
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect single-nucleotide resolution sequencing of DNA 5-methylcytosine using engineered DNA methyltransferase-mediated CMD-seq†
Wei
Liu‡
ab,
Zhao-Cheng
Ma‡
a,
Shan
Zhang
a,
Fang-Yin
Gang
a,
Tong-Tong
Ji
a,
Yao-Hua
Gu
ac,
Neng-Bin
Xie
a,
Shu-Yi
Gu
a,
Xia
Guo
a,
Tian
Feng
a,
Yu
Liu
*ad,
Jun
Xiong
*a and
Bi-Feng
Yuan
 *abef
*abef
aDepartment of Occupational and Environmental Health, School of Public Health, Wuhan University, Department of Radiation and Medical Oncology, Zhongnan Hospital of Wuhan University, Wuhan 430071, China. E-mail: liuyu97@whu.edu.cn; jxiong@whu.edu.cn; bfyuan@whu.edu.cn
bResearch Center of Public Health, Renmin Hospital of Wuhan University, Wuhan University, Wuhan 430060, China
cSchool of Nursing, Wuhan University, Wuhan 430071, China
dHubei Key Laboratory of Tumor Biological Behaviors, Cancer Clinical Study Center, Zhongnan Hospital of Wuhan University, Wuhan 430071, China
eState Key Laboratory of Metabolism and Regulation in Complex Organisms, College of Life Sciences, Wuhan University, Wuhan 430072, China
fHubei Key Laboratory of Biomass Resource Chemistry and Environmental Biotechnology, Wuhan University, Wuhan 430071, China
First published on 16th April 2025
Abstract
5-Methylcytosine (5mC) is a crucial epigenetic modification in the mammalian genome, primarily occurring at CG dinucleotides. Accurate localization of 5mC is essential for understanding its functional significance. In this study, we discovered a novel DNA methyltransferase, designated M.MedI, from the bacterium Mycoplasmopsis edwardii. M.MedI exhibits carboxymethylation activity towards cytosines in CG sites in DNA. We further engineered a variant of M.MedI by mutating its critical active site residue 377 asparagine (N) to lysine (K), resulting in M.MedI-N377K. This engineered M.MedI-N377K enzyme demonstrated superior carboxymethylation activity towards cytosines in CG sites in both unmethylated and hemi-methylated DNA. Utilizing the newly identified M.MedI-N377K methyltransferase, we developed a novel method, engineered DNA methyltransferase-mediated carboxymethylation deamination sequencing (CMD-seq), for the stoichiometric detection of 5mC in DNA at single-nucleotide resolution. In CMD-seq, M.MedI-N377K efficiently transfers a carboxymethyl group to cytosines in CG sites in the presence of carboxy-S-adenosyl-L-methionine (caSAM), generating 5-carboxymethylcytosine (5camC). Subsequent treatment with the deaminase A3A deaminates 5mC to form thymine (T), which pairs with adenine (A) and is read as T, while 5camC remains unchanged, pairing with guanine (G) and being read as cytosine (C) during sequencing. We successfully applied CMD-seq to quantify 5mC sites in the promoters of tumor suppressor genes RASSF1A and SHOX2 in human lung cancer tissue and adjacent normal tissue. The quantification results were highly comparable to those obtained using traditional bisulfite sequencing. Overall, CMD-seq provides a valuable tool for bisulfite-free, single-nucleotide resolution, and quantitative detection of 5mC in limited DNA samples.
Introduction
DNA methylation (5-methylcytosine, 5mC) is a fundamental epigenetic modification conserved across most eukaryotes.1,2 The formation of 5mC is catalyzed by DNA methyltransferases, which transfer a methyl group from S-adenosyl-L-methionine (SAM) to cytosine.3 In mammals, DNA methylation primarily occurs at palindromic CG dinucleotides.4,5 As the “fifth base” of DNA, 5mC plays a crucial role in regulating gene expression and is implicated in various diseases.6–12 The genome-wide 5mC landscape has revealed the significant potential of 5mC as a diagnostic and therapeutic target, highlighting its importance in advancing disease management.13–15To fully understand the biological functions of 5mC in genomes, precise quantification analysis at specific genomic sites is crucial.16–18 Several methods have been developed to achieve this goal, including methylated DNA immunoprecipitation sequencing (MeDIP-seq),19 methyl-CpG binding domain sequencing (MBD-seq),20 and methylation-sensitive restriction enzyme sequencing (MRE-seq).21 However, DNA immunoprecipitation-based sequencing methods lack the ability to detect 5mC at single-base resolution, while restriction enzyme-based methods are limited to specific sequences due to the properties of endonucleases and suffer from incomplete cleavage.22 Bisulfite sequencing (BS-seq) has been considered the gold standard for detecting 5mC at single-base resolution.23 This method uses bisulfite to convert cytosine to uracil, resulting in a C-to-T transition in sequencing results, while 5mC remains unchanged and is read as C during sequencing.24 However, BS-seq has several limitations that hinder accurate quantification analysis of 5mC. One major limitation is the harsh reaction conditions required for bisulfite treatment, which can lead to the degradation of over 99% of the input DNA.25 Furthermore, the C-to-U conversion inherent in BS-seq reduces the sequence complexity, leading to an imbalance of nucleobases in the input DNA.22
Several new methods have been developed to detect 5mC at single-base resolution in recent years. One such method is TET-assisted pyridine borane sequencing (TAPS), which utilizes the enzymatic activity of TET proteins to oxidize 5mC, producing 5caC. This intermediate is then reduced to dihydrouracil (DHU) by pyridine borane.26,27 During subsequent PCR amplification, DHU pairs with adenine, resulting in a 5mC-to-T transition that can be detected through sequencing. However, the presence of DHU in DNA can hinder the amplification efficiency of DNA polymerase, potentially compromising the quantitative detection of 5mC.28 Additionally, other sequencing techniques, such as single molecule-real-time (SMRT) sequencing and nanopore sequencing, have been proposed for detecting 5mC in DNA.29–31 While these methods present promising alternatives for identifying modified nucleobases, they may be associated with relatively high false-positive rates.22,32,33 Enzymatic methylation sequencing (EM-seq) and engineered APOBEC3C (A3C) sequencing (EAC-seq) are two enzymatic methods developed for the detection of 5mC.34–37 Both approaches employ deaminases to convert cytosine (C) to uracil (U). EM-seq utilizes the APOBEC3A (A3A) protein, while EAC-seq employs an engineered A3C (eA3C) protein. Although these methods provide a milder alternative to bisulfite treatment, they still lead to nucleobase imbalances similar to those observed in BS-seq.
Our group and the Kohli group recently developed methyltransferase-directed labeling combined with A3A deamination (MLAD-seq) and direct methylation sequencing (DM-Seq) for 5mC detection.28,38 These methods employ carboxymethylation to protect unmodified cytosine, allowing only 5mC to be deaminated and read as thymine. We utilized M.MpeI-N374K, a CG site-specific DNA methyltransferase from Mycoplasma penetrans, for carboxymethylation of cytosines. M.MpeI-N374K can transfer a carboxymethyl group from carboxy-S-adenosyl-L-methionine (caSAM) to cytosines at CG sites, generating 5-carboxymethylcytosine (5camC).39 The formation of 5camC prevents its deamination by A3A, while 5mC is readily deaminated to T, thereby enabling the sequencing of 5mC. However, M.MpeI-N374K has limited carboxymethylation activity, specifically targeting hemi-methylated DNA.28 This limitation introduces additional complexity, highlighting the need for DNA methyltransferases with high carboxymethylation efficiency for both unmethylated and hemi-methylated DNA.
In this study, we discovered a novel DNA methyltransferase, designated M.MedI, from the bacterium Mycoplasmopsis edwardii. We engineered a variant of M.MedI by mutating its critical active site residue 377 asparagine (N) to lysine (K), resulting in M.MedI-N377K. This engineered enzyme exhibited superior carboxymethylation activity towards cytosines in CG sites in both unmethylated and hemi-methylated DNA. Leveraging the newly discovered M.MedI-N377K methyltransferase, we developed a novel method, engineered DNA methyltransferase-mediated carboxymethylation deamination sequencing (CMD-seq), for the stoichiometric detection of 5mC in DNA at single-nucleotide resolution.
