
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric total synthesis of penicilfuranone A through an NHC-catalyzed umpolung strategy†
Yiming Ding‡
ab,
Xianwen Long‡b,
Jingwei Zhangb,
Chunlei Qub,
Peng Wangb,
Xiaodong Yang a,
Pema-Tenzin Punoc and
Jun Deng*a
a,
Pema-Tenzin Punoc and
Jun Deng*a
aKey Laboratory of Medicinal Chemistry for Natural Resources, Ministry of Education, Yunnan Provincial Center for Research & Development of Natural Products, School of Chemical Science and Technology, Yunnan University, Kunming, 650091, China
bState Key Laboratory and Institute of Elemento-Organic Chemistry, Frontiers Science Center for New Organic Matter, College of Chemistry, Nankai University, Tianjin 300071, China. E-mail: dengjun@nankai.edu.cn
cState Key Laboratory of Phytochemistry and Natural Medicines, Yunnan Key Laboratory of Natural Medicinal Chemistry, Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, China
First published on 8th April 2025
Abstract
The first asymmetric total synthesis of penicifuranone A was accomplished in eight steps through an NHC-catalyzed umpolung strategy. Key features of the synthesis include an Al-Salen catalyzed asymmetric cyanosilylation to install the tertiary alcohol of gregatin A, and an NHC catalyzed Stetter–Aldol cascade reaction. The umpolung strategy of the benzyl aldehyde fragment facilitated a convergent formal [4 + 2] annulation with gregatin A, ultimately leading to the formation of penicifuranone A.
Introduction
Furancarboxylic acids are a class of naturally occurring phytotoxins isolated from fungi, known for their broad application in polymer synthesis1 and diverse bioactivities, including phytotoxic and antibiotic effects.2 These compounds have garnered significant attention within the synthetic community due to their complex structures and pharmacological potential.3 In 2016, penicilfuranone A (1), a new furancarboxylic acid, was isolated from an endophytic strain of Penicillium sp. sh18 by Puno and co-workers.4 Structurally, penicilfuranone A (1) possesses a tricyclic framework and is classified as an aromatic polyketide (Scheme 1A). It features four stereogenic centers, including three consecutive stereogenic centers and two quaternary chiral centers. Biologically, penicilfuranone A (1) exhibited a significant antifibrotic effect in activated hepatic stellate cells, suggesting its potential as a lead compound for the treatment of hepatic fibrosis. In addition to penicilfuranone A (1), several structurally divergent gregatin A heterodimers have been discovered,5 such as citrifuran A (2), asperone A (3), and the recently reported penidaleodiolide A (4). | ||
| Scheme 1 Penicilfuranone A and its proposed biosynthetic pathway. | ||
One of the major challenges in the asymmetric synthesis of penicilfuranone A (1) lies in the stereo-controlled synthesis of the fully decorated benzocyclohexanone (B ring, highlighted in red), which is highly oxidized and enriched in stereocenters. The presence of C-19 tertiary alcohol renders this structure prone to aromatization upon exposure to acidic conditions, further complicating its synthesis. The biosynthesis of penicilfuranone A (1) is hypothesized to involve gregatin A (8) and phenol 9 through an intermolecular Michael addition, an intramolecular aldol addition and subsequent C16 benzylic oxidation (Scheme 1B). Wang and Matsuda showed that gregatin A (8) is derived from polyketide 5 through enzyme-catalyzed oxidative cyclization and methylation.6 An intriguing aspect of this biosynthetic pathway is the formation of furanone through a vinylogous internal nucleophilic substitution (SNi′), where the conjugated double bond migrates to an unconjugated position, forming a quaternary carbon center in an enantioselective manner.
As shown in Scheme 2, in order to make the synthesis convergent and efficient, we conducted a retrosynthetic analysis of penicilfuranone A (1) by late-stage formal [4 + 2] annulation to install this stereochemically intricate B ring. The target molecule could be derived from C15 epimerization of 12, which could be traced back either to gregatin A (8) and benzoisofuran (13) through an intermolecular Diels–Alder reaction, or to gregatin A (8) and benzaldehyde 14 through an intermolecular Stetter/Aldol cascade reaction. The key intermediate, gregatin A (8), might be synthesized through linear precursor 15 through a bioinspired vinylogous internal nucleophilic substitution (SNi′).
