
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning intermediate binding enables selective electroreduction of carbon dioxide to carbon monoxide on a copper–indium catalyst†
Shengzhou
Xu
 a,
Chenglong
Wang
a,
Chunjing
Ran
a,
Hexing
Yang
a,
Wangjiang
Gao
a,
Bitao
Dong
*b,
Yuhang
Liu
c and
Dan
Ren
*a
a,
Chenglong
Wang
a,
Chunjing
Ran
a,
Hexing
Yang
a,
Wangjiang
Gao
a,
Bitao
Dong
*b,
Yuhang
Liu
c and
Dan
Ren
*a
aSchool of Chemical Engineering and Technology, Xi'an Jiaotong University, West Xianning Road 28, Xi'an 710049, China. E-mail: dan.ren@xjtu.edu.cn
bDepartment of Materials Sciences and Engineering, Angstrom Laboratory, Uppsala University, Uppsala, SE-75105, Sweden. E-mail: dongbitao0023@outlook.com
cSchool of Materials Science and Engineering, Xi'an Jiaotong University, West Xianning Road 28, Xi'an 710049, China
First published on 21st April 2025
Abstract
Electrosynthesis of carbon monoxide (CO) from carbon dioxide (CO2) and water driven by renewable electricity represents a sustainable route to carbon upgrading, but the lack of cost-effective catalysts hinders its scaling-up. Here, we judiciously designed a bimetallic Cu–In catalyst via in situ electroreduction of In-coated CuO nanowires. This facilely-prepared Cu–In catalyst delivers an excellent performance towards CO production in a flow cell, with a faradaic efficiency of CO of up to 91% at a geometric current density of −69 mA cm−2. Different from previous studies suggesting that In-modified Cu strengthens the adsorption of *COOH and/or weakens the binding of *H, we discovered that In acts as the active site. The modification of In by Cu weakens the adsorption of *CO. This facilitates a faster desorption of *CO, thus inhibiting the C–C coupling process. As a result, the formation of multi-carbon products is suppressed. This conclusion was drawn through a rigorous analysis of the electrochemical reduction of CO, the electrochemical adsorption of *CO and in situ Raman spectroscopy. Finally, we wired our CuIn-based electrolyzer to an efficient triple-junction solar cell for the demonstration of solar-driven CO2 conversion and achieved a solar-to-chemical energy conversion efficiency of greater than 10% for CO.
Introduction
The global climate change caused by the excessive emission of carbon dioxide (CO2) into the atmosphere is a potential threat to human beings. Electrochemical reduction of CO2 to fuels and chemicals, driven by renewable electricity, could potentially reduce the atmospheric concentration of CO2.1,2 Among over 20 carbonaceous products reported from the electroreduction of CO2,3–5 carbon monoxide (CO) is a vital raw material for the Fischer–Tropsch synthesis of a range of chemical feedstocks.6 The global market size of CO, which is mainly derived from fossil fuels, reached over 3 billion USD in 2023.7 Synthesis of CO from the electrochemical reduction of CO2 serves as a promising alternative to the generation of this feedstock. However, the lack of cost-effective, efficient and stable electrocatalysts hinders its large-scale deployment.8,9Extensive research efforts have been devoted to designing efficient electrocatalysts based on Au,10–12 Ag13–15 or Pd16–18 for the electrochemical reduction of CO2 to CO. For example, Sun and co-workers investigated Au nanoparticles of different sizes for CO2 reduction to CO and found that 8-nm Au nanoparticles exhibited a faradaic efficiency (FE) of 90% for CO at −0.67 V vs. RHE (reversible hydrogen electrode).12 Liu et al. synthesized triangular silver nanoplates, which delivered a FE of 96.8% for the production of CO at −0.855 V vs. RHE.14
Interestingly, bimetallic catalysts, such as copper-indium (Cu–In)19–22 and copper-tin (Cu-Sn)23–26 have shown potential as a cost-effective alternative to Au or Ag for CO production. Takanabe and co-workers first reported the selective production of CO with a FE of 95% on a Cu–In alloy.19 By density functional theory (DFT) modelling, they proposed that the presence of In weakens the binding of *H on Cu and strengthens the adsorption of *CO2 on Cu, thus making Cu sites suitable for CO formation. Later, Pérez-Ramírez and co-workers found that the presence of In(OH)3 in their Cu–In electrocatalysts is crucial in catalyzing the formation of CO.20 Züttel and co-workers proposed that the Cu–In interface enhances the adsorption strength of *CO and destabilizes the adsorption of *H through DFT modelling.21 Shao and co-workers suggested that the binding energy of key intermediates is tuned through Cu–In interaction.22
To recapitulate, although the selective formation of CO has been achieved on Cu–In catalysts, the exact synergistic mechanism of Cu and In for CO production is still under debate. Controversial statements on the active site within CuIn by different research groups have been made. According to the early study by Hori. et al.27 as well as the computational modeling by Rossmeisl et al.,28 Cu and the formate-producing group metal (including Sn and In) are located on the opposite side of the CO-producing group metals (including Ag and Au) in terms of the binding strength of key intermediates, such as *CO. This seems to indicate that the mixture of Cu and In might lead to the characteristic of CO selectivity.29 A deeper understanding of this effect is urgently needed for an understanding of this type of bimetallic synergy.
Here, we develop a facile method to prepare Cu–In bimetallic catalysts and investigate their electrocatalytic activity for the reduction of CO2. Namely, In is sputtered onto a CuO nanowire substrate to form CuO–In, where the thickness of In is systematically varied by altering the time of sputtering. The Cu–In catalyst, formed by electrochemical reduction of CuO–In, exhibits good selectivity and activity towards the production of CO. The synergy between Cu and In was thoroughly investigated through the analysis of electrochemical CO reduction, electrochemical CO adsorption, and surface-enhanced in situ Raman spectroscopy. In contrast to the previous studies, which suggest that In-modified Cu enhances the adsorption of *COOH and/or weakens the binding of *H, we experimentally reveal that the modification of In on Cu weakens the adsorption of *CO and facilitates rapid desorption of *CO from Cu, thus inhibiting the C–C coupling process hence the formation of multi-carbon products. The catalyst we developed is further used as the cathode for a CO2 electrolyzer that is powered by a triple-junction solar cell, delivering a decent solar-to-chemical conversion efficiency.
Results and discussions
Synthesis and characterization of Cu and Cu–In catalysts
The schematic flow of the preparation of the CuO/GDE and CuO–In/GDE films is depicted in Fig. 1a. Briefly, a sputtered Cu film with a thickness of 900 nm on a GDE (Fig. S1, S2 and Table S1†) was first anodized in 3 M KOH solution to form Cu(OH)2 nanowires. Cu(OH)2 was annealed at 150 °C for 1 h to form CuO on the GDE (CuO/GDE).30 | ||
| Fig. 1 Structural and chemical characterizations of the GDE, CuO, CuO–In, Cu and Cu–In. (a) Schematic route for the synthesis of CuO and CuO–In. (b and e) Scanning electron micrograph (SEM), (c and f) transmission electron micrograph and (d, g) high-resolution transmission electron micrograph of (b–d) CuO–In-30 and (e–g) CuIn-30. (h) High-angle annular dark-field image and energy dispersive mapping of CuO–In-30. (i) X-ray photoelectron spectra of In 3d and Cu 2p of CuO–In-30 and CuIn-30. (j) X-ray diffractograms of the GDE, Cu, CuO, CuO–In-30 and CuIn-30. (k) Ex situ Raman spectra of CuO and CuO–In-30. | ||
This film was subsequently coated with In of varying thicknesses ranging from 0.5 to 60 nm via sputtering. It is noted that here the spontaneous exchange reaction between In and CuO, as given below, is expected to take place.
2In(s) + 3CuO(s) ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) In2O3(s) +3Cu(s) In2O3(s) +3Cu(s) | (1) |
| ΔG0 = −442.8 kJ mol−1 at 298 K |
Thus, the resulting film may exhibit various oxidation states of Cu and In. For simplification, these films are denoted based on the precursors as CuO–In-X (where X represents the thickness of In).
The as-prepared CuO–In-X and CuO were characterized by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). CuO consists of wires with a length of a few micrometers and a diameter of <100 nm (Fig. S3†). After the coating of In, CuO–In maintains the nanowire structure (Fig. 1b). The transmission electron micrograph of the CuO–In wire shows a uniform coating of In, with a distinct boundary between the two layers (Fig. 1c). The thickness of the In coating with the sputtering time of 628 s is estimated to be 29.91 ± 9.61 nm by quantitative analysis of multiple TEM images (Fig. S4 and Table S2†), hence being named as CuO–In-30. A lattice spacing of 0.271 nm, which belongs to the diffraction of the (101) spacing of the In lattice (I4/mmm space group),31 is observed in the HR-TEM of CuO–In-30 (Fig. 1d).
CuO and CuO–In-30 were reduced at a constant geometric current density of −69 mA cm−2 for 600 s. Both CuO and CuO–In-30 are expected to be reduced to metallic films (as will be discussed later),21,32,33 hence being named as Cu and CuIn-30. SEM shows that Cu exhibits a rougher texture with nanoparticles of around 500 nm covered on the surface as compared to CuO (Fig. S5†). The roughness factor of Cu was determined to be 136, based on the estimation using double layer capacitance (Fig. S6 and Table S3†).
CuIn-30 maintains a wire-like structure, with agglomerates of particles present on the surface (Fig. 1e–f and S7†). SEM-EDX analysis reveals a dramatic increase in the atomic ratio of Cu/In on CuIn-30 (Cu/In = 3.32) as compared to CuO–In-30 (Cu/In = 1.36) in the bulk, indicating the possible leaching of In species (Fig. S8 and Table S4†), which is further demonstrated by ICP-MS analysis of the electrolyte (Table S5†). HR-TEM reveals that CuIn-30 particles show lattice spacings of 0.251 nm and 0.286 nm, corresponding to diffractions from the (002) spacing of CuO (C2/c space group) and (222) spacing of the In2O3 lattice (I213 space group), respectively (Fig. 1g).34,35 High-angle annular dark-field imaging with energy dispersive X-ray spectroscopy on CuO–In-30 reveals the coverage of In over the CuO nanowires (Fig. 1h). After reduction, In is homogeneously distributed across the surface of the CuIn-30 nanowires (Fig. S9†). It is evident that Cu atoms within the inner shell migrate to the surface during reduction, leading to a mixture of Cu with In on the surface.
CuO–In-30 and CuIn-30 were also characterized by X-ray photoelectron spectroscopy (XPS). For In 3d3/2 and 3d5/2, CuO–In-30 only shows features of In3+, while CuIn-30 merely consists of In0 species (Fig. 1i). For Cu 2p1/2 and Cu 2p3/2, peaks at 954.1 eV and 934.1 eV, respectively demonstrate that the surface of CuO–In-30 and CuIn-30 contains Cu(II) species. This is further corroborated by the appearance of satellite features at 942.9 eV and 962.5 eV. The peaks at 932.6 and 952.3 eV are attributed to Cu+/Cu0 species (Fig. 1i). The presence of Cu0/Cu+ on the surface of CuO–In-30 demonstrates that the surface CuO has been partially reduced. We also analyzed the surface Cu/In atomic ratio of the CuO–In-30 and CuIn-30 catalysts to be 0.27 and 2.13, respectively (Table S6†), indicating the migration of Cu species to the surface during electrochemical reduction.
In the X-ray diffractograms, Cu exhibits visible peaks at 2θ = 43.3°, 50.4°, and 74.1°, which could be assigned to the diffractions from the (111), (200), and (220) facets of Cu (Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group), respectively (Fig. 1j).36 For the CuO and CuO–In-30 films, the diffractograms show additional peaks at 2θ = 35.5°, 38.7°, and 48.7°, corresponding to the diffractions from the (002), (111), and (
m space group), respectively (Fig. 1j).36 For the CuO and CuO–In-30 films, the diffractograms show additional peaks at 2θ = 35.5°, 38.7°, and 48.7°, corresponding to the diffractions from the (002), (111), and (![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 02) facets of CuO (C2/c space group).26 The peaks corresponding to metallic Cu in the two diffractograms could be partially attributed to the residual sputtered Cu underneath. The diffractogram of CuO–In-30 also exhibits visible peaks at 2θ = 32.9°, 36.3°, 39.2°, and 63.2° that are assigned to diffractions from the (101), (002), (110), and (103) facets of In (I4/mmm space group),37 respectively. This demonstrates the successful coating of In on the substrate. The diffractogram of the CuIn-30 catalyst still exhibits the diffractions of In, together with the diffractions of CuO.
02) facets of CuO (C2/c space group).26 The peaks corresponding to metallic Cu in the two diffractograms could be partially attributed to the residual sputtered Cu underneath. The diffractogram of CuO–In-30 also exhibits visible peaks at 2θ = 32.9°, 36.3°, 39.2°, and 63.2° that are assigned to diffractions from the (101), (002), (110), and (103) facets of In (I4/mmm space group),37 respectively. This demonstrates the successful coating of In on the substrate. The diffractogram of the CuIn-30 catalyst still exhibits the diffractions of In, together with the diffractions of CuO.
CuO and CuO–In-30 were further characterized by ex situ Raman spectroscopy (Fig. 1k). The Raman peak at 155–157 cm−1 corresponds to the Γ−15 phonon of the Cu2O lattice, which is observed in both the CuO NWs and CuO–In-30 catalyst.38 The Raman peaks at 286 and 600 cm−1 for the CuO NWs correspond to Cu–O vibrations in the CuO lattice.38 The Raman peaks at 472 and 560 cm−1 for the CuO–In-30 catalyst correspond to the phonons associated with the bcc-structured indium oxide.39
From TEM, XPS, XRD, and Raman analysis of CuO and CuO–In-30, it is evident that Cu species exist in a mixed state of Cu0 and Cu2+ after the coating of In, which is consistent with the expected reaction as shown in eqn (1). However, only metallic In species are observed in the XRD and TEM of CuO–In-30, but XPS shows that the surface of CuO–In-30 only contains In3+, indicating that the formed In2O3 is likely to be amorphous or the quantity of In2O3 is too small. We thus conclude that the coated In film of CuO–In-30 contains both metallic and oxide In species. TEM analysis of Cu and CuIn-30 shows the existence of oxides, which is likely due to the quick oxidation of the surface in air. Nevertheless, these catalysts are named as CuIn-X in the following context.
Improved activity of CO2 reduction to CO on the Cu–In catalyst
Eight different electrocatalysts, including Cu, CuIn-0.5, CuIn-5, CuIn-15, CuIn-20, CuIn-30, CuIn-60, and In, were individually used as the cathode for CO2 reduction in 0.5 M KHCO3 using a customized flow cell (Fig. S10†).40 Linear sweep voltammograms show that CuIn-30 exhibits a larger cathodic geometric current density compared to Cu (Fig. 2a). Moreover, the cathodic current density increases under CO2 as compared to the one under Ar, if the potential reaches <−0.6 V vs. RHE. The catalytic behavior was then assessed under chronopotentiometric bias, with the gaseous products being analyzed using online gas chromatography and the liquid products being quantified through high-performance liquid chromatography after electrolysis (Fig. S11 and S12†). The steady state potential under different current density and j–V plots of all the catalysts under a CO2 atmosphere are presented in Fig. S13.† The values of FE are tabulated in Tables S7–S15.† | ||
| Fig. 2 Electrocatalytic carbon dioxide reduction performance of Cu, In and the CuIn-X catalysts. (a) Total geometric current density (jtotal) of the Cu and CuIn catalysts as a function of applied potential under CO2 or Ar atmosphere. FE of different products on the (b) Cu, (c) In, (d) CuIn-30 and (e) CuIn-60 catalysts as a function of cathodic current density. (f) FE of different products on the CuIn-X catalysts with different In thicknesses at about −70 mA cm−2. (g) FE of the produced CO and the detected half-cell potential at −70 mA cm−2 over 450-min of electrolysis on the CuIn-30 catalyst. Each data point in (b)–(f) corresponds to the average of multiple independent measurements and the error bar represents the standard deviation of these measurements. Electrolyte: 0.5 M KHCO3. Flow rate of CO2 or Ar: 15 cm3 min−1. | ||
Cu catalyzes the formation of a mixture of carbonaceous products, including CO, formate, ethylene, ethanol, acetate and n-propanol at different current densities, from −42 to −250 mA cm−2 (Fig. 2b and Table S7†). When the cathodic current density reaches 250 mA cm−2, the FE for the C2+ products peaks at 60%. The selectivity of the C2+ species rises along with a decline in the FE of CO, which corroborates that CO is the intermediate towards the formation of C2+ products.41,42 On the contrary, the In catalyst delivers formate as the major product in the electrochemical reduction of CO2, which is in line with previous studies using In-based catalysts (Fig. 2c).43,44 Interestingly, the CuIn-X catalysts show drastically different activity (Fig. S14–17†). Specifically, the CuIn-30 catalyst exhibits a decent selectivity in catalyzing electrochemical CO2 reduction to CO. The FE for CO remains above 83% within a wide current density range from −41 to −80 mA cm−2. At −69 mA cm−2, the FE for CO production reaches up to 91 ± 3% (Fig. 2d), with a trace amount of hydrogen and hydrocarbons being detected. Our catalyst is among the best reported literature values in terms of the selectivity towards CO (Tables S16 and S17†). Compared to recently-emerged single atom catalysts and molecular catalysts, the ease of preparation of our catalyst offers advantage over those materials that require solvents or high-temperature treatment.
The effect of the thickness of In on the catalytic performance of CuIn-X was also investigated. Upon the thickness of coated In increasing to 60 nm, only a FE of 62.7% for CO is achieved and the FE of formate increases to 16.5% at −70 mA cm−2 (Fig. 2e). It is clear that excessive loading of In leads to the formation of formate. Formate ions could migrate from the catholyte to the anolyte through the anion exchange membrane (AEM) and its oxidation at the Pt anode (Fig. S18†) leads to the underestimated quantification of formate and hence a relatively low total FE value, especially on In and CuIn-60 (Table S14†). Once the cathodic current density exceeds −100 mA cm−2, C2+ products are generated from the electrochemical reduction of CO2 on CuIn-60, in accordance with the one on CuIn-30. This is likely due to the improved CO coverage on CuIn-60 at large current density, facilitating the C–C coupling process.
The catalytic performance of different CuIn-X catalysts at a current density of about −70 mA cm−2 is compared in Fig. 2f. For CuIn-5 and CuIn-15, C2+ products are formed, with a FE of 3.8% and 0.6%, respectively. Hence, an optimal value for the thickness of In being coated on the CuO NWs is needed for maximized suppression of C2+ products.
The long-term electrolysis shows an appreciable stability of CuIn-30 in CO production (Fig. 2g). It is noted that the half-cell potential fluctuates periodically, likely due to salt precipitation, which hinders the mass transport of CO2 and reactants, and subsequently results in the accumulation and release of bubbles. The faradaic efficiency of CO remains relatively stable over a 450-min electrolysis at a current density of −69 mA cm−2, with the average FE for CO remaining nearly 80%. After 450-min electrolysis, the ratio of Cu/In shows a decrease from 3.32 to 2.29 (Fig. S19, S20 and Table S18†). This might be ascribed to the dissolution of both Cu and In, which is verified by the ICP-MS analysis (Table S19†). More importantly, the CuIn-30 surface suffers from severe aggregation of the In particles (Fig. S21 and S22†). Both the structural change and the dissolution of Cu could lead to the degradation of the catalytic performance of CuIn-30 after 450-min of electrolysis.
Active sites of CuIn-30 for CO2 reduction to CO
To reveal the underlying reason for the catalytic activity of CuIn-30 towards the production of CO, we first investigated the surface characteristics of CuIn-30 as well as those of Cu and In. The cyclic voltammograms of Cu, In, and CuIn-30 under CO flow are shown in Fig. 3a. It is noted that the slightly skewed baseline observed is due to the inevitable reduction of residual oxygen within the pores of the GDE substrate. | ||
| Fig. 3 Investigation of the synergy between Cu and In. (a) Cyclic voltammograms of CuIn-30 as well as the Cu and In catalysts measured in a flow cell at a scan rate of 50 mV s−1 under a CO atmosphere. (b–d) Cyclic voltammograms of the Cu, In and CuIn-30 catalysts measured within a narrow potential range at a scan rate of 50 mV s−1, with N2 (dash line) or CO (solid line) being flowed at a rate of 50 cm3 min−1. FE of different products in the electrochemical reduction of CO on the (e) Cu and (f) CuIn-30 catalysts. (g) Catalytic performance of the CuIn-X catalysts in the electrochemical reduction of CO2 at a current density of about −140 mA cm−2. Each data point in (e)–(g) corresponds to the average of multiple independent measurements and the error bar represents the standard deviation of these measurements. Electrolyte: 0.5 M KHCO3. | ||
Cu exhibits two reduction peaks at −0.16 V and −0.73 V, which represent the reduction of copper oxides.45,46 In shows a cathodic peak at −0.26 V and an anodic peak at 0.83 V, which are likely due to the redox reactions between In and In2O3/In(OH)2+.47 CuIn-30 shows both Cu/CuOx redox peaks and In-related redox peaks, indicating a mixture of Cu and In features on the surface, consistent with the TEM analysis (Fig. 1f–h). In addition, the cyclic voltammogram of CuIn-30 in 1 M KOH clearly exhibits the adsorption of *OH on Cu (100), demonstrating the presence of Cu species on the surface of CuIn-30 (Fig. S23†).
Further, *CO adsorption on CuIn-30 catalyst was characterized to reveal the binding strength between the active sites and *CO. As a comparison, the CO adsorption behaviours on Cu metal and In metal were also characterized (Fig. 3b–d). The peak observed at −0.07 V vs. RHE on Cu could be attributed to the desorption of CO from Cu,23,48 while the peak observed at −0.16 V vs. RHE on In is likely due to the desorption of CO from In. Interestingly, only a cathodic peak at −0.07 V is observed on CuIn-30. This identical feature to the one observed on Cu thereby indicates that Cu sites in CuIn-30 are capable of adsorbing CO. Conversely, CuIn-30 shows no *CO adsorption/desorption peak on In, demonstrating that the presence of Cu modifies surface In species, making them not capable of binding CO strongly. The temperature-programmed desorption (TPD) behaviour of Cu and CuIn-30 in a CO atmosphere further verifies that the interaction between Cu and In results in a relatively weak adsorption of CO on the Cu–In catalyst (Fig. S24†). The weak binding between In and CO favors the release of CO from CuIn at low overpotential.
The above analysis raises the question of why the formation of C2+ products is suppressed on CuIn-30, as Cu sites within CuIn-30 show the capability of adsorbing CO. Therefore, we further investigated the electrochemical reduction of carbon monoxide on the Cu and CuIn-30 catalysts, as shown in Fig. 3e–f (Tables S20, S21, Fig. S25 and S26†). Interestingly, CO reduction on Cu leads to the generation of C2+ products, with the FE of C2+ products reaching around 40% in a wide current density range from −34 to −94 mA cm−2. Interestingly, CuIn-30 also exhibits the generation of C2+ products in CO reduction at a current density from −40 mA cm−2 to −95 mA cm−2. The FE of C2+ products reaches approximately 44% at −77 mA cm−2. This value is significantly higher than the one observed in CO2 reduction on CuIn-30. It is also noted that H2 is produced with a FE of 18–44%, indicating the non-optimal binding strength of CO on CuIn.
As suggested by theoreticians, the key step towards the formation of C2+ product is the coupling of two single-carbon (C1) intermediates. This not only requires overcoming the reaction energy barrier for carbon–carbon coupling but also ensures adequate adsorption of the C1 intermediate on the catalyst surface.8 Combined with electrochemical *CO adsorption and the performance of the CO reduction on Cu and CuIn-30, we conclude that Cu sites in CuIn-30 are capable of adsorbing *CO as well as further reducing adsorbed *CO to C2+ products at the tested potential/current. However, CuIn-30 hardly catalyzes the production of C2+ products in CO2 reduction at low overpotential. Thus, the limited production of C2+ from CO2 on CuIn-30 could only be attributed to the insufficient availability of *CO on Cu at low overpotential. This is further corroborated by electrochemical CO2 reduction on CuIn-30 at higher cathodic current density, where Cu sites become active for CO2-to-CO conversion to produce CO intermediates. At −140 mA cm−2, the FE of C2+ products on CuIn-30 is 10%, and the value on CuIn-15 reaches around 27% (Fig. 3g and Table S15†).
In situ Raman spectroscopy
The reconstruction of the catalysts during pre-reduction and the binding behavior of key species was further investigated by in situ Raman spectroscopy at various current densities on representative films (Fig. 4).40 A custom-built spectro-electrochemical Raman flow cell (Fig. 4a and S27†) was used to perform CO2 electrolysis in 0.5 M KHCO3, with the catalytic interfaces being simultaneously probed by a Raman spectrometer. The catalyst-loaded GDE was positioned upward, between the cathodic gas chamber and the catholyte chamber. To ensure a better signal-to-noise ratio, we employed a water immersion objective, featuring a small working distance and a large numerical aperture, to collect the scattered light. | ||
| Fig. 4 In situ Raman spectra on Cu and CuIn catalysts. (a) Exploded view of the in situ Raman electrolysis flow cell. In situ Raman spectra and corresponding chronoamperograms (inset) of the (b) CuO–In-5, (c) CuO–In-30 and (d) CuO–In-60 catalysts at −30 mA cm−2. In situ Raman spectra of the (e) Cu, (f) CuIn-0.5, (g) CuIn-1.25 and (h) CuIn-30 catalysts during electrochemical reduction of CO2 at different current densities. CO2 was continuously flowed to the cathodic gas chamber during measurement. A near-infrared laser (785 nm) was used as the excitation source. The spectra were collected at a range of 100−2200 cm−1 after sufficient electrolysis time to ensure the reduction of oxides. A representative spectrum of multiple measurements at steady state of the electrolysis is shown under each condition. Electrolyte: 0.5 M KHCO3. The spikes in some spectra are due to cosmic rays. | ||
We first investigated the time-dependent surface change during pre-reduction using CuO–In-5, CuO–In-30, and CuO–In-60, at a current density of −30 mA cm−2 (Fig. 4b–d). At open-circuit potential, multiple Raman peaks at about 470 and 570 cm−1 correspond to the phonons associated with the bcc-structured indium oxide.39,49 The Raman peak at about 620 cm−1 corresponds to the Γ−15 phonon of the Cu2O lattice.50 Once a negative bias is applied, the reduction of oxides is demonstrated by the gradual disappearance of these peaks.38 The spectrum becomes almost featureless after 2 min, proving that the majority of the oxides have been reduced to their metallic forms during the pre-reduction process. Recently, it has been proposed that Cu+ species could be favorable for the C–C coupling process.51–55 However, the reduction process leads to a change of the bulk oxides to their metallic forms on our catalysts.
Interestingly, when the negative bias is removed, under open-circuit potential, the oxide emerges on the surface within tens of seconds. It can be explicitly stated that both Cu and In were mainly metallic during the CO2 reduction process. The oxides observed in the TEM might be ascribed to the rapid oxidation of the catalyst after the cathodic bias is removed (Fig. 1g).
The Raman spectra of four representative catalysts during CO2 reduction were then collected (Fig. 4e–h). The peak at ∼1070 cm−1, observed on Cu, CuIn-0.5, CuIn-1.25, and CuIn-30 during CO2 electrolysis, corresponds to the vibration of CO32−.56 *CO adsorbed on Cu exhibits distinct Raman peaks at 280, 360, and 2100 cm−1, representing the restricted rotation of Cu–C, Cu–C stretching, and ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O stretching of atop-adsorbed *CO on Cu, respectively (Fig. 4e).57 These peaks are still visible on thin-In-coated catalysts, such as CuIn-0.5 and CuIn-1.25 (Fig. 4f–g), demonstrating the adsorption of *CO on Cu. This phenomenon is consistent with the observed formation of C2+ products in CO2 reduction on CuIn-0.5 and CuIn-1.25 (Fig. 2f). Interestingly, no peak related to adsorbed *CO is observed on the CuIn-30 catalyst (Fig. 4h), which agrees well with the suppressed formation of C2+ products on CuIn-30. It is noted that complementary Infrared spectroscopy might provide more insights into CO vibration signals but engineering a flow-type spectro-electrochemical cell for in situ infrared spectroscopy is technically challenging. In addition, a Raman peak at 1248 cm−1 is observed on CuIn-30, which might correspond to the symmetric stretching of adsorbed *CO2−.58 The appearance of the *CO2− adsorption peak indicates the enhanced activation of CO2 on CuIn-30.
O stretching of atop-adsorbed *CO on Cu, respectively (Fig. 4e).57 These peaks are still visible on thin-In-coated catalysts, such as CuIn-0.5 and CuIn-1.25 (Fig. 4f–g), demonstrating the adsorption of *CO on Cu. This phenomenon is consistent with the observed formation of C2+ products in CO2 reduction on CuIn-0.5 and CuIn-1.25 (Fig. 2f). Interestingly, no peak related to adsorbed *CO is observed on the CuIn-30 catalyst (Fig. 4h), which agrees well with the suppressed formation of C2+ products on CuIn-30. It is noted that complementary Infrared spectroscopy might provide more insights into CO vibration signals but engineering a flow-type spectro-electrochemical cell for in situ infrared spectroscopy is technically challenging. In addition, a Raman peak at 1248 cm−1 is observed on CuIn-30, which might correspond to the symmetric stretching of adsorbed *CO2−.58 The appearance of the *CO2− adsorption peak indicates the enhanced activation of CO2 on CuIn-30.
Mechanism of CO2 reduction to CO on the CuIn catalyst
We then carried out DFT calculations through the Vienna Ab initio Simulation Package (VASP) model (Fig. 5a–c and S28–S30†). CO2 is first converted into the key adsorbed intermediate *COOH. Subsequently, *COOH undergoes proton-coupled electron transfer to form *CO, which then desorbs from the catalyst surface to yield CO. Compared with Cu and In, the ΔG required for the formation of *COOH on the surface of CuIn-30 is lower, indicating that the surface of the CuIn catalyst is more prone to the formation of *COOH (Fig. 5a). Additionally, the competing hydrogen evolution reaction and formate production processes exhibit higher energy barriers compared to those of Cu and In (Fig. 5b and c).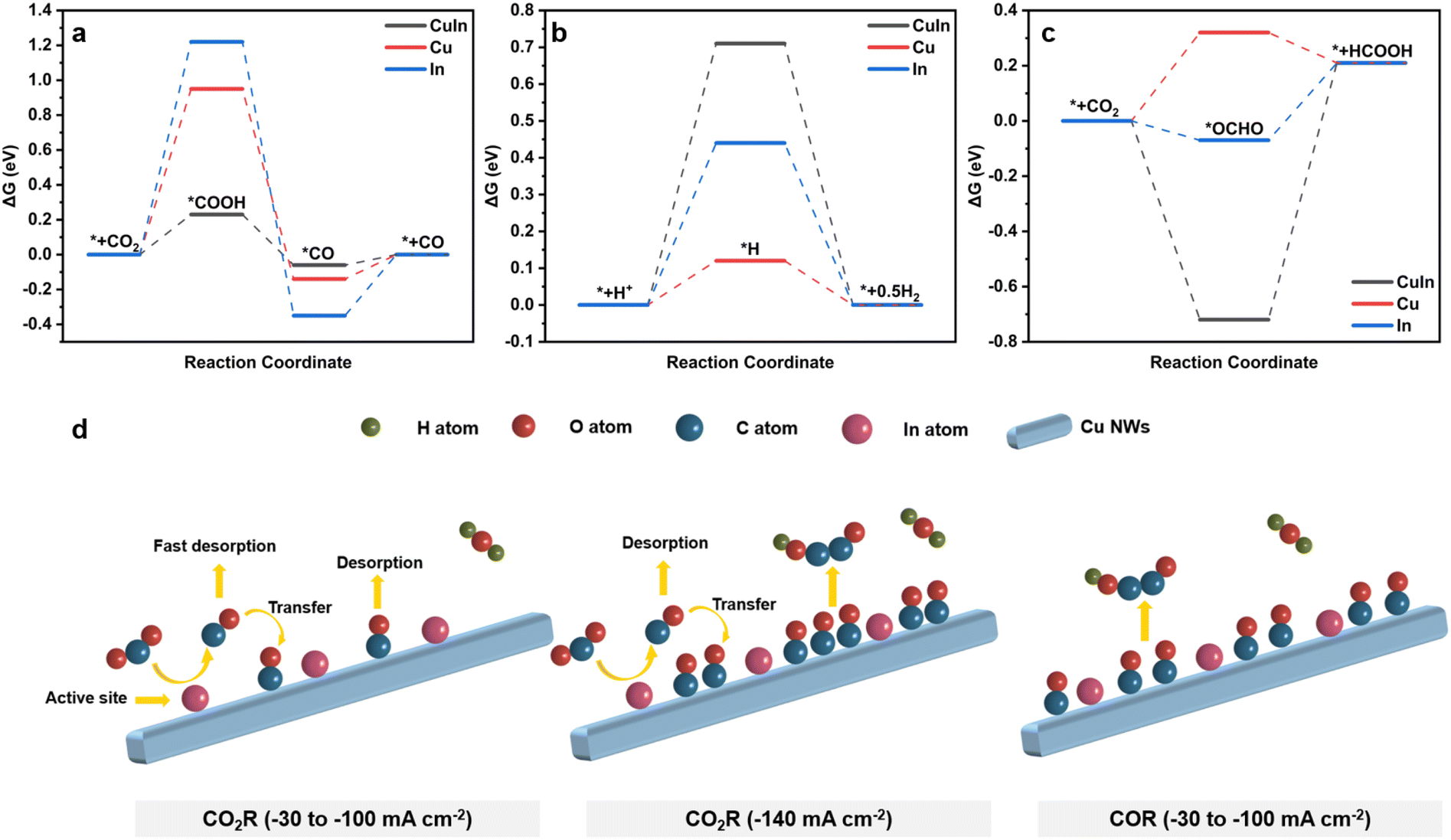 | ||
| Fig. 5 Free-energy diagrams for (a) CO2 to CO, (b) H2 evolution, and (c) CO2 to HCOOH on CuIn, Cu, and In surfaces. (d) Proposed mechanism for the formation of CO and C2+ products on the CuIn-30 catalyst under different conditions. | ||
Combining electrochemical analysis and DFT modelling, we suggest the reaction mechanism of CO2 or CO reduction on CuIn-30 (Fig. 5d). At a current density from −30 mA cm−2 to −100 mA cm−2, In is the main active site. Due to the modification of Cu, In could not only actively transform CO2 to CO but also release the weakly-bonded CO (Fig. 3b) to form CO gas, with a small amount of CO being transferred to adjacent Cu sites, which hinders efficient C–C coupling (Fig. 2d). Consistently, the signal of adsorbed *CO is invisible in the Raman spectrum (Fig. 4h). When the current density reaches −140 mA cm−2, the coverage of CO on Cu is enhanced, either by the spillover of CO from the In site or the activated CO2-to-CO conversion on the Cu site. The improved surface coverage of CO on Cu will trigger C–C coupling to generate C2+ products (Fig. 3e). We note here that the Raman features of adsorbed *CO under this condition are hardly acquired, as the Raman signal is adversely affected by the gas bubbles (Fig. 4h). Nevertheless, the enhanced production of C2+ products at higher current density on CuIn indicates that the surface coverage of CO is high. Additionally, the surface coverage of CO is greatly enhanced during CO reduction at a current density from −30 mA cm−2 to −100 mA cm−2, leading to the generation of C2+ products.
Photosynthesis of CO from CO2 on the CuIn catalyst
With the CuIn catalyst, we further constructed a two-electrode electrolyzer with electrodeposited InOx as the anode (Fig. S31†).59 We deliberately eliminated the AEM and employed 2 M KOH instead of 0.5 M KHCO3 as the electrolyte to minimize the voltage loss. The electrolyzer is further powered by a trip-junction solar cell under illumination, as a proof-of-concept of an artificial photosynthesis system (Fig. 6a). The solar-driven CO2 reduction system was engineered by wiring the solar cell with the electrolyzer. The solar cell employed here is a triple-junction InGaP2/InGaAs/Ge photovoltaic cell, with an effective illumination area of 0.92 cm2. The full system was also illuminated continuously, with the generated gas products periodically sampled into GC for quantification. | ||
| Fig. 6 Photosynthesis of CO from solar-driven CO2 reduction. (a) A schematic of the solar-driven system, which consists of a III–V triple-junction solar cell and a two-electrode flow reactor. (b) Representative linear sweep voltammograms of the solar cell and electrolysis cell. (c) Photocurrent density of the unassisted photovoltaic-driven electrolysis under standard AM 1.5 G illumination. (d) FE of the generated CO and solar-to-CO energy conversion efficiency in the unassisted photovoltaic-driven electrolysis under standard AM 1.5 G illumination. The current density in (b) and (c) is normalized against the working area of the solar cell (0.92 cm2). MPP denotes the maximum power point of the solar cell. | ||
The voltage–current behaviour of the solar cell under global air mass (AM) 1.5 G illumination was first characterized. The open-circuit photovoltage (Voc) is 2.42 V and the short-circuit current density (jsc) is 15.40 mA cm−2, with a fill factor of 82.0%, yielding a maximum power conversion efficiency (PCE) of 30.6% (Fig. 6b). The initial linear sweep voltametric scan of the electrolyzer shows a peak at about 1.6 V, but this peak disappears in the subsequent scan. This is likely due to the reduction of surface oxides on the cathode. The intersection of the j–V curves of the electrolyzer and solar cell shows that the predicted operating voltage and current density are about 1.9 V and 11.5 mA cm−2, respectively (Fig. 6b). It is noted that the electrolysis current is also normalized against the effective illuminated area of the solar cell (0.92 cm2).
The full system was then operated under continuous AM 1.5 G illumination for 105 min. The average current density is recorded to be 10.59 mA cm−2 (Fig. 6c). The FE of CO reaches lower than the value in the three-electrode system, likely due to the change of electrolyte from 0.5 M KHCO3 to 2 M KOH. The high alkalinity of the electrolyte, though it reduces voltage loss, affects the activity of the CuIn catalysts. Over a 105 min electrolysis period, our solar-driven system shows a peak solar-to-fuel energy conversion efficiency of 10.3% for solar-to-CO (Fig. 6d), with an average value of about 9.3% during a 105-min test (Table S22†). This efficiency value stands high among the previously-reported energy efficiencies (Table S23†).
The deployment of efficient solar-driven CO2 reduction remains as a vital step towards the industrialization of a CO2 reduction system. This not only requires a fundamental advancement in the understanding of the structure–performance relationship, but also depends on system engineering. Here, we proposed the usage of a CuIn catalyst as an alternative to Ag or Au for the efficient electrosynthesis of CO from CO2. This proof-of-concept demonstration of solar-driven CO2 reduction makes a step forward towards the utilization of excess solar electricity for carbon upgrading.
Conclusions
In summary, we developed a CuIn catalyst for highly efficient CO synthesis through CO2 electroreduction. In a custom-designed flow cell, CuIn-30 delivers a high FE for CO production, reaching up to 91% at −69 mA cm−2. By electrochemical CO adsorption and CO reduction, we propose that the In serves as the active site for CO generation with rapid desorption of CO. In situ Raman spectroscopic investigation on the key binding species further supports our hypothesis. We also employed a triple-junction photovoltaic cell to power our electrolyzer, with a solar-to-fuel energy conversion efficiency exceeding 10%.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
D. R. conceptualized and supervised the project. S. X. synthesized the catalysts, conducted the catalytic experiments and related data processing, and performed material characterization and analysis. C. W. and C. R. assisted with the electrochemical adsorption of *CO and *OH experiments. H. Y. and W. G. assisted with the double layer capacitance measurement experiments. B. D. and Y. L. assisted with the solar-driven electrochemical CO2 reduction experiments. S. X. wrote the manuscript. D. R. revised the manuscript. All authors approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Young Scientist Funding (22409158, D. R.) and Excellent Young Scientist (Overseas) Funding (GYKP042, D. R.) from the National Natural Science Foundation of China, start-up funding from Xi'an Jiaotong University (HG6J002, D. R.), a Qin Chuang Yuan Grant (QCYRCXM-2022-122, D.R.) from Shaanxi Ministry of Science and Technology, and Fundamental Research Funding for Central Universities from Xi'an Jiaotong University (xzd012022005, D. R.) for financial support. We thank Ms. Xiaojing Zhang at School of Physics, Xi'an Jiaotong University for her help with Transmission Electron Microscopy.References
- J. Qiao, Y. Liu, F. Hong and J. Zhang, A review of catalysts for the electroreduction of carbon dioxide to produce low-carbon fuels, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- G. Wang, J. Chen, Y. Ding, P. Cai, L. Yi, Y. Li, C. Tu, Y. Hou, Z. Wen and L. Dai, Electrocatalysis for CO2 conversion: from fundamentals to value-added products, Chem. Soc. Rev., 2021, 50, 4993–5061 RSC.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, New insights into the electrochemical reduction of carbon dioxide on metallic copper surfaces, Energy Environ. Sci., 2012, 5, 7050–7059 RSC.
- J. Gao, A. Bahmanpour, O. Krocher, S. M. Zakeeruddin, D. Ren and M. Gratzel, Electrochemical synthesis of propylene from carbon dioxide on copper nanocrystals, Nat. Chem., 2023, 15, 705–713 CrossRef CAS PubMed.
- R. Kortlever, I. Peters, C. Balemans, R. Kas, Y. Kwon, G. Mul and M. T. M. Koper, Palladium-gold catalyst for the electrochemical reduction of CO2 to C1–C5 hydrocarbons, Chem. Commun., 2016, 52, 10229–10232 RSC.
- W. Gao, Y. Xu, L. Fu, X. Chang and B. Xu, Experimental evidence of distinct sites for CO2-to-CO and CO conversion on Cu in the electrochemical CO2 reduction reaction, Nat. Catal., 2023, 6, 885–894 CrossRef CAS.
- Grand View Research, Carbon Monoxide Market Size, Share & Trends Analysis Report By Application (Chemical, Metal Fabrication, Electronics, Pharma & Biotechnology, Meat & Coloring Preservative), By Region (North America, Europe), And Segment Forecasts 2025 - 2030, https://www.grandviewresearch.com/industry-analysis/carbon-monoxide-market-report Search PubMed.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Progress and Perspectives of Electrochemical CO2 Reduction on Copper in Aqueous Electrolyte, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- Y. Y. Birdja, E. Perez-Gallent, M. C. Figueiredo, A. J. Gottle, F. Calle-Vallejo and M. T. M. Koper, Advances and challenges in understanding the electrocatalytic conversion of carbon dioxide to fuels, Nat. Energy, 2019, 4, 732–745 Search PubMed.
- S. Li, A. V. Nagarajan, D. R. Alfonso, M. Sun, D. R. Kauffman, G. Mpourmpakis and R. Jin, Boosting CO2 Electrochemical Reduction with Atomically Precise Surface Modification on Gold Nanoclusters, Angew. Chem., Int. Ed., 2021, 60, 6351–6356 CrossRef CAS.
- R. Shi, J. Guo, X. Zhang, G. I. N. Waterhouse, Z. Han, Y. Zhao, L. Shang, C. Zhou, L. Jiang and T. Zhang, Efficient wettability-controlled electroreduction of CO2 to CO at Au/C interfaces, Nat. Commun., 2020, 11, 3028 CrossRef CAS.
- W. Zhu, R. Michalsky, O. Metin, H. Lv, S. Guo, C. J. Wright, X. Sun, A. A. Peterson and S. Sun, Monodisperse Au Nanoparticles for Selective Electrocatalytic Reduction of CO2 to CO, J. Am. Chem. Soc., 2013, 135, 16833–16836 CrossRef CAS PubMed.
- S. Liu, C. Sun, J. Xiao and J.-L. Luo, Unraveling Structure Sensitivity in CO2 Electroreduction to Near-Unity CO on Silver Nanocubes, ACS Catal., 2020, 10, 3158–3163 CrossRef CAS.
- S. Liu, H. Tao, L. Zeng, Q. Liu, Z. Xu, Q. Liu and J.-L. Luo, Shape-Dependent Electrocatalytic Reduction of CO2 to CO on Triangular Silver Nanoplates, J. Am. Chem. Soc., 2017, 139, 2160–2163 CrossRef CAS PubMed.
- Q. Lu, J. Rosen, Y. Zhou, G. S. Hutchings, Y. C. Kimmel, J. G. Chen and F. Jiao, A selective and efficient electrocatalyst for carbon dioxide reduction, Nat. Commun., 2014, 5, 3242 CrossRef PubMed.
- D. Gao, H. Zhou, J. Wang, S. Miao, F. Yang, G. Wang, J. Wang and X. Bao, Size-Dependent Electrocatalytic Reduction of CO2 over Pd Nanoparticles, J. Am. Chem. Soc., 2015, 137, 4288–4291 CrossRef CAS PubMed.
- H. Huang, H. Jia, Z. Liu, P. Gao, J. Zhao, Z. Luo, J. Yang and J. Zeng, Understanding of Strain Effects in the Electrochemical Reduction of CO2: Using Pd Nanostructures as an Ideal Platform, Angew. Chem., Int. Ed., 2017, 56, 3594–3598 Search PubMed.
- W. Sheng, S. Kattel, S. Yao, B. Yan, Z. Liang, C. J. Hawxhurst, Q. Wu and J. G. Chen, Electrochemical reduction of CO2 to synthesis gas with controlled CO/H2 ratios, Energy Environ. Sci., 2017, 10, 1180–1185 Search PubMed.
- S. Rasul, D. H. Anjum, A. Jedidi, Y. Minenkov, L. Cavallo and K. Takanabe, A Highly Selective Copper–Indium Bimetallic Electrocatalyst for the Electrochemical Reduction of Aqueous CO2 to CO, Angew. Chem., Int. Ed., 2015, 54, 2146–2150 CrossRef CAS PubMed.
- G. O. Larrazabal, A. J. Martin, S. Mitchell, R. Hauert and J. Perez-Ramirez, Enhanced Reduction of CO2 to CO over Cu-In Electrocatalysts: Catalyst Evolution is the Key, ACS Catal., 2016, 6, 6265–6274 CrossRef CAS.
- W. Luo, W. Xie, R. Mutschler, E. Oveisi, G. L. De Gregorio, R. Buonsanti and A. Zuttel, Selective and Stable Electroreduction of CO2 to CO at the Copper/Indium Interface, ACS Catal., 2018, 8, 6571–6581 CrossRef CAS.
- C. Shen, P. Wang, L. Li, X. Huang and Q. Shao, Phase and structure modulating of bimetallic Cu/In nanoparticles realizes efficient electrosynthesis of syngas with wide CO/H2 ratios, Nano Res., 2022, 15, 528–534 CrossRef CAS.
- J. Gao, J. Li, Y. H. Liu, M. Xia, Y. Z. Finfrock, S. M. Zakeeruddin, D. Ren and M. Grätzel, Solar reduction of carbon dioxide on copper-tin electrocatalysts with energy conversion efficiency near 20%, Nat. Commun., 2022, 13, 5898 CrossRef CAS PubMed.
- P. Wang, M. Qiao, Q. Shao, Y. Pi, X. Zhu, Y. Li and X. Huang, Phase and structure engineering of copper tin heterostructures for efficient electrochemical carbon dioxide reduction, Nat. Commun., 2018, 9, 4933 CrossRef PubMed.
- M. Zhang, Z. Zhang, Z. Zhao, H. Huang, D. H. Anjum, D. Wang, J.-h. He and K.-W. Huang, Tunable Selectivity for Electrochemical CO2 Reduction by Bimetallic Cu–Sn Catalysts: Elucidating the Roles of Cu and Sn, ACS Catal., 2021, 11, 11103–11108 CrossRef CAS.
- L. C. P. Perez, A. Arndt, S. Stojkovikj, I. Y. Ahmet, J. T. Arens, F. Dattila, R. Wendt, A. G. Buzanich, M. Radtke, V. Davies, K. Hoeflich, E. Koehnen, P. Tockhorn, R. Golnak, J. Xiao, G. Schuck, M. Wollgarten, N. Lopez and M. T. Mayer, Determining Structure–Activity Relationships in Oxide Derived Cu–Sn Catalysts During CO2 Electroreduction Using X-Ray Spectroscopy, Adv. Energy Mater., 2022, 12, 2103328 CrossRef.
- Y. Hori, H. Wakebe, T. Tsukamoto and O. Koga, Electrocatalytic process of co selectivity in electrochemical reduction of CO2 at metal-electrodes in aqueous-media, Electrochim. Acta, 1994, 39, 1833–1839 CrossRef CAS.
- A. Bagger, W. Ju, A. Sofia Varela, P. Strasser and J. Rossmeisl, Electrochemical CO2 Reduction: A Classification Problem, ChemPhysChem, 2017, 18, 3266–3273 CrossRef CAS PubMed.
- D. Ren, J. Gao, S. M. Zakeeruddin and M. Graetzel, Bimetallic Electrocatalysts for Carbon Dioxide Reduction, Chimia, 2019, 73, 928–935 CrossRef CAS PubMed.
- D. Ren, J. Gao, L. Pan, Z. Wang, J. Luo, S. M. Zakeeruddin, A. Hagfeldt and M. Grätzel, Atomic Layer Deposition of ZnO on CuO Enables Selective and Efficient Electroreduction of Carbon Dioxide to Liquid Fuels, Angew. Chem., Int. Ed., 2019, 58, 15036–15040 CrossRef CAS PubMed.
- J. F. Smith and V. L. Schneider, Anisotropic thermal expansion of indium, J. Less-Common Met., 1964, 7, 17–22 CrossRef CAS.
- J. Kim, W. Choi, J. W. Park, C. Kim, M. Kim and H. Song, Branched Copper Oxide Nanoparticles Induce Highly Selective Ethylene Production by Electrochemical Carbon Dioxide Reduction, J. Am. Chem. Soc., 2019, 141, 6986–6994 CrossRef CAS PubMed.
- X. Wang, K. Klingan, M. Klingenhof, T. Moeller, J. F. de Araujo, I. Martens, A. Bagger, S. Jiang, J. Rossmeisl, H. Dau and P. Strasser, Morphology and mechanism of highly selective Cu(II) oxide nanosheet catalysts for carbon dioxide electroreduction, Nat. Commun., 2021, 12, 794 CrossRef CAS PubMed.
- S. Asbrink and A. Waskowska, CuO-X-ray single-crystal structure determination at 196k and room-temperature, J. Phys.:Condens. Matter, 1991, 3, 8173–8180 CrossRef.
- M. Marezio, Refinement of the crystal structure of In2O3 at two wavelengths, Acta Crystallogr., 1966, 20, 723–728 CrossRef CAS.
- I. K. Suh, H. Ohta and Y. Waseda, High-temperature thermal-expansion of 6 metallic elements measured by dilatation method and X-ray-diffraction, J. Mater. Sci., 1988, 23, 757–760 CrossRef CAS.
- K. Adaikalam, S. Valanarasu, A. M. Ali, M. A. Sayed, W. Yang and H.-S. Kim, Photosensing effect of indium-doped ZnO thin films and its heterostructure with silicon, J. Asian Ceram. Soc., 2022, 10, 108–119 CrossRef.
- Y. Deng, A. D. Handoko, Y. Du, S. Xi and B. S. Yeo, In Situ Raman Spectroscopy of Copper and Copper Oxide Surfaces during Electrochemical Oxygen Evolution Reaction: Identification of CuIII Oxides as Catalytically Active Species, ACS Catal., 2016, 6, 2473–2481 Search PubMed.
- W. B. White and V. G. Keramidas, Vibrational-spectra of oxides with C-type rare-earth oxide structure, Spectrochim. Acta, Part A, 1972, A 28, 501–509 CrossRef.
- D. Ren, J. Gao, S. M. Zakeeruddin and M. Grätzel, New Insights into the Interface of Electrochemical Flow Cells for Carbon Dioxide Reduction to Ethylene, J. Phys. Chem. Lett., 2021, 12, 7583–7589 CrossRef CAS PubMed.
- J. Gao, H. Zhang, X. Guo, J. Luo, S. M. Zakeeruddin, D. Ren and M. Grätzel, Selective C–C Coupling in Carbon Dioxide Electroreduction via Efficient Spillover of Intermediates As Supported by Operando Raman Spectroscopy, J. Am. Chem. Soc., 2019, 141, 18704–18714 CrossRef CAS PubMed.
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. M. Koper, Catalysts and Reaction Pathways for the Electrochemical Reduction of Carbon Dioxide, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS.
- B. Wulan, X. Cao, D. Tan, J. Ma and J. Zhang, To Stabilize Oxygen on In/In2O3 Heterostructure via Joule Heating for Efficient Electrocatalytic CO2 Reduction, Adv. Funct. Mater., 2023, 33, 2209114 CrossRef CAS.
- Y. Huang, X. Mao, G. Yuan, D. Zhang, B. Pan, J. Deng, Y. Shi, N. Han, C. Li, L. Zhang, L. Wang, L. He, Y. Li and Y. Li, Size-Dependent Selectivity of Electrochemical CO2 Reduction on Converted In2O3 Nanocrystals, Angew. Chem., Int. Ed., 2021, 60, 15844–15848 CrossRef CAS PubMed.
- W. Tang, A. A. Peterson, A. S. Varela, Z. P. Jovanov, L. Bech, W. J. Durand, S. Dahl, J. K. Norskov and I. Chorkendorff, The importance of surface morphology in controlling the selectivity of polycrystalline copper for CO2 electroreduction, Phys. Chem. Chem. Phys., 2012, 14, 76–81 RSC.
- C. S. Chen, A. D. Handoko, J. H. Wan, L. Ma, D. Ren and B. S. Yeo, Stable and selective electrochemical reduction of carbon dioxide to ethylene on copper mesocrystals, Catal. Sci. Technol., 2015, 5, 161–168 RSC.
- Y.-H. Chung and C.-W. Lee, Electrochemical behaviors of Indium, J. Electrochem. Sci. Technol, 2012, 3, 1–13 CrossRef CAS.
- Y. Hori, O. Koga, H. Yamazaki and T. Matsuo, Infrared-spectroscopy of adsorbed CO and intermediate species in electrochemical reduction of CO2 to hydrocarbons on a Cu electrode, Electrochim. Acta, 1995, 40, 2617–2622 CrossRef CAS.
- M. Jothibas, C. Manoharan, S. J. Jeyakumar and P. Praveen, Study on structural and optical behaviors of In2O3 nanocrystals as potential candidate for optoelectronic devices, J. Mater. Sci.-Mater. Electron., 2015, 26, 9600–9606 CrossRef CAS.
- P. Y. Yu and Y. R. Shen, Resonance raman studies in Cu2O.II. yellow and green excitonic series, Phys. Rev. B: Condens. Matter Mater. Phys., 1978, 17, 4017–4030 CrossRef CAS.
- H. Li, Y. Jiang, X. Li, K. Davey, Y. Zheng, Y. Jiao and S.-Z. Qiao, C2+ Selectivity for CO2 Electroreduction on Oxidized Cu-Based Catalysts, J. Am. Chem. Soc., 2023, 145, 14335–14344 CrossRef CAS PubMed.
- Y. Zhou, F. Che, M. Liu, C. Zou, Z. Liang, P. De Luna, H. Yuan, J. Li, Z. Wang, H. Xie, H. Li, P. Chen, E. Bladt, R. Quintero-Bermudez, T.-K. Sham, S. Bals, J. Hofkens, D. Sinton, G. Chen and E. H. Sargent, Dopant-induced electron localization drives CO2 reduction to C2 hydrocarbons, Nat. Chem., 2018, 10, 974–980 CrossRef CAS PubMed.
- X. He, L. Lin, X. Li, M. Zhu, Q. Zhang, S. Xie, B. Mei, F. Sun, Z. Jiang, J. Cheng and Y. Wang, Roles of copper(I) in water-promoted CO2 electrolysis to multi-carbon compounds, Nat. Commun., 2024, 15, 9923 CrossRef CAS PubMed.
- H. Mistry, A. S. Varela, C. S. Bonifacio, I. Zegkinoglou, I. Sinev, Y.-W. Choi, K. Kisslinger, E. A. Stach, J. C. Yang, P. Strasser and B. Roldan Cuenya, Highly Selective Plasma-activated Copper Catalysts for Carbon Dioxide Reduction to Ethylene, Nat. Commun., 2016, 7, 12123 CrossRef PubMed.
- T. Zhang, S. L. Xu, D. L. Chen, T. Luo, J. L. Zhou, L. C. Kong, J. J. Feng, J. Q. Lu, X. X. Weng, A. J. Wang, Z. Q. Li, Y. Q. Su and F. Yang, Selective Increase in CO2 Electroreduction to Ethanol Activity at Nanograin-Boundary-Rich Mixed Cu(I)/Cu(0) Sites via Enriching Co-Adsorbed CO and Hydroxyl Species, Angew. Chem., Int. Ed., 2024, 63, e202407748 CrossRef CAS PubMed.
- B. Smith, D. Irish, P. Kedzierzawski and J. Augustynski, A surface enhanced roman scattering study of the intermediate and poisoning species formed during the electrochemical reduction of CO2 on copper, J. Electrochem. Soc., 1997, 144, 4288 CrossRef CAS.
- I. Oda, H. Ogasawara and M. Ito, Carbon monoxide adsorption on copper and silver electrodes during carbon dioxide electroreduction studied by infrared reflection absorption spectroscopy and surface-enhanced raman spectroscopy, Langmuir, 1996, 12, 1094–1097 CrossRef CAS.
- J. de Ruiter, H. An, L. Wu, Z. Gijsberg, S. Yang, T. Hartman, B. M. Weckhuysen and W. van der Stam, Probing the Dynamics of Low-Overpotential CO2-to-CO Activation on Copper Electrodes with Time-Resolved Raman Spectroscopy, J. Am. Chem. Soc., 2022, 144, 15047–15058 CrossRef CAS PubMed.
- D. Ren, N. W. X. Loo, L. Gong and B. S. Yeo, Continuous Production of Ethylene from Carbon Dioxide and Water Using Intermittent Sunlight, ACS Sustain. Chem. Eng., 2017, 5, 9191–9199 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc01110h |
| This journal is © The Royal Society of Chemistry 2025 |