
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTailoring dual-hydrophobic microenvironment for tandem CO2/CO feedstock to enhance CO2 electroreduction on Cu-based catalysts†
Tete
Zhao
a,
Ziyao
Zhou
a,
Xiao
Wang
a,
Kuiming
Liu
a,
Jialei
Chen
a,
Kang
Liu
a,
Yue
Li
a,
Meng
Yu
 *a,
Yan
Xu
*b,
Zhenhua
Yan
a,
Lifang
Jiao
a and
Fangyi
Cheng
*a
*a,
Yan
Xu
*b,
Zhenhua
Yan
a,
Lifang
Jiao
a and
Fangyi
Cheng
*a
aState Key Laboratory of Advanced Power Sources, Key Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), Engineering Research Center of High-Efficiency Energy Storage (Ministry of Education), Frontiers Science Center for New Organic Matter, College of Chemistry, Nankai University, Tianjin 300071, China. E-mail: nkyu2023@nankai.edu.cn; fycheng@nankai.edu.cn
bCollege of Energy Storage Technology, Shandong University of Science and Technology, Qingdao, Shandong 266590, China. E-mail: xuyan2020@sdust.edu.cn
First published on 27th June 2025
Abstract
Achieving high selectivity for value-added products in the electrochemical reduction of CO2 remains challenging due to severe hydrogen evolution, sluggish CO2 mass transport and low *CO coverage. Herein, we integrate aerophilic SiO2 and polymer-functionalized copper nanoparticles (Cu-poly) to construct a hierarchical-hydrophobic Cu-poly/SiO2 composite, which limits the accessibility of H2O, improves the local concentration of CO2 and enhances the dimerization of *CO–*CO. Comprehensive investigation using X-ray absorption spectroscopy, in situ infrared spectroscopy and molecular dynamics simulations indicates that the polymer and SiO2 elevate the oxidation state of Cu species, enhance the CO2 diffusion coefficients (from 5.27 × 10−7 on Cu to 8.81 × 10−7 cm2 s−1 on Cu-poly/SiO2) and enrich the local *CO concentration. The Cu-poly/SiO2 electrode delivers an enhanced faradaic efficiency of 60.54% for C2+ products, compared to 46.1% of Cu at 600 mA cm−2. Notably, a high FE of 36.91% and partial current density of 221.46 mA cm−2 are achieved for C2H4 generation in membrane electrode assembly devices adopting an aqueous bicarbonate electrolyte. This work provides a valuable insight into designing catalytic microenvironments of electrocatalysts for enhancing carbonaceous products by facilitating the co-electrolysis of CO2 and in situ-generated *CO.
Introduction
Electrochemical CO2 reduction reaction (CO2RR) to value-added chemicals and fuels using renewable electricity on Cu-based electrocatalysts provides a promising strategy to achieve carbon neutrality.1–5 However, severe flooding of the gas diffusion layer (GDL), poor *CO–*CO coupling and sluggish CO2 mass transport impede the high-efficiency formation of multicarbon (C2+) products.6–9 An appealing method is to regulate the reaction microenvironment of catalysts, including the ratio of H2O and CO2, the local coverage of *CO, and the concentration of cations through molecular engineering, functionalized polymers, ionomers, and similar modifications. This limits H2O accessibility, enhances CO2 diffusion dynamics and stabilizes key intermediates, ultimately improving the formation rate of carbonaceous products.10–17 Although these strategies accelerate the generation of value-added products in a three-electrode system, they still confront obstacles when employed as a membrane electrode assembly (MEA) electrolyzer to achieve a commercial current density for CO2RR. Moreover, the hydrophobicity of the catalytic layer (CL) and CO2 mass transfer channel in GDL are susceptible to destruction due to the organically modified layers peeling off, caused by rapid evolution of product bubbles.Furthermore, an alternative strategy to tailor the reaction microenvironments involves the direct incorporation of hydrophobic additives, such as polytetrafluoroethylene (PTFE), polyvinylidene fluoride (PVDF), pyridine-containing microgels, graphene, metal–organic frameworks (MOFs) and covalent organic frameworks (COFs), into the vicinity of electrocatalysts, which improves the mass transfer of CO2, the local pH and coverage of CO, thus enhancing the efficiency of CO2 electroreduction.18–22 Although these additives could optimize the microenvironments of catalysts, the electrochemical-adsorption/desorption behavior of reactant and intermediates on these additional promoters is still poorly understood, leading to undesirable reaction pathways, decreasing the selectivity for C2+ products. More importantly, the organic backbone of additives can become deactivated and gradually dissolve into the electrolyte under an electric field during prolonged electrolysis. This mainly occurs due to the polarity of H2O through the anion exchange membrane (AEM) from the anolyte, reducing the organic fluorine content and thus increasing hydrophilicity.23,24 Therefore, it is challenging to achieve the hydrophobic requirements of the CL of gas diffusion electrodes (GDEs) by relying solely on additives under industrial-grade current electrolysis.
Herein, we report the integration of inorganic SiO2 with robust hydrophobicity into the CL of polymer-functionalized Cu nanoparticles (Cu-poly) towards efficient CO2RR. Dual-hydrophobic catalytic characteristics are designed for Cu-poly/SiO2 composites by considering both the surface and interface. This approach combines modification and additive engineering strategies to enhance CO2 electroreduction. Our results demonstrate that the production rate of C2H4 is significantly improved on the dual-hydrophobic Cu-poly/SiO2 electrodes compared to bare Cu, surface-modified Cu-poly and Cu/SiO2 electrodes in MEA devices by suppressing the hydrogen evolution reaction (HER). Meanwhile, we investigate the effect of polymer and SiO2 on the surface oxidation state of Cu and the composition of CO2RR products. The positive effect of the local concentration of CO2/H2O and in situ-generated CO on the production of C2H4 is investigated by a comprehensive combination of spectroscopic analysis, electrochemical performance evaluation and molecular dynamics (MD) simulation results. The SiO2 and polymer components are favorable for enriching and accelerating CO2 mass transfer, limiting H2O accessibility and elevating local CO concentration. This work elucidates the engineering of dual-hydrophobic microenvironments via integration of surface and interface modifications on electrocatalysts, providing further perspectives on the co-electrolysis mechanism of CO2 and CO in modified-Cu electrodes towards C2H4.