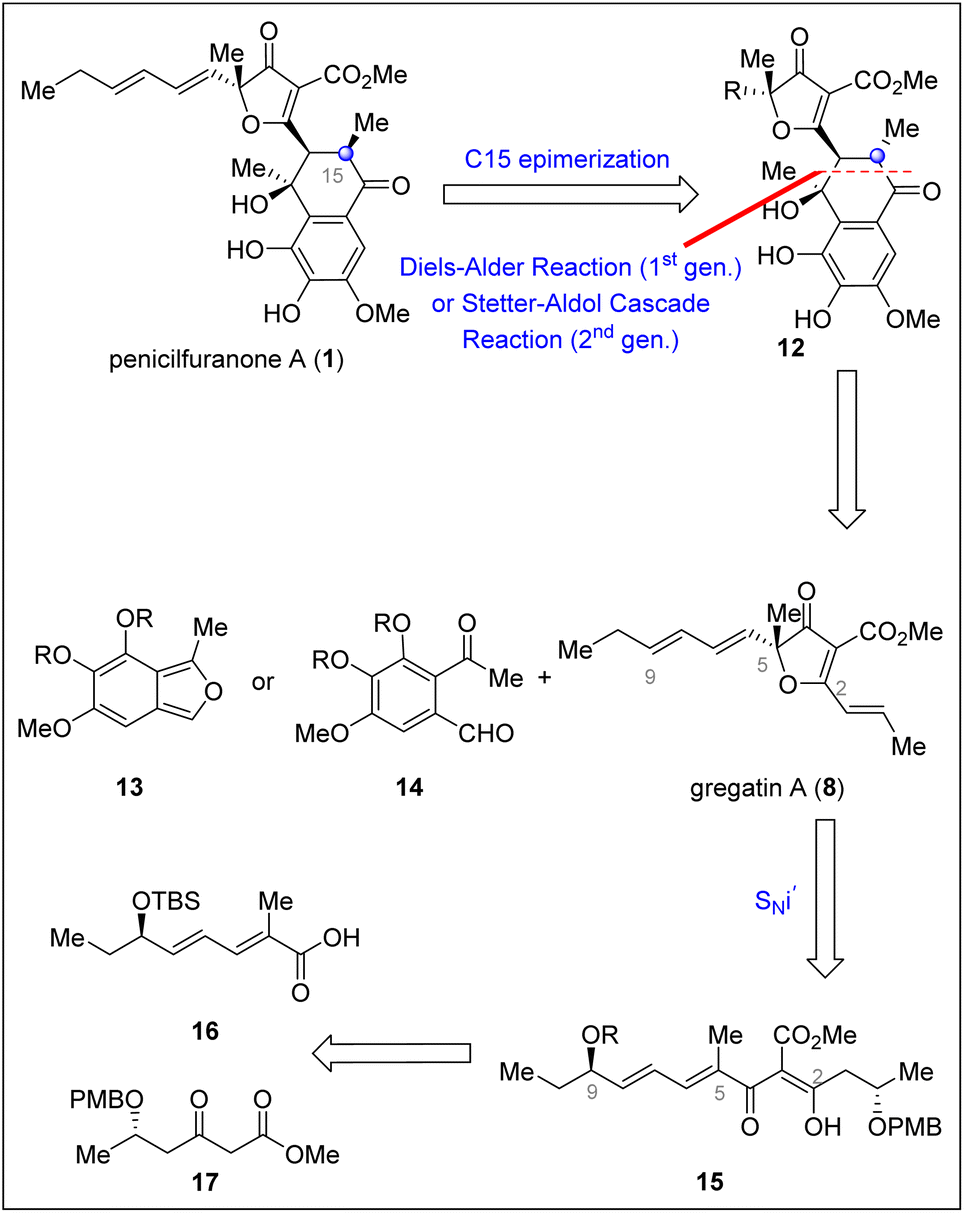 | ||
| Scheme 2 Retrosynthetic analysis of penicilfuranone A. | ||
Results and discussion
Asymmetric synthesis of key fragments
We initiated our synthesis with the preparation of hemiacetal 20, benzaldehyde 14a and gregatin A (8) (Scheme 3).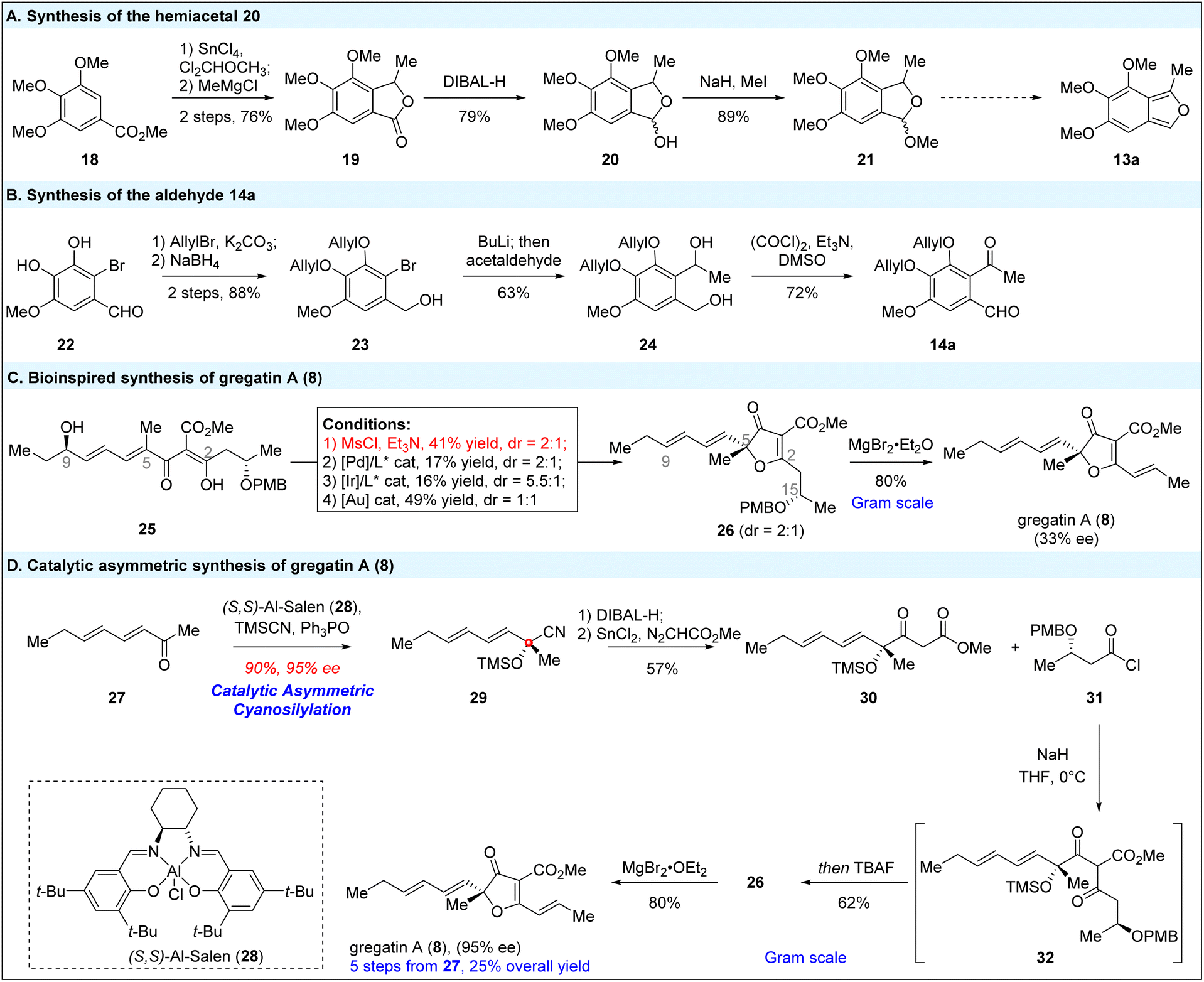 | ||
| Scheme 3 Asymmetric synthesis of gregatin A (8), hemiacetal 20 and benzaldehyde 14a. | ||
Hemiacetal 20 was synthesized in a three-step sequence. First, commercially available methyl 3,4,5-trimethoxybenzoate 18 was treated with SnCl4 and 1,1-dichlorodimethyl ether, followed by the addition of methylmagnesium chloride to the resulting aldehyde, leading to the formation of a secondary alcohol. This alcohol was in situ trapped by the methyl ester to afford lactone 19 in 76% yield. Reduction of the lactone with DIBAL-H gave hemiacetal 20 in 79% yield. This hemiacetal could then be methylated to provide methyl acetal 21 in 89% yield. Subsequently, diphenol 22 was protected with allyl bromide, and the aldehyde group was reduced, converting aldehyde 22 to benzylic alcohol 23. After halide-lithium exchange, the resulting aryl lithium was trapped with acetaldehyde to yield diol 24 in 63% yield. The diol was then oxidized using Swern oxidation to give benzaldehyde 14a in 72% yield. Notably, diol 24 proved unstable under acidic conditions, and treatment with other oxidants (e.g., PIFA or DMP) led to the formation of undesired isobenzodihydrofuran or styrene by-products.
Next, we turned our attention to the asymmetric synthesis of gregatin A (8) (Scheme 3C). In our ongoing efforts to chemically emulate the biosynthetic pathway of natural products,7 our initial strategy was to replicate the internal nucleophilic substitution reaction proposed in gregatin A (8) biosynthesis (Scheme 1B) and hopefully realize the enantioselective synthesis of gregatin A (8) through the C9 → C5 chiral relay strategy. Starting from chiral allylic alcohol,8 we synthesized cyclization precursor 25 (90% ee) in five steps (see the ESI†). To our delight, activation of the allylic alcohol with MsCl successfully initiated the vinylogous SNi′ reaction, yielding the desired furanone in 41% yield. However, the 2° → 3° stereocenter chiral relay did not result in sufficient diastereocontrol at the newly formed tertiary C–O bond, leading to a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereomeric ratio (dr). Gregatin A (8) was obtained in only 33% ee after elimination of the C15 PMBOH with MgBr2·OEt2.9 The use of MgBr2·OEt2 was critical to secure the high yield of gregatin A (8), because basic conditions led to side-chain cleavage of gregatin A (8) through retro-Aldol addition (see the ESI†). Despite previous success with chiral relay strategies in SNi′ reactions,10 vinylogous SNi′ reactions remain challenging.11
:1 diastereomeric ratio (dr). Gregatin A (8) was obtained in only 33% ee after elimination of the C15 PMBOH with MgBr2·OEt2.9 The use of MgBr2·OEt2 was critical to secure the high yield of gregatin A (8), because basic conditions led to side-chain cleavage of gregatin A (8) through retro-Aldol addition (see the ESI†). Despite previous success with chiral relay strategies in SNi′ reactions,10 vinylogous SNi′ reactions remain challenging.11
We were unable to identify conditions that guaranteed both high yield and diastereoselectivity, even with the use of chiral ligands and Pd or Ir catalysts (see ESI Table S1† for details). Since both gregatin A (8) and its enantiomer aspertetronin A, are natural products, enantiopure gregatin A (8) is essential for the subsequent formal [4 + 2] annulation to avoid the formation of undesired diastereomers.
