damage for overcoming the chemotherapeutic resistance of Pt drugs and achieving self-augmented synergistic photodynamic-chemo therapy. The PEGylation of Pt–CDPEN (named LET-9) reduced the systemic toxicity of Pt drugs and enhanced tumor accumulation through systemic administration. This work provides a meaningful guide to address the issues of chemotherapeutic resistance and systemic toxicity associated with Pt-based drugs, suggesting future directions towards efficient synergistic antitumor therapy.
 |
| | Scheme 1 An illustration of photoactive lysosomal escape of Pt(II)–cyanine dye (Pt–CDPEN) with enhanced intersystem crossing (ISC) for cellular nucleus and mitochondria dual-targeted synergistic photodynamic-chemo antitumor therapy. | |
Results and discussion
Characterization of LET-9
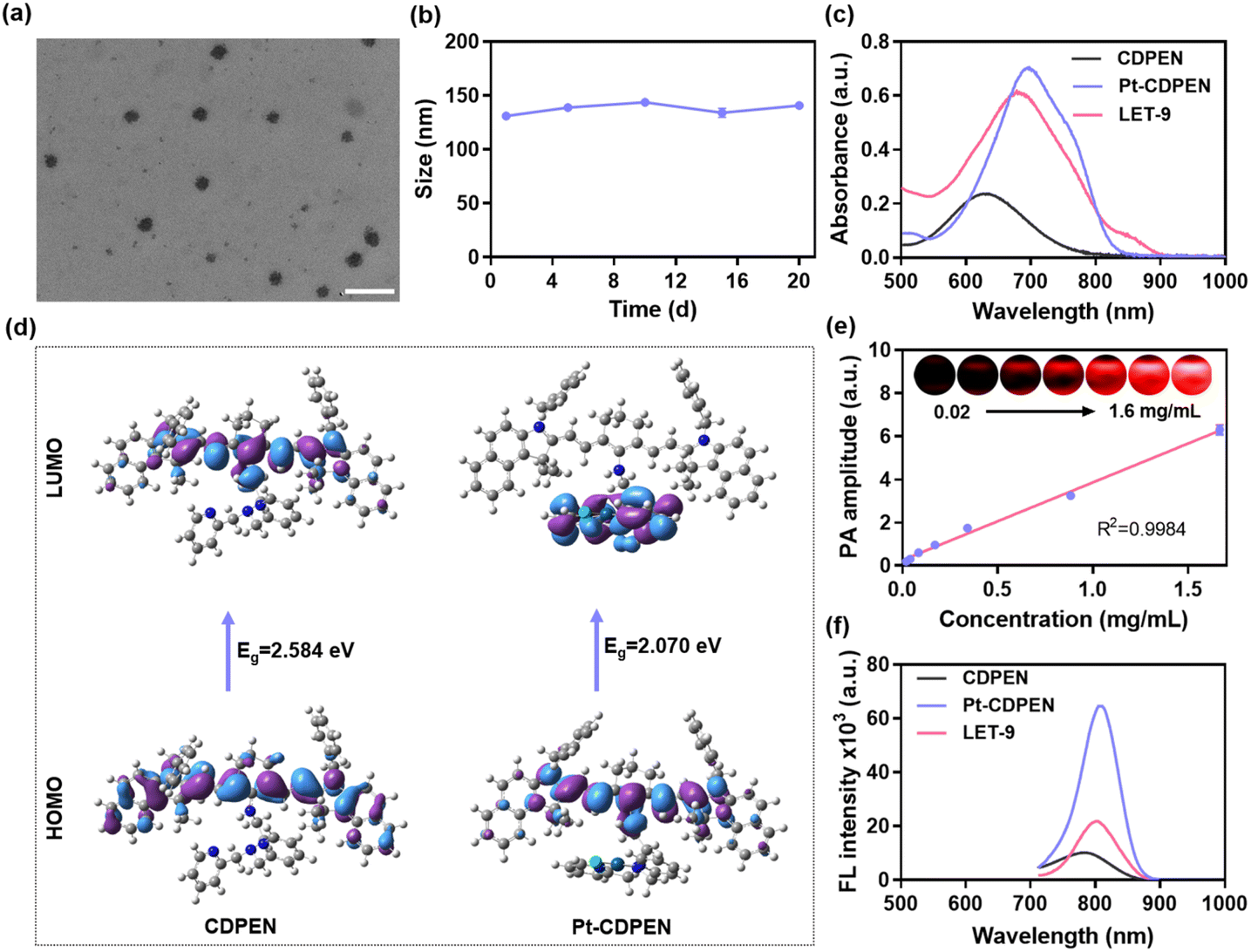
The monofunctional Pt–CDPEN complex was synthesized via one-step reaction of potassium tetrachloroplatinate and dipicolylethylenediamine-modified cyanine dye (CDPEN) (Fig. S1–S6†).46,47 Its chemical structure was well characterized by 1H NMR, 13C NMR and 195Pt NMR.48–50 To reduce the systematic toxicity as well as to improve the feasibility of Pt–CDPEN for biomedical applications, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine–N-[methoxy(polyethylene glycol)]-2000 (DSPE–PEG2000) was employed to encapsulate Pt–CDPEN through nanoprecipitation, thus obtaining PEGylated Pt–CDPEN (denoted as LET-9). To obtain a uniform size distribution, different ratios of Pt–CDPEN and DSPE–PEG2000 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, 1:4, 1:6, and 1:8) were investigated to prepare LET-9. Based on transmission electron microscopy (TEM) observation and dynamic light scattering (DLS) measurement, the LET-9 obtained from the ratio of 1:4 exhibited a well-defined spherical structure (Fig. 1a) with a hydrodynamic diameter of 131 ± 18 nm, polydispersity index (PDI) of 0.116 (Fig. S7†), and zeta potential of −24.46 mV (Fig. S8†) and was used for further studies. As shown in Fig. 1b, the hydrodynamic diameter of LET-9 exhibited negligible change after 20 days of storage, which indicated its good stability. After introducing the Pt atom, the absorption bands for both Pt–CDPEN and LET-9 showed a bathochromic shift from 630 to 700 nm, with significantly increased intensity (Fig. 1c). To better understand the physicochemical properties, density functional theory (DFT) calculations were conducted for CDPEN and Pt–CDPEN. The results showed that the energy gaps (Eg) between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of CDPEN narrowed from 2.584 to 2.070 eV after the introduction of Pt in CDPEN (Fig. 1d and S9†), which results in the redshift phenomenon of the absorption spectra.51–53 The HOMO distribution patterns of both CDPEN and Pt–CDPEN in the ground state show electron delocalization over the whole π-conjugated