Materials and methods
Chemicals and reagents
2′-Deoxyadenosine (dA), 2′-deoxycytidine (dC), 2′-deoxyguanosine (dG), thymidine (T), 2′-deoxynucleoside-5′-triphosphates (dATP, dCTP, dGTP, and TTP), phosphodiesterase I, ethylene diamine tetraacetic acid (EDTA), tris(hydroxymethyl)aminomethane (Tris), and 2-morpholinoethanesulphonic acid (MES) were purchased from Sigma-Aldrich (St. Louis, MO, USA). 5-Methyl-2′-deoxycytidine was purchased from Berry & Associates (Dexter, MI, USA). 5-Methyl-2′-deoxycytidine-5′-triphosphate (5mdCTP) was purchased from TriLink BioTechnologies (San Diego, CA, USA). S1 nuclease, DNase I, alkaline phosphatase (CIAP), and Klenow Fragment were purchased from Takara Biotechnology Co. Ltd (Dalian, China). S-Adenosyl-L-methionine (SAM), dithiothreitol (DTT), HpaII, EpiMark Hot Start Taq DNA polymerase, and Taq DNA polymerase were obtained from New England Biolabs (Ipswich, MA, USA). Carboxy-S-adenosyl-L-methionine (caSAM) was synthesized according to a previously described method.38Expression and purification of proteins
The coding sequences of the DNA methyltransferases M.MedI-WT (NCBI accession: WP_270161023.1), M.MedI-N377K, M.MpeI-N374K, and M.MpeI-E45D/N374K (Table S1 in the ESI†) were individually cloned into the pET-28a(+) plasmid. The plasmids of DNA methyltransferases included a His-tag and were transformed into E. coli BL21 (DE3) pLysS competent cells (Sangon). After the cells were grown to an optical density (OD600) of 0.4–0.6 at 37 °C, the protein expression was induced with the addition of 1 mM IPTG for 16 h at 25 °C. The His-tagged recombinant proteins (M.MedI-WT, M.MedI-N377K, M.MpeI-N374K, and M.MpeI-E45D/N374K) were purified using HisSep Ni-NTA agarose resin (Yeasen Biotechnology Co. Ltd) according to the manufacturer's protocol and then eluted with 150 mM imidazole in the elution buffer (20 mM Tris–HCl, pH 8.0, 150 mM NaCl). The purified proteins were further concentrated by ultrafiltration (Merck Millipore Ltd). The A3A protein was expressed and purified according to previously published protocols.40–43 The details for the expression and purification of A3A are available in the ESI.† All the purified proteins were analyzed by SDS-PAGE. The protein concentrations were quantified with a BCA protein assay kit (Beyotime, Shanghai, China) and the proteins were stored at −80 °C.Evaluation of the activity of DNA methyltransferases by LC-MS/MS analysis
Two 28-mer oligonucleotides (28-CG-F and 28-CG-R, Table S2 in the ESI†) were synthesized (Sangon) and annealed to form duplex DNA. A 5 pmol aliquot of the duplex DNA was reacted with DNA methyltransferases (M.MedI-WT and M.MedI-N377K) in a 5 μL reaction containing 10 mM Tris–HCl (pH 7.9), 50 mM NaCl, 1 mM DTT, 1 mM EDTA, and 160 μM SAM or caSAM at 37 °C for 1 h. The reaction was stopped by heating at 95 °C for 5 min. The resulting DNA was enzymatically digested and analyzed by LC-MS/MS according to a previously reported method.37 Nucleosides were detected in multiple reaction monitoring (MRM) mode using a Shimadzu 8045 mass spectrometer (Tokyo, Japan) equipped with a Shimadzu LC-30AD UPLC system. The MRM mass transitions and optimized mass spectrometry parameters are listed in Table S3 in the ESI.† Detailed procedures for DNA digestion and LC-MS/MS analysis can be found in the ESI.†Evaluation of activities of DNA methyltransferases by restriction endonuclease digestion
A fluorescein (FAM)-labeled 49-mer oligonucleotide containing a CG site (49-CG-FAM-F, Table S2 in the ESI†) was annealed with its complementary oligonucleotide (49-CG-R, Table S2 in the ESI†). The resulting dsDNA (49-CG-FAM, 5 pmol) was treated with four different DNA methyltransferases in the presence of 160 μM SAM or caSAM, 10 mM Tris–HCl (pH 7.9), 50 mM NaCl, 1 mM DTT, and 1 mM EDTA at 37 °C for 1 h, followed by Proteinase K digestion at 55 °C for 15 min. The resulting DNA was purified using a Nucleotide Removal kit (Qiagen) and digested with HpaII restriction endonuclease at 37 °C for 1 h. The digested DNA was analyzed on a 20% denaturing polyacrylamide gel electrophoresis (PAGE) and visualized using a Tanon 4600SF gel imaging system (Shanghai, China).Steady-state kinetics study
The carboxymethylation activities of various DNA methyltransferases (M.MedI-WT, M.MedI-N377K, M.MpeI-N374K, and M.MpeI-E45D/N374K) were also assessed through steady-state kinetic studies using duplex DNA (49-CG-FAM, 400 nM). The concentration and reaction time for each enzyme were optimized to ensure that the carboxymethyl labeling efficiency did not exceed 20% (Table S4 in the ESI†). The concentration of caSAM was varied from 10 to 160 μM. The carboxymethylation efficiency was calculated as follows:| Efficiency = substrate/(substrate + cleaved product) |
The kinetic parameters kcat and KM were determined using nonlinear regression fitting to the Michaelis–Menten equation:
| Efficiency = (kcat)([S])/(KM + [S]) |
Comparison of the carboxymethylation by sequencing
Two distinct workflows were employed to compare the carboxymethylation of cytosine by various methyltransferases through sequencing, utilizing the 216-bp C-DNA (Table S2 in the ESI†) as a template. Workflow 1 involves direct carboxymethylation labeling through the combined action of DNA methyltransferase and A3A deamination. In contrast, Workflow 2 incorporates an additional DNA extension step prior to carboxymethylation labeling by DNA methyltransferase and A3A deamination. In Workflow 2, a total volume of 17.5 μL containing 60 ng of 216-bp C-DNA and 0.4 μM extension primer (216-EXT, Table S5 in the ESI†) was heated at 95 °C for 5 min and chilled in ice water for 5 min. Then the DNA extension step was carried out in a 25 μL reaction containing 40 μM 5mdCTP, 40 μM dATP, 40 μM dGTP, 40 μM TTP, 1× Klenow Fragment buffer (10 mM Tris–HCl, pH 7.5, 7 mM MgCl2, 1 mM DTT), 10 U of Klenow Fragment (Takara) at 37 °C for 3 h. The reaction was terminated by heating at 65 °C for 5 min. The resulting DNA was then purified using 1.5× KAPA Pure beads (Roche) to yield hemi-methylated 241-bp dsDNA. Carboxymethylation labeling was subsequently performed in both Workflow 1 and Workflow 2 using the four DNA methyltransferases, following the procedures described above. The labeled DNA was then treated with A3A according to a previously established method.38,44 The deamination products were subsequently amplified via PCR, using a reaction mixture consisting of 10 μL of 5× EpiMark reaction buffer, 0.625 U of EpiMark Hot Start Taq DNA polymerase, 0.2 mM dNTPs, and 0.4 μM primers (specifically, 216-MF and 216-A3A-down for Workflow 1, and 216-MF and 216-Edown for Workflow 2, Table S5 in the ESI†). Finally, the resulting products from both Workflow 1 and Workflow 2 were subjected to Sanger sequencing (TsingKe). The sequencing data were then analyzed by comparing the peak heights of C and T, allowing for the determination of the relative efficiencies of the four DNA methyltransferases.Quantitative analysis of 5mC in synthesized DNA
The 216-bp C-DNA (Table S2†) was mixed with varying proportions of 5mC-DNA (0%, 33%, 66%, and 100%). These mixtures were then subjected to carboxymethylation labeling by M.MedI-N377K and A3A deamination, following the protocol outlined in Workflow 1. The resulting DNA was amplified by PCR and subsequently analyzed by Sanger sequencing (TsingKe). The peak heights of C and T at NCG sites (TCG, CCG, GCG, and ACG) were used to calculate the 5mC levels, enabling the assessment of the assay's sensitivity and accuracy.Quantitative determination of 5mC in specific sites of lung tissue DNA by CMD-Seq
The human non-small cell lung carcinoma (NSCLC) tissue and the matched tumor-adjacent normal tissue without preoperative target therapy/chemotherapy were collected from Zhongnan Hospital of Wuhan University. The study was conducted with the approval of the Ethics Committee of Wuhan University and in accordance with the principles outlined in the Declaration of Helsinki. All experimental procedures were performed in strict adherence to the guidelines and regulations set forth by the Ethics Committee of Wuhan University.The genomic DNA of NSCLC tissue and tumor-adjacent tissue was extracted using the Tissue DNA Kit (Omega). DNA was fragmented into the size of 300–500 bp by using an ultrasonic homogenizer (Scientz). The fragmented DNA was treated with M.MedI-N377K in a 5 μL volume consisting of 50 ng of fragmented DNA, 15 μM M.MedI-N377K, 160 μM caSAM, and 1× carboxymethylation buffer (10 mM Tris–HCl, pH 7.9, 50 mM NaCl, 1 mM DTT, and 1 mM EDTA) at 37 °C for 1 h. The resulting DNA was denatured at 95 °C for 10 min after adding 2 μL of DMSO and chilled in ice water for 5 min. Then, the deamination reaction was performed by adding 2 μL of A3A (10 μM) and 2 μL of 10× deamination buffer (200 mM MES, pH 6.5, and 0.1% Triton X-100) using the following gradient: 4 °C for 5 min, linear ramping temperature from 4–37 °C at + 0.1 °C/13 s, 37 °C for 2 h, and linear ramping temperature from 37–50 °C at + 0.1 °C/13 s. The reaction was terminated by heating at 95 °C for 10 min and subsequently deaminated by A3A. PCR amplification was performed in a 50 μL volume including 0.625 U of EpiMark Hot Start Taq DNA polymerase, 0.2 mM dNTPs, and 0.4 μM site-specific primers for the promoter regions of tumor suppressor genes ras association domain family 1 isoform A (RASSF1A) and short stature homeobox 2 (SHOX2) (Table S5 in the ESI†) in 1× EpiMark reaction buffer. The resulting PCR products were subjected to Sanger sequencing. As for BS-seq, 500 ng of the fragmented DNA was processed using the Hieff DNA Bisulfite Conversion kit (Yeasen).
Results and discussion
Principle of the carboxymethylation deamination sequencing (CMD-seq)
In this study, we identified a novel DNA methyltransferase, M.MedI, from the bacterium Mycoplasmopsis edwardii. We further engineered a variant of M.MedI, termed M.MedI-N377K, by substituting the critical active site residue 377 asparagine (N) with lysine (K). This engineered M.MedI-N377K exhibits enhanced carboxymethylation activity towards cytosines in CG sites in both unmethylated and hemi-methylated DNA. Harnessing the unique properties of M.MedI-N377K, we developed a novel method, carboxymethylation deamination sequencing (CMD-seq), for the precise detection of 5mC in DNA at single-nucleotide resolution. The CMD-seq workflow involves the carboxymethylation of cytosines in CG sites by M.MedI-N377K, resulting in the formation of 5-carboxymethylcytosine (5camC). Subsequent treatment with the deaminase A3A converts 5mC to T, which base pairs with A and is read as T during sequencing. In contrast, original cytosines are converted to 5camC, which resists A3A-mediated deamination and base pairs with G, being read as C during sequencing. Ultimately, CMD-seq enables the distinction between original C and 5mC at CG sites, which are read as C and T, respectively, thereby providing a high-resolution map of 5mC in DNA (Fig. 1). | ||
| Fig. 1 Principle of CMD-seq. In the presence of caSAM as a cofactor, M.MedI-N377K specifically carboxymethylates cytosines at CG sites, resulting in the formation of 5camC. Subsequently, treatment with A3A deaminates 5mC to produce T, which base pairs with A. In contrast, 5camC is resistant to deamination by A3A and continues to base pair with G. Consequently, C and 5mC will be read as C and T, respectively, in subsequent sequencing. This allows for the differentiation of C and 5mC at CG sites in CMD-seq. | ||
Rationale for the screening and engineering of DNA methyltransferases
Previous studies have shown that the M.MpeI mutant, in which asparagine (N) is replaced by lysine (K) at position 374 (M.MpeI-N374K), exhibits carboxymethylation activity towards cytosine residues within CG sites.38,39 The N374K mutant with positively charged lysine forms a potential salt bridge with the negatively charged carboxylate group of caSAM and establishes direct hydrogen bonds with the lysine side chain.45 This significantly enhances the binding stability between the enzyme and caSAM, conferring carboxymethyltransferase activity on the enzyme. Additionally, glutamic acid (E) at position 45 (E45) of M.MpeI is crucial for caSAM binding.45 When E45 is replaced by aspartic acid (D), the smaller side chain expands the enzyme's active site. Simultaneous introduction of both E45D and N374K mutations in M.MpeI (M.MpeI-E45D/N374K) expands the bidentate interaction with the ribosyl sugar of caSAM, further strengthening the enzyme–caSAM interaction. However, although the carboxymethylation activity of these mutants is improved, these mutants exhibit limited ability to achieve complete carboxymethylation of unmodified dsDNA.28To address this limitation, we employed the Basic Local Alignment Search Tool (BLAST) to identify proteins with high sequence homology to M.MpeI. Our search yielded several candidates, including DNA methyltransferase of M. edwardii (designated as M.MedI), which exhibits high sequence similarity to M.MpeI. Alignment of the amino acid sequences of these two DNA methyltransferases revealed that they share highly conserved motifs (Fig. 2A, S1, and Table S1 in the ESI†). Based on this sequence similarity, we hypothesized that the mutant protein M.MedI-N377K may possess carboxymethylation activity, analogous to M.MpeI-N374K.
 | ||
| Fig. 2 Evaluation of the carboxymethylation activities of M.MedI-WT and M.MedI-N377K by LC-MS/MS. (A) Schematic representation of the amino acid sequences of the four DNA methyltransferases with or without mutations. (B) Analysis of the purified M.MedI-N377K protein by SDS-PAGE. (C) Diagram illustrating the methylation and carboxymethylation of cytosine by DNA methyltransferases. (D) Schematic representation of the evaluation procedures by LC-MS/MS analysis. (E) Extracted-ion chromatogram of 5mC following treatment with M.MedI-WT and SAM cofactor. (F) Extracted-ion chromatogram of 5mC following treatment with M.MedI-N377K and SAM cofactor. (G) Extracted-ion chromatogram of 5camC following treatment with M.MedI-WT and caSAM cofactor. (H) Extracted-ion chromatogram of 5camC following treatment with M.MedI-N377K and caSAM cofactor. 100 ng of the 28-CG DNA was used for the assay. | ||
Assessment of carboxymethylation activities of M.MedI-WT and M.MedI-N377K by LC-MS/MS
To evaluate the carboxymethylation activities of M.MedI-WT and M.MedI-N377K, we expressed and purified these enzymes (Fig. 2B, S2 and S3 in the ESI†). We then used LC-MS/MS to assess their methylation and carboxymethylation activities (Fig. 2C and D). A 28-bp DNA substrate containing a CG site (Table S2 in the ESI†) was used, along with SAM or caSAM as cofactor (Fig. 2C and D).The results showed that both M.MedI-WT and M.MedI-N377K catalyzed the formation of 5mC when SAM was used as the cofactor (Fig. 2E and F). In contrast, when caSAM was used as the cofactor, M.MedI-N377K exhibited enhanced carboxymethylation activity, resulting in the formation of 5camC, whereas M.MedI-WT showed only weak carboxymethylation activity (Fig. 2G and H). These results suggest that M.MedI is a novel DNA methyltransferase and its variant, M.MedI-N377K, has the potential for highly effective carboxymethylation activity towards cytosine residues within CG sites.
Validation of carboxymethylation activities of DNA methyltransferases by restriction enzyme cleavage
To further assess the carboxymethylation efficiencies of M.MedI-WT and M.MedI-N377K, we employed restriction enzyme cleavage assay using a FAM-labeled 49-bp DNA substrate (49-CG-FAM, Table S2 in the ESI†) containing a CCGG site. HpaII, a 5mC-sensitive restriction endonuclease, cleaves unmodified CCGG motifs, whereas 5mC or 5camC at the second cytosine of the CCGG motif blocks its cleavage (Fig. 3A).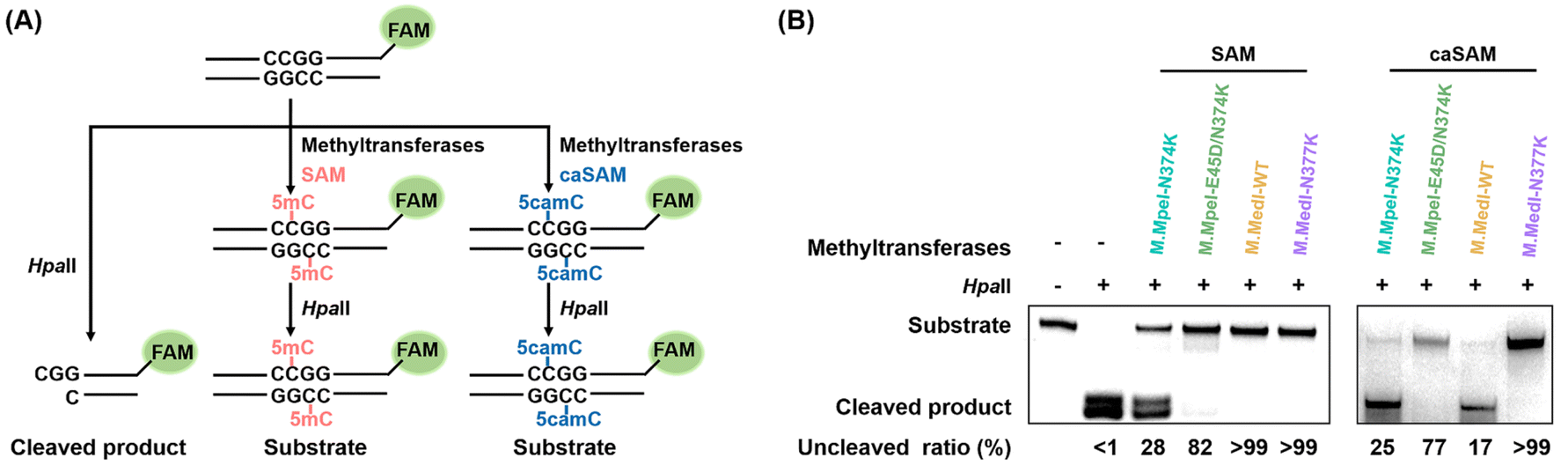 | ||
| Fig. 3 Comparative analysis of carboxymethylation activities of DNA methyltransferases using restriction enzyme cleavage. (A) Schematic representation of the restriction enzyme cleavage assay. (B) PAGE analysis of the cleavage products resulting from treatment of DNA with different methyltransferases and cofactors (SAM or caSAM). In this assay, the 49-CG-FAM DNA substrate was incubated with various DNA methyltransferases in the presence of SAM or caSAM to form 5mC or 5camC, respectively. The restriction enzyme HpaII was then used to cleave the resulting DNA. HpaII specifically recognizes and cleaves unmodified dsDNA at the CCGG site, whereas dsDNA containing 5mC or 5camC is resistant to HpaII digestion. | ||
First, we evaluated the methylation efficiencies of M.MedI-WT and M.MedI-N377K using SAM as the cofactor. The results showed that HpaII specifically cleaved the unmodified 49-CG-FAM, while the methylated DNA produced by both M.MedI-WT and M.MedI-N377K was fully protected against HpaII digestion (>99%, Fig. 3B). For comparison, we also expressed M.MpeI-N374K and M.MpeI-E45D/N374K (Fig. S4 and S5 in the ESI†) and evaluated their methylation efficiencies. The results revealed lower methylation efficiencies for M.MpeI-N374K (28%) and M.MpeI-E45D/N374K (82%) (Fig. 3B).
Next, we assessed the carboxymethylation efficiencies of these four DNA methyltransferases using caSAM as the cofactor. The results showed that over 99% of the DNA remained uncleaved after M.MedI-N377K treatment, whereas only 17%, 25%, and 77% of the DNA remained uncleaved after treatment with M.MedI-WT, M.MpeI-N374K, and M.MpeI-E45D/N374K, respectively (Fig. 3B). These results suggest that M.MedI-N377K can more efficiently carboxymethylate cytosine within CG sites than M.MpeI-N374K and M.MpeI-E45D/N374K, two previously identified carboxymethylation methyltransferases.
Quantitative evaluation of carboxymethylation efficiencies of DNA methyltransferases
To further investigate the carboxymethylation efficiencies of these different DNA methyltransferases, we conducted a steady-state kinetic study (Fig. S6†). The results revealed that M.MedI-N377K exhibited the highest carboxymethylation activity, with a kcat/KM value of 14.52 nM−1 min−1. In contrast, M.MpeI-E45D/N374K and M.MpeI-N374K displayed significantly lower activities, with kcat/KM values of 0.63 nM−1 min−1 and 0.07 nM−1 min−1, respectively. M.MedI-WT showed the lowest activity, with a kcat/KM value of 0.04 nM−1 min−1 (Table 1). Notably, the activity of M.MedI-N377K was 207-fold higher than that of M.MpeI-N374K (Table 1).| DNA methyltransferases | K M (μM) | k cat (min−1) | k cat/KM (nM−1 min−1) | Relative activity |
|---|---|---|---|---|
| M.MpeI-N374K | 25.34 ± 3.45 | 0.0018 | 0.07 | 1.00 |
| M.MpeI-E45D/N374K | 22.99 ± 8.65 | 0.0145 | 0.63 | 9.00 |
| M.MedI-WT | 8.23 ± 2.26 | 0.0003 | 0.04 | 0.57 |
| M.MedI-N377K | 47.17 ± 8.12 | 0.6851 | 14.52 | 207.43 |
In summary, the results demonstrate that M.MedI is a novel DNA methyltransferase and its variant M.MedI-N377K can catalyze the carboxymethylation of cytosine within CG sites with higher efficiency than the previously reported M.MpeI-N374K and M.MpeI-E45D/N374K. In addition, most 5mC DNA methyltransferases recognize sequences containing four or more nucleobases. Since the majority of 5mC sites in mammalian genomes are located within the CG context,46 the CG-specific methyltransferase M.MedI-N377K offers superior sequence coverage compared to non-CG-specific methyltransferases that require four or more nucleobases for recognition. These results highlight the potential of M.MedI-N377K as a valuable tool for the development of the CMD-seq method.
Comparison of carboxymethylation efficiencies of DNA methyltransferases in unmethylated and hemi-methylated DNA
Previous studies by our group and the Kohli group have shown that M.MpeI-N374K exhibits higher carboxymethylation efficiency when the DNA template is hemi-methylated.28,38 To further investigate this phenomenon, we compared the carboxymethylation efficiencies of the four DNA methyltransferases using unmethylated DNA and hemi-methylated DNA. We designed two workflows: Workflow 1, which directly carboxymethylates unmethylated DNA, and Workflow 2, which includes an additional DNA extension step before carboxymethylation (Fig. 4A). | ||
| Fig. 4 Comparative analysis of carboxymethylation efficiencies of DNA methyltransferases in unmethylated and hemi-methylated DNA. (A) Schematic representation of the two workflows used in this study. Workflow 1 and Workflow 2 differ in that Workflow 2 includes an additional extension step prior to DNA methyltransferase treatment. (B) Sequencing results of DNA treated with different DNA methyltransferases, followed by A3A deamination and sequencing according to the Workflow 1 protocol. (C) The calculated C/(C + T) ratio in various CG contexts for different DNA methyltransferases, as determined from (B). (D) Sequencing results of DNA treated with different DNA methyltransferases, followed by A3A deamination and sequencing according to the Workflow 2 protocol. (E) The calculated C/(C + T) ratio in various CG contexts for different DNA methyltransferases, as determined from (D). A 216-bp C-DNA substrate was used to evaluate the carboxymethylation efficiencies of the DNA methyltransferases. | ||
To assess the carboxymethylation efficiency, we subjected the carboxymethylated DNA to A3A deamination in both workflows (Fig. S7†). A3A protein is known to efficiently deaminate C and 5mC in DNA to produce uracil (U) and thymine (T), respectively.34,47,48 However, our previous study demonstrated that A3A protein cannot deaminate 5camC.38 Therefore, carboxymethylated cytosine is read as C in sequencing, while 5mC is deaminated by A3A and read as T. We first evaluated the A3A deamination activity by directly deaminating 216-bp C-DNA and 216-bp 5mC-DNA. The sequencing results showed complete C-to-T and 5mC-to-T conversion, indicating the strong deaminase activity of A3A (Fig. S8†). We then subjected the C-DNA to Workflow 1 and Workflow 2.
For M.MpeI-N374K and M.MpeI-E45D/N374K, the cytosines in the CG context in the C-DNA were partially read as T and partially read as C in Workflow 1 (Fig. 4B and C), indicating that both methyltransferases only partially carboxymethylate cytosines in CG sites in unmethylated DNA. In Workflow 2, the proportions of cytosines in CG sites read as C increased for both M.MpeI-N374K and M.MpeI-E45D/N374K (Fig. 4D and E), indicating that both methyltransferases exhibit higher carboxymethylation efficiency for hemi-methylated DNA strands. In contrast, M.MedI-WT showed weak carboxymethylation activity for both unmethylated and hemi-methylated DNA strands, with most cytosines in CG sites read as T in both Workflow 1 and Workflow 2 (Fig. 4B–E). However, M.MedI-N377K exhibited superior carboxymethylation efficiency, with all cytosines in the CG context in the C-DNA read as C in both Workflow 1 and Workflow 2 (Fig. 4B–E). These results indicate that M.MedI-N377K is an ideal enzyme for developing the CMD-seq method, as it can highly efficiently carboxymethylate cytosines in CG sites in both unmethylated and hemi-methylated DNA strands.
Assessing the performance of CMD-seq for 5mC detection in DNA with multiple C and 5mC in different sequence contexts
Using the characterized M.MedI-N377K enzyme, we developed the CMD-seq method for direct, single-nucleotide resolution sequencing of 5mC in DNA. To validate the specificity and efficiency of CMD-seq, we employed a 400-bp DNA template containing multiple C or 5mC sites (Table S2 in the ESI†). As anticipated, all cytosines in the CG context within the 400-bp C-DNA were accurately read as C, whereas all 5mC sites in the 400-bp 5mC-DNA were correctly read as T (Fig. 5A and B). However, cytosines in non-CG contexts were not carboxymethylated and were deaminated to uracil, which was read as T during sequencing (Fig. 5A). These results demonstrate that M.MedI-N377K is a CG site-specific DNA carboxymethyltransferase and that CMD-seq can effectively distinguish between C and 5mC in DNA containing multiple C and 5mC sites across diverse sequence contexts. | ||
| Fig. 5 Assessment of CMD-seq performance using DNA containing cytosines in various sequence contexts. (A) Sequencing results of DNA with unmethylated cytosines across different sequence contexts. A 400-bp C-DNA without 5mC was utilized. (B) Sequencing results of DNA with methylated cytosines in different sequence contexts. A 400-bp 5mC-DNA was used, with all cytosines methylated. | ||
Evaluation of the quantitative capability of CMD-seq
Next, we quantitatively assessed the 5mC levels at individual CG sites using the CMD-seq method. We prepared DNA mixtures with varying proportions of unmethylated DNA (216-bp C-DNA) and methylated DNA (216-bp 5mC-DNA), with molar ratios of 216-bp C-DNA/(216-bp C-DNA + 216-bp 5mC-DNA) of 0, 33, 66, and 100%. In CMD-seq, the sequencing reads of C and T correspond to the original unmethylated C and 5mC in CG sites, respectively. Therefore, the peak height ratio of T/(C + T) reflects the 5mC level at each CG site. Sanger sequencing revealed that the measured T/(C + T) values for four CG contexts (TCG, CCG, GCG, and ACG) were highly proportional to the theoretical 5mC levels (TCG, slope = 0.96, R2 = 0.9999; CCG, slope = 0.99, R2 = 0.9988; GCG, slope = 0.97, R2 = 0.9997; ACG: slope = 0.99, R2 = 0.9997, Fig. 6). These results demonstrate that CMD-seq can accurately quantify 5mC levels at individual CG sites.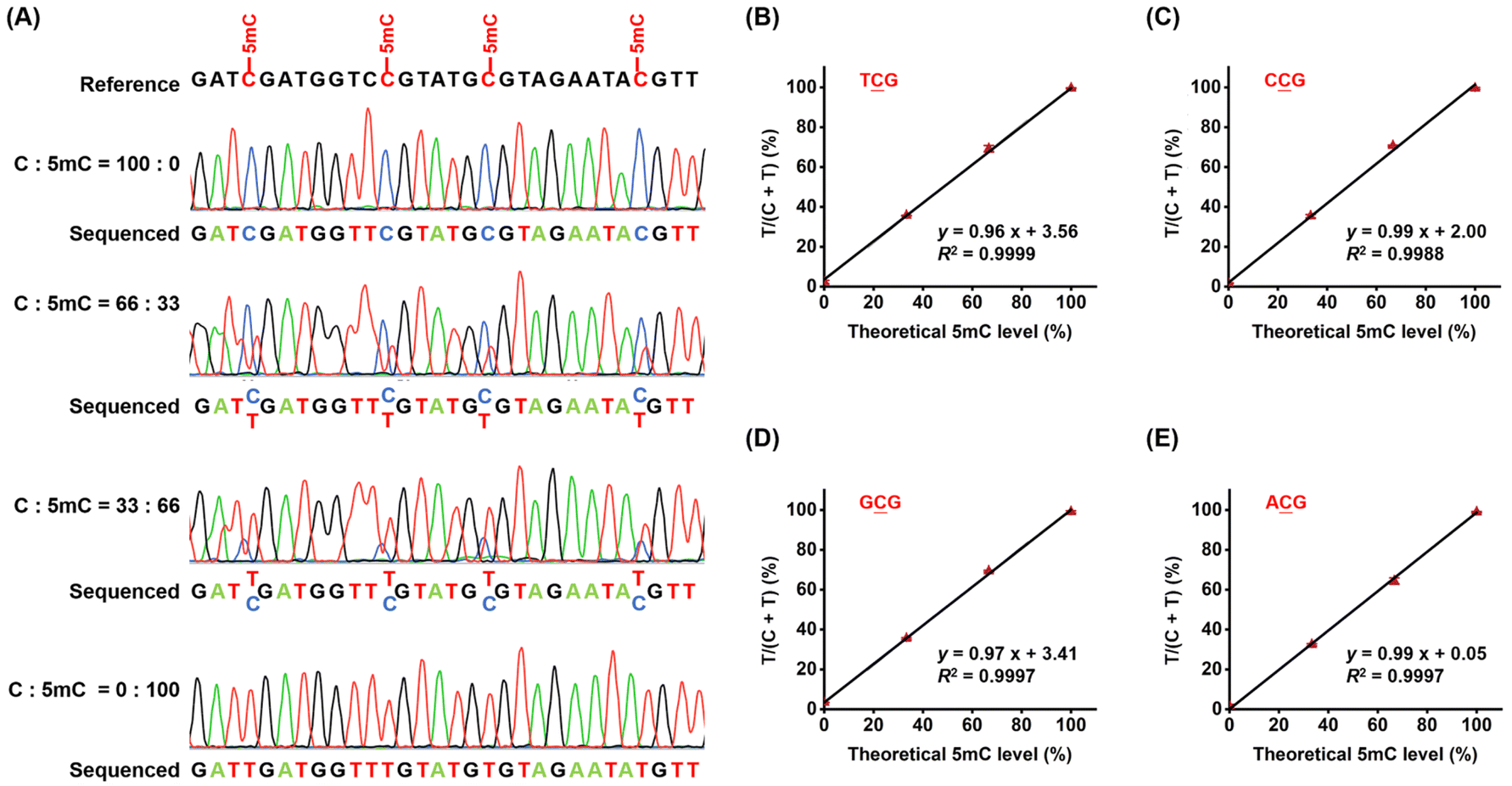 | ||
Fig. 6 Evaluation of the quantitative capability of CMD-seq. (A) A mixture of 216-bp C-DNA and 5mC-DNA was prepared at different ratios (100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0, 66:33, 33:66, and 0:100) and used as substrates for CMD-seq. (B–E) The ratios of T/(C + T) in the original TCG, CCG, GCG, and ACG sequences obtained from Sanger sequencing were plotted against the theoretical percentages of 5mC-DNA/(C-DNA + 5mC-DNA). :0, 66:33, 33:66, and 0:100) and used as substrates for CMD-seq. (B–E) The ratios of T/(C + T) in the original TCG, CCG, GCG, and ACG sequences obtained from Sanger sequencing were plotted against the theoretical percentages of 5mC-DNA/(C-DNA + 5mC-DNA). | ||
Locus-specific analysis of 5mC in genomic DNA by CMD-seq
DNA methylation is a common epigenetic phenomenon that often leads to the inactivation of tumor suppressor genes, contributing to tumor development.6,49 Lung cancer remains one of the leading causes of cancer-related death worldwide.50,51 The global burden of lung cancer is increasing, emphasizing the need for early screening to improve diagnosis and reduce mortality.52,53RASSF1A and SHOX2 are well-characterized tumor suppressor genes frequently methylated in lung cancer.54–56 We applied CMD-seq to quantify 5mC at specific loci in the promoter regions of RASSF1A and SHOX2 in lung tumor and tumor-adjacent normal tissues.The results revealed that the 5mC site (chr3: 50378230) in the RASSF1A promoter exhibited a mixed methylation pattern in tumor tissue, with both C and T reads detected, whereas in tumor-adjacent normal tissue, this site was predominantly read as C (Fig. 7A). Quantification revealed that the 5mC levels in the RASSF1A promoter (chr3: 50378230) were 40.4% in tumor tissue and 5.8% in tumor-adjacent normal tissue, closely comparable to those measured by BS-seq (44.2% and 1.1% in tumor and tumor-adjacent normal tissue, respectively, Fig. 7A).