Results and discussion
To design the dual-hydrophobic catalytic microenvironments, bare Cu nanoparticles were synthesized by a facile liquid-phase reduction method using sodium borohydride.25 The fluorine-polymer-functionalized Cu (Cu-poly) was prepared by in situ radical polymerization under the initiation of azobisisobutyronitrile with precursor molecules of trifluoromethyl methacrylate and 3-(methacryloyloxy)propyltrimethoxysilane.26 Afterwards, the obtained Cu-poly and hydrophobic SiO2 were ultrasonically mixed to carefully fabricate the Cu-poly/SiO2 composites. Fig. 1a and b comparatively illustrate the different catalytic microenvironments of bare Cu and Cu-poly/SiO2 composites. For hydrophilic GDEs, the surface of Cu is prone to being wetted by an aqueous electrolyte, resulting in sluggish CO2 mass transfer, excessive H2O supply and low *CO coverage (Fig. 1a). It is reasonable that the HER would be more favourable on hydrophilic Cu than the CO2RR.20 In contrast, the GDEs covered by Cu-poly/SiO2 consist of aerophilic SiO2 and polymer-functionalized Cu, forming abundant gas–liquid–solid three-phase interfaces and benefiting the mass transfer of reactants (Fig. 1b). SiO2 and polymer endow Cu-poly/SiO2 with robust hydrophobicity from the interfaces and surface, respectively. These characteristics play multiple roles in facilitating the mass transfer of CO2 and decreasing the local concentration of H2O, due to the interconnected gas diffusion networks and strong hydrophobicity. More importantly, the dual-hydrophobicity reaction microenvironment originating from the polymer and SiO2 could accelerate the in situ generation of CO during CO2RR, owing to the absence of H2O on the surface of Cu. Consequently, Cu-poly/SiO2 promotes the local concentration of CO on Cu and then achieves the co-electrolysis of CO2 generating CO, which would contribute to the highly efficient generation of C2+ products.27 | ||
| Fig. 1 Catalyst design, preparation and characterization. Schematic illustration of the hydrophilic microenvironments of (a) Cu and (b) the dual-hydrophobic microenvironment of Cu-poly/SiO2. (c and d) SEM images of (c) Cu and (d) Cu-poly/SiO2. (e) CO2 adsorption/desorption isotherm curves of Cu and Cu-poly/SiO2. (f) FTIR spectra of Cu and Cu-poly/SiO2. | ||
As shown in scanning electron microscopy (SEM, Fig. 1c) and high-resolution transmission electron microscopy (HRTEM, Fig. S1a, ESI†) images, the prepared Cu presents an irregular spherical morphology, accompanied by the minor facets of (111) and (220) of Cu2O and the major facets of (111), (200) and (220) of Cu.28 The phase structure and corresponding facets are also revealed by the X-ray diffraction (XRD) patterns, HRTEM, and selected-area electron diffraction (SAED) images (Fig. S1b–f and S2, ESI†). Furthermore, high-angle annular dark-field STEM (HADDF-STEM, Fig. S3a–c, ESI†) images also reveal the irregular morphology and exposed facets. The corresponding elemental mapping images show that Cu and O are uniformly distributed (Fig. S3d–f, ESI†). To evaluate the stability of hydrophobicity for Cu and Cu-poly/SiO2, we employed the ultrasonication method to compare their behaviours. The results show that the Cu powder quickly disperses in H2O after 30 min, whereas due to the combined effect of the polymer and SiO2, Cu-poly/SiO2 remains floating on the surface of H2O after 30 min, demonstrating stable hydrophobicity (Fig. S4, ESI†). Benefiting from the durable hydrophobic characteristics, Cu-poly/SiO2 offers an abundant reaction vicinity to adsorb and activate CO2 molecules. To reveal the synergistic effects of hydrophobic SiO2 and polymer on CO2 adsorption, we conducted the measurement of CO2 adsorption–desorption isotherms under ice-water mixtures, and the results show that Cu-poly/SiO2 possesses an enhanced adsorption capacity of CO2 compared to Cu according to the normalized adsorption value (0.108 on Cu-poly/SiO2vs. 0.075 on Cu) by the surface area at the fixed pressure of P/P0 = 1 (Fig. 1e and S5, ESI†).
FTIR spectra of Cu and Cu-poly/SiO2 were recorded to further investigate the surface composition and bonding information. As depicted in Fig. 1f, the broad peaks corresponding to the stretching (3436 cm−1) and bending vibrations (1632 cm−1) of H2O are clearly observed on bare Cu.29 In comparison, these signals are not obvious on Cu-poly/SiO2, which indicates that the polymer and SiO2 could limit the adsorption of H2O. Notably, there is a sharp peak located at 617 cm−1, which could be attributed to the stretching vibration of the Cu–O bond in Cu2O, which further confirms the oxidation of Cu.30 However, the wavenumber of the Cu–O bond shifts from 617 to 635 cm−1 and the stretching vibration of Cu–O is stronger in Cu-poly/SiO2 than that in Cu, implying that Cu-poly/SiO2 is more readily oxidized due to polymerization. This result is further supported by the vibration of the Cu–O bond in the Raman spectra of Cu and Cu-poly/SiO2 (Fig. S6, ESI†). Furthermore, the vibrations located at 467 and 1099 cm−1 are attributed to the Si–O bonds, confirming the incorporation of SiO2 into Cu-poly/SiO2.31 More importantly, the peak located at 1742 cm−1 is attributed to the vibrations of –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O in the polymer that coats the surface of Cu.32
O in the polymer that coats the surface of Cu.32
To examine the microstructure, aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC-HAADF-STEM) images were collected on Cu-poly/SiO2. The resulting images reveal that the catalysts exhibit a polycrystalline structure containing both Cu and Cu2O phases (Fig. S2, ESI†), along with abundant pores generated by the aggregation of Cu-poly and SiO2. These pores facilitate the efficient transport of CO2 while limiting the accessibility of H2O (Fig. 2a and S7a, b, ESI†). Furthermore, the high-resolution AC-HAADF-STEM image clearly identifies well-defined interfaces between Cu2O, the polymer and SiO2. Notably, the oxidized Cu particles are tightly encapsulated by the polymer with the interface, indicating that the polymer-coated Cu interacts with SiO2 (Fig. 2b). In addition, the corresponding annular bright field (ABF)-STEM images further confirm these observations, suggesting that this structure may inhibit hydrogen production while activating inert CO2 molecules.33 The observed lattice fringes exhibit an interplanar spacing of 0.24 nm, which is ascribed to the (111) facet of Cu2O (Fig. 2c and S7c, d, ESI†).34 Energy dispersive spectrometer (EDS) mapping analysis reveals a homogeneous distribution of Cu, Si, F, C, and O across Cu-poly/SiO2 (Fig. 2d–i and S8, ESI†).
 | ||
| Fig. 2 Structural characterization of Cu-poly/SiO2. (a and b) HAADF-STEM images. (c) Annular bright field (ABF)-STEM image. (d–i) Elemental mapping images of Cu-poly/SiO2. Colors in (d–i): Cu green, O yellow, Si pink, C red, F cyan. | ||
Additionally, we also measured the solid nuclear magnetic resonance (NMR) spectrum of 13C, 19F and 1H to analyze the surface elemental composition after polymerization in Cu-poly. The 13C spectra show five sharp peaks located at chemical shifts of 176.7, 125.5, 61.5, 45.8, and 18.5 ppm, corresponding to the functional groups of –O–CO, –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N, –O–C, and the carbon in the main chains of polymer (–CH2/–CH/–C) and –CH3, respectively (Fig. 3a). Furthermore, the 19F and 1H spectra were also recorded on Cu-poly (Fig. S9, ESI†). These NMR results indicate that the polymer successfully coated the surface of Cu, a conclusion also corroborated by HADDF-STEM images and Fourier-transform infrared (FTIR) spectra, as shown in Fig. 1f and 2a–d. In addition, the XPS surveys of Si and F spectra also indicate that the polymer is coated on the surface of Cu (Fig. S10a–c, ESI†).
N, –O–C, and the carbon in the main chains of polymer (–CH2/–CH/–C) and –CH3, respectively (Fig. 3a). Furthermore, the 19F and 1H spectra were also recorded on Cu-poly (Fig. S9, ESI†). These NMR results indicate that the polymer successfully coated the surface of Cu, a conclusion also corroborated by HADDF-STEM images and Fourier-transform infrared (FTIR) spectra, as shown in Fig. 1f and 2a–d. In addition, the XPS surveys of Si and F spectra also indicate that the polymer is coated on the surface of Cu (Fig. S10a–c, ESI†).
 | ||
| Fig. 3 Surface oxidation and chemical valence of Cu. (a) Solid NMR spectrum of 13C. (b) Cu Auger LMM spectra of Cu and Cu-poly/SiO2. (c and d) Normalized Cu K-edge XANES spectra of Cu, Cu-poly, Cu-poly/SiO2 and Cu-foil, Cu2O, CuO standard. (e) Average oxidation state of Cu in Cu, Cu-poly and Cu-poly/SiO2 based on Cu foil, Cu2O and CuO references. (f) Cu K-edge Fourier transform (FT) EXAFS spectra of Cu, Cu-poly, Cu-poly/SiO2 and Cu-foil, Cu2O, CuO standard. (g) Morlet wavelet transforms contour plots for the k3-weighted EXAFS spectra of Cu foil, Cu, Cu-poly and Cu-poly/SiO2. | ||
Previous studies have highlighted the key role of the surface oxidation state of Cu in regulating selectivity towards C2+ products.35 To investigate the effect of SiO2 and polymer on this feature, we comparatively analyzed the chemical valence of Cu species in Cu, Cu-poly and Cu-poly/SiO2 using X-ray photoelectron spectroscopy (XPS) and X-ray absorption spectroscopy (XAS). Auger LMM spectra of Cu exhibit an obvious shift in binding energy from 569.9 eV of bare Cu to 570.2 eV of Cu-poly/SiO2, indicating a higher oxidation state of Cu in Cu-poly/SiO2 (Fig. 3b and S10d, ESI†). The X-ray absorption near-edge structure (XANES) spectra of Cu K-edge demonstrate that the absorption edge positions of Cu, Cu-poly, Cu-poly/SiO2 and all standard samples are in alignment with that of Cu foil (Fig. 3c). Further analysis of the locally enlarged XANES spectra show that the absorption edges of Cu in Cu, Cu-poly and Cu-poly/SiO2 are all located between those of Cu foil, Cu2O and CuO reference samples, indicating that the Cu species are in a positive valence between 0 and +2.0 (Fig. 3d). Based on the linear correlation depicted in Fig. 3e, which relates the energy position of standard samples (Cu foil, Cu2O, and CuO) to their oxidation states, the Cu valences in Cu, Cu-poly and Cu-poly/SiO2 are estimated to be +0.64, +0.72, and +1.10, respectively. The higher oxidation of Cu in Cu-poly/SiO2, compared to Cu and Cu-poly can be primarily attributed to oxygenated interface of SiO2.36
The Fourier-transformed extended X-ray absorption fine structure (FT-EXAFS) spectra consistently reveal the Cu–Cu and Cu–O scattering paths for Cu, Cu-poly, and Cu-poly/SiO2, exhibiting patterns analogous to those observed in Cu foil (Fig. 3f). Notably, the intensity of the Cu–Cu shell in Cu-poly is higher than that observed in Cu, indicating an increase in the coordination number (CN) of the Cu atom after polymerization. After introducing SiO2, whose surface promotes the enrichment of oxidized species, the signal intensity of the Cu–O shell layer in Cu-poly/SiO2 increases further, closely approaching the level of CuO. These variations in CN and bond length suggest that Cu coordination can be modulated through the incorporation of polymer and SiO2. The scattering paths of Cu–O in Cu exhibit the same trend as those in Cu2O, indicating that the Cu–O bond length in Cu is consistent with that in Cu2O. However, the bond length in Cu-poly and Cu-poly/SiO2 is notably increased, approaching that of the CuO reference, pointing to a more pronounced oxidation state, resulting from polymerization and the addition of SiO2. In addition to the FT-EXAFS analysis, we also employed the Morlet Wavelet Transform (WT) to simultaneously examine the information in both R-space and k-space. As shown in Fig. 3g, the WT-EXAFS contour map for Cu foil, Cu, Cu-poly, and Cu-poly/SiO2 clearly differentiate peaks corresponding to Cu–O and Cu–Cu bonds in R-space. The peak position of the Cu–Cu bond in Cu and Cu-poly are lower than that in Cu foil, indicating the involvement of O atoms in coordination, leading to a shift in k-space to lower values. Notably, the peak position in k-space for Cu is even lower than those of Cu-poly composites, while the Cu–Cu peak in Cu-poly/SiO2 appears at a higher k-space position than Cu foils, Cu and Cu-poly. This difference implies that, in addition to oxygen, heavier silicon atoms also coordinate with Cu.33
Apart from the information about the physical and chemical structures of catalysts, we also evaluated the electrocatalytic performances on Cu and Cu-poly/SiO2 electrodes with the different reaction microenvironments using an MEA cell, benefiting from the characteristics of the zero-gap construction, freedom from catholyte and CO2 direct electroreduction.37 Fig. S11 and S12, ESI† illustrate the schematic design of the MEA device, along with a depiction of its operational conditions, including the detailed parameters for the GDL, AEM, electrolyte and anode. Cyclic voltammetry (CV) analysis reveals that the current density of HER on the exposed Cu (−10.85 mA cm−2) is obviously higher than that of Cu-poly/SiO2 (−2.67 mA cm−2) under the same applied voltage of 2.0 V (Fig. 4a). This suggests that the dual-hydrophobic Cu-poly/SiO2 effectively stops H2O from reaching the surface of Cu-poly. Corresponding results obtained in an argon atmosphere for Cu, Cu-poly/SiO2, Cu-poly, and Cu/SiO2 further support that polymer modification and SiO2 could suppress the rate of HER (Fig. S13, ESI†). On the one hand, hydrophobic SiO2 repels H2O from the bulk electrolyte, preventing it from contacting the catalyst surface, thereby reducing the coverage of H2O. On the other hand, the polymer also repels H2O through the organic silane functional groups in its structure, further lowering the H2O content on the surface of Cu-poly/SiO2. However, introducing SiO2 and polymer inevitably leads to partial coverage of the active sites, which consequently diminishes the reduction current of CO2RR (Fig. 4b and S14, ESI†). These findings indicate the dual-hydrophobic microenvironments of Cu-poly/SiO2 reduces the local concentration of H2O, restricting its interaction with catalytic sites, which in turn reduces the hydrogen production rate. A similar trend is also observed on Cu and Cu-poly/SiO2 when using traditional H-type electrolytic cells (Fig. S15 and S16, ESI†). As depicted in comparative linear sweep voltammetry (LSV) curves of Cu and Cu-poly/SiO2, an obvious decrease in total current density of CO2RR is observed on Cu-poly/SiO2 in contrast to bare Cu at the same cell potentials (Fig. 4b). This reduction is attributed to a decrease in electrochemically active surface area (Fig. S17, ESI†).
 | ||
| Fig. 4 Electrocatalytic performance of Cu and Cu-poly/SiO2. (a) CV and (b) LSV curves of Cu and Cu-poly/SiO2 in 1 M KHCO3. (c and d) Faradaic efficiency (FE) for (c) Cu and (d) Cu-poly/SiO2. (e and f) FE of CO and C2H4 on (e) Cu and (f) Cu-poly/SiO2 at various current densities. | ||
To determine the composition of gaseous products, an online gas chromatograph (GC) equipped with a thermal conductivity detector (TCD) and flame ionization detector (FID) was employed. The composition of gaseous products was studied on the different electrodes every 11 min under chronopotentiometry with the current density ranging from 200 to 600 mA cm−2. The primary gas products are H2, CO, and C2H4, over the four main catalysts. The faradaic efficiency (FE) of H2 over Cu increases from 40.56% at 200 mA cm−2 to 51.18% at 600 mA cm−2, while the FE of C2+ products rises from 34.08% to 46.11%, accompanied by a partial current density of C2H4 from 48.78 to 140.52 at the same current density. Notably, the FE of CO exhibits a lesser extent of variation, fluctuating between 4.07% and 12.88% from 300 to 600 mA cm−2. Unfortunately, the total FEs of products for both Cu and Cu-poly/SiO2 fluctuate around 100%, due to variations in the volume of the liquid electrolyte during collection procedures (Fig. 4c). For Cu-poly and Cu/SiO2, the FE of H2 remains relatively stable at lower current densities but increases at high current densities (Fig. S18, ESI†), due to the individual polymer or SiO2 incompletely limiting the accessibility of H2O and enhancing the local concentration of CO2. In comparison, the FE of H2 over Cu-poly/SiO2 ranges from 17.74% at 300 to 20.50% at 200 mA cm−2, while the FE of C2+ products of Cu-poly/SiO2 increases from 46.64% at 200 to 60.54% at 600 mA cm−2, with a partial current density of C2H4 ranging from 28.28 to 221.46 mA cm−2, exceeding those of Cu and moderately hydrophobic Cu-poly and Cu/SiO2 electrodes (Fig. 4d and S18, ESI†). It should be noted that the FE of CO on Cu-poly/SiO2 fluctuates from 17.57% at 600 mA cm−2 to 37.61% at 200 mA cm−2, exceeding that of Cu.
It is worth noting that the FE of C2H4 and CO on Cu exhibits a similar trend, with both reaching a peak at 300 mA cm−2. However, the FE of C2H4 and CO on Cu-poly/SiO2 are inversely proportional, and the FE of C2H4 reaches its maximum at 600 mA cm−2 (Fig. 4e and f), while CO production is significantly reduced. This observation suggests that the enhanced FE of C2H4 on Cu-poly/SiO2 could be correlated with in situ CO, which is more pronounced than that on Cu, Cu-poly and Cu/SiO2, and plays a critical role in increasing the local CO coverage, thus facilitating the co-electrolysis of CO2 and generating CO.38 Moreover, the polymer and SiO2 appear to reduce the surface H2O content, thereby limiting the protonation of CO and improving the selectivity of CO product. Similarly, the findings are observed on carbon-paper-supported Cu and Cu-poly/SiO2 electrodes (Fig. S19, ESI†), with results from the H-type cells aligning with those from the MEA devices (Fig. S20, ESI†). These electrochemical results indicate that a strong correlation between the generation of C2H4 and CO, suggesting that modulation of the reaction pathway can be achieved by regulating the catalytic microenvironments.
To gain deep insights into the reaction mechanism, in situ attenuated total reflection surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS) was employed to monitor the adsorption behavior, coverage, and bonding information of molecules, ions, and intermediates on Cu and Cu-poly/SiO2 during CO2RR.39 A schematic illustration and digital images of the ATR electrolytic cell are presented in Fig. S21, ESI.† Real-time FTIR spectra were recorded during the CO2RR over Cu and Cu-poly/SiO2 within the potential range from the open circuit potential (OCP) to −2.2 V (V vs. Ag/AgCl, throughout unless otherwise stated). Two distinct broad adsorption bands at wavenumbers of ∼1650 cm−1 and ∼3400 cm−1 were observed and assigned to the bending (δ(OH)) and stretching vibration (ν(OH)) of adsorbed H2O, respectively (Fig. 5 and S22 ESI†).40 Notably, the larger peak area of the δ(OH) signal observed on Cu relative to Cu-poly/SiO2 suggests a higher coverage of H2O and thus more favorable hydrogen evolution (Fig. S23a†).15
 | ||
| Fig. 5 In situ FTIR and synergistic catalytic mechanism. ATR-SEIRAS spectra on (a) Cu NPs and (b) Cu-poly/SiO2 electrode. | ||
A small downward peak emerged at 2100 cm−1 on Cu with a potential of −1.1 V, which is attributed to the linear adsorbed CO ν(COL),41 and this band shifts towards a lower wavenumber of 2080 cm−1 due to the Stark effect (Fig. 5a and S24a, ESI†).42 Similarly, the wavenumber of ν(COL) on Cu-poly/SiO2 appears at 2090 cm−1 and exhibits higher intensity compared to that of Cu (Fig. 5b and S24b, ESI†). The larger area of ν(COL) than that of Cu should be noted, indicating that Cu-poly/SiO2 could enhance the coverage of *CO and achieve the synergistic electroreduction of reactant CO2 and in situ-generated CO (Fig. S23b†).29 Notably, a distinct band is located at 1240 cm−1, which is ascribed to the vibration of Si–O–Si due to the Si substrates of both Cu and Cu-poly/SiO2 (Fig. 5).43–47
To elucidate the influence of aerophilic SiO2 and polymer on the diffusion behavior of CO2 molecules across the surface of Cu, molecular dynamics (MD) simulations were employed to unravel the underlying interactions. The simulation results indicate that the Cu-poly/SiO2 system could promptly capture and absorb CO2 molecules from the surrounding bulk phase within 6 ns, as evidenced by the time sequence of representative snapshots (Fig. 6a). The rapid CO2 uptake can be attributed to the hydrophobic GDL formed by SiO2 and polymer components. In a similar fashion, CO2 molecules also exhibit rapid diffusion on Cu-poly (Fig. 6b) within 6 ns. In contrast, the Cu electrode repels gaseous CO2, preventing diffusion from the bulk phase (Fig. 6c). Furthermore, the Cu-poly/SiO2 presents more negative system energy (−67.04 eV at 100 ns) compared to the Cu (−0.003 eV at 100 ns), indicating that the dual-hydrophobic catalytic microenvironments present a lower system energy due to the fast CO2 diffusion. Notably, Cu-poly exhibits the lowest system energy (−190.82 eV at 100 ns) compared to the other electrodes, indicating that the polymer facilitates stronger adsorption of CO2 (Fig. 6d).
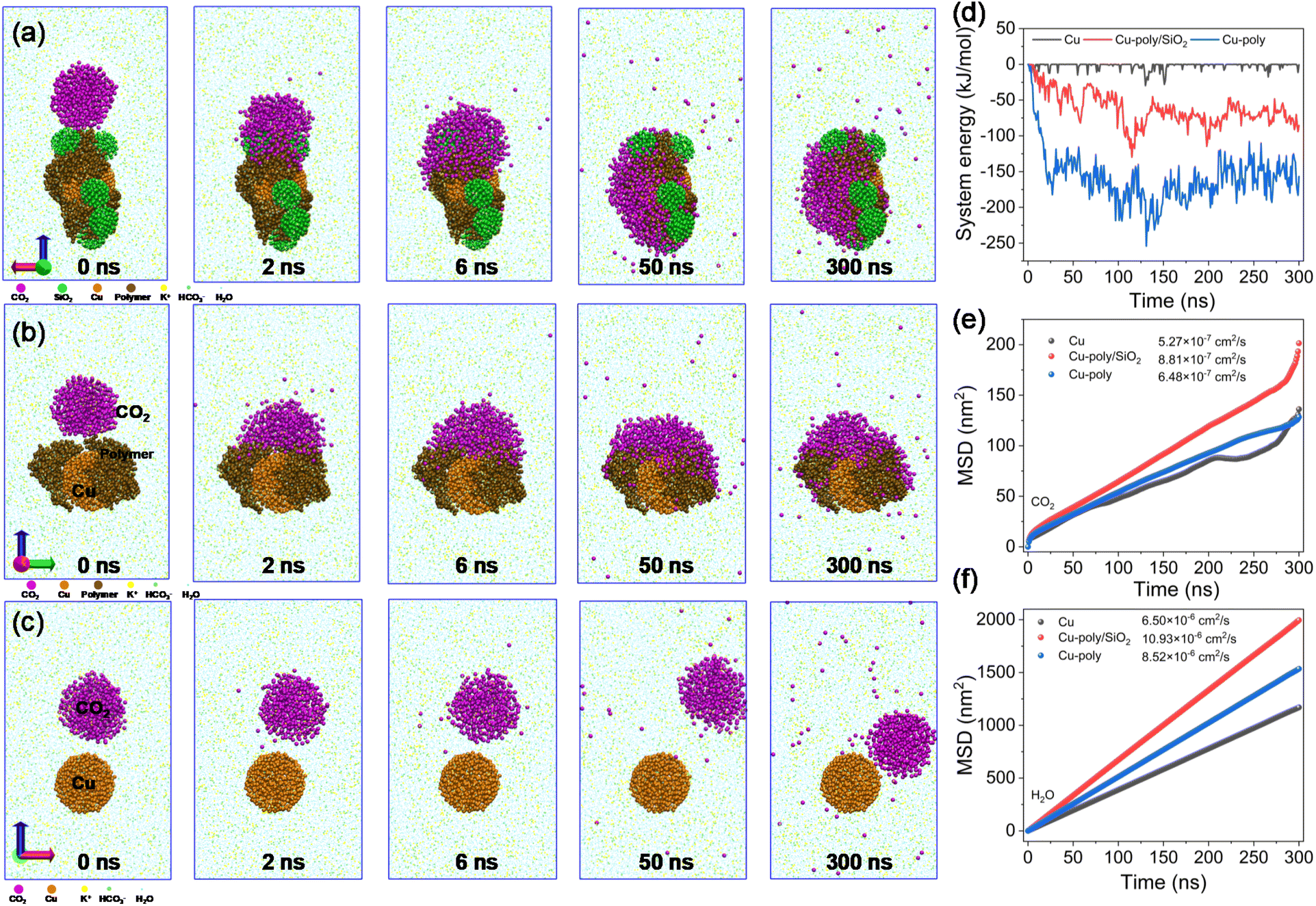 | ||
| Fig. 6 CO2/H2O diffusion dynamics. Evolution of typical snapshots of CO2 diffusion dynamics on (a) Cu-poly/SiO2 and (b) Cu-poly and (c) Cu NPs. (d) Time-dependent change in system energies of Cu and CO2 molecules on Cu, Cu-poly and Cu-poly/SiO2. (e and f) Mean square displacement (MSD) versus time for (d) CO2 and (e) H2O over Cu, Cu-poly and Cu-poly/SiO2. Diffusion coefficients of CO2 and H2O are calculated by the MSD. | ||
We further calculated the diffusion coefficients of CO2 and H2O over Cu, Cu-poly and Cu-poly/SiO2 by the mean square displacement (MSD) derived from the MD simulations. These simulations were conducted at a constant temperature of 300 K, with a fixed number of molecules and volume within a duration of 300 ns. The simulation shows that the CO2 diffusion coefficient increases from 5.27 × 10−7 cm2 s−1 on Cu to 6.48 × 10−7 cm2 s−1 on Cu-poly and reaching 8.81 × 10−7 cm2 s−1 on Cu-poly/SiO2 (Fig. 6e), demonstrating the fast CO2 diffusion from the bulk phase to the vicinity of Cu, mainly due to the reduction in diffusion resistance of CO2 created by the robust hydrophobicity of polymer modification and SiO2. Correspondingly, the H2O diffusion coefficient increases from 6.50 × 10−6 cm2 s−1 on Cu to 8.52 × 10−6 cm2 s−1 on Cu-poly and further to 10.93 × 10−6 cm2 s−1 on Cu-poly/SiO2 (Fig. 6f). This enhancement arises from disruption of the hydrogen-bonding network by nonpolar functional groups (–CF3, (CH3O)3Si–, and –CH3) in the hydrophobic composite, which promotes the formation of isolated H2O molecules or small clusters, thereby reducing intermolecular friction. Furthermore, the minimized H2O-substrate interactions facilitate rapid interfacial water displacement. Consequently, the presence of hydrophobic SiO2 and polymer largely expedites the diffusion of CO2 and does not impede the accessibility of H2O from the bulk electrolyte to the surface of Cu, resulting in simultaneous improvement of the CO2RR.
Conclusions
In conclusion, we propose a useful strategy for regulating dual-hydrophobic catalytic microenvironments by incorporating SiO2 into polymer-functionalized Cu, thereby enriching the local concentration of CO2 and in situ-generated CO product. The optimized Cu-poly/SiO2 electrode displays an improved current density and selectivity of C2H4 compared to bare Cu. Combined in situ ATR-SEIRAS, XAS and MD simulations demonstrate that these enhanced performances can be ascribed to the systematic electrocatalysis of CO2 and CO, accelerated CO2 mass transfer dynamics and elevated oxidation state of Cu species. Compared to Cu, Cu-poly/SiO2 optimizes the local concentration of CO2, CO, and H2O, facilitating the co-feeding modes by enhancing the hydrophobicity of GDEs in MEA devices. This work highlights the importance of interfacial and surficial engineering for constructing dual-hydrophobic catalytic microenvironments for Cu and its derivatives, significantly promoting the formation rate of C2+ products.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
T. Z. and F. C. conceived the idea and designed the experiments. Y. X. performed MD simulations. Z. Z., X. W., K. L., J. C., K. L., Y. L., M. Y., Z. Y., L. J. analyzed the experimental data. M. Y. and F. C. supervised the project. All authors participated in discussing the results and preparing the manuscript.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was financially supported by NSFC (22121005 and 32000922), the Fundamental Research Funds for the Central Universities, the Shandong Province Natural Science Foundation (ZR2024QC122). T. Z. acknowledges the support of the Project funded by China Postdoctoral Science Foundation (2024M760079). We also thank Yan Zhang for the help in data collection of in situ ATR-SEIRAS (Nicolet is50).References
- H. Xu, D. Rebollar, H. He, L. Chong, Y. Liu, C. Liu, C.-J. Sun, T. Li, J. V. Muntean, R. E. Winans, D.-J. Liu and T. Xu, Nat. Energy, 2020, 5, 623–632 CrossRef CAS.
- M. Fan, J. E. Huang, R. K. Miao, Y. Mao, P. Ou, F. Li, X.-Y. Li, Y. Cao, Z. Zhang, J. Zhang, Y. Yan, A. Ozden, W. Ni, Y. Wang, Y. Zhao, Z. Chen, B. Khatir, C. P. O'Brien, Y. Xu, Y. C. Xiao, G. I. N. Waterhouse, K. Golovin, Z. Wang, E. H. Sargent and D. Sinton, Nat. Catal., 2023, 6, 763–772 CrossRef CAS.
- G. Wang, J. Chen, Y. Ding, P. Cai, L. Yi, Y. Li, C. Tu, Y. Hou, Z. Wen and L. Dai, Chem. Soc. Rev., 2021, 50, 4993–5061 RSC.
- X. Wu, S. Zhang, S. Ning, C. Yang, L. Li, L. Tang, J. Wang, R. Liu, X. Yin, Y. Zhu, S. Chen and J. Ye, Chem. Sci., 2025, 16, 4568–4594 RSC.
- Y. Wang, J. Liu and G. Zheng, Adv. Mater., 2021, 18, 2005798 CrossRef PubMed.
- N. Zhang and Y. Zhang, J. Mater. Chem. A, 2025, 13, 2902–2910 RSC.
- M. Zhuansun, Y. Liu, R. Lu, F. Zeng, Z. Xu, Y. Wang, Y. Yang, Z. Wang, G. Zheng and Y. Wang, Angew. Chem., Int. Ed., 2023, 62, e202309875 CrossRef CAS PubMed.
- Z. Wei, W. Wang, T. Shao, S. Yang, C. Liu, D. Si, R. Cong and M. Cao, Angew. Chem., Int. Ed., 2024, 64, e202417066 CrossRef PubMed.
- K. Yang, R. Kas, W. A. Smith and T. Burdyny, ACS Energy Lett., 2020, 6, 33–40 CrossRef.
- C. Chen, H. Jin, P. Wang, X. Sun, M. Jaroniec, Y. Zheng and S.-Z. Qiao, Chem. Soc. Rev., 2024, 53, 2022–2055 RSC.
- M. S. Xie, B. Y. Xia, Y. Li, Y. Yan, Y. Yang, Q. Sun, S. H. Chan, A. Fisher and X. Wang, Energy Environ. Sci., 2016, 9, 1687–1695 RSC.
- C. Kim, J. C. Bui, X. Luo, J. K. Cooper, A. Kusoglu, A. Z. Weber and A. T. Bell, Nat. Energy, 2021, 6, 1026–1034 CrossRef CAS.
- D. Wakerley, S. Lamaison, F. Ozanam, N. Menguy, D. Mercier, P. Marcus, M. Fontecave and V. Mougel, Nat. Mater., 2019, 18, 1222–1227 CrossRef CAS PubMed.
- X. Tong, P. Zhang, P. Chen, Z. He, X. Kang, Y. Yin, Y. Cheng, M. Zhou, L. Jing, G. Wang, B. Xu, L. Zheng, X. Xing, Z. Wu and B. Han, Angew. Chem., Int. Ed., 2024, 64, e202413005 CrossRef PubMed.
- T. Deng, S. Jia, C. Chen, J. Jiao, X. Chen, C. Xue, W. Xia, X. Xing, Q. Zhu, H. Wu, M. He and B. Han, Angew. Chem., Int. Ed., 2023, 63, e202313796 CrossRef.
- K. Xu, J. Li, F. Liu, X. Chen, T. Zhao and F. Cheng, Angew. Chem., Int. Ed., 2023, 62, e202311968 CrossRef CAS.
- D. Wang, J. Mao, C. Zhang, J. Zhang, J. Li, Y. Zhang and Y. Zhu, eScience, 2023, 3, 100119 CrossRef.
- H. Rabiee, M. Li, P. Yan, Y. Wu, X. Zhang, F. Dorosti, X. Zhang, B. Ma, S. Hu, H. Wang, Z. Zhu and L. Ge, Adv. Sci., 2024, 11, 2402964 CrossRef CAS.
- D. H. Nam, O. Shekhah, A. Ozden, C. McCallum, F. Li, X. Wang, Y. Lum, T. Lee, J. Li, J. Wicks, A. Johnston, D. Sinton, M. Eddaoudi and E. H. Sargent, Adv. Mater., 2022, 34, e2207088 CrossRef PubMed.
- H.-Q. Liang, S. Zhao, X.-M. Hu, M. Ceccato, T. Skrydstrup and K. Daasbjerg, ACS Catal., 2021, 11, 958–966 CrossRef CAS.
- Y. Wu, M. N. Idros, D. Feng, W. Huang, T. Burdyny, B. Wang, G. Wang, M. Li and T. E. Rufford, ACS Appl. Mater. Interfaces, 2024, 16, 56967–56974 CrossRef CAS.
- M. Zhang, M. Lu, M.-Y. Yang, J.-P. Liao, Y.-F. Liu, H.-J. Yan, J.-N. Chang, T.-Y. Yu, S.-L. Li and Y.-Q. Lan, eScience, 2023, 3, 100116 CrossRef.
- R. B. Kutz, Q. Chen, H. Yang, S. D. Sajjad, Z. Liu and I. R. Masel, Energy Technol., 2017, 5, 929–936 CrossRef CAS.
- Y. S. Cho, S. C. Lin, C. C. Wang, Y. T. Yang, Y. R. Ho and J. J. Huang, Appl. Phys. A, 2024, 130, 663 CrossRef CAS.
- B. Zhang, J. Zhang, M. Hua, Q. Wan, Z. Su, X. Tan, L. Liu, F. Zhang, G. Chen, D. Tan, X. Cheng, B. Han, L. Zheng and G. Mo, J. Am. Chem. Soc., 2020, 142, 13606–13613 CrossRef CAS.
- M. Ding and H.-L. Jiang, CCS Chem., 2020, 2, 2740–2748 Search PubMed.
- Y. Xu, W. Gao, Z. Chen, Z. Yin, Z. Chen, X. Chang, M. Cheng and B. Xu, ACS Catal., 2024, 14, 10829–10838 CrossRef CAS.
- M. Wang, Z. Wang, Z. Huang, M. Fang, Y. Zhu and L. Jiang, ACS Nano, 2024, 18, 15303–15311 CrossRef CAS.
- X. Wei, Z. Yin, K. Lyu, Z. Li, J. Gong, G. Wang, L. Xiao, J. Lu and L. Zhuang, ACS Catal., 2020, 10, 4103–4111 CrossRef CAS.
- L. Wan, Q. Zhou, X. Wang, T. E. Wood, L. Wang, P. N. Duchesne, J. Guo, X. Yan, M. Xia, Y. F. Li, A. A. Jelle, U. Ulmer, J. Jia, T. Li, W. Sun and G. A. Ozin, Nat. Catal., 2019, 2, 889–898 CrossRef CAS.
- Y. Xu, X. Li, J. Cao, J. Wang, G. Ma, X. Wen, Y. Yang, Y. Li and M. Ding, Science, 2021, 371, 610–613 CrossRef CAS PubMed.
- T. Zhao, X. Zong, J. Liu, J. Chen, K. Xu, X. Wang, X. Chen, W. Yang, F. Liu, M. Yu and F. Cheng, Appl. Catal., B, 2024, 340, 123281 CrossRef CAS.
- W.-F. Xiong, D.-H. Si, H.-F. Li, X. Song, T. Wang, Y.-B. Huang, T.-F. Liu, T. Zhang and R. Cao, J. Am. Chem. Soc., 2023, 146, 289–297 CrossRef.
- B. Deng, M. Huang, K. Li, X. Zhao, Q. Geng, S. Chen, H. Xie, X. Dong, H. Wang and F. Dong, Angew. Chem., Int. Ed., 2021, 61, e202114080 CrossRef PubMed.
- X. Yuan, S. Chen, D. Cheng, L. Li, W. Zhu, D. Zhong, Z. J. Zhao, J. Li, T. Wang and J. Gong, Angew. Chem., Int. Ed., 2021, 60, 15344–15347 CrossRef CAS PubMed.
- T. Zhao, A. Kumar, X. Xiong, M. Ma, Y. Wang, Y. Zhang, S. Agnoli, G. Zhang and X. Sun, ACS Appl. Mater. Interfaces, 2020, 12, 25832–25842 CrossRef CAS.
- B. Ó. Joensen, J. A. Zamora Zeledón, L. Trotochaud, A. Sartori, M. Mirolo, A. B. Moss, S. Garg, I. Chorkendorff, J. Drnec, B. Seger and Q. Xu, Joule, 2024, 8, 1–18 CrossRef.
- C. Wei, Y. Yang, H. Ma, G. Sun, X. Wang, Y. Cheng, C. Zhang, B. Yeo, C. He and A. Wong, Adv. Funct. Mater., 2023, 33, 2214992 CrossRef CAS.
- H. Li, K. Jiang, S.-Z. Zou and W.-B. Cai, Chin. J. Catal., 2022, 43, 2772–2791 CrossRef CAS.
- H. Wang and H. D. Abruña, J. Am. Chem. Soc., 2023, 145, 18439–18446 CrossRef CAS.
- Z. Li, Y. Yang, Z. Yin, X. Wei, H. Peng, K. Lyu, F. Wei, L. Xiao, G. Wang, H. D. Abruña, J. Lu and L. Zhuang, ACS Catal., 2021, 11, 2473–2482 CrossRef CAS.
- K. Jiang, X.-Y. Ma, S. Back, J. Zhao, F. Jiang, X. Qin, J. Zhang and W.-B. Cai, CCS Chem., 2021, 3, 241–251 CrossRef CAS.
- V. T. T. Phan, Q. P. Nguyen, B. Wang and I. J. Burgess, J. Am. Chem. Soc., 2024, 146, 4830–4841 CrossRef CAS.
- K. Jiang, K. Xu, S. Zou and W.-B. Cai, J. Am. Chem. Soc., 2014, 136, 4861–4864 CrossRef CAS PubMed.
- R.-L. Wei, Y. Liu, Z. Chen, W.-S. Jia, Y.-Y. Yang and W.-B. Cai, J. Electroanal. Chem., 2021, 896, 115254 CrossRef CAS.
- L. Wang, S.-F. Hung, S. Zhao, Y. Wang, S. Bi, S. Li, J.-J. Ma, C. Zhang, Y. Zhang, L. Li, T.-Y. Chen, H.-Y. Chen, F. Hu, Y. Wu and S. Peng, Nat. Commun., 2025, 16, 3502 CrossRef CAS PubMed.
- D. Enders, T. Nagao, A. Pucci, T. Nakayama and M. Aono, Phys. Chem. Chem. Phys., 2011, 13, 4935–4941 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc01226k |
| This journal is © The Royal Society of Chemistry 2025 |