system, while the LUMO distribution pattern of Pt–CDPEN is located at the Pt moiety acceptor side. The electron density concentrated on the donating triphenylamine moiety of Pt–CDPEN showed more asymmetry, which could cause the increased HOMO and the narrowed Eg.51,52 The PA intensity at 710 nm (PA710) of LET-9 showed a good linear relationship with its concentration in the range of 0.02–1.6 mg mL−1 (Fig. 1e). The FL emission band of Pt–CDPEN exhibited a bathochromic shift from 785 to 805 nm, with significantly increased intensity (Fig. 1f), which may be attributed to the altered intramolecular charge transfer (ICT) effect.41,54 However, due to the aggregation-induced quenching effect, the FL intensity of LET-9 was partly quenched compared with that of free Pt–CDPEN.
:2, 1:4, 1:6, and 1:8) were investigated to prepare LET-9. Based on transmission electron microscopy (TEM) observation and dynamic light scattering (DLS) measurement, the LET-9 obtained from the ratio of 1:4 exhibited a well-defined spherical structure (Fig. 1a) with a hydrodynamic diameter of 131 ± 18 nm, polydispersity index (PDI) of 0.116 (Fig. S7†), and zeta potential of −24.46 mV (Fig. S8†) and was used for further studies. As shown in Fig. 1b, the hydrodynamic diameter of LET-9 exhibited negligible change after 20 days of storage, which indicated its good stability. After introducing the Pt atom, the absorption bands for both Pt–CDPEN and LET-9 showed a bathochromic shift from 630 to 700 nm, with significantly increased intensity (Fig. 1c). To better understand the physicochemical properties, density functional theory (DFT) calculations were conducted for CDPEN and Pt–CDPEN. The results showed that the energy gaps (Eg) between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of CDPEN narrowed from 2.584 to 2.070 eV after the introduction of Pt in CDPEN (Fig. 1d and S9†), which results in the redshift phenomenon of the absorption spectra.51–53 The HOMO distribution patterns of both CDPEN and Pt–CDPEN in the ground state show electron delocalization over the whole π-conjugated system, while the LUMO distribution pattern of Pt–CDPEN is located at the Pt moiety acceptor side. The electron density concentrated on the donating triphenylamine moiety of Pt–CDPEN showed more asymmetry, which could cause the increased HOMO and the narrowed Eg.51,52 The PA intensity at 710 nm (PA710) of LET-9 showed a good linear relationship with its concentration in the range of 0.02–1.6 mg mL−1 (Fig. 1e). The FL emission band of Pt–CDPEN exhibited a bathochromic shift from 785 to 805 nm, with significantly increased intensity (Fig. 1f), which may be attributed to the altered intramolecular charge transfer (ICT) effect.41,54 However, due to the aggregation-induced quenching effect, the FL intensity of LET-9 was partly quenched compared with that of free Pt–CDPEN.
 |
| | Fig. 1 Physicochemical properties of LET-9. (a) TEM image of LET-9. Scale bar: 200 nm. (b) Size change of LET-9 during 20 days of storage. (c) UV-vis-NIR spectra for CDPEN, Pt–CDPEN, and LET-9. (d) The HOMO and LUMO distribution patterns for CDPEN and Pt–CDPEN calculated using DFT (Gaussian 16/6-31G for C, H, N, and O; B3LYP/LanL2DZ for Pt). (e) The PA710 intensity and PA images (inset) of LET-9 solutions as a function of concentration (0.02–1.6 mg mL−1). (f) FL emission spectra of CDPEN, Pt–CDPEN, and LET-9. | |
1O2 QY of LET-9
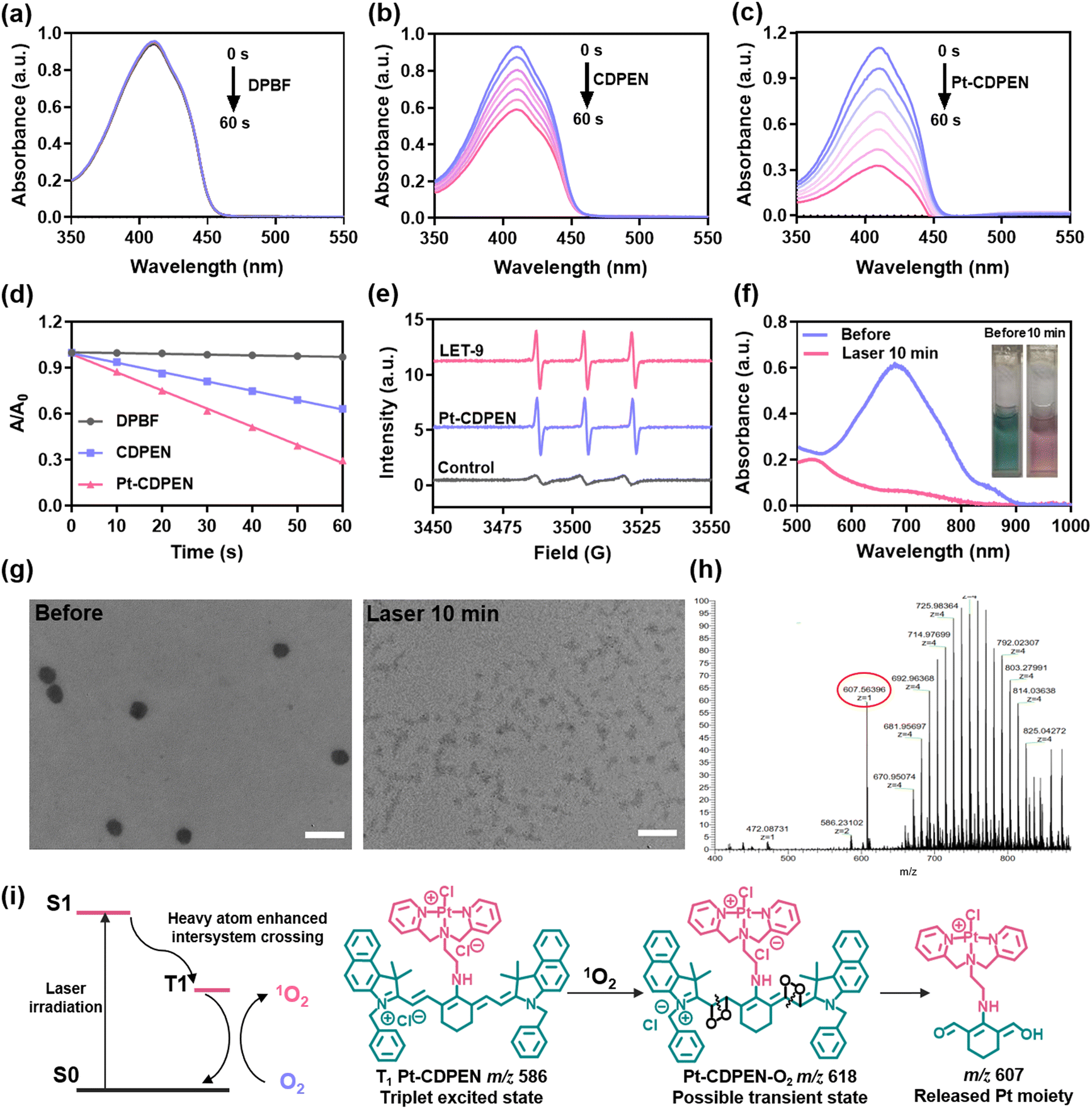
The heavy atom introduced into the dye could enhance the 1O2 QY.34 Therefore, the 1O2 generation can be evaluated using the 1,3-diphenylisobenzofuran (DPBF) degradation reaction.53 As shown in Fig. 2a–c, the absorption intensity at 415 nm of DPBF treated with Pt–CDPEN was significantly decreased compared with that of DPBF treated with CDPEN under laser irradiation. The 1O2 QY of Pt–CDPEN was determined to be 0.21 (Fig. 2d),33,34 which was 2.1-fold higher than that of CDPEN. Moreover, electron spin resonance (ESR) spectra exhibited a strong 1O2 signal of with triplet peaks (1:1:1) using 2,2,6,6- tetramethylpiperidine (TEMP) as a radical trapping agent in the presence of either Pt–CDPEN or LET-9 (Fig. 2e). It was obvious that the absorption intensity of LET-9 was decreased after 10 min of laser irradiation, and the color of the LET-9 solution changed from green to pink (Fig. 2f), indicating the severe photobleaching of cyanine.55 TEM images of LET-9 showed that the nanostructures degraded into tiny dots after 10 min of laser irradiation (Fig. 2g). Upon laser irradiation, LET-9 was destroyed due to the C–C cleavage of the polyene chain of the cyanine structure. The laser-triggered degradation of LET-9 was proved by high-resolution mass spectroscopy (HRMS). A new peak at m/z 607 was observed after irradiation (Fig. 2h) compared with that without laser irradiation (Fig. S10†), corresponding to the fragment of the cyanine moiety including the dipicolylethylenediamine part. Hence, we propose a mechanism for the laser-triggered release of the Pt moiety from LET-9 (Fig. 2i). Pt–CDPEN was excited from the ground state to the lowest excited singlet state (S1) and then moved to the lowest excited triplet state (T1) by the strong ISC process, followed by energy transfer to the surrounding O2 to produce 1O2. The active 1O2 cleaves the polyene chain of Pt–CDPEN via the dioxetane intermediate to generate carbonyl products.55 Due to the destruction of the molecular skeleton, the strong π–π interactions of LET-9 were eliminated, resulting in the dissociation of LET-9 and the decomposition of Pt–CDPEN. More interestingly, an abundance of Pt fragments was observed, indicating that the Pt drug moiety was not affected even after laser irradiation. These results suggest that LET-9 could be used for photoactivated chemotherapy.