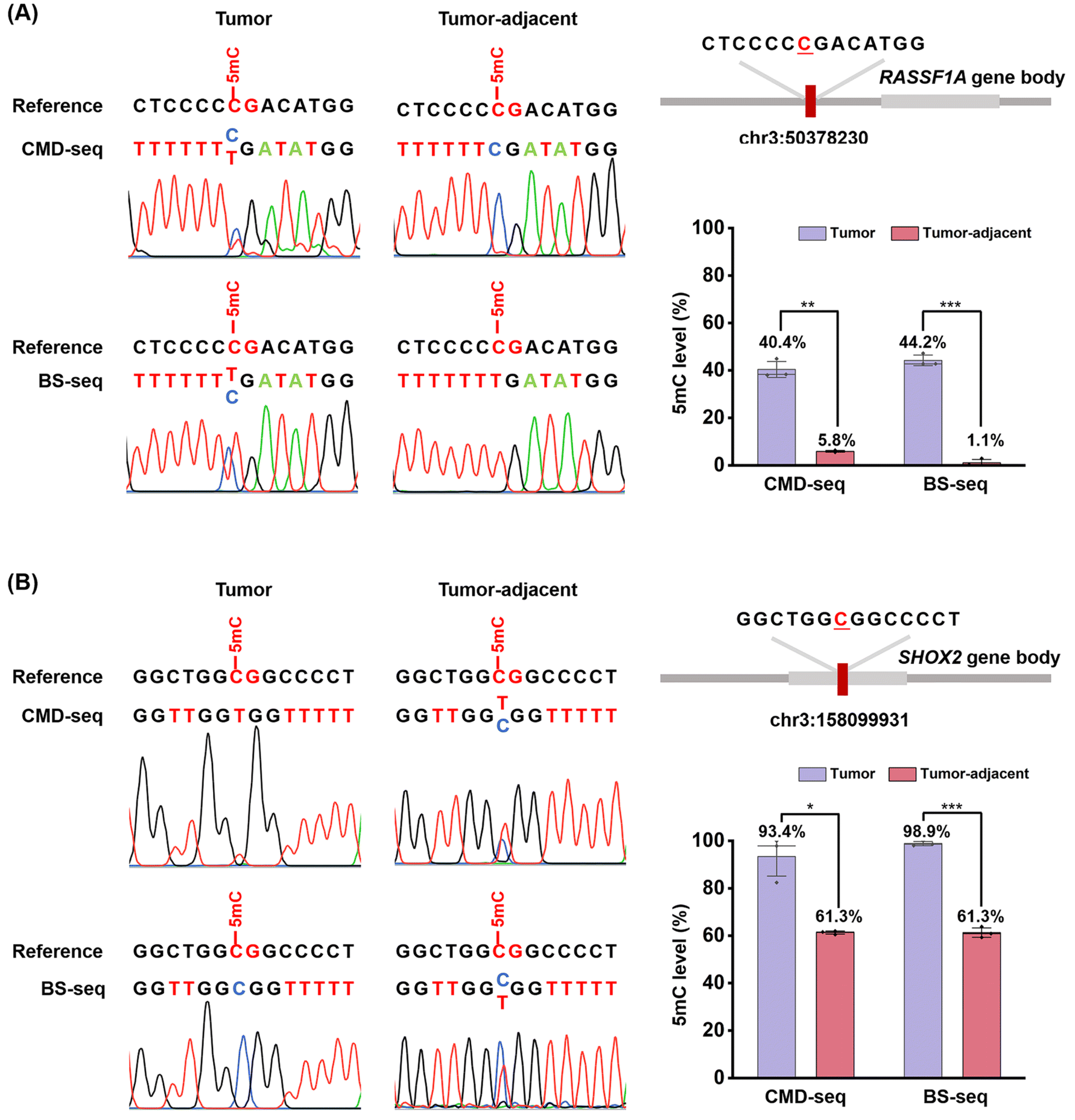 | ||
| Fig. 7 Locus-specific analysis of 5mC in genomic DNA from human lung cancer tissue and adjacent normal tissue using CMD-seq and BS-seq. (A) Sequencing results of the 5mC site (chr3: 50378230) in the promoter region of the RASSF1A gene from human lung tissue and adjacent normal tissue, analyzed by CMD-seq and BS-seq. (B) Sequencing results of the 5mC site (chr3: 158099931) in the promoter region of the SHOX2 gene from human lung tissue and adjacent normal tissue, analyzed by CMD-seq and BS-seq. | ||
Similarly, for the 5mC site (chr3: 158099931) in the SHOX2 promoter, the results showed that it was almost completely read as T in tumor tissue but partially read as C and partially as T in tumor-adjacent normal tissue (Fig. 7B). Quantification results indicated that the 5mC levels at the SHOX2 locus (chr3: 158099931) were 93.4% in tumor tissue and 61.3% in tumor-adjacent normal tissue, consistent with BS-seq measurements (98.9% and 61.3%, respectively; Fig. 7B). These results confirm that 5mC levels in the promoter regions of RASSF1A and SHOX2 are significantly higher in lung tumor tissues, aligning with previous studies.57,58
The high consistency between CMD-seq and BS-seq validates the reliability of CMD-seq for quantitative, single-nucleotide resolution 5mC detection. It should be noted that BS-seq relies on the chemical deamination of cytosines but not 5mC, enabling the detection of 5mC at arbitrary cytosine locations. M.MedI-N377K exhibits CG-specific carboxymethylation activity and CMD-seq primarily detects 5mC at CG sites. In mammalian genomes, 5mC is predominantly established and maintained by the CG-specific methyltransferases DNMT1, DNMT3A, and DNMT3B, suggesting that 5mC primarily functions by forming 5mC at CG sites.59 Indeed, the majority of 5mC sites in the mammalian genomes are found within the CG context.46 Therefore, the CG site specificity of M.MedI-N377K has a relatively limited impact on whole-genome sequencing.
Compared to existing methods, CMD-seq offers several advantages. Firstly, CMD-seq is a direct sequencing approach, deaminating only 5mC to T while leaving the majority of cytosines in CG sites intact and read as C. Secondly, the high carboxymethylation efficiency of M.MedI-N377K enables direct and complete carboxymethylation of genomic DNA without the need for a DNA extension step, simplifying library construction. Thirdly, CMD-seq is a bisulfite-free and non-destructive method that minimizes DNA degradation. The EM-seq method is also an enzymatic approach for mapping 5mC.35,36 In EM-seq, 5mC is oxidized by TET2, resulting in various products (5hmC, 5fC, and 5caC), with 5 fC accounting for approximately 10%.35,36 However, 5fC is readily deaminated by A3A and read as T.60 Consequently, around 10% of 5mC is incorrectly identified as T, compromising the method's accuracy. Additionally, the in vitro expression of recombinant TET2 protein requires mammalian or insect cells, which can be challenging.26,35,36 In contrast, the M.MedI-N377K protein can be readily expressed in E. coli cells, making its expression and purification relatively easy. The intrinsic nature of CMD-seq provides a strong foundation for future applications, enabling reliable mapping of 5mC at the single-nucleotide level on a genome-wide scale while in combination with high-throughput sequencing. In summary, CMD-seq offers a direct, accurate, and reliable method for 5mC detection at single-nucleotide resolution, providing a valuable tool for epigenetic research and clinical applications.
Conclusion
In summary, we developed a novel method, CMD-seq, for the quantitative detection of 5mC at single-nucleotide resolution without the need for bisulfite treatment. This approach leverages the superior carboxymethylation activity of the newly discovered DNA methyltransferase M.MedI-N377K, which selectively carboxymethylates cytosines in CG sites in both unmethylated and hemi-methylated DNA strands. By employing M.MedI-N377K in conjunction with the A3A protein, CMD-seq enables the differential deamination of 5camC and 5mC, allowing for precise quantification of 5mC in genomic DNA. We successfully applied CMD-seq to quantify 5mC sites in the promoters of tumor suppressor genes RASSF1A and SHOX2 in human lung cancer tissue and adjacent normal tissue, with results that are highly comparable to those obtained using traditional BS-seq. Notably, CMD-seq offers several advantages over BS-seq, including the elimination of harsh chemical conditions that can lead to significant DNA degradation. As a bisulfite-free and non-destructive method, CMD-seq preserves DNA integrity, making it well-suited for analyzing 5mC in limited DNA samples. In conclusion, CMD-seq offers a bisulfite-free, single-nucleotide resolution, and DNA-friendly approach that holds considerable promise for epigenetic research and clinical applications.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Bi-Feng Yuan: writing – review & editing, investigation, supervision, conceptualization, funding acquisition. Jun Xiong: writing – review & editing, investigation, conceptualization, funding acquisition. Yu Liu: writing – review & editing, investigation, supervision. Wei Liu: writing – original draft, methodology, formal analysis, data curation, conceptualization. Zhao-Cheng Ma: methodology, formal analysis, data curation, conceptualization. Shan Zhang: formal analysis, data curation, conceptualization. Fang-Yin Gang: formal analysis, data curation. Tong-Tong Ji: formal analysis, data curation. Yao-Hua Gu: formal analysis, data curation. Neng-Bin Xie: formal analysis, data curation. Shu-Yi Gu: data curation. Xia Guo: data curation. Tian Feng: data curation.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22207090 and 22277093), the Fundamental Research Funds for the Central Universities (no. 2042024kf1045), and the Key Research and Development Project of Hubei Province (2023BCB094).References
- A. de Mendoza, R. Lister and O. Bogdanovic, J. Mol. Biol., 2020, 432, 1687–1705 CrossRef CAS PubMed.
- Y. Feng, J. J. Chen, N. B. Xie, J. H. Ding, X. J. You, W. B. Tao, X. Zhang, C. Yi, X. Zhou, B. F. Yuan and Y. Q. Feng, Chem. Sci., 2021, 12, 11322–11329 RSC.
- S. Adam, V. Klingel, N. E. Radde, P. Bashtrykov and A. Jeltsch, Nucleic Acids Res., 2023, 51, 6622–6633 CrossRef CAS PubMed.
- E. Li and Y. Zhang, Cold Spring Harb. Perspect. Biol., 2014, 6, a019133 CrossRef PubMed.
- A. D. Hay, N. J. Kessler, D. Gebert, N. Takahashi, H. Tavares, F. K. Teixeira and A. C. Ferguson-Smith, Nat. Commun., 2023, 14, 5336 CrossRef CAS PubMed.
- Z. D. Smith, S. Hetzel and A. Meissner, Nat. Rev. Genet., 2025, 26, 7–30 CrossRef CAS PubMed.
- X. Xue, Z. Wang, Y. Wang and X. Zhou, ACS Chem. Biol., 2023, 18, 2114–2127 CrossRef CAS PubMed.
- M. V. C. Greenberg and D. Bourc'his, Nat. Rev. Mol. Cell Biol., 2019, 20, 590–607 CrossRef CAS PubMed.
- S. Yeou, J. Hwang, J. Yi, C. Kim, S. K. Kim and N. K. Lee, Chem. Sci., 2022, 13, 7516–7525 RSC.
- Z. Lin, Y. Lu, G. Yu, H. Teng, B. Wang, Y. Yang, Q. Li, Z. Sun, S. Xu, W. Wang and P. Tian, Sci. China Life Sci., 2023, 66, 2354–2369 CrossRef CAS PubMed.
- S. Y. Gu, T. Feng, F. Y. Gang, S. Y. Yu, W. Chan, Z. C. Ma, Y. H. Gu and B. F. Yuan, Chin. Chem. Lett., 2025, 36, 110957 CrossRef.
- T. Feng, Y. L. Gao, D. Hu, K. Y. Yuan, S. Y. Gu, Y. H. Gu, S. Y. Yu, J. Xiong, Y. Q. Feng, J. Wang and B. F. Yuan, Chin. Chem. Lett., 2024, 35, 109259 CrossRef CAS.
- A. P. Feinberg, N. Engl. J. Med., 2018, 378, 1323–1334 CrossRef CAS PubMed.
- V. M. Sales, A. C. Ferguson-Smith and M. E. Patti, Cell Metab., 2017, 25, 559–571 CrossRef CAS PubMed.
- A. P. Feinberg and A. Levchenko, Science, 2023, 379, eaaw3835 CrossRef CAS PubMed.
- Y. F. Du, Y. Tang, B. Q. Lin, X. C. Xue, Y. F. Wang and Y. B. Liu, Sci. China Chem., 2023, 66, 3044–3053 CrossRef CAS.
- Y. Dai, B. F. Yuan and Y. Q. Feng, RSC Chem. Biol., 2021, 2, 1096–1114 RSC.
- F. Y. Gang, N. B. Xie, M. Wang, S. Zhang, T. T. Ji, W. Liu, X. Guo, S. Y. Gu and B. F. Yuan, Anal. Chem., 2024, 96, 20559–20567 CrossRef CAS PubMed.
- M. Weber, J. J. Davies, D. Wittig, E. J. Oakeley, M. Haase, W. L. Lam and D. Schubeler, Nat. Genet., 2005, 37, 853–862 CrossRef CAS PubMed.
- D. Serre, B. H. Lee and A. H. Ting, Nucleic Acids Res., 2010, 38, 391–399 CrossRef CAS PubMed.
- A. K. Maunakea, R. P. Nagarajan, M. Bilenky, T. J. Ballinger, C. D'Souza, S. D. Fouse, B. E. Johnson, C. Hong, C. Nielsen, Y. Zhao, G. Turecki, A. Delaney, R. Varhol, N. Thiessen, K. Shchors, V. M. Heine, D. H. Rowitch, X. Xing, C. Fiore, M. Schillebeeckx, S. J. Jones, D. Haussler, M. A. Marra, M. Hirst, T. Wang and J. F. Costello, Nature, 2010, 466, 253–257 CrossRef CAS PubMed.
- Y. Kong, E. A. Mead and G. Fang, Nat. Rev. Genet., 2023, 24, 363–381 CrossRef CAS PubMed.
- Q. Dai, C. Ye, I. Irkliyenko, Y. Wang, H. L. Sun, Y. Gao, Y. Liu, A. Beadell, J. Perea, A. Goel and C. He, Nat. Biotechnol., 2024, 42, 1559–1570 CrossRef CAS PubMed.
- Y. Li and T. O. Tollefsbol, Methods Mol. Biol., 2011, 791, 11–21 CrossRef CAS PubMed.
- K. Tanaka and A. Okamoto, Bioorg. Med. Chem. Lett., 2007, 17, 1912–1915 CrossRef CAS PubMed.
- Y. Liu, P. Siejka-Zielinska, G. Velikova, Y. Bi, F. Yuan, M. Tomkova, C. Bai, L. Chen, B. Schuster-Bockler and C. X. Song, Nat. Biotechnol., 2019, 37, 424–429 CrossRef CAS PubMed.
- J. Chen, J. Cheng, X. Chen, M. Inoue, Y. Liu and C. X. Song, Nucleic Acids Res., 2022, 50, e104 CrossRef CAS PubMed.
- T. Wang, J. M. Fowler, L. Liu, C. E. Loo, M. Luo, E. K. Schutsky, K. N. Berrios, J. E. DeNizio, A. Dvorak, N. Downey, S. Montermoso, B. Y. Pingul, M. Nasrallah, W. S. Gosal, H. Wu and R. M. Kohli, Nat. Chem. Biol., 2023, 19, 1004–1012 CrossRef CAS PubMed.
- S. Ardui, A. Ameur, J. R. Vermeesch and M. S. Hestand, Nucleic Acids Res., 2018, 46, 2159–2168 CrossRef CAS PubMed.
- Y. Wang, Y. Zhao, A. Bollas, Y. Wang and K. F. Au, Nat. Biotechnol., 2021, 39, 1348–1365 CrossRef CAS PubMed.
- Y. Han, D. M. Yang, S. Jiang, S. L. Zhao, F. Ma and C. Y. Zhang, Trac. Trends Anal. Chem., 2024, 172, 117553 CrossRef CAS.
- Z. K. O'Brown, K. Boulias, J. Wang, S. Y. Wang, N. M. O'Brown, Z. Hao, H. Shibuya, P. E. Fady, Y. Shi, C. He, S. G. Megason, T. Liu and E. L. Greer, BMC Genom., 2019, 20, 445 CrossRef PubMed.
- M. Jain, H. E. Olsen, B. Paten and M. Akeson, Genome Biol., 2016, 17, 239 CrossRef PubMed.
- E. K. Schutsky, C. S. Nabel, A. K. F. Davis, J. E. DeNizio and R. M. Kohli, Nucleic Acids Res., 2017, 45, 7655–7665 CrossRef CAS PubMed.
- R. Vaisvila, V. K. C. Ponnaluri, Z. Sun, B. W. Langhorst, L. Saleh, S. Guan, N. Dai, M. A. Campbell, B. S. Sexton, K. Marks, M. Samaranayake, J. C. Samuelson, H. E. Church, E. Tamanaha, I. R. Correa Jr, S. Pradhan, E. T. Dimalanta, T. C. Evans Jr, L. Williams and T. B. Davis, Genome Res., 2021, 31, 1280–1289 CrossRef CAS PubMed.
- Z. Y. Sun, R. Vaisvila, L. M. Hussong, B. Yan, C. Baum, L. Saleh, M. Samaranayake, S. X. Guan, N. Dai, I. R. Correa, S. Pradhan, T. B. Davis, T. C. Evans and L. M. Ettwiller, Genome Res., 2021, 31, 291–300 CrossRef PubMed.
- M. Wang, N. B. Xie, K. K. Chen, T. T. Ji, J. Xiong, X. Guo, S. Y. Yu, F. Tang, C. Xie, Y. Q. Feng and B. F. Yuan, Anal. Chem., 2023, 95, 1556–1565 CAS.
- J. Xiong, K. K. Chen, N. B. Xie, T. T. Ji, S. Y. Yu, F. Tang, C. Xie, Y. Q. Feng and B. F. Yuan, Anal. Chem., 2022, 94, 15489–15498 CrossRef CAS PubMed.
- T. Wang and R. M. Kohli, Cell Chem. Biol., 2021, 28, 97–104 CrossRef CAS PubMed.
- N. B. Xie, M. Wang, T. T. Ji, X. Guo, J. H. Ding, B. F. Yuan and Y. Q. Feng, Chem. Sci., 2022, 13, 7046–7056 RSC.
- N. B. Xie, M. Wang, T. T. Ji, X. Guo, F. Y. Gang, Y. Hao, L. Zeng, Y. F. Wang, Y. Q. Feng and B. F. Yuan, Chem. Sci., 2024, 15, 10073–10083 RSC.
- N. B. Xie, M. Wang, W. Chen, T. T. Ji, X. Guo, F. Y. Gang, Y. F. Wang, Y. Q. Feng, Y. Liang, W. Ci and B. F. Yuan, ACS Cent. Sci., 2023, 9, 2315–2325 CrossRef CAS PubMed.
- S. Zhang, Y. Liang, T. Feng, X. Guo, M. Wang, T. T. Ji, J. Xiong, X. Xiao, Y. Liu, Y. Liu, W. Ci, N. B. Xie and B. F. Yuan, CCS Chem., 2025 DOI:10.31635/ccschem.025.202405023.
- J. H. Ding, G. Li, J. Xiong, F. L. Liu, N. B. Xie, T. T. Ji, M. Wang, X. Guo, Y. Q. Feng, W. Ci and B. F. Yuan, Anal. Chem., 2024, 96, 4726–4735 CrossRef CAS PubMed.
- C. E. Loo, M. A. Hix, T. Wang, G. A. Cisneros and R. M. Kohli, ACS Chem. Biol., 2023, 18, 2224–2232 CrossRef CAS PubMed.
- R. Lister, M. Pelizzola, R. H. Dowen, R. D. Hawkins, G. Hon, J. Tonti-Filippini, J. R. Nery, L. Lee, Z. Ye, Q. M. Ngo, L. Edsall, J. Antosiewicz-Bourget, R. Stewart, V. Ruotti, A. H. Millar, J. A. Thomson, B. Ren and J. R. Ecker, Nature, 2009, 462, 315–322 CrossRef CAS PubMed.
- X. Guo, J. Wu, T. T. Ji, M. Wang, S. Zhang, J. Xiong, F. Y. Gang, W. Liu, Y. H. Gu, Y. Liu, N. B. Xie and B. F. Yuan, Chem. Sci., 2025, 16, 3953–3963 RSC.
- J. Xiong, P. Wang, W. X. Shao, G. J. Li, J. H. Ding, N. B. Xie, M. Wang, Q. Y. Cheng, C. H. Xie, Y. Q. Feng, W. M. Ci and B. F. Yuan, Chem. Sci., 2022, 13, 9960–9972 RSC.
- Y. Feng, Y. Q. Tian, Y. Q. Zhao, S. J. Chen and B. F. Yuan, Chin. Chem. Lett., 2024, 35, 109656 CrossRef CAS.
- M. Klutstein, D. Nejman, R. Greenfield and H. Cedar, Cancer Res., 2016, 76, 3446–3450 CrossRef CAS PubMed.
- F. Drakopanagiotakis, E. Krauss, I. Michailidou, V. Drosos, S. Anevlavis, A. Günther and P. Steiropoulos, Cancers, 2024, 16, 25 CrossRef PubMed.
- G. Hardavella, A. Frille, R. Chalela, K. B. Sreter, R. H. Petersen, N. Novoa and H. J. de Koning, Eur. Respir. Rev., 2024, 33, 17 Search PubMed.
- D. R. Aberle, Clin. Radiol., 2017, 72, 401–406 CrossRef CAS PubMed.
- D. Raos, M. Ulamec, A. Katusic Bojanac, F. Bulic-Jakus, D. Jezek and N. Sincic, Bosn. J. Basic Med. Sci., 2021, 21, 386–397 CAS.
- B. Schmidt, V. Liebenberg, D. Dietrich, T. Schlegel, C. Kneip, A. Seegebarth, N. Flemming, S. Seemann, J. Distler, J. Lewin, R. Tetzner, S. Weickmann, U. Wille, T. Liloglou, O. Raji, M. Walshaw, M. Fleischhacker, C. Witt and J. K. Field, BMC Cancer, 2010, 10, 600 CrossRef CAS PubMed.
- G. Weiss, A. Schlegel, D. Kottwitz, T. Konig and R. Tetzner, J. Thorac. Oncol., 2017, 12, 77–84 CrossRef PubMed.
- D. C. Lu, Y. P. Chen, L. F. Ke, Z. F. Huang, Y. D. Lu and J. Wang, Chem. Eng. J., 2025, 503, 158491 CrossRef CAS.
- M. Ren, C. Wang, D. Sheng, Y. Shi, M. Jin and S. Xu, Ann. Diagn. Pathol., 2017, 27, 57–61 CrossRef PubMed.
- Z. Chen and Y. Zhang, Annu. Rev. Biochem., 2020, 89, 135–158 CrossRef CAS PubMed.
- M. Wang, N. B. Xie, F. Y. Gang, S. Zhang, L. Zeng, T. T. Ji, J. Xiong, X. Guo, Y. Hao, Y. Liu and B. F. Yuan, Sci. China Life Sci., 2025 DOI:10.1007/s11427-024-2702-8.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis of carboxy-S-adenosyl-L-methionine (caSAM); expression and purification of A3A protein; enzymatic digestion of DNA; LC-MS/MS analysis; preparation of double-strand DNA with C and 5 mC; evaluation of the deamination activity of A3A by sequencing; Tables S1–S5 and Fig. S1–S8. See DOI: https://doi.org/10.1039/d5sc01211b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |