
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulti-STEM MePCR: a bisulfite-free, multiplex, highly sensitive and highly specific assay to measure DNA methylation†
Hao
Yang
a,
Jiani
Qiu
a,
Yaping
Xu
a,
Wei
Ren
a,
LinQing
Zhen
a,
Gaolian
Xu
*a,
Hongchen
Gu
 *ab and
Hong
Xu
*ab
*ab and
Hong
Xu
*ab
aSchool of Biomedical Engineering/Med-X Research Institute, Shanghai Jiao Tong University, Shanghai 200030, China. E-mail: gaolian.xu@sicii.org.cn; hcgu@sjtu.edu.cn; xuhong@sjtu.edu.cn
bHefei Cancer Early Screening Innovation Technology Institute, Anhui Province, China
First published on 10th June 2025
Abstract
Aberrant CpG island methylation serves as a pivotal biomarker for cancer diagnosis, with accuracy substantially enhanced by analyzing multiple loci. Current techniques, such as bisulfite conversion or restriction enzyme-based methods, often fall short in delivering efficient multiplexed genomic methylation analysis using standard PCR platforms. Herein, we introduce an innovative bisulfite-free, multiplex assay—multiple specific terminal mediated methylation PCR (multi-STEM MePCR). This assay integrates a methylation-dependent restriction endonuclease (MDRE) with a novel multiplex PCR, leveraging innovative stem-loop structured assays for simultaneous detection of multiple CpG sites. As a proof-of-concept, the multi-STEM MePCR platform simultaneously achieved quantification of three methylation model sites down to ten copies per tube, accompanied by a broader linear dynamic range, and attained a sensitivity of 0.1% against a background of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 unmethylated gene copies. Crucially, by markedly minimizing cross-reactivity and reducing competition among targets, this technique adeptly distinguishes between sites exhibiting significant variations in methylation abundance. Additionally, this method effectively detects digestion products of various sizes, demonstrating clinical precision comparable to bisulfite sequencing, yet with simpler operation, less time, and lower cost. This multi-STEM PCR technology pioneers an advanced strategy for multiplexed methylation analysis, which is essential for epigenetic research and clinical DNA methylation diagnostics.
000 unmethylated gene copies. Crucially, by markedly minimizing cross-reactivity and reducing competition among targets, this technique adeptly distinguishes between sites exhibiting significant variations in methylation abundance. Additionally, this method effectively detects digestion products of various sizes, demonstrating clinical precision comparable to bisulfite sequencing, yet with simpler operation, less time, and lower cost. This multi-STEM PCR technology pioneers an advanced strategy for multiplexed methylation analysis, which is essential for epigenetic research and clinical DNA methylation diagnostics.
Introduction
Methylation status changes in CpG islands are widely recognized as biomarkers for cancer diagnosis and early screening.1–3 Increasing evidence indicates that different CpG targets contribute variably, and often complementarily, to cancer diagnostics.4–6 Moreover, due to tumor heterogeneity, reliance on a single methylated target often fails to meet the clinical requirements for accurate cancer diagnosis. Consequently, panels of methylated targets have been developed that combine multiple biomarkers to enhance the diagnostic sensitivity and specificity, particularly for early-stage cancers.7–11 However, methylation detection methods are often ineffective for real clinical samples, particularly bodily fluids, where methylated genes are present in low abundance against a high background of unmethylated genes.12,13Unfortunately, sequential partitioning of limited clinical samples into independent reaction compartments for multiplex detection of ultralow-abundance methylated genes inevitably dilutes the already scarce target molecules, thereby lowering their effective concentration per reaction and significantly compromising both the sensitivity and reliability of clinical diagnoses. Therefore, the development of a highly sensitive, multiplexed platform for simultaneous and accurate quantification of DNA methylation status in a single reaction is urgently needed for wide clinical applicability.
DNA methylation information is not retained by standard molecular biology methods, in which methylation-dependent pre-treatment is necessary for accurate identification and analysis.14,15 Currently, the gold standard for methylation analysis is bisulfite pre-treatment, which converts unmethylated cytosines to uracils, while leaving methylated cytosines unchanged.16 Thus far, sequencing-based detection with bisulfite pre-treatment is the most powerful strategy to obtain multi-loci CpG information, in which massive methylated targets can be simultaneously detected.17–19 However, the time-consuming protocols of sequencing-based methods with their high cost and expensive equipment would greatly limit the clinical application of DNA methylation.20 Hence, a PCR-based technology with multiplex detection property, fast and easy operation, low cost, and inexpensive equipment is the optimal strategy for methylation detection in daily clinical practice,21,22 especially for primary hospitals.
Bisulfite-based PCR methods, such as methylation-specific PCR (MSP)23 and MethyLight,24 are currently the most widely used in clinical practice, and several extended methods, such as quantitative allele-specific real-time target and signal amplification (QuARTS) and quantitative multiplex methylation-specific PCR (QM-MSP) can also achieve multiplexed detection by increasing the number of primer pairs and probes.25,26 However, the ability of these methods to detect multiple targets remains insufficient due to decreased sequence diversity caused by bisulfite treatment, and the addition of primer pairs may lead to problems, such as primer-dimer formation and cross-reactions among different targets.
To overcome the primer complexity of multiplexed detection in bisulfite-treated samples, a multiplexed ligation-dependent assay was developed, in which the methylated site triggered a specific linking reaction, and then was amplified by the universal primer.27,28 However, the sensitivity of the method was restricted by the ligase efficiency,29 and false-positive signals cannot be avoided due to the non-specific ligation among massive probes.30 More significantly, the methylation ratios differ across various loci, even within a high dynamic range where the amplification of loci with low-abundance methylation would be inhibited by that of the loci with heavy methylation, due to the competing reactions.31,32 Furthermore, there are severe drawbacks to bisulfite-based methods in clinical application: tedious and complex pre-treatment, template degradation, unavailable UDG, and false signal caused by incomplete conversion or overtreatment.33,34
Methylation-sensitive restriction enzyme (MSRE)-qPCR is a bisulfite-free method that is also used in methylation detection, in which the methylated templates are enriched by digesting unmethylated templates and subsequently performing a qPCR process.35,36 Multiplexed detection was also achieved by integration with a microarray37,38 and nanotechnology-based sensors.39–41 However, the incomplete digestion of MSREs on unmethylated templates would cause false-positive results, and ultimately decrease the sensitivity and specificity of the results. Additionally, multiplexed detection depends on a hybridization process, in which considerable variability exists for specificity and efficiency.42
Fortunately, the false-positive signal can be avoided by using methylation-dependent restriction endonucleases (MDREs) to directly digest the methylated templates, such as MDRE-exponential amplification reaction (EXPAR) and helper-dependent chain reactions.43 However, reliable detection sensitivity is difficult to obtain with these methods due to the difficulty in signal interpretation caused by the low efficiency and high background signal, which results in difficulty in meeting clinical testing requirements.44
In this study, we developed a bisulfite-free, multiplex-specific terminal mediated methylation PCR (multi-STEM MePCR) with high sensitivity and specificity. In the multi-STEM MePCR system, various methylated templates are simultaneously treated in one tube and first digested by an MDRE, and then, the products are captured by corresponding tailored-foldable primers (TFPs). This setup facilitates a specialized multiplex PCR using a universal primer (UP) and various terminal-specific primers (TSPs).
Owing to the high specificity of MDRE digestion and STEM-MePCR,45 no non-specific amplification occurs on the unmethylated templates, and no false-positives are generated. Moreover, the ingenious primer design and strategic primer combinations effectively mitigate cross-reactions and competitive interactions among sites with significant methylation level variations. Therefore, the multi-STEM MePCR system is expected to overcome the challenges previously faced in multiplex methylation detection.
Results and discussion
The basic principles of the multi-STEM-MePCR system for methylation detection
The principle of the multi-STEM-MePCR system is shown in Fig. 1. The system consists of three stages, namely, MDRE cutting, TFP-mediated intramolecular folding, and multiplexed amplification. At the first stage, various methylated DNA templates were cut by an MDRE at specific sites to produce the specific 5′-end products while keeping the unmethylated templates intact. At the next stage, the TFPs with tailored designs were specifically bound with the treated templates and extended by Taq DNA polymerase to generate linear extension products. | ||
| Fig. 1 Schematic illustration of the multi-STEM-MePCR system to detect multi-loci of DNA methylation. | ||
For the methylated targets, the elongation reactions were stopped at the cutting sites to form products with a definite sequence at the 3′ end. Then, the extension products self-folded to form partial stem-loop structures (HP1) via intramolecular binding, and further extended to form complete hairpin structures (HP2). However, for the unmethylated templates, the extension of TFPs will not terminate at the precise location, which then results in the addition of a long sequence to the 3′ end. The 3′ end of subsequent HP1 remains in an unbound state, leading to the failure of HP2 generation. Thus, each of the generated HP2 structures contains two universal regions (UR1 and UR2) that can be further used as a primer region to trigger an exponential PCR amplification, resulting in the efficient detection of methylated targets.
Finally, at the multiplexed amplification stage, different HP2s that were formed from multi-methylated targets were opened by a unique TSP, which consisted of several specific bases on the 3′ end and universal bases on the 5′ region. The TSPs preferentially hybridize to the stem of HP2s due to the high concentration, resulting in stem-loop unwinding and the generation of a linear template via TSP extension, which ends at an extension blocker on HP2. The linear templates, generated from different HP2s, can be amplified by UP (as in UR1) and specific TSPs to generate a PCR signal, thereby enabling the multiplexed detection of DNA methylation.
Design philosophy and optimization of the multi-STEM-MePCR system
Specifically, the key to achieving multiplexed detection of methylated targets is the TFP with a unique design pattern. The design philosophy of the multi-STEM-MePCR system with defined parameters is shown in Fig. 2A. The reaction is initiated by the cleavage of methylated templates, producing digested fragments with different lengths from 28 bp to 150 bp that mainly consist of three regions: the capture region (CR), the probe region, and the folding region (FR). Then, the cutting product is captured by the TFP, which consists of five regions (5′ – 3′): universal region 2 (UR2), FR, extension blocker, universal region 1 (UR1), and capture complementary region (CRc). | ||
| Fig. 2 (A) Multi-STEM-PCR amplification primer pattern. (B) The thermodynamic process of the TFP during template extension and folding initiated by the extension product. (C) Effect of different FR lengths from 7 to 17 nt with the different primer sets. Ct value as a function of template concentration (10-fold serial dilutions of methylated plasmids from 106 copies per reaction to 102 copies per reaction) for different lengths of the FR. The data are the average of three repeats, and error bars represent the standard deviation. (D) A histogram of the melting temperature (Tm) of HP1 and curve of the final amplification efficiency at different lengths on the FR. The efficiency was calculated by the equation E = 2−(Ct17−Ctx), and data were normalized by setting the value of 17t to 1. (E) The modification of the 2′Ome base on the FR of the TFP. The histogram of Ct values at TFPs with different numbers of 2′Ome bases on the FR. 1000, 250, and 50 copies per reaction of methylated Jurkat genomic DNA were tested. Ct = 40 denotes no amplification; N/17: N denotes the number of 2′Ome bases and 17 denotes the total length of the FR. (F) The mechanism of inhibition of non-specific amplification using the 2′Ome modified bases on the FR of the TFP. (G) Agarose gel electrophoresis map of different 2′Ome-modified TFPs at 50 copies per reaction of methylated genomic DNA. | ||
As shown in Fig. 2B, the extension of the TFP ends at the cutting location, which subsequently generates a linear extension product with the complementary sequence of FR (FRc). The FR and FRc of the extension product participate in intramolecular binding with high affinity to generate a partial stem-loop structure (HP1), which then initiates self-priming to create a complete stem-loop structure (HP2), with a sequence complementary to the TSP. Although the intramolecular hairpin conformation of HP2 exhibits a higher thermodynamic stability than its intermolecular hybridization with the TSP, the significantly higher concentration of TSP induces a concentration-dependent conformational transition, promoting strand displacement and providing the thermodynamic impetus for initiating the downstream amplification process.
The TFP extension is a linear reaction; therefore, the TFPs can operate with high efficiency at low target concentrations, reducing the total amount of primers required and further avoiding cross-reactions among various TFPs through the use of an extension blocker. During the amplification stage, multiple generated HP2s are amplified by a single UP and multiple TSPs with high specificity.
All the TSPs share the same sequence at the 5′ end with only a few specific bases at the 3′ end. Therefore, each HP2 can only be selectively opened and amplified by its corresponding TSP, avoiding competition among various targets in multiplexed PCR. Ingeniously, the combination of a UP and TSPs greatly reduces the sequence diversity and cross-reaction of primers in the multiplexed PCR, providing an excellent and clinically accessible platform for multiplexed methylation detection.
According to the principle of multi-STEM-MePCR, the critical step of the reaction is the self-folding of the linear extension product of TFP (HP1), which acts as the initiator for subsequent exponential amplification. The efficiency of the self-folding process depends on the thermodynamic stability of HP1, which is related to the length and GC content of the FR. To clarify the relationship between the detection performance and different folding lengths of the FR, methylated Septin9 plasmids (shown in Fig. S1†), which are related to colorectal cancer (CRC),46 were used as model methylated templates. TFPs with different lengths of the FR from 7 to 17 nt were designed to study the amplification efficiency using the Ct value with different template concentrations (102 to 106 copies per reaction).
As shown in Fig. 2C, the methylated templates were amplified by each TFP with varying efficiencies. As the FR length increased from 7 to 17 nt, the Ct values decreased by approximately ten at 106 copies per reaction of plasmids, indicating that longer FR lengths lead to higher amplification efficiency. Moreover, there is no amplification at low concentrations (100 copies) when the length is less than 12 nt, indicating that a long FR plays a critical role in ensuring high sensitivity.
To further explore the thermodynamic parameters of HP1, the melting temperature (Tm) and Gibbs free energy (ΔG) of the HP1s with different FR lengths were calculated using the IDT OligoAnalyzer Tool (Table S2†). The HP1 with 17 bp has a low ΔG (−6.16 kcal mol−1, 65 °C), which can maintain the thermostability under reaction conditions, but the ΔG gradually increased in the shorter FR length, and the ΔG > 0 when the length was decreased to nine bp, in which the P1 tended to form a linear structure, resulting in low efficiency of the reaction. As shown in Fig. 2D, the amplification efficiency of the system exhibited an exponential correlation with the length of the FR. Furthermore, the Tm of HP1 decreased markedly when the FR length was reduced to nine base pairs or fewer, which rendered it insufficient to support stable self-priming at the PCR annealing temperature. As a result, amplification efficiency decreased by more than 100-fold compared to that with a 17 bp FR, underscoring the critical role of FR length in achieving high detection sensitivity.
As mentioned above, the binding affinity of the FR is crucial for maintaining the efficiency of the amplification reaction. However, for TFPs with a high Tm of HP1, the amplification of genomic DNA at low concentrations was completely inhibited, and strong non-specific bands were observed (Fig. S2A†). It was observed that excessively long lengths and high GC content of the FR could trigger non-specific amplification. This occurs through the random binding of the CRc and FR on the TFP, as illustrated in the schematic diagram at the lower left corner of Fig. 2G. This leads to self-folding of the TFP, thereby triggering non-specific amplification bands (Fig. S2C†), which closely resemble the sequence of PCR products from specific amplification (Fig. S2B†).
In detail, to achieve multiplexed detection in the design of the multi-STEM-MePCR, both the sequences of the UP and TSP introduced into TFP. This results in the self-folding of the TFP and production of an analogous HP2, which can then be further amplified by the pair of primers, resulting in non-specific amplification products. This phenomenon is attributable to the significantly high concentration of the TFP (5–10 nM), which increases the probability of non-specific amplification despite the inherently low affinity of random binding (as shown in Fig. S3†). It was indicated that the sensitivity of the system would be reduced by competitive inhibition from non-specific amplification, and more importantly, the risk of non-specific amplification would be further enhanced in a multiplexed reaction (more TFPs), which would ultimately deteriorate the multiplexed property of the system.
To avoid non-specific amplification, 2′-O-methyl (2′Ome)-modified bases that can block the extension of Taq polymerase47 were introduced into the FR to inhibit the self-priming of the TFP (shown in Fig. 2F). The effect of these modifications on the FR was systematically studied using TFPs (FR of 17 bp) with different numbers of 2′Ome-modified bases. As shown in Fig. 2E, the Ct values of TFPs with different modification patterns (0/17, 7/17, 10/17, 12/17, 14/17, 17/17, 2′Ome bases/total bases) were compared in a histogram. When the FR was fully modified by 2′Ome bases (17/17), the amplification was completely inhibited, demonstrating the ability of 2′Ome bases to block primer extension by Taq polymerase. Decreasing the number of 2′Ome bases to ten resulted in a gradual improvement in amplification efficiency. However, further reduction of 2′Ome bases to zero led to a decline in efficiency, especially at the low template concentration of 50 copies per reaction. These findings indicate that an appropriate pattern of 2′Ome modification on the FR is critical for achieving optimal amplification efficiency.
To further explore the amplification status at low concentrations, the PCR products of all TFPs at 50 copies per reaction were analyzed by agarose gel electrophoresis. As shown in Fig. 2G, TFPs without 2′Ome bases exhibited only non-specific bands, while the intensity of the specific target bands increased with the addition of 2′Ome bases. Non-specific amplification was totally inhibited at 10/17 TFP. However, as the number of 2′Ome bases further increased, the intensity of specific bands decreased, and even disappeared, which suggested that maintaining at least seven normal bases is essential for ensuring the extension properties of Taq polymerase.
Overall, the addition of 2′Ome bases to the FR region of TFPs inhibited the extension of Taq polymerase on the FR, prevented the formation of analogous HP2 byproducts by the self-folding of TFP, and eliminated non-specific amplification. However, excessive 2′Ome bases on the FR can also inhibit the normal self-priming of HP1. Thus, a certain design pattern of the FR (seven normal bases + ten 2′Ome bases) should be maintained to ensure the sensitivity of the system.
Sensitivity and detection performance of the multi-STEM-MePCR system
The TFP, optimized for parameters, was selected to validate the detection performance of methylation using the Septin9 gene as a model. As shown in Fig. 3A, methylated templates were quantitatively detected across a range from 10 to 5000 copies per reaction, with Ct values showing a linear correlation with the logarithm of the number of templates (logarithmic correlation coefficient R2 = 0.994). The PCR products were further analyzed via agarose gel electrophoresis assay. An excepted single band can be seen for the positive groups, while no band in the negative group (Fig. 3B) indicates the high specificity of the primer groups on whole genomic sequences. | ||
| Fig. 3 (A) Real-time amplification curves of the CpG site in the Septin9 gene with serially diluted methylated Jurkat genomic DNA at 5000, 1250, 250, 50, and 10 (copies per reaction), with free water as a negative control (NC). Standard curve of Ct values and template concentrations from 5000 to 10 (copies per reaction). The data are the average of three repeats, and the error bars represent the standard deviation. The data were fitted by linear regression (R2 = 0.994). (B) Agarose gel electrophoresis map of PCR products from different concentrations of methylated DNA and the NC. (C) Ct values of 20 replicates and the positive rates of STEM-PCR with 5, 10, 20, and 40 copies per reaction of input methylated DNA. (D) Real-time amplification curves of the CpG site in the Septin9 gene with different ratios of methylated DNA (50%, 12.5%, 2.5%, 0.5%, 0.1%, and 0%) in a background of 10000 copies of unmethylated DNA (Jurkat DNA). Standard curve of Ct values and methylation ratios from 50% to 0.1%. The data were fitted by linear regression (R2 = 0.996). (E) Agarose gel electrophoresis map of PCR products from different ratios of methylated DNA and the NC. (F) Standard curves of methylation sites on the RASS-F1 (R2 = 0.991) and SDC2 genes (R2 = 0.999), respectively. (G) Line chart of Ct values (STEM PCR system) of 10 ng genomic DNA, and methylation ratios measured by NGS of the CpG site in the Septin9 gene from 14 colorectal cancer (CRC) samples. (H) The digestion of the MDRE on each of two adjacent methylation sites would produce DNA fragments of varying lengths, and fragments of each length, even as low as 28 bp, can be captured and amplified by the TFP. (I) Agarose gel electrophoresis map of PCR products from templates with different lengths, from 28 bp to 75 bp. | ||
To further explore the detection limit of this system, 20 replicates with various copy numbers (40, 20, 10, and 5 copies per reaction) were analyzed. As shown in Fig. 3C, 100% of the replicates were positive, with 10 to 40 copies per reaction, while 90% were positive at five copies per reaction. These results indicate that the multi-STEM-MePCR system robustly performs with rare templates, achieving a limit of detection of at least ten copies per reaction, which is comparable to digital PCR and represents the ultimate sensitivity of real-time fluorescence quantitative PCR technology. Furthermore, to simulate actual sample conditions with a high background of unmethylated DNA, methylated genomic DNA was serially diluted with 10000 copies per reaction of unmethylated DNA.
All samples contained different ratios of methylated templates ranging from 50% to 0.1%, and demonstrated good linear correlation between Ct values and the logarithm of methylation ratios (R2 = 0.996). The system reliably detected methylation ratios as low as 0.1%, with satisfactory repeatability and no cross-reaction on unmethylated templates, as evidenced by amplification curves (Fig. 3D) and gel electrophoresis (Fig. 3E). Additionally, the multi-STEM-MePCR system performed well using genomic DNA from CRC tissue samples, with detection accuracy matching that of bisulfite sequencing (Fig. 3G and Table S3†).
To demonstrate the universality of the developed multi-STEM-MePCR system, methylated sites in the RASSF1 and SDC2 genes, associated with lung cancer48 and CRC,49 respectively, were employed to establish distinct detection assays. Methylated templates, ranging from 10 to 5000 copies per reaction, were successfully detected in RASSF1 and SDC2 through amplification curves and gel electrophoresis, yielding the expected specific sequences (Fig. S4 and S5†). As shown in Fig. 3F, the quantitative performance at two CpG sites was further evaluated by constructing standard curves correlating Ct values with the logarithm of copy numbers, revealing robust linear relationships for RASSF1 (R2 = 0.991) and SDC2 (R2 = 0.999). These results, characterized by low standard deviation in repetitions and high correlation coefficients, underscore the multi-STEM-MePCR system's superior repeatability and quantitative accuracy across different methylation targets, confirming its universality and laying a strong foundation for subsequent multi-target detection.
Notably, the methylation detection was initiated by MDRE digestion, in which many methylated CpG sites on templates could be cut to ultimately generate a large number of fragments with various lengths. For some MDREs, such as MspJI and FspEI, the resulting template lengths were notably short, even as low as 28 bp,50 which are typically unsuitable for PCR. To assess the performance of the multi-STEM-MePCR system with these challenging conditions, templates ranging from 75 bp to 28 bp were synthesized to simulate digestion products with different sizes (Fig. 3H).
As shown in Fig. 3I, the target bands in the gel after electrophoresis were observable across all template lengths (approximately 100 copies), with the variation trend in the length of the amplified products precisely mirroring that of the templates. This indicates the exceptional size flexibility in multi-STEM-MePCR-based methylation detection, and its ability to process a wide range of fragment lengths. This not only ensures the detection of multiple targets but also offers a potential amplification method for nucleic acids with extremely short lengths.
Multiplexed detection performance of the multi-STEM-MePCR system
The multiplexed detection of methylation sites in one tube has great clinical significance, as mentioned above, especially for samples with a low abundance of genomic DNA. The design of the TFP is a key factor in maintaining the high-efficiency multiplex amplification performance of the multi-STEM MePCR system. The TFP incorporates sequences from TSP and UP1. However, undesired cross-hybridization between the TFP and either UP1 or TSP can trigger significant non-specific amplification.To mitigate this risk, we introduced Spacer 18 and 2′Ome modification at the central and FRs of the TFP, respectively, to block unintended primer extension and cross-amplification. To verify the effectiveness of these modifications in inhibiting Taq polymerase activity, three single-stranded DNA templates were designed: unmodified, Spacer 18-modified, and 2′Ome modified. As shown in Fig. 4A, the unmodified template underwent efficient amplification, whereas no amplification was observed in the templates containing either Spacer 18 or 2′Ome. These results demonstrate that both modifications effectively inhibit polymerase extension and reduce the risk of cross-amplification in subsequent multiplex reactions.
 | ||
| Fig. 4 (A) Mechanism of TFP-mediated suppression of primer cross-amplification and validation of the extension-blocking performance by Spacer 18 and 2′Ome modifications. (B) Triple real-time amplification curves of multi-STEM-PCR on methylated sites of the Septin9, RASSF1, and SDC2 genes. Serially diluted methylated Jurkat genomic DNA at 1000, 250, and 50 copies per reaction were tested in the multi-STEM-PCR system, with free water as a negative control (NC). (C) Capillary electrophoresis of PCR products in the single and triple multi-STEM-PCR systems. (D) Comparison of Ct values between single (smooth bar) and triple (diagonal bar) reactions of the multi-STEM-PCR system at 1000, 250, and 50 copies per reaction. The data are expressed as the mean ± SD (n = 3). (E) Multiplex amplification results of different CpG sites on the SDC2 gene. | ||
For a proof-of-concept, the properties of the multi-STEM-MePCR system at multiplexed targets, including three methylated targets from the Septin9, RASSF1, and SDC2 genes, were examined in one tube using 1000, 250, and 50 copies per reaction of methylated genomic DNA with a triple repeat. As shown in Fig. 4B, the three methylation sites can all be detected from 1000 to 50 copies per reaction with good repeatability via the amplification curves, demonstrating the strong feasibility of multi-STEM-MePCR for simultaneous multiplexed methylation detection. Further analysis via capillary electrophoresis (Fig. 4C) revealed that the peak values for each CpG site in the triplex system closely matched those from single-plex systems, underscoring its excellent multiplexing efficiency.
To further evaluate the efficiency of the multiplexed system, the amplification results of the triplex detection system were compared with those from a single-plex reaction system. As shown in Fig. 4D, there is no significant difference in Ct values between the single-plex and triple-plex systems from 1000 to 50 copies per reaction at each site. The triplex and single-plex amplification systems demonstrate uniform amplification efficiency at each site, suggesting that primers can reach the optimal amplification performance without cross-interference from the components of the multiplex reaction.
The excellent performance in multiplexed detection by the multi-STEM-MePCR system was obtained by relying on the primers TFP, UP, and TSP. The function of TFPs involves a linear reaction, in which TFP molecules operate with high and uniform efficiency at lower concentrations. Modifications, such as extension blockers and 2′Ome bases assist in minimizing cross-reactions among different TFP molecules, as well as TFPs with other primers. The exponential amplification of multiplex targets is facilitated by UPs and TSPs, where the complexity of the primers is significantly reduced through the use of universal bases. This reduction in complexity further reduces cross-reactivity among primer pairs, thereby ensuring robust multiplex-detection capability of the multi-STEM-MePCR system.
Multiplex detection of multiple methylation sites within a single gene offers significant clinical utility for the diagnosis of specific cancer types. Accordingly, SDC2, a gene frequently associated with CRC, was selected as the target for further analysis. Three adjacent methylation sites within the SDC2 promoter region were simultaneously analyzed using the established multi-STEM-MePCR system. As illustrated in Fig. 4E, all three methylation sites were consistently and sensitively detected at input levels of 1000, 250, and 50 copies per test. These results demonstrate the system's robustness in detecting multiple epigenetic markers within a single gene and underscore its potential for clinical application in methylation-based diagnostics.
More importantly, unlike the traditional multiplexed detection of genomic DNA, where the copy number of different loci is the same, the methylation ratio significantly varies from site to site, sometimes by more than 100-fold. This variation in concentration can impact the performance of multiplexed detection (competitive inhibition) due to competition among targets (Fig. 5A).
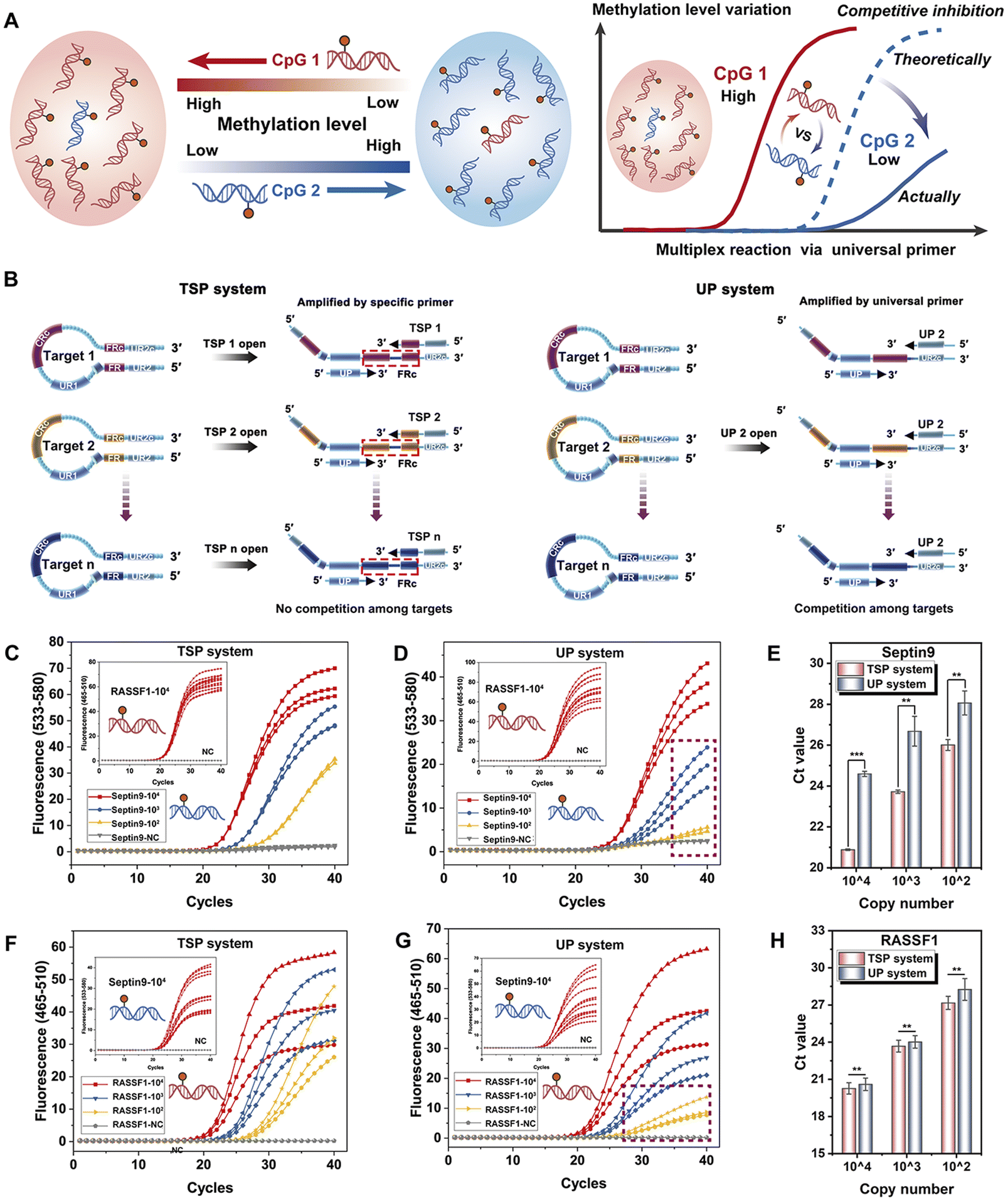 | ||
| Fig. 5 (A) Diagram illustrating the competitive reactions arising from differential methylation at various sites within a multiplex system. (B) The schema of the TSP system: the different HPs were opened by specific TSPs, and further amplified by TSPs and the UP. The UP system: the different HPs were all opened by UP1, and further amplified by UP1 and UP2. The amplification curves of serially diluted Septin9 methylated plasmid at 104, 103, and 102 copies per reaction with a background of 104 copies of RASS-F1 methylated plasmid in the (C) TSP system and the (D) UP system. (E) Comparison of Ct values from Septin9 at 104, 103, and 102 (copies per reaction) within 104 of RASSF1 between the TSP and UP systems. The amplification curves of serially diluted RASSF1 methylated plasmid at 104, 103, and 102 (copies per reaction) with a background of 104 copies of the Septin9 methylated plasmid in the (F) TSP system and the (G) UP system. (H) Comparison of Ct values from RASS-F1 at 104, 103, and 102 (copies per reaction) within 104 of Septin9 between the TSP and UP systems. The data are expressed as the mean ± SD (n = 3), p-values: **p < 0.01, ***p < 0.001. | ||
To address this, we propose a TSP with a tailored design to reduce the competition in the multiplexed reaction by introducing considerable differences in the methylation ratio. There are several specific bases from the FR at the 3′ end of the TSP. In the multiplexed reaction, different targets are independently amplified by the corresponding TSP and UP, thus reducing competition among different loci. To validate the theoretical assumptions and the irreplaceability of TSPs in the multiplexed system, a control primer without 3′ specific bases (UP2) was designed, where all targets were amplified by the two universal primers, similar to conventional universal primer designs (Fig. 5B). Specifically, the methylated plasmids containing the Septin9 gene were diluted from 104 copies to 102 copies against a background of RASSF1 plasmids (104 copies) to simulate samples with varying concentration gaps between targets.
As shown in Fig. 5C, all curves of the RASSF1 gene almost overlap, indicating the same background genes, and the S-standard amplification curves of the Septin9 gene can be obtained from 104 to 102 copies per reaction in the TSP system, demonstrating the excellent amplification efficiency of targets at low concentrations. However, in the system with two universal primers (the UP system) shown in Fig. 5D, the amplification curves of methylated Septin9 exhibit a suppressed state at low concentrations, where the amplification of 100 copies is nearly completely inhibited under the RASSF1 gene background, indicating the strong competitive reactions induced by high-concentration targets.
Further comparison of Ct values (Fig. 5E) reveals that across all concentrations, the Ct values of the TSP system were consistently lower than those of the UP system. This suggests that the design strategy of the TSP system not only mitigates competitive inhibition in multiplex reactions but also significantly improves the amplification efficiency. Similarly, when Septin9 plasmids were used as the background and the RASSF1 plasmids were diluted as the target, the same results were also obtained (Fig. 5F and G).
In Fig. 5H, under a concentration gradient, the TSP system demonstrates a reduction in Ct values as compared to the UP system, further corroborating the enhanced performance of the system. In the UP system, the universal primers are preferentially exhausted by the targets with high concentration, which ultimately inhibits the amplification of other low-abundance targets. However, in the TSP system, different targets were amplified by specific TSPs and a UP, without any interaction among the targets at the multiplexed amplification stage, thus avoiding the inhibition of targets at low concentration and ensuring the sensitivity of the system for different methylated targets. The multi-STEM-MePCR demonstrates excellent performance in methylation detection, especially for multi-targets with various methylation levels, and provides a powerful tool for the clinical application of methylation detection.
Conclusions
In this study, we describe a bisulfite-free, multiplexed methylation detection strategy, termed multi-STEM MePCR, which offers significant advantages over previous bisulfite-based methods. This technique demonstrates excellent quantitative capability, eliminates the need for cumbersome bisulfite pre-treatment, and reduces overall the reaction time to under three hours. Additionally, UDG, an antifouling reagent, can be used without any hesitation to overcome the limitations of bisulfite-conversion-based methylation methods in this regard, which is highly favored in clinical applications.Remarkably, this method can reliably detect as few as ten copies of rare analytes and 0.1% of methylated targets against a background of 10000 copies of unmethylated alleles, performing comparably to or even better than the most sensitive methods currently available. The system's flexibility with size allows it to virtually cover the entire range of MDRE digestion products, demonstrating excellent design universality. More importantly, the multi-STEM MePCR system serves as a robust multiplexed detection platform, where the cross-reaction and competition among different targets in one tube are effectively eliminated, enabling the highly efficient detection of multiple methylated targets even with varying methylation levels. This developed multi-STEM PCR technology paves a new and promising path for multiplexed methylation detection and is critical for basic research in epigenetics and the clinical application of DNA methylation.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
H. Yang, H. Xu, and G. Xu conceived the idea; H. Yang, H. Xu, and G. Xu designed the study; H. Yang, J. Qiu, Y. Xu, and W. Ren performed the experiments; H. Yang, H. Xu, G. Xu, and H. Gu analyzed and interpreted the results; L. Zhen tested the clinical FFPE samples; and H. Yang, H. Xu, G. Xu, and H. Gu were involved in the writing, review and editing of the manuscript. All authors reviewed and approved the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Key Projects of Special Development funds for the Shanghai Zhangjiang National Innovation Demonstration Zone (ZJ2021-ZD-007); Fundamental Research Funds for the Central Universities (20190201); Research Fund of Hefei Cancer Early Screening Innovation Technology Institute; and the China Postdoctoral Science Foundation under Grant Number 2023M742398.References
- P. D. Yousefi, M. Suderman, R. Langdon, O. Whitehurst, G. Davey Smith and C. L. Relton, DNA methylation-based predictors of health: applications and statistical considerations, Nat. Rev. Genet., 2022, 23, 369–383 CrossRef CAS PubMed
.
- S.-C. Kim, D.-W. Kim, E. J. Cho, J.-Y. Lee, J. Kim, C. Kwon, J. Kim-Ha, S. K. Hong, Y. Choi and N.-J. Yi, A circulating cell-free DNA methylation signature for the detection of hepatocellular carcinoma, Mol. Cancer, 2023, 22, 164 CrossRef CAS PubMed
- E. Post, 98P Translating cancer tissue methylation to cell-free DNA methylation for minimally invasive cancer detection, Ann. Oncol., 2024, 35, S253 CrossRef
- A. Koch, S. C. Joosten, Z. Feng, T. C. de Ruijter, M. X. Draht, V. Melotte, K. M. Smits, J. Veeck, J. G. Herman, L. Van Neste, W. Van Criekinge, T. De Meyer and M. van Engeland, Analysis of DNA methylation in cancer: location revisited, Nat. Rev. Clin. Oncol., 2018, 15, 459–466 CrossRef CAS PubMed
- H. Y. Luo, Q. Zhao, W. Wei, L. H. Zheng, S. H. Yi, G. Li, W. Q. Wang, H. Sheng, H. Y. Pu, H. Y. Mo, Z. X. Zuo, Z. X. Liu, C. F. Li, C. B. Xie, Z. L. Zeng, W. M. Li, X. K. Hao, Y. Y. Liu, S. M. Cao, W. L. Liu, S. Gibson, K. Zhang, G. L. Xu and R. H. Xu, Circulating tumor DNA methylation profiles enable early diagnosis, prognosis prediction, and screening for colorectal cancer, Sci. Transl. Med., 2020, 12, 11 Search PubMed
- Q. Gao, Y. Lin, B. Li, G. Wang, L. Dong, B. Shen, W. Lou, W. Wu, D. Ge and Q. Zhu, Unintrusive multi-cancer detection by circulating cell-free DNA methylation sequencing (THUNDER): development and independent validation studies, Ann. Oncol., 2023, 34, 486–495 CrossRef CAS PubMed
- C. Senore and N. Segnan, Multitarget stool DNA testing for colorectal-cancer screening, N. Engl. J. Med., 2014, 371, 187–188 Search PubMed
- C. Chen, X. Huang, W. Yin, M. Peng, F. Wu, X. Wu, J. Tang, M. Chen, X. Wang and A. Hulbert, Ultrasensitive DNA hypermethylation detection using plasma for early detection of NSCLC: a study in Chinese patients with very small nodules, Clin. Epigenet., 2020, 12, 39 CrossRef CAS
- D. A. Ahlquist, H. Zou, M. Domanico, D. W. Mahoney, T. C. Yab, W. R. Taylor, M. L. Butz, S. N. Thibodeau, L. Rabeneck and L. F. Paszat, Next-Generation Stool DNA Test Accurately Detects Colorectal Cancer and Large Adenomas, Gastroenterology, 2012, 142, 248–256 CrossRef CAS
- J. Ibrahim, M. Peeters, G. Van Camp and K. O. de Beeck, Methylation biomarkers for early cancer detection and diagnosis: current and future perspectives, Eur. J. Cancer, 2023, 178, 91–113 CrossRef CAS PubMed
- T. Wang, P. Li, Q. Qi, S. Zhang, Y. Xie, J. Wang, S. Liu, S. Ma, S. Li and T. Gong, A multiplex blood-based assay targeting DNA methylation in PBMCs enables early detection of breast cancer, Nat. Commun., 2023, 14, 4724 CrossRef CAS
- E. Y. Rykova, A. A. Ponomaryova, I. A. Zaporozhchenko, V. V. Vlassov, N. V. Cherdyntseva and P. P. Laktionov, Circulating DNA-based lung cancer diagnostics and follow-up: looking for epigenetic markers, Transl. Cancer Res., 2018, 7, S153–S170 CrossRef CAS
- D. W. Cescon, S. V. Bratman, S. M. Chan and L. L. Siu, Circulating tumor DNA and liquid biopsy in oncology, Nat. Cancer, 2020, 1, 276–290 CrossRef CAS PubMed
- P. W. Laird, Principles and challenges of genomewide DNA methylation analysis, Nat. Rev. Genet., 2010, 11, 191–203 CrossRef CAS PubMed
- M. Berdasco and M. Esteller, Clinical epigenetics: seizing opportunities for translation, Nat. Rev. Genet., 2019, 20, 109–127 CrossRef CAS
- H. Hayatsu, Y. Wataya and K. Kazushige, The addition of sodium bisulfite to uracil and to cytosine, J. Am. Chem. Soc., 1970, 92, 724–726 CrossRef CAS PubMed
- N. Liang, B. Li, Z. Jia, C. Wang, P. Wu, T. Zheng, Y. Wang, F. Qiu, Y. Wu, J. Su, J. Xu, F. Xu, H. Chu, S. Fang, X. Yang, C. Wu, Z. Cao, L. Cao, Z. Bing, H. Liu, L. Li, C. Huang, Y. Qin, Y. Cui, H. Han-Zhang, J. Xiang, H. Liu, X. Guo, S. Li, H. Zhao and Z. Zhang, Ultrasensitive detection of circulating tumour DNA via deep methylation sequencing aided by machine learning, Nat. Biomed. Eng., 2021, 5, 586–599 CrossRef CAS PubMed
- Q. Zhang, S. Ma, Z. Liu, B. Zhu, Z. Zhou, G. Li, J. J. Meana, J. González-Maeso and C. Lu, Droplet-based bisulfite sequencing for high-throughput profiling of single-cell DNA methylomes, Nat. Commun., 2023, 14, 4672 CrossRef CAS
- Q. Dai, C. Ye, I. Irkliyenko, Y. Wang, H.-L. Sun, Y. Gao, Y. Liu, A. Beadell, J. Perea and A. Goel, Ultrafast bisulfite sequencing detection of 5-methylcytosine in DNA and RNA, Nat. Biotechnol., 2024, 1–12 Search PubMed
- M. J. Ellington, O. Ekelund, F. M. Aarestrup, R. Canton, M. Doumith, C. Giske, H. Grundman, H. Hasman, M. T. G. Holden, K. L. Hopkins, J. Iredell, G. Kahlmeter, C. U. Koser, A. MacGowan, D. Mevius, M. Mulvey, T. Naas, T. Peto, J. M. Rolain, O. Samuelsen and N. Woodford, The role of whole genome sequencing in antimicrobial susceptibility testing of bacteria: report from the EUCAST Subcommittee, Clin. Microbiol. Infect., 2017, 23, 2–22 CrossRef CAS
- R. C. Fitzgerald, A. C. Antoniou, L. Fruk and N. Rosenfeld, The future of early cancer detection, Nat. Med., 2022, 28, 666–677 CrossRef CAS
- C. Liu, J. Zhao, F. Tian, L. Cai, W. Zhang, Q. Feng, J. Chang, F. Wan, Y. Yang, B. Dai, Y. Cong, B. Ding, J. Sun and W. Tan, Low-cost thermophoretic profiling of extracellular-vesicle surface proteins for the early detection and classification of cancers, Nat. Biomed. Eng., 2019, 3, 183–193 CrossRef CAS PubMed
- J. G. Herman, J. R. Graff, S. Myohanen, B. D. Nelkin and S. B. Baylin, Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 9821–9826 CrossRef CAS
- C. A. Eads, K. D. Danenberg, K. Kawakami, L. B. Saltz, C. Blake, D. Shibata, P. V. Danenberg and P. W. Laird, MethyLight: a high-throughput assay to measure DNA methylation, Nucleic Acids Res., 2000, 28, E32 CrossRef CAS PubMed
- H. Zou, A. Hatim, X. Cao, D. Mike, H. Jonathan, W. R. Taylor, Y. Tracy, D. A. Ahlquist and L. Graham, Quantification of methylated markers with a multiplex methylation-specific technology, Clin. Chem., 2012, 375–383 CrossRef CAS
-
M. J. Fackler and S. Sukumar, Quantitation of DNA Methylation by Quantitative Multiplex Methylation-Specific PCR (QM-MSP) Assay, DNA Methylation Protocols, 2018 Search PubMed
- A. Nygren, A. Najim, H. Duarte, R. Vijzelaar, W. Quinten, C. J. Hess, J. P. Schouten and E. Abdellatif, Methylation-Specific MLPA (MS-MLPA): simultaneous detection of CpG methylation and copy number changes of up to 40 sequences, Nucleic Acids Res., 2005, 33, e128 CrossRef PubMed
- C. Dahl and P. Guldberg, A ligation assay for multiplex analysis of CpG methylation using bisulfite-treated DNA, Nucleic Acids Res., 2007, 35, e144 CrossRef PubMed
- Z. X. Xiao, H. M. Cao, X. H. Luan, J. L. Zhao and J. H. Xiao, Effects of additives on efficiency and specificity of ligase detection reaction, Mol. Biotechnol., 2007, 35, 129–133 CrossRef CAS PubMed
- Y. Y. Sun, X. H. Lu, F. X. Su, L. M. Wang, C. H. Liu, X. R. Duan and Z. P. Li, Real-time fluorescence ligase chain reaction for sensitive detection of single nucleotide polymorphism based on fluorescence resonance energy transfer, Biosens. Bioelectron., 2015, 74, 705–710 CrossRef CAS PubMed
- W. Y. Huang, S. D. Hsu, H. Y. Huang, Y. M. Sun, C. H. Chou, S. L. Weng and H. D. Huang, MethHC: a database of DNA methylation and gene expression in human cancer, Nucleic Acids Res., 2015, 43, D856–D861 CrossRef CAS PubMed
- N. G. Xie, M. X. Wang, P. Song, S. Mao, Y. Wang, Y. Yang, J. Luo, S. Ren and D. Y. Zhang, Designing highly multiplex PCR primer sets with simulated annealing design using dimer likelihood estimation (SADDLE), Nat. Commun., 2022, 13, 1881 CrossRef CAS PubMed
- D. P. Genereux, W. C. Johnson, A. F. Burden, S. Reinhard and C. D. Laird, Errors in the bisulfite conversion of DNA: modulating inappropriate- and failed-conversion frequencies, Nucleic Acids Res., 2008, 36, e150 CrossRef PubMed
- T. Gong, H. Borgard, Z. Zhang, S. Chen, Z. Gao and Y. Deng, Analysis and performance assessment of the whole genome bisulfite sequencing data workflow: currently available tools and a practical guide to advance DNA methylation studies, Small Methods, 2022, 6, 2101251 CrossRef PubMed
- D. Hua, Y. Hu, Y.-Y. Wu, Z.-H. Cheng, J. Yu, X. Du and Z.-H. Huang, Quantitative methylation analysis of multiple genes using methylation-sensitive restriction enzyme-based quantitative PCR for the detection of hepatocellular carcinoma, Exp. Mol. Pathol., 2011, 91, 455–460 CrossRef CAS
- S. Yegnasubramanian, X. Lin, M. C. Haffner, A. M. DeMarzo and W. G. Nelson, Combination of methylated-DNA precipitation and methylation-sensitive restriction enzymes (COMPARE-MS) for the rapid, sensitive and quantitative detection of DNA methylation, Nucleic Acids Res., 2006, 34, e19 CrossRef PubMed
- R. Viswanathan, E. Cheruba, P.-M. Wong, Y. Yi, S. Ngang, D. Q. Chong, Y.-H. Loh, I. B. Tan and L. F. Cheow, DARESOME enables concurrent profiling of multiple DNA modifications with restriction enzymes in single cells and cell-free DNA, Sci. Adv., 2023, 9, eadi0197 CrossRef CAS PubMed
- P. Pataer, P. Zhang and Z. Li, Single Methylation Sensitive Restriction Endonuclease-Based Cascade Exponential Amplification Assay for Visual Detection of DNA Methylation at Single-Molecule Level, Anal. Chem., 2024, 96, 13335–13343 CrossRef CAS PubMed
- X. Chen, J. Huang, S. Zhang, F. Mo, S. Su, Y. Li, L. Fang, J. Deng, H. Huang, Z. Luo and J. Zheng, Electrochemical Biosensor for DNA Methylation Detection through Hybridization Chain-Amplified Reaction Coupled with a Tetrahedral DNA Nanostructure, ACS Appl. Mater. Interfaces, 2019, 11, 3745–3752 CrossRef CAS PubMed
- J. Cai and Q. Zhu, New advances in signal amplification strategies for DNA methylation detection in vitro, Talanta, 2024, 273, 125895 CrossRef CAS
- X. Liu, J. Zhang, Y. Cai, S. Zhang, K. Ma, K. Hua and Y. Cui, A novel DNA methylation biosensor by combination of isothermal amplification and lateral flow device, Sens. Actuators, B, 2021, 333, 129624 CrossRef CAS
- J. Goris, K. T. Konstantinidis, J. A. Klappenbach, T. Coenye and J. M. Tiedje, DNA-DNA hybridization values and their relationship to whole-genome sequence similarities, Int. J. Syst. Evol. Microbiol., 2007, 57, 81 CrossRef CAS PubMed
- K. N. Rand, G. P. Young, T. Ho and P. L. Molloy, Sensitive and selective amplification of methylated DNA sequences using helper-dependent chain reaction in combination with a methylation-dependent restriction enzymes, Nucleic Acids Res., 2013, 41, e15 CrossRef CAS PubMed
- Y. Sun, Y. Sun, W. Tian, C. Liu, K. Gao and Z. Li, A novel restriction endonuclease GlaI for rapid and highly sensitive detection of DNA methylation coupled with isothermal exponential amplification reaction, Chem. Sci., 2018, 9, 1344–1351 RSC
- G. Xu, H. Yang, J. Qiu, J. Reboud, L. Zhen, W. Ren, H. Xu, J. M. Cooper and H. Gu, Sequence terminus dependent PCR for site-specific mutation and modification detection, Nat. Commun., 2023, 14, 1169 CrossRef CAS PubMed
- J. D. Warren, W. Xiong, A. M. Bunker, C. P. Vaughn, L. V. Furtado, W. L. Roberts, J. C. Fang, W. S. Samowitz and K. A. Heichman, Septin 9 methylated DNA is a sensitive and specific blood test for colorectal cancer, BMC Med., 2011, 9, 9 CrossRef
- B. H. Yoo, E. Bochkareva, A. Bochkarev, T. C. Mou and D. M. Donald, 2’-O-methyl-modified phosphorothioate antisense oligonucleotides have reduced non-specific effects in vitro, Nucleic Acids Res., 2004, 32(6), 2008–2016 CrossRef CAS PubMed
- H. Kanzaki, H. Hanafusa, H. Yamamoto, Y. Yasuda, K. Imai, M. Yano, M. Aoe, N. Shimizu, K. Nakachi, M. Ouchida and K. Shimizu, Single nucleotide polymorphism at codon 133 of the RASSF1 gene is preferentially associated with human lung adenocarcinoma risk, Cancer Lett., 2006, 238, 128–134 CrossRef CAS PubMed
- J. Chen, H. P. Sung, W. S. Tang, L. Zhou, X. Xie, Z. Qu, M. F. Chen, S. Y. Wang, T. Yang, Y. Dai, Y. L. Wang, T. J. Gao, Q. Zhou, Z. Song, M. M. Liao and W. D. Liu, DNA methylation biomarkers in stool for early screening of colorectal cancer, J. Cancer, 2019, 10, 5264–5271 CrossRef CAS PubMed
- D. Cohen-Karni, D. Xu, L. Apone, A. Fomenkov, Z. Y. Sun, P. J. Davis, S. R. M. Kinney, M. Yamada-Mabuchi, S. Y. Xu, T. Davis, S. Pradhan, R. J. Roberts and Y. Zheng, The MspJI family of modification-dependent restriction endonucleases for epigenetic studies, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11040–11045 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc01429h |
| This journal is © The Royal Society of Chemistry 2025 |