 |
| | Fig. 2 Laser-triggered degradation of LET-9. The UV spectra of (a) DPBF, (b) DPBF + CDPEN and (c) DPBF + Pt–CDPEN upon laser irradiation (λex = 635 nm, 0.2 W cm−2). (d) The corresponding absorption decay rates. (e) ESR spectra of TEMP solution in the presence of Pt–CDPEN (1 μM) or LET-9 (1 μM) irradiated with 635 nm laser for 1 min. (f) The UV-vis-NIR spectra and color (inset) change of LET-9 before and after laser irradiation. (g) TEM images of LET-9 before and after irradiation. Scale bar: 200 nm. (h)The HRMS of LET-9 after irradiation. (i) Proposed photodegradation mechanism of Pt–CDPEN. | |
Cytotoxicity of Pt–CDPEN and LET-9 against A549DDP
The cytotoxicity of Pt–CDPEN and LET-9 was assessed on different cell lines, using Cis-Pt as a positive control. Compared with Cis-Pt-treated cells, cells treated with Pt–CDPEN exhibited lower cell viability, suggesting that Pt–CDPEN is more cytotoxic (Fig. S11a†). Compared with Pt–CDPEN-treated cells, cells treated with LET-9 exhibited higher cell viability, suggesting that PEGylation could reduce the cytotoxicity of Pt–CDPEN. Furthermore, LET-9 exhibited similar cell viability toward normal cells (Fig. S12†). For cells treated with Pt–CDPEN or LET-9 plus laser irradiation (Fig. S11b†), the cell viability was markedly decreased compared with the dark control. The half-maximal inhibitory concentration (IC50) values of Pt–CDPEN were 1.130, 1.380, and 1.144 μM against 4T1, A549, and Cis-Pt-resistant A549DDP cells in the dark, respectively (Table 1), which were 1-, 1.7-, and 17.5-fold lower than those of Cis-Pt (Fig. S13†), indicating that Pt–CDPEN has a better chemotherapeutic effect, even for drug-resistant cells. Notably, the A549 cells treated with Pt–CDPEN plus laser irradiation showed a 1.7-fold lower IC50 value compared with non-laser irradiation, indicating the synergistic effect of photodynamic-chemo therapies. The IC50 values of LET-9 upon laser irradiation were determined to be 0.361, 1.109, and 1.241 against 4T1, A549, and A549DDP cell lines, respectively, which are 3.6-, 2.1-, and 16-fold lower than those of Cis-Pt. The cell viability of LET-9 also showed laser-irradiation-time-dependence (Fig. S14†). All these results indicate that LET-9 has higher cytotoxicity under laser irradiation and lower cytotoxicity in the dark compared with Cis-Pt.
Table 1 The IC50 values of compounds incubated with 4T1, A549 and A549DDP cell lines
| Cell lines |
IC50 in dark (μM) |
IC50 + laser (μM) |
| Cis-Pt |
Pt–CDPEN |
LET-9 |
Cis-Pt |
Pt–CDPEN |
LET-9 |
| 4T1 |
1.298 |
1.130 |
>20 |
1.233 |
0.352 |
0.361 |
| A549 |
2.293 |
1.380 |
4.251 |
>20 |
0.799 |
1.109 |
| A549DDP |
>20 |
1.144 |
2.107 |
>20 |
0.998 |
1.241 |
ROS promoted Pt lysosome escape for dual nucleus and mitochondria targeting
The action mechanism of monofunctional Pt(II) is distinctly different from that of traditional Cis-Pt. Monofunctional Pt(II) adducts on DNA cannot distort the duplex like Cis-Pt but demonstrated faster binding kinetics than that of Cis-Pt,56,57 which inhibited transcription and prevented the DNA repair.7,58 Some cationic Pt(II) complexes show the ability to impair the structure and function of mitochondria.8,12,59 Thus, a series of cell studies were conducted to investigate the mechanism of action for LET-9. First, the cellular uptake of LET-9 was tested by flow cytometry. An increase in the FL intensity was observed over time, which indicated that LET-9 can be rapidly taken up by cells (Fig. 3a and S15†). Afterwards, the laser-induced ROS generation was evaluated by using 2′,7′-dichlorofluorescein diacetate (DCFH-DA) as an indicator. As shown in Fig. 3b, the 4T1 cells incubated with LET-9 plus laser irradiation showed a stronger green FL signal for DCFH-DA than other groups. These results proved that LET-9 can generate ROS upon laser irradiation. The generated ROS could help Pt drugs escape from lysosomes, which is the main trap for most nanomedicines. The lysosomal colocalization results showed that a red FL signal for LET-9 appeared in the nucleus after laser irradiation, whereas it was trapped in the periphery of the nucleus in the dark group (Fig. 3c). The colocalization curves for the green/red FL signals were well-matched even after 24 h incubation in the dark groups, but were obviously separated in the laser irradiation groups. Furthermore, compared to that in the 5 min laser-treated group, the better colocalization of the red/blue FL signals in the 10 min laser-treated group indicated the higher ROS generation, which promoted the escape of more Pt moieties from the lysosomes to the nucleus and mitochondria. The positive charge of the monofunctional Pt complex means that it preferentially accumulates in the mitochondria. The intracellular distribution of LET-9 was investigated using MitoTracker. The colocalization results showed that the Pearson's correlation coefficient between the green FL signal of LET-9 and the red FL signal of MitoTracker was about 0.86 (Fig. 3d), suggesting the mitochondrial targeting capability of LET-9.
 |
| | Fig. 3 The cytotoxicity mechanism of LET-9. (a) The cell uptake of LET-9 detected by flow cytometry. (b) ROS generation FL images of 4T1 cells incubated with LET-9 (20 μM) in the dark or under laser irradiation. (c) Confocal FL images of 4T1 cells incubated with LET-9 (20 μM) for 1 or 24 h, then irradiated for 5 or 10 min, and their corresponding lysosomal colocalization curves. (d) The mitochondrial localization images of LET-9-treated 4T1 cells with MitoTracker (red) and the corresponding scatter plot. (e) DNA damage marker immunofluorescence images of 4T1 and A549DDP cells with different treatments. The corresponding FL intensity analysis of (f) 4T1 and (g) A549DDP cells. (h) The DNA platination in A549 cells after 6 h treatment with PBS, Cis-Pt, and LET-9, with or without laser irradiation. Scale bar: 20 μm. **p < 0.01, ****p < 0.0001. | |
Furthermore, DNA damage assessment was also carried out to evaluate the nucleus targeting. The phosphorylated histone protein, γ-histone 2AX (γ-H2AX), an indicator of DNA double-strand breaks, was used as the marker of DNA damage in different treatments.60–62 Upon laser irradiation, the green FL signals of γ-H2AX appeared in the LET-9-treated group (Fig. 3e). Notably, the signal was observed across the whole nucleus compared with the Cis-Pt- or LET-9-treated groups, which only lit the periphery of the nucleus in 4T1 cells. Quantitative analysis showed that the green FL intensity was significantly higher than that of other groups in both 4T1 (Fig. 3f) and A549DDP (Fig. 3g) cells. This phenomenon further confirms that LET-9 can escape from the lysosomes to the nucleus and mitochondria with the help of laser irradiation. Moreover, the Pt-species-induced DNA platination was evaluated by quantitative analysis of the Pt content in DNA (Fig. 3h). The LET-9 + laser-treated groups contained much higher concentrations of Pt than the Cis-Pt- and LET-9-treated groups, reaching 137.67 ng mg−1 of DNA in the 10 min laser irradiation group. The longer the laser irradiation, the higher the Pt content in the nucleus, which strongly supports the proposed mechanism of photoactivated lysosomal escape of Pt species.
In vivo PA and FL duplex imaging-guided therapy
In vivo PA and FL duplex imaging were conducted on 4T1 tumor-bearing mice. As shown in Fig. 4a, the PA intensities of the tumors gradually increased after the intravenous (i.v.) injection of LET-9 and plateaued within 4–8 h (Fig. 4b). Based on PA imaging, 4–8 h p.i. is the optimal time period for conducting PDT. In vivo FL images showed the maximum FL intensity of LET-9 at 24 h p.i. (Fig. 4c and d), which was attributed to the recovery of the FL signal during nanostructure disassembly. Conversely, no FL signal was observed for the tumor tissues in free Pt–CDPEN-treated mice, indicating low tumor accumulation of Pt–CDPEN only. This may be mainly due to the rapid elimination of small molecules from the body.63 The in vivo tumor inhibition effect of LET-9 was further evaluated on 4T1 tumor-bearing mice. Guided by PA imaging, laser irradiation (635 nm, 0.2 W cm−2, two 10 min sessions) was initiated at 4–8 h p.i. of LET-9. The laser irradiation treatment was repeated for the (LET-9 + L) × 2 group at day 7. As shown in Fig. 5a and b, the (LET-9 + L) × 2 group achieved the best tumor suppression, while the others showed limited tumor inhibition effects. All mice treated with LET-9 + L survived for the full 30 days of the observation period, whereas the other treated groups exhibited greater mortality (Fig. 5c). The Cis-Pt-treated group showed a certain degree of therapeutic efficacy, but the mice underwent severe weight loss during the treatment (Fig. 5d), whereas no obvious weight loss was observed in the other groups. Hematoxylin and eosin (H&E) staining images of the tumor sections in the LET-9 + L group clearly showed apoptosis and damage compared with other treatments (Fig. 5e). More importantly, the blood biochemical indicators (Fig. 5f), hemolysis assay (Fig. S16†) and H&E staining images of the major organs (Fig. S17†) showed no obvious changes. In short, all these results demonstrate that LET-9 has good biocompatibility and overcomes the toxicity limitations of other Pt drugs, exhibiting better therapeutic effects in vivo.
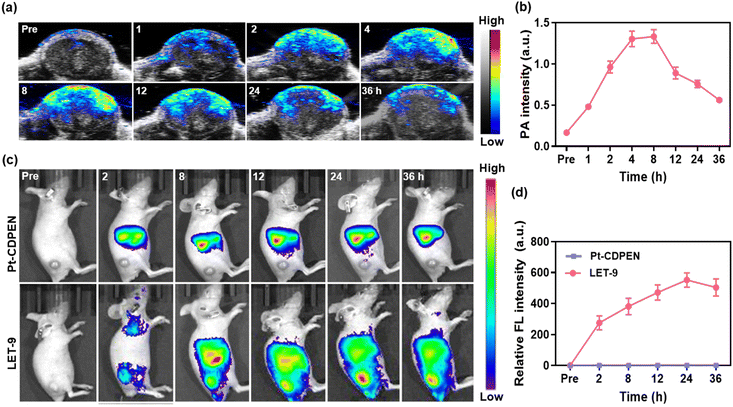 |
| | Fig. 4
In vivo imaging of LET-9. (a) In vivo PA images of 4T1 tumor-bearing mice at 0, 1, 2, 4, 8, 12, 24, and 36 h post-injection with LET-9, and (b) corresponding quantitative analysis of (a). (c) In vivo FL images of 4T1 tumor-bearing mice at 0, 2, 8, 12, 24, and 36 h post-injection with LET-9 or Pt–CDPEN, and (d) corresponding relative FL intensity quantitative analysis of (c). | |
 |
| | Fig. 5 The in vivo treatment evaluation of LET-9 on 4T1-tumor-bearing mice. (a) Tumor growth curves for all groups (PBS, Cis-Pt, Pt–CDPEN, LET-9, LET-9 + L, and (LET-9 + L) × 2) after i.v. injection with a dose of 10 mg kg−1. L: 635 nm laser. (b) Statistical results for tumor volume. *p < 0.05, ***p < 0.001. (c) Percent survival after different treatments. (d) Body weight changes of 4T1 tumor-bearing mice after different treatments. (e) H&E staining images for all groups after treatment. Scale bar: 100 μm. (f) The blood biochemical analysis of LET-9. | |
Conclusions
We developed a photoactivated lysosomal escape monofunctional modified Pt(II)–cyanine dye (Pt–CDPEN) for synergistic cellular nucleus and mitochondria dual-targeted photodynamic-chemo antitumor therapy. The as-prepared Pt–CDPEN exhibited enhanced 1O2 generation due to the heavy atom effect, which induced the lysosomal escape of Pt–CDPEN under laser irradiation, thus accessing the cellular nucleus and mitochondria effectively and causing DNA damage, overcoming the chemotherapeutic resistance of traditional Pt drugs. Both in vitro and in vivo experimental results suggested that the PEGylated Pt–CDPEN further reduced the systemic toxicity of Pt drugs and enhanced tumor accumulation after systemic administration, thereby giving a good tumor suppression effect. Our findings provide a new strategy to simultaneously improve the therapeutic effect of monofunctional Pt complex-based chemotherapy and cyanine-based PDT, showing great potential to alleviate the side effects of chemotherapy.
Ethical statement
All animal experiments were approved by the Animal Ethical and Welfare Committee of Shenzhen University (AEWC-SZU) and complied with all relevant policies and regulations.
Data availability
The data supporting this article have been included in the ESI.†
Author contributions
Ting He, Qiaoju Ren and Yu Zhang: investigation, data curation and writing original draft. Qinan Tang, Ziguang Wang and Chao Jiang: investigation and data curation. Yurong Liu, Shan Lei, Yifan Zhang and Peng Huang: review and editing, Jing Lin: conceptualization, supervision, funding acquisition, and review and editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work is financially supported by the Shenzhen Medical Research Fund (B2302047), the Shenzhen Science and Technology Program (JCYJ20200109105620482, KQTD20190929172538530), the National Natural Science Foundation of China (82372116, 22107073) and the Research Team Cultivation Program of Shenzhen University (2023QNT019). The authors thank the Instrumental Analysis Center of Shenzhen University.
References
- S. Zhang, X. Zhong, H. Yuan, Y. Guo, D. Song, F. Qi, Z. Zhu, X. Wang and Z. Guo, Chem. Sci., 2020, 11, 3829–3835 RSC.
- Y. Wang, Y. Jiang, D. Wei, P. Singh, Y. Yu, T. Lee, L. Zhang, H. K. Mandl, A. S. Piotrowski-Daspit, X. Chen, F. Li, X. Li, Y. Cheng, A. Josowitz, F. Yang, Y. Zhao, F. Wang, Z. Zhao, A. Huttner, R. S. Bindra, H. Xiao and W. Mark Saltzman, Nat. Biomed. Eng., 2021, 5, 1048–1058 CrossRef CAS PubMed.
- X. Zeng, Y. Wang, J. Han, W. Sun, H. J. Butt, X. J. Liang and S. Wu, Adv. Mater., 2020, 32, e2004766 CrossRef PubMed.
- J. Chen, X. Wang, Y. Yuan, H. Chen, L. Zhang, H. Xiao, J. Chen, Y. Zhao, J. Chang, W. Guo and X. J. Liang, Sci. Adv., 2021, 7, eabc5269 Search PubMed.
- Y. Li, Y. Deng, X. Tian, H. Ke, M. Guo, A. Zhu, T. Yang, Z. Guo, Z. Ge, X. Yang and H. Chen, ACS Nano, 2015, 9, 9626–9637 CrossRef CAS PubMed.
- H. Xia, F. Li, X. Hu, W. Park, S. Wang, Y. Jang, Y. Du, S. Baik, S. Cho, T. Kang, D. H. Kim, D. Ling, K. M. Hui and T. Hyeon, ACS Cent. Sci., 2016, 2, 802–811 CrossRef CAS PubMed.
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, Chem. Rev., 2016, 116, 3436–3486 CrossRef CAS PubMed.
- Y. Guo, S. Jin, H. Yuan, T. Yang, K. Wang, Z. Guo and X. Wang, J. Med. Chem., 2021, 65, 520–530 CrossRef PubMed.
- K. Kuang, C. Li, F. Maksut, D. Ghosh, R. Vinck, M. Wang, J. Poupon, R. Xiang, W. Li, F. Li, Z. Wang, J. Du, M. P. Teulade-Fichou, G. Gasser, S. Bombard and T. Jia, J. Biomed. Sci., 2024, 31, 50–71 CrossRef CAS PubMed.
- S. E. Weinberg and N. S. Chandel, Nat. Chem. Biol., 2015, 11, 9–15 CrossRef CAS PubMed.
- S. Jin, Y. Guo, Z. Guo and X. Wang, Pharmaceuticals, 2021, 14, 133–156 CrossRef CAS.
- Z. Zhu, Z. Wang, C. Zhang, Y. Wang, H. Zhang, Z. Gan, Z. Guo and X. Wang, Chem. Sci., 2019, 10, 3089–3095 RSC.
- X. Xue, C. Qian, H. Fang, H. K. Liu, H. Yuan, Z. Guo, Y. Bai and W. He, Angew. Chem., Int. Ed., 2019, 131, 12791–12796 CrossRef.
- Y. Zong, H. Li, P. Liao, L. Chen, Y. Pan, Y. Zheng, C. Zhang, D. Liu, M. Zheng and J. Gao, Signal Transduct. Targeted Ther., 2024, 9, 124 CrossRef PubMed.
- C. Zhang, C. Xu, X. Gao and Q. Yao, Theranostics, 2022, 12, 2115–2132 CrossRef CAS PubMed.
- M. Leo, L. I. Schmitt, P. Kusterarent, A. Kutritz, T. Rassaf, C. Kleinschnitz, U. B. Hendgen-Cotta and T. Hagenacker, Int. J. Mol. Sci., 2020, 21, 8636 CrossRef CAS.
- Y. Yang, Z. Mai, Y. Zhang, Z. Yu, W. Li, Y. Zhang, F. Li, P. Timashev, P. Luan, D. Luo, X. J. Liang and Z. Yu, ACS Nano, 2023, 17, 1275–1286 CrossRef CAS PubMed.
- J. Yang, Z. Yang, H. Wang, Y. Chang, J. F. Xu and X. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202402291 CrossRef CAS PubMed.
- S. Rottenberg, C. Disler and P. Perego, Nat. Rev. Cancer, 2021, 21, 37–50 CrossRef CAS PubMed.
- X. Zhao, J. Liu, J. Fan, H. Chao and X. Peng, Chem. Soc. Rev., 2021, 50, 4185–4219 RSC.
- J. Xie, Y. Wang, W. Choi, P. Jangili, Y. Ge, Y. Xu, J. Kang, L. Liu, B. Zhang, Z. Xie, J. He, N. Xie, G. Nie, H. Zhang and J. S. Kim, Chem. Soc. Rev., 2021, 50, 9152–9201 RSC.
- P. Agostinis, K. Berg, K. A. Cengel, T. H. Foster, A. W. Girotti, S. O. Gollnick, S. M. Hahn, M. R. Hamblin, A. Juzeniene, D. Kessel, M. Korbelik, J. Moan, P. Mroz, D. Nowis, J. Piette, B. C. Wilson and J. Golab, Ca-Cancer J. Clin., 2011, 61, 250–281 CrossRef PubMed.
- Y. Zhang, Y. Liao, Q. Tang, J. Lin and P. Huang, Angew. Chem., Int. Ed., 2021, 60, 10647–10653 CrossRef CAS PubMed.
- J. F. Lovell, T. W. Liu, J. Chen and G. Zheng, Chem. Rev., 2010, 110, 2839–2857 CrossRef CAS PubMed.
- W. Fan, P. Huang and X. Chen, Chem. Soc. Rev., 2016, 45, 6488–6519 RSC.
- B. M. Luby, C. D. Walsh and G. Zheng, Angew. Chem., Int. Ed., 2019, 58, 2558–2569 CrossRef CAS PubMed.
- W. Zhang, X. F. Du, B. Liu, C. Li, J. Long, M. X. Zhao, Z. Yao, X. J. Liang and Y. Lai, ACS Nano, 2021, 16, 1421–1435 CrossRef PubMed.
- Y. Zhang, Y. Wan, Y. Chen, N. T. Blum, J. Lin and P. Huang, ACS Nano, 2020, 14, 5560–5569 CrossRef CAS PubMed.
- G. C. Hoover and D. S. Seferos, Chem. Sci., 2019, 10, 9182–9188 RSC.
- A. Gorman, J. Killoran, C. O'Shea, T. Kenna, W. M. Gallagher and D. F. O'Shea, J. Am. Chem. Soc., 2004, 126, 10619–10631 CrossRef CAS PubMed.
- J. Cao, J. Chi, J. Xia, Y. Zhang, S. Han and Y. Sun, ACS Appl. Mater. Interfaces, 2019, 11, 25720–25729 CrossRef CAS PubMed.
- Z. Guo, H. He, Y. Zhang, J. Rao, T. Yang, T. Li, L. Wang, M. Shi, M. Wang, S. Qiu, X. Song, H. Ke and H. Chen, Adv. Mater., 2021, 33, e2004225 CrossRef PubMed.
- X. Zhao, Q. Yao, S. Long, W. Chi, Y. Yang, D. Tan, X. Liu, H. Huang, W. Sun, J. Du, J. Fan and X. Peng, J. Am. Chem. Soc., 2021, 143, 12345–12354 CrossRef CAS PubMed.
- J. Atchison, S. Kamila, H. Nesbitt, K. A. Logan, D. M. Nicholas, C. Fowley, J. Davis, B. Callan, A. P. McHale and J. F. Callan, Chem. Commun., 2017, 53, 2009–2012 RSC.
- Y. Liu, J. Zhang, X. Zhou, Y. Wang, S. Lei, G. Feng, D. Wang, P. Huang and J. Lin, Angew. Chem., Int. Ed., 2024, 136, e202408064 CrossRef.
- K. Wen, H. Tan, Q. Peng, H. Chen, H. Ma, L. Wang, A. Peng, Q. Shi, X. Cai and H. Huang, Adv. Mater., 2021, 33, 2108146 Search PubMed.
- H. Ma, Y. Lu, Z. Huang, S. Long, J. Cao, Z. Zhang, X. Zhou, C. Shi, W. Sun, J. Du, J. Fan and X. Peng, J. Am. Chem. Soc., 2022, 144, 3477–3486 CrossRef CAS PubMed.
- Q. Yao, J. Fan, S. Long, X. Zhao, H. Li, J. Du, K. Shao and X. Peng, Chem, 2022, 8, 197–209 CAS.
- Z. Guo, Y. Zou, H. He, J. Rao, S. Ji, X. Cui, H. Ke, Y. Deng, H. Yang, C. Chen, Y. Zhao and H. Chen, Adv. Mater., 2016, 28, 10155–10164 CrossRef CAS PubMed.
- P. Zhang, J. Ke, D. Tu, J. Li, Y. Pei, L. Wang, X. Shang, T. Guan, S. Lu, Z. Chen and X. Chen, Angew. Chem., Int. Ed., 2022, 61, e202112125 CrossRef CAS PubMed.
- X. Peng, F. Song, E. Lu, Y. Wang, W. Zhou, J. Fan and Y. Gao, J. Am. Chem. Soc., 2005, 127, 4170–4171 CrossRef CAS PubMed.
- P. Huang, Y. Gao, J. Lin, H. Hu, H. S. Liao, X. Yan, Y. Tang, A. Jin, J. Song, G. Niu, G. Zhang, F. Horkay and X. Chen, ACS Nano, 2015, 9, 9517–9527 CrossRef CAS PubMed.
- Y. Hu, C. Chi, S. Wang, L. Wang, P. Liang, F. Liu, W. Shang, W. Wang, F. Zhang, S. Li, H. Shen, X. Yu, H. Liu and J. Tian, Adv. Mater., 2017, 29, 1700448 CrossRef.
- L. Jiang, S. Zhou, X. Zhang, C. Li, S. Ji, H. Mao and X. Jiang, Nat. Commun., 2021, 12, 2390 CrossRef CAS PubMed.
- H. Bian, D. Ma, X. Zhang, K. Xin, Y. Yang, X. Peng and Y. Xiao, Small, 2021, 17, e2100398 CrossRef PubMed.
- L. Zeng, G. Ma, H. Xu, J. Mu, F. Li, X. Gao, Z. Deng, J. Qu, P. Huang and J. Lin, Small, 2019, 15, e1803866 CrossRef PubMed.
- T. He, Q. Tang, Q. Ren, Y. Liu, G. He, Y. Pan, Z. Wang, P. Huang and J. Lin, ACS Nano, 2024, 18, 5434–5445 CAS.
- L. He, W. Lin, Q. Xu, M. Ren, H. Wei and J. Y. Wang, Chem. Sci., 2015, 6, 4530–4536 RSC.
- L. Stackova, P. Stacko and P. Klan, J. Am. Chem. Soc., 2019, 141, 7155–7162 CrossRef CAS PubMed.
- J. R. L. Priqueler, I. S. Butler and F. D. Rochon, Appl. Spectrosc. Rev., 2006, 41, 185–226 CrossRef CAS.
- S. Zhu, Z. Hu, R. Tian, B. C. Yung, Q. Yang, S. Zhao, D. O. Kiesewetter, G. Niu, H. Sun, A. L. Antaris and X. Chen, Adv. Mater., 2018, 30, e1802546 CrossRef PubMed.
- A. K. Narsaria, J. Poater, C. F. Guerra, A. W. Ehlers, T. A. Hamlin, K. Lammertsma and F. M. Bickelhaupt, Chem. Eur. J., 2020, 26, 2080–2093 CrossRef CAS PubMed.
- X. Zhao, K. C. Zhao, L. J. Chen, Y. S. Liu, J. L. Liu and X. P. Yan, Chem. Sci., 2020, 12, 442–452 RSC.
- R. M. Exner, F. Cortezon-Tamarit and S. I. Pascu, Angew. Chem., Int. Ed., 2021, 60, 6230–6241 CrossRef CAS PubMed.
- A. P. Gorka, R. R. Nani, J. Zhu, S. Mackem and M. J. Schnermann, J. Am. Chem. Soc., 2014, 136, 14153–14159 CrossRef CAS PubMed.
- D. Veclani, A. Melchior, M. Tolazzi and J. P. Ceron-Carrasco, J. Am. Chem. Soc., 2018, 140, 14024–14027 CrossRef CAS PubMed.
- A. A. Almaqwashi, W. Zhou, M. N. Naufer, I. A. Riddell, O. H. Yilmaz, S. J. Lippard and M. C. Williams, J. Am. Chem. Soc., 2019, 141, 1537–1545 CrossRef CAS PubMed.
- K. S. Lovejoy, R. C. Todd, S. Zhang, M. S. McCormick, J. A. D'Aquino, J. T. Reardon, A. Sancar, K. M. Giacomini and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 8902–8907 CrossRef CAS PubMed.
- Y. Guo, Y. He, S. Wu, S. Zhang, D. Song, Z. Zhu, Z. Guo and X. Wang, Inorg. Chem., 2019, 58, 13150–13160 CrossRef CAS PubMed.
- Q. Wang, M. Xiao, D. Wang, X. Hou, J. Gao, J. Liu and J. Liu, Adv. Funct. Mater., 2021, 31, 2101826 CrossRef CAS.
- M. Kaushik Tiwari, D. A. Colon-Rios, H. C. R. Tumu, Y. Liu, E. Quijano, A. Krysztofiak, C. Chan, E. Song, D. T. Braddock, H. W. Suh, W. M. Saltzman and F. A. Rogers, Nat. Biotechnol., 2022, 40, 325–334 CrossRef CAS PubMed.
- S. Sheng, F. Liu, M. Meng, C. Xu, H. Tian and X. Chen, CCS Chem., 2021, 3, 2299–2310 Search PubMed.
- X. Jiang, Q. Zhou, B. Du, S. Li, Y. Huang, Z. Chi, W. M. Lee, M. Yu and J. Zheng, Sci. Adv., 2021, 7, eabd9847 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc01616a |
| ‡ Ting He, Qiaoju Ren, Yu Zhang contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence and
Jing
Lin
and
Jing
Lin