In the asymmetric synthesis of gregatin A (8), the enantioselective installation of the C5 tertiary C–O bond posed a major challenge. Brückner and co-workers achieved the first asymmetric synthesis of gregatin A (8) using the Seebach–Fráter “self-reproduction of stereocenters” methodology, starting from enantiomerically pure lactic esters.3h However, our approach aimed to install this chiral tertiary alcohol via the catalytic asymmetric addition of methyl ketone 27. While significant progress has been made in the catalytic asymmetric addition of aryl-substituted ketones,12 such conditions have not been suitable for alkyl-substituted ketones, such as 27 in this case. After extensive exploration, we were pleased to find that Zhou's asymmetric cyanosilylation strategy was highly effective for aliphatic ketones.13 As shown in Scheme 3D, the catalytic asymmetric cyanosilylation of methyl ketone 27 using Al-Salen as the catalyst proceeded efficiently under standard conditions, delivering an excellent outcome with 90% yield and 95% enantiomeric excess at the decagram scale. Following cyanide reduction with DIBAL-H and formal C–H insertion with methyl diazoacetate (via Roskamp homologation),14 methyl acetate 30 was obtained in 57% yield over two steps. A subsequent Claisen condensation with acetyl chloride 31, along with desilylation, etherification, and β-elimination of PMBOH, provided gregatin A (8) in an overall yield of 25% from methyl ketone 27. Notably, the use of β-PMBO substituted acetyl chloride 31, rather than a conjugated alkene, was critical for achieving high yield (see ESI Table S2† for more details).
Synthetic attempts to form the B ring of penicilfuranone A (1)
With these three fragments in hand, we focused on constructing the 4-hydroxytetralone skeleton (B ring) of penicilfuranone A (1) via a formal [4 + 2] annulation (Scheme 4). Initially, we attempted to construct the B ring through an intermolecular Diels–Alder reaction between gregatin A (8) and benzoisofuran 13a. However, despite exploring various acidic and basic conditions, we were unable to detect the formation of benzoisofuran 13a. Next, we explored trapping the in situ generated benzoisofuran 13a with different dienophiles, including gregatin A (8). Unfortunately, this approach also proved unsuccessful, as the conditions required for the formation of isobenzofuran were incompatible with hemiacetal 20. Specifically, acidic conditions (e.g., AcOH, PTSA, and Cu(OTf)2) led to the decomposition of hemiacetal 20.15 Additionally, attempts to employ an elimination strategy for methyl acetal 21 with lithium diisopropylamide (LDA) or Cu(OTf)2 were also unsuccessful (see ESI Table S3† for more details).16 At this point, we hypothesized that this highly functionalized 4-hydroxytetralone skeleton could be synthesized through an NHC-catalyzed formal [4 + 2] annulation.17 Stetter reactions, as a well-established type of umpolung reaction,18 are commonly used to couple benzaldehyde and electron-deficient alkene fragments. We anticipated that the enolate generated from the Stetter reaction could be effectively trapped by the methyl ketone. To quickly verify the feasibility of this approach, we tested model substrates electron-deficient alkene 34 and benzaldehyde 35 for the Stetter–Aldol cascade reaction (Scheme 4B).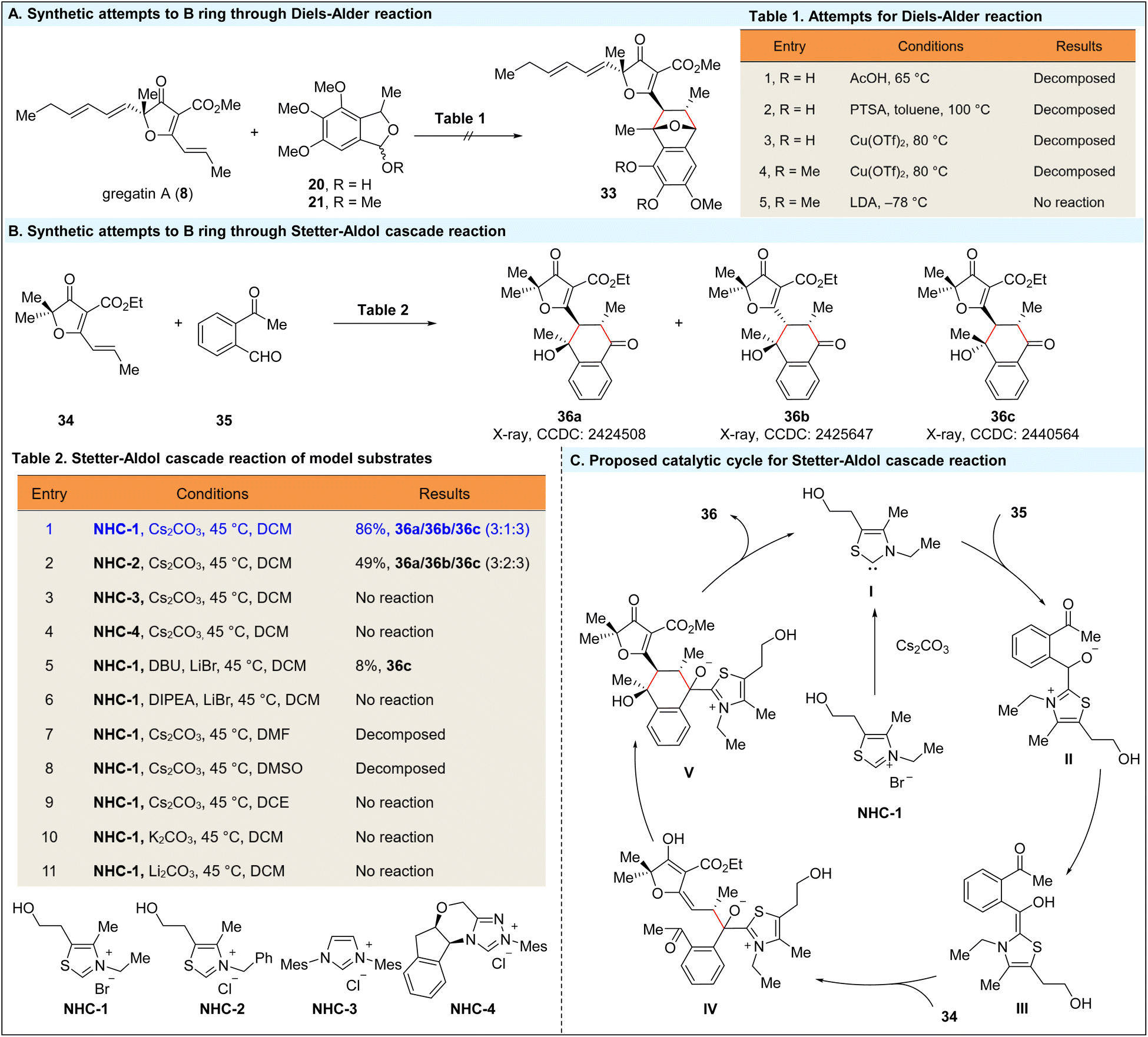 | ||
| Scheme 4 Synthetic attempts to form the B ring through the Diels–Alder or Stetter–Aldol cascade reaction. | ||
We employed an NHC-catalyzed intermolecular Stetter–Aldol cascade reaction to synthesize the 4-hydroxytetralone skeleton. To prevent homo-benzoin condensation of benzaldehyde 35, we utilized an activated Michael acceptor, electron-deficient alkene 34 (Table 2). To our delight, treatment of benzaldehyde 35 and alkene 34 with a carbene catalyst, generated by deprotonating the thiazolium salt NHC-1 with Cs2CO3, resulted in the desired 4-hydroxytetralone skeleton, which existed as three diastereoisomers—36a, 36b, and 36c in a 3:1:3 ratio, achieving an 86% overall yield (entry 1). The structure of 36a, 36b, and 36c was confirmed through X-ray crystallography.19 NHCs derived from thiazolium salts were effective in promoting the reaction (entry 2),20 while NHCs derived from imidazolium and triazolium salts were ineffective (entries 3 and 4), leading to the homo-benzoin condensation product of benzaldehyde 35.21 Additional screening revealed that Cs2CO3 was crucial for the success of the Stetter–Aldol cascade reaction, while other bases, such as K2CO3 and Li2CO3, and organic bases such as DBU and DIPEA, were ineffective (entries 5–11) or resulted in low yields (entry 5). Furthermore, only DCM was effective in promoting the reaction, while other solvents caused decomposition of both alkene 34 and benzaldehyde 35 (entries 7–8). While Ye and other groups have reported intermolecular Stetter–Aldol cascade reactions between benzaldehydes and electron-deficient alkenes,22 to the best of our knowledge, there is no precedent for trapping the enolate with ketones. This represents the first example of an intermolecular Stetter–Aldol cascade reaction with a ketone. As depicted in the proposed catalytic cycle for this Stetter–Aldol cascade reaction (Scheme 4C), NHC catalysis proves to be highly effective in this umpolung formal [4 + 2] annulation, forming two C–C bonds and generating three stereocenters in a single process.
Asymmetric total synthesis of penicilfuranone A (1)
With the optimized conditions in hand, we proceeded with the asymmetric total synthesis of penicilfuranone A (1) (Scheme 5). Under the catalysis of NHC-1, benzaldehyde 14a and gregatin A (8) underwent dimerization, yielding three diastereoisomers—37a, 37b, and 37c—in a 1:1:1 ratio, with a 67% overall yield. Although three diastereoisomers were formed, all could be converted to the desired compound 39. For instance, after C15 epimerization with Cs2CO3, 37a could be transformed into 39; both 37b and 37c could be converted to 39 through a retro-aldol/aldol/C15 epimerization cascade, yielding the precursor to penicilfuranone A (1), compound 39, in a 33% overall yield from gregatin A (8). Interestingly, C15 epimerization, which converts the C14/C15 trans configuration to the cis configuration, initially appeared abnormal. However, preliminary DFT calculation and our experimental results suggested that the cis configuration is thermodynamically favored, likely due to the steric repulsion between the C15 methyl group and C14 dihydrofuranone ring fragments (37b and 39 versus 37a and 37c) (see the ESI† for more details). After identifying these intermediates, we were also able to synthesize compound 39 in one step in a 31% yield from benzaldehyde 14a and gregatin A (8) using excess Cs2CO3 (2.0 equiv.).
 | ||
| Scheme 5 Asymmetric total synthesis of penicilfuranone A (1). | ||
The crystal structure of penicilfuranone A (1) further revealed an intramolecular hydrogen bond between the C19 OH group and the C20 O, which likely plays a crucial role in stabilizing the axial configuration of the dihydrofuranone ring. The final step in the synthesis of penicilfuranone A (1) involved deallylation. Under standard conditions (Pd/C, HCO2NH4), this step could lead to the formation of penicilfuranone A (1) in good yield. However, this condition was not reproducible in some instances. Ultimately, we found that the Ru-catalyzed deallylation method developed by Kitamura23 proved robust and efficient for this substrate, yielding penicilfuranone A (1) in 86% yield. The synthetic penicilfuranone A (1) was fully characterized, and its spectral data were identical to those reported by Puno.4
Conclusions
In conclusion, we have successfully achieved the first asymmetric total synthesis of penicilfuranone A (1) in the longest linear sequence of eight steps, starting from methyl ketone 27. Moreover, we have uncovered a novel NHC-catalyzed intermolecular Stetter–Aldol cascade reaction between benzaldehydes and electron-deficient alkenes, enabling the efficient synthesis of the 4-hydroxytetralone skeleton. This new synthetic approach not only facilitates the synthesis of penicilfuranone A (1) but also offers opportunities for the synthesis of other 4-hydroxytetralone containing natural products with potential pharmacological activities, such as rishirilide B (5)24 and tetracycline (6).25Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Y. D., X. L. and J. D. designed the research. Y. D., X. L. and J. Z. performed the synthetic work. J. Z. performed the quantum-molecular calculations. P. W. performed the crystallographic analysis. J. D. wrote the manuscript. X. Y., and P.-T. P. contributed to discussions. J. D. supervised the research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (no. 22222105, 22371142, and 22301146), the Frontiers Science Center for New Organic Matter, Nankai University (no. 63181206), a Key project at the central government level: the ability establishment of sustainable use for valuable Chinese medicine resources (no. 2060302), the Fundamental Research Funds for the Central Universities (no. 23JCYBJC01410), and the China Postdoctoral Science Foundation (no. 332608) is gratefully acknowledged.Notes and references
- J. Iglesias, I. Martínez-Salazar, P. Maireles-Torres, D. M. Alonso, R. Mariscal and M. L. Granados, Chem. Soc. Rev., 2020, 49, 5704–5771 RSC.
- (a) M. Alizadeh, M. Jalal, K. Hamed, A. Saber, S. Kheirouri, F. Pourteymour Fard Tabrizi and N. Kamari, J. Inflamm. Res., 2020, 13, 451–463 Search PubMed; (b) C. Almeida, N. El Aouad, J. Martín, I. Pérez-Victoria, V. González-Menéndez, G. Platas, M. de la Cruz, M. C. Monteiro, N. de Pedro, G. F. Bills, F. Vicente, O. Genilloud and F. Reyes, J. Antibiot., 2014, 67, 421–423 CrossRef CAS PubMed.
- (a) N. G. Clemo and G. Pattenden, Tetrahedron Lett., 1982, 23, 585–588 CrossRef CAS; (b) K. Takeda, H. Kubo, T. Koizumi and E. Yoshii, Tetrahedron Lett., 1982, 23, 3175–3178 CrossRef CAS; (c) O. Miyata and R. R. Schmidt, Tetrahedron Lett., 1982, 23, 1793–1796 CrossRef CAS; (d) A. Takaiwa and K. Yamashita, Agric. Biol. Chem., 1982, 46, 1721–1722 CAS; (e) A. Takaiwa and K. Yamashita, Agric. Biol. Chem., 1984, 48, 2061–2065 CAS; (f) N. G. Clemo and G. Pattenden, J. Chem. Soc. Perkin Trans. I, 1985, 2407–2411 RSC; (g) H. Burghart-Stoll and R. Brückner, Org. Lett., 2011, 13, 2730–2733 CrossRef CAS PubMed; (h) H. Burghart-Stoll and R. Brückner, Eur. J. Org Chem., 2012, 2012, 3978–4017 CrossRef CAS; (i) F. Weber and R. Brückner, Org. Lett., 2014, 16, 6428–6431 CrossRef CAS PubMed; (j) G. Dai, Q. Shen, Y. Zhang and X. Bian, J. Fungi., 2022, 8, 320 CrossRef CAS PubMed; (k) Y. Ding, X. Zhao, C. Qu, X. Long, Y. Zhao and J. Deng, Org. Biomol. Chem., 2025 10.1039/d4ob02108h.
- W.-G. Wang, A. Li, B.-C. Yan, S.-B. Niu, J.-W. Tang, X.-N. Li, X. Du, G. L. Challis, Y. Che, H.-D. Sun and J.-X. Pu, J. Nat. Prod., 2016, 79, 149–155 CrossRef CAS PubMed.
- (a) G.-P. Yin, Y.-R. Wu, M.-H. Yang, T.-X. Li, X.-B. Wang, M.-M. Zhou, J.-L. Lei and L.-Y. Kong, Org. Lett., 2017, 19, 4058–4061 CrossRef CAS PubMed; (b) G.-P. Yin, Y.-R. Wu, C. Han, X.-B. Wang, H.-L. Gao, Y. Yin, L.-Y. Kong and M.-H. Yang, Org. Chem. Front., 2018, 5, 2432–2436 RSC; (c) Q.-Y. Wang, H.-P. Chen, H. Tao, X. Li, Q. Zhao and J.-K. Liu, Org. Lett., 2024, 26, 7632–7637 CrossRef CAS PubMed.
- W.-G. Wang, H. Wang, L.-Q. Du, M. Li, L. Chen, J. Yu, G.-G. Cheng, M.-T. Zhan, Q.-F. Hu, L. Zhang, M. Yao and Y. Matsuda, J. Am. Chem. Soc., 2020, 142, 8464–8472 CrossRef CAS PubMed.
- (a) X. W. Long, Y. M. Ding and J. Deng, Angew. Chem., Int. Ed., 2018, 57, 14221–14224 CrossRef CAS PubMed; (b) Y. Long, Y. M. Ding, H. Wu, C. L. Qu, H. Liang, M. Zhang, X. L. Zhao, X. W. Long, S. Wang, P. T. Puno and J. Deng, Angew. Chem., Int. Ed., 2019, 58, 17552–17557 CrossRef CAS PubMed; (c) X. W. Long, H. Wu, Y. M. Ding, C. L. Qu and J. Deng, Chem, 2021, 7, 212–223 CrossRef CAS; (d) X. Long, J. Li, F. Gao, H. Wu and J. Deng, J. Am. Chem. Soc., 2022, 144, 16292–16297 CrossRef CAS PubMed; (e) X. W. Long, M. Zhang, X. D. Yang and J. Deng, Org. Lett., 2022, 24, 1303–1307 CrossRef CAS PubMed.
- (a) R. M. Moslin and T. F. Jamison, Org. Lett., 2006, 8, 455–458 CrossRef CAS PubMed; (b) R. M. Moslin, K. M. Miller and T. F. Jamison, Tetrahedron, 2006, 62, 7598–7610 CrossRef CAS.
- T. Kikuchi, K. Narita, K. Saijo, C. Ishioka and T. Katoh, Eur. J. Org Chem., 2016, 34, 5659–5666 CrossRef.
- For selected examples of SNi′ reaction in natural product synthesis: (a) S. D. Burke and L. Jiang, Org. Lett., 2001, 3, 1953–1955 CrossRef CAS PubMed; (b) S. Tanaka, T. Seki and M. Kitamura, Angew. Chem., Int. Ed., 2009, 48, 8948–8951 CrossRef CAS PubMed; (c) A. Guérinot, A. Serra-Muns, C. Bensoussan, S. Reymond and J. Cossy, Tetrahedron, 2011, 67, 5024–5033 CrossRef; (d) X. Wang, G. Huang, Y. Wang and J. Gui, J. Am. Chem. Soc., 2023, 145, 9354–9363 CrossRef CAS PubMed.
- P.-S. Wang, P. Liu, Y.-J. Zhai, H.-C. Lin, Z.-L. Han and L.-Z. Gong, J. Am. Chem. Soc., 2015, 137, 12732–12735 CrossRef CAS PubMed.
- (a) L.-X. Ruan, B. Sun, J.-M. Liu and S.-L. Shi, Science, 2023, 379, 662–670 CrossRef CAS PubMed; (b) M. Cao, H. Wang, F. Hou, Y. Zhu, Q. Liu, C.-H. Tung and L. Liu, J. Am. Chem. Soc., 2024, 146, 18396–18406 Search PubMed; (c) H. An, S. Liu, S.-J. Wang, X. Yu, C. Shi, H. Lin, S. B. Poh, H. Yang, M. W. Wong, Y. Zhao, Z. Tu and S. Lu, Org. Lett., 2024, 26, 702–707 CrossRef CAS PubMed.
- (a) X.-P. Zeng, Z.-Y. Cao, X. Wang, L. Chen, F. Zhou, F. Zhu, C.-H. Wang and J. Zhou, J. Am. Chem. Soc., 2016, 138, 416–425 CrossRef CAS; (b) X.-P. Zeng and J. Zhou, J. Am. Chem. Soc., 2016, 138, 8730–8733 CrossRef CAS; (c) W.-B. Wu, X. Yu, J.-S. Yu, X. Wang, W.-G. Wang and J. Zhou, CCS Chem., 2022, 4, 2140–2152 CrossRef CAS.
- Roskamp Homologation: C. R. Holmquist and E. J. Roskamp, J. Org. Chem., 1989, 54, 3258–3260 CrossRef CAS.
- Acid-promoted isobenzofuran formation: (a) J. G. Smith, P. W. Dibble and R. E. Sandborn, J. Org. Chem., 1986, 51, 3762–3768 CrossRef CAS; (b) P. Nandhikonda and M. D. Heagy, Org. Lett., 2010, 12, 4796–4799 CrossRef CAS PubMed.
- Base-promoted isobenzofuran formation: (a) D. Tobia and B. Rickborn, J. Org. Chem., 1986, 51, 3849–3858 CrossRef CAS; (b) C. Martin, P. Mailliet and J. Maddaluno, J. Org. Chem., 2001, 66, 3797–3805 CrossRef CAS PubMed.
- For selected reviews and examples of NHC catalyzed Umpolung reactions: (a) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606–5655 CrossRef CAS PubMed; (b) C. Grondal, M. Jeanty and D. Enders, Nat. Chem., 2010, 2, 167–178 CrossRef CAS PubMed; (c) X. Bugaut and F. Glorius, Chem. Soc. Rev., 2012, 41, 3511–3522 RSC; (d) D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Chem. Rev., 2015, 115, 9307–9387 CrossRef CAS PubMed; (e) X. Chen, H. Wang, Z. Jin and Y. R. Chi, Chin. J. Chem., 2020, 38, 1167–1202 CrossRef CAS; (f) X. Fang, K. Jiang, C. Xing, L. Hao and Y. R. Chi, Angew. Chem., Int. Ed., 2011, 50, 1910–1913 CrossRef CAS PubMed; (g) S. Zhuo, T. Zhu, L. Zhou, C. Mou, H. Chai, Y. Lu, L. Pan, Z. Jin and Y. R. Chi, Angew. Chem., Int. Ed., 2019, 58, 1784–1788 CrossRef CAS PubMed; (h) P. Zheng, S. Wu, C. Mou, W. Xue, Z. Jin and Y. R. Chi, Org. Lett., 2019, 21, 5026–5029 CrossRef CAS PubMed; (i) Y. Nakano and D. W. Lupton, Angew. Chem., Int. Ed., 2016, 55, 3135–3139 CrossRef CAS PubMed; (j) M. H. Wang and K. A. Scheidt, Angew. Chem., Int. Ed., 2016, 55, 14912–14922 CrossRef CAS PubMed; (k) C. Guo, M. Fleige, D. Janssen-Müller, C. G. Daniliuc and F. Glorius, J. Am. Chem. Soc., 2016, 138, 7840–7843 CrossRef CAS PubMed.
- For reviews of Umpolung reactions: (a) D. Seebach and E. J. Corey, J. Org. Chem., 1975, 40, 231–237 CrossRef CAS; (b) D. Seebach, Angew. Chem., Int. Ed., 1979, 18, 239–258 CrossRef; (c) A. B. Smith and C. M. Adams, Acc. Chem. Res., 2004, 37, 365–377 CrossRef CAS PubMed; (d) X.-J. Dai, C.-C. Li and C.-J. Li, Chem. Soc. Rev., 2021, 50, 10733–10742 RSC; (e) B. Shen, D. M. Makley and J. N. Johnston, Nature, 2010, 465, 1027–1032 CrossRef CAS PubMed; (f) Y. Wu, L. Hu, Z. Li and L. Deng, Nature, 2015, 523, 445–450 CrossRef CAS PubMed; (g) Y. Chen, M. Duan, S. L. Lin, Y. W. Liu, J. Cheng, S.-H. Xiang, P. Yu, K. N. Houk and B. Tan, Nat. Chem., 2024, 16, 408–416 CrossRef CAS PubMed.
- Deposition numbers 2424508 (for 36a), 2425647 (for 36b), and 2440564 (for 36c) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- (a) E. Sánchez-Larios and M. Gravel, J. Org. Chem., 2009, 74, 7536–7539 CrossRef PubMed; (b) E. Sánchez-Larios, J. M. Holmes, C. L. Daschner and M. Gravel, Org. Lett., 2010, 12, 5772–5775 CrossRef PubMed; (c) P. Liu, M. Lei, L. Ma and L. Hu, Synlett, 2011, 1133–1136 CAS; (d) B.-C. Hong, N. S. Dange, C.-S. Hsu and J.-H. Liao, Org. Lett., 2010, 12, 4812–4815 CrossRef CAS PubMed.
- (a) T. Ema, Y. Oue, K. Akihara, Y. Miyazaki and T. Sakai, Org. Lett., 2009, 11, 4866–4869 CrossRef CAS PubMed; (b) S. Kankala, R. Edulla, S. Modem, R. Vadde and C. S. Vasam, Tetrahedron Lett., 2011, 52, 3828–3831 CrossRef CAS; (c) T. Ema, K. Akihara, R. Obayashi and T. Sakai, Adv. Synth. Catal., 2012, 354, 3283–3290 CrossRef CAS; (d) M. Sharique and U. K. Tambar, Chem. Sci., 2020, 11, 7239–7243 RSC.
- For selected examples of NHC catalyzed Stetter–Aldol cascade reaction: (a) F.-G. Sun, X.-L. Huang and S. Ye, J. Org. Chem., 2010, 75, 273–276 CrossRef CAS PubMed; (b) E. Sánchez-Larios, J. M. Holmes, C. L. Daschner and M. Gravel, Org. Lett., 2010, 12, 5772–5775 Search PubMed; (c) D. Barman, T. Ghosh, K. Show, S. Debnath, T. Ghosh and D. K. Maiti, Org. Lett., 2021, 23, 2178–2182 CrossRef CAS PubMed.
- (a) S. Tanaka, H. Saburi, Y. Ishibashi and M. Kitamura, Org. Lett., 2004, 6, 1873–1875 CrossRef CAS PubMed; (b) D. E. Ward and M. M. Zahedi, Org. Lett., 2012, 14, 6246–6249 CrossRef CAS PubMed.
- (a) H. Iwaki, Y. Nakayama, M. Takahashi, S. Uetsuki, M. Kido and Y. Fukuyama, J. Antibiot., 1984, 37, 1091–1093 CrossRef CAS PubMed; (b) J. G. Allen and S. J. Danishefsky, J. Am. Chem. Soc., 2001, 123, 351–352 CrossRef CAS PubMed; (c) M. Odagi, K. Furukori, K. Takayama, K. Noguchi and K. Nagasawa, Angew. Chem., Int. Ed., 2017, 56, 6609–6612 CrossRef CAS PubMed.
- (a) M. G. Charest, C. D. Lerner, J. D. Brubaker, D. R. Siegel and A. G. Myers, Science, 2005, 308, 395–398 CrossRef CAS PubMed; (b) C. Sun, Q. Wang, J. D. Brubaker, P. M. Wright, C. D. Lerner, K. Noson, M. Charest, D. R. Siegel, Y. M. Wang and A. G. Myers, J. Am. Chem. Soc., 2008, 130, 17913–17927 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2424508, 2425647 and 2440564. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc01508a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |