
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Redox non-innocent bis-silylene aluminium complexes with a carborane backbone†
Artemis
Saddington
a,
Shenglai
Yao
a,
Christian
Lorent
 b and
Matthias
Driess
*a
b and
Matthias
Driess
*a
aDepartment of Chemistry: Metalorganics and Inorganic Materials, Technische Universität Berlin, Strasse des 17. Juni 115, Sekr. C2, Berlin, 10623, Germany. E-mail: matthias.driess@tu-berlin.de
bDepartment of Chemistry: Physical and Biophysical Chemistry, Technische Universität Berlin, Strasse des 17. Juni 135, Sekr. PC14, Berlin, 10623, Germany
First published on 7th March 2025
Abstract
The redox non-innocent bis-silylenyl ortho-carborane ligands [SiII(CCcage)SiII] (CCcage = o-C2B10H10, SiII = ArC(NtBu)2Si; Ar = C6H5, p-tBuC6H4), with their particular chelating and electronic properties, have been employed for the synthesis of new donor-stabilized SiII → AlIII complexes, potential precursors to low oxidation state aluminium complexes. Due to the redox non-innocence of the carborane backbone, [AlI2+] complexes with three ligand oxidation states were characterized: with neutral and radical anionic closo- as well as dianionic nido-C2B10 cores. Reduction at the aluminium center could also be enacted with potassium/naphthalene leading to {K[SiII(CCcage)SiII]Al(C10H8)} derivatives from [1 + 4] cycloaddition reaction. The mechanism of this dearomatization reaction is proposed to occur via the formation of transient low oxidation state aluminium intermediates (radicals and/or aluminylenes) that are trapped by naphthalene.
Introduction
Strong σ-donors are an important tool in the synthesis of zero-valent main group element complexes, which have garnered significant interest in the last decade.1–4 Complexes of isolobal molecular ions supported by neutral ligands also rely on strong σ-donors for stabilization. Not only do such low valent compounds have interesting structures but they also possess unique potential applications. For example, they can act as ligands with unique bridging coordination modes, form otherwise difficult-to-realize molecules through oxidation, and have potential as novel single-source precursors for the synthesis of functional materials.5–9 Among the group 13 elements, the first zero oxidation state Bn (n = 1, 2, 4) compounds were isolated in recent years, mainly with carbene ligands.10–14 Of these, the rather π-accepting cAACs have also been applied to the synthesis of donor-stabilized aluminium compounds, such as an extraordinary mononuclear AlI hydride reported by Braunschweig and coworkers, that also features significant diradical character at the carbene-C centers.15–19 Whilst no zero oxidation state aluminium compounds have been proposed yet, the first AlI (aluminylene) cation I was synthesized in 2022 by Krossing and co-workers, with only neutral AlCp* substituents.20 Related GaI ion complexes with various donor ligands (e.g. PPh3, NHC) have been accessed from the useful salt complex [Ga(C6H5Me)2]+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) [Al(OC(CF3)3)4]− since 2010.21,22 In 2017, the first BI (borylene) cation II (X = CO) was reported by Xie and Lin using a chelating bis-silylene ligand.23 We have demonstrated that bis-silylenes can act as effective σ-donor ligands, enabling access to monoatomic zero oxidation state group 14 and 15 complexes.5,24–26 Such compounds are capable of facile activation of small molecules such as H2, NH3, 9-BBN, CO and CO2.27–30 Silylenes are defined as divalent silicon species, isoelectronic with singlet carbenes.31,32 Amidinato-silylenes in particular can be fixed to many spacer molecules, establishing a family of highly adaptable σ-donating chelating ligands.33–36
[Al(OC(CF3)3)4]− since 2010.21,22 In 2017, the first BI (borylene) cation II (X = CO) was reported by Xie and Lin using a chelating bis-silylene ligand.23 We have demonstrated that bis-silylenes can act as effective σ-donor ligands, enabling access to monoatomic zero oxidation state group 14 and 15 complexes.5,24–26 Such compounds are capable of facile activation of small molecules such as H2, NH3, 9-BBN, CO and CO2.27–30 Silylenes are defined as divalent silicon species, isoelectronic with singlet carbenes.31,32 Amidinato-silylenes in particular can be fixed to many spacer molecules, establishing a family of highly adaptable σ-donating chelating ligands.33–36
The area of low oxidation state silicon–aluminium chemistry is still in its infancy with the first examples of such compounds published in 2022–2023.37–40 Last year, we reported the transient pincer bis-silylene-supported aluminylene III (Ar = Ph), that dimerizes to give an Si2Al2 heterocycle (Fig. 1).41 Compound III could also be trapped as an iron(0) complex, from which we inferred its structure using density functional theory (DFT) calculations. Later, the isolable aluminylene III (Ar = Mes) was characterized by Mo and coworkers and agreed with our calculations.42 We also investigated the neutral bis-silylene [SiII(Xant)SiII] (Xant = 9,9-dimethylxanthene) for the synthesis of aluminylene complexes, but found it unsuitable for the synthesis of aluminylene compounds except as an Fe0 adduct IV.41,43
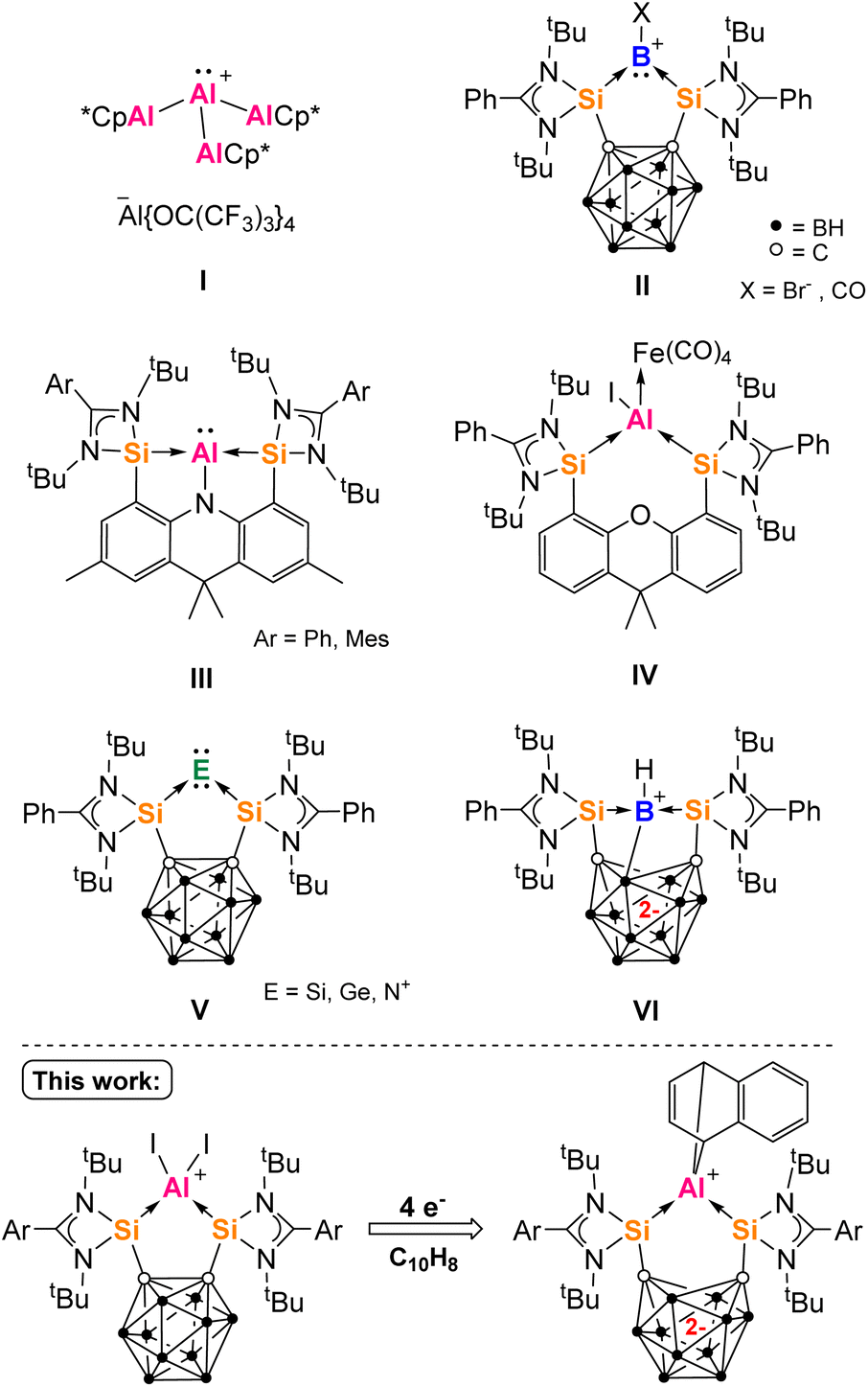 | ||
| Fig. 1 An Al(I) complex supported only by neutral ligands (I), examples of group 13–15 bis-silylene complexes (II–VI) and the new bis-silylene aluminium complexes of this work. | ||
We turned our interest towards the bis-silylene [SiII(CCcage)SiII] 1a based on an ortho-icosahedral carborane spacer [CCcage = o-C2B10H10, SiII = LSi, L = PhC(NtBu)2].44,451a is a stronger σ-donor and π-acceptor ligand than [SiII(Xant)SiII], which has been demonstrated experimentally by their differing reactivity towards CO and P4, and relates to the inductive withdrawing effect of carborane substituents.27,46–491a is also highly rigid, has a fixed smaller bite angle and generates more stable five-membered rings on complexation to a single atom. Furthermore, (C2B10)-functionalized compounds are capable of reversibly accepting one or two electrons to give radical anion or dianion complexes respectively.50,51 Accordingly, the two-electron reduction product {(K+(THF)4)2[SiII(CCcage)SiII]2−}n has been isolated.52 We have used 1a to furnish some exceptional redox non-innocent complexes of Si0, Ge0 and NIVII, with other chelating carboranyl tetrylenes since being investigated.48,52–58 Complexes II and V (Fig. 1) are capable of redox-induced valence tautomerism or electron transfer (ET), rare for main group element complexes. This manifests as the unexpected flow of electrons in or out of the carborane cage away or towards the element center, often accompanied by E–E coupling.59,60 For example, upon two-electron reduction, II (X = Br−) becomes VI (Fig. 1).61 In this work, we investigated the suitability of 1 for the synthesis of primarily donor-stabilized aluminium compounds, that could potentially act as precursors to low oxidation state aluminium complexes. We were first able to isolate AlIII complexes of 1 with three different ligand oxidation states. Further reduction with K(C10H8) leads to 1,4-naphthalene derivatives of AlIII, which points to the existence of largely unknown low-valent aluminium intermediates.
Results and discussion
Generation of bis-silylenyl carborane aluminium complexes
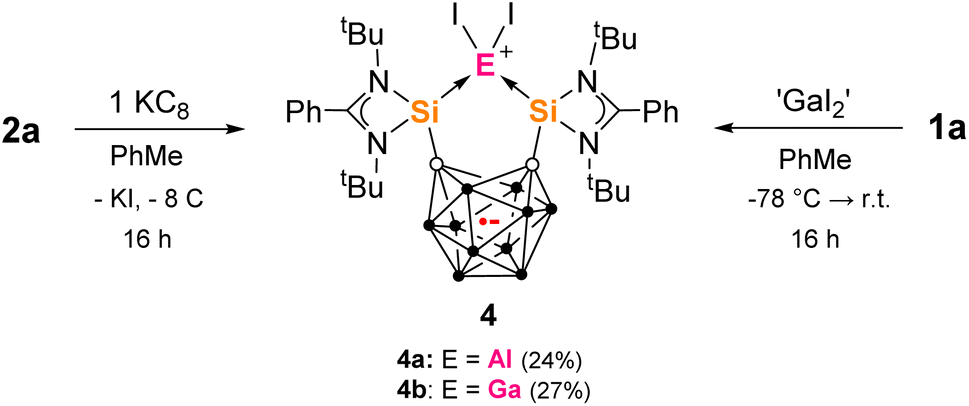
Many metallacarborane derivatives containing aluminium have been described since the 1970s.62–65 Despite chelating ligands with carborane scaffolds being well known, only recently did a report of an aluminium complex bearing a carborane backbone appear, specifically a nido-C2B9-based salen [NOON] ligand.66,67 We were able to prepare a series of closo-C2B10-based aluminium complexes straightforwardly using the bis-silylenes 1a and 1b (Scheme 1). Thus, adduct 2a was produced as colorless precipitate in 79% yield from the reaction of one molar equiv of 1a with one molar equiv AlI3 in toluene at low temperature. 2a exhibits very poor solubility in common aprotic solvents and decomposes instantly when dissolved in THF at room temperature. Its 1H and 1H/13C-coupled NMR spectra could therefore only be recorded in o-dichlorobenzene-D4. The marginally more soluble ion pair 2b was prepared similarly in 85% yield using two molar equivs of AlI3. A few colorless crystals of 2b were grown from Et2O solutions that were suitable for an scXRD analysis. Dissolution of 2b in THF led to an orange solution and almost clean reformation of the free bis-silylene 1a detected by 1H NMR. | ||
| Scheme 1 Synthesis of complexes 2a–c from bis-silylenes 1a-b and AlI3 and subsequent reaction of 2c with NaBPh4 to give 2d. | ||
The cation of 2b contains a symmetrical C2Si2Al five-membered ring with an Si–Al–Si angle of 89.07(8)°, as determined by the rigid geometry of 1a, and with an average Si–Al bond length 2.493(3) Å (Fig. 2); the carborane C–C bond measures 1.729(8) Å. Compared to 2b, the isostructural complex [{(SiII(Xant)SiII)AlI2}I] has a much larger Si–Al–Si bond angle of 123.01(5)° but a similar average Si–Al bond length of 2.463(13) Å.41 Additionally, we later prepared the tBu-functionalized analogue 1b ([SiII(CCcage)SiII] with SiII = L′Si, L′ = p-tBuC6H4C(NtBu)2).68 The aluminium iodide adduct 2c was prepared in the same way as 2a and isolated as a pale-yellow precipitate in 76% yield (Scheme 1). 2c was characterized by multinuclear NMR spectroscopy in dichloromethane (DCM) before slowly decomposing overnight. Akin to 2a, 2c is also unstable in THF and still poorly soluble in acetonitrile. Where anion exchange reaction of 2a had failed using NaBPh4 and AgOTf when attempted in various solvents, we conveniently found that the iodide counterion of 2c could be exchanged simply in DCM. The reaction of one molar equiv 2c with one equiv NaBPh4 in DCM gives 2d as a pale-yellow solid in 91% yield. 2d is stable in DCM solution for weeks, whilst 2c is not. This indicates that the iodide counterion is not purely a spectator ion and potentially aids the relatively slower decomposition process to give free bis-silylene (compared to 2a in THF) which would go on to react with DCM (as known for silylenes).69
 | ||
| Fig. 2 Molecular structures of the AlI2+-containing complexes of 2b (top, AlI4− counterion omitted), 4a (middle) and [3(Et2O)4] (bottom) as determined by scXRD. Thermal ellipsoids have been set at 50% probability. Hydrogens and solvent molecules have been omitted and selected groups are shown in wireframe for clarity. | ||
Redox chemistry of the o-C2B10-cage in bis-silylenyl carborane MIII complexes (M = Al, Ga)
We first probed KC8 as a reducing agent, which has been successfully applied in the reduction steps to produce both II and III (Fig. 1). Reaction of one molar equiv 2a with a large excess KC8 (5 equivs) in toluene for 36 h leads to a bright red solution (Scheme 2). Dark pink needle-shaped crystals grew from the filtrate and were revealed by scXRD to be the zwitterionic dipotassium salt [3(C6H5Me)2], isolated in 72% yield. 3 was characterized by multinuclear NMR spectroscopy in THF but decomposed significantly within 2 hours. 11B{1H} NMR spectroscopy confirmed a change of environment for the B10 cage atoms with four signals at δB = −3.2, −7.2, −16.6 and −32.6 ppm [for 2cδB = 1.9, −3.5, −8.7 and −13.2 ppm (CD2Cl2)]. | ||
| Scheme 2 Two-electron reduction of 2a with KC8 to give dimer 3 and the reverse oxidation reaction with I2 to re-form 2a. | ||
Crystals of [3(Et2O)4] suitable for scXRD analysis could be grown from Et2O solutions at low temperature (Fig. 2). In the ‘back-to-back’ dimeric structure of 2, the two K+ ions sit in-between the two carborane cages with the AlI2+ moieties facing outward. The reduction of the o-carborane moiety is apparent from the increased C⋯C distance. 3 features a nido-C2B10 cluster core with a C⋯C distance of 2.59 Å compared to 1.730(2) in 2b. Accompanying this change, the Si–Al–Si angle increases slightly from 89.08(7) to 101.99(6) . Other metal complexes of 1a, such as the SiI–SiI and GeI–GeI dimers (derived from V, Fig. 1) or dipotassium compound, have one-dimensional chain structures with K+ ions also bridging the dianionic nido-carborane moieties.52,54 The scXRD and 11B NMR data indicate that during reduction, two electrons are accepted by the C2B10 cage whilst the oxidation state of aluminium is unchanged.
. Other metal complexes of 1a, such as the SiI–SiI and GeI–GeI dimers (derived from V, Fig. 1) or dipotassium compound, have one-dimensional chain structures with K+ ions also bridging the dianionic nido-carborane moieties.52,54 The scXRD and 11B NMR data indicate that during reduction, two electrons are accepted by the C2B10 cage whilst the oxidation state of aluminium is unchanged.
3 can also be formed in an alternate synthesis, where 1a is reduced first with two equivs KC8 in THF (to give the dipotassium salt complex in situ) then reacted with one molar equiv AlI3 at low temperature. We propose that in the reaction of 2a with KC8, reduction begins with one electron being accepted by the unsaturated amidinato ligand which then moves to the carborane backbone to give a carborane radical anion intermediate. A similar mechanism for reduction of ligand 1a was demonstrated by DFT calculations investigating the reduction of silylone V.52 The initial formation of a transient aluminium radical intermediate is also possible. The formation of 3 contrasts with the formation of the related complexes of BIII and Si0, Ge0V, demonstrating how the closo-C2B10 cluster is reduced preferentially over AlIII but not over SiII, GeII or BIII under these reaction conditions.
We then investigated oxidation reactions of 3 that would release the two electrons from the nido-carborane cage. Test reactions with one or two equivs of AgOTf or FcPF6 (Fc = ferrocenium) in THF led to complex NMR spectra of unknown decomposition products. This can at least be in part explained by the instability of 2 in THF. 3 also decomposed in o-difluorobenzene (o-DFB), exhibited by a color change to dark brown (due to the formation of dark green precipitate). With polar solvents unsuitable, we used elemental iodine as an oxidizing agent. Thus, 0.5 molar equivs of dimer 3 were allowed to react with one molar equiv I2 in toluene, re-forming 2a in 65% isolated yield (Scheme 2). Attempts to determine the redox potentials of 2c in DCM by cyclic voltametric measurement were unsuccessful. Additionally, no K+ sequestration of 3 could be achieved with 18-crown-6 (18-c-6) or [2.2.2]-cryptand in THF. 3 also showed no reactivity towards NaBPh4 or CsBPh4. This lack of reactivity for 3 demonstrates that the K+ and I− ions are strongly bound within the complex, to the carborane dianions and cationic aluminium centers respectively. In contrast, the structurally related NI cation complex {K[SiII(CCcage)SiII]N} can form an ion-separated pair with 18-c-6 which is stable and undergoes one- and two-electron oxidation of the carborane cage with AgOTf.53
From the reaction of one molar equiv of 2a with a smaller excess of KC8 (2.5 equivs), the radical anion 4a, proposed as an intermediate in the formation of 3, was isolated in 24% yield (Scheme 3). The dark red crystals of 4a were suitable for scXRD analysis. The paramagnetic nature of 4a was evident from its NMR silence. EPR measurements at room temperature and below identified a strong isotropic radical signal at g = 2.007 and line width of 23.4 G, which suggests a delocalized carborane-based radical; no hyperfine coupling could be resolved (Fig. 3). In the solid-state structure of 4a, the C⋯C carborane distance is increased similarly to 3, at 2.43 Å (Fig. 2). The smaller carborane core opening agrees with the acceptance of one electron by the cluster to give a carboranyl radical anion. The geometry and bond distances around the Al center in 4a are generally similar to those in 3. Reduction of 4a with excess of KC8 (2.4 molar equivs) in Et2O also produces 3, additionally confirming 4a as a likely intermediate in the formation of 3 from 2a.
 | ||
| Scheme 3 Formation of the bis-silylenyl-carborane radical complexes 4a and 4b from 2a and KC8 or 1a and ‘GaI2’ respectively. | ||
 | ||
| Fig. 3 EPR data of the radical complexes 4a (top, left) and 4b (top, right) at room temperature. Cryogenic EPR spectra at 120 K (bottom, left) and the corresponding power saturation of 4a and 4b (bottom, right). | ||
We were also able to isolate the gallium analogue 4b from the reaction of 1a with 1.4 molar equivs of ‘GaI2’ in toluene at low temperature (Scheme 3). ‘GaI2’ is a solid prepared from the reaction of elemental gallium and iodine in a 1:2 ratio.70,714b could be isolated as a mixture of dark red and yellow crystals from red-orange toluene solutions in 27% yield. Radical 4b has an almost identical EPR signature to 4a with a strong isotropic radical signal at g = 2.008 and line width of 23.1 G, with no hyperfine coupling (Fig. 3). 4b and its water adduct [4b + (H2O)5] were additionally detected as molecular ions in mass spectrometry.
The scXRD analyses confirmed that both crystal types of 4b contain the same molecular structure. The yellow-colored crystals of 4b gave better metric data and the experimental bond lengths and angles were found to be almost identical to those of 4a (Fig. S36†). The different colored crystals of 4b result from different crystal packing and systems [yellow – orthorhombic, red – monoclinic]. The darker-colored crystals form predominantly from more concentrated solutions. In this two-fold reaction, the bis-silylenyl o-carborane accepts one electron and becomes bound to a GaI2+ fragment in the final product. The exact mechanism of how this might happen is unclear but overall, the ‘GaI2’ acts as a source of the GaI2˙ radical.
The EPR spectra of 4a and 4b recorded at 120 K verified that their isotropic radical signals are conserved at 120 K, exhibiting a small but significant difference in power saturation. The line shape, power and temperature dependency of 4a and 4b determined by EPR strongly suggest that the spin is delocalized in the C2B10 core. Whilst the spin of other paramagnetic aluminium complexes is typically ligand-based, such compounds tend to exhibit hyperfine coupling.16,72–75
With 2a, 3 and 4a in hand, we have established an interconvertible redox series of carborane-derived aluminium complexes, with three oxidation states characterized. This series based on 2a is unique for featuring an inorganic (carborane) electron storage unit (rather than aromatic/organic) that is not directly coupled or bonded to the Al center. The aluminum cation center is additionally only attached to the compound through donor bonds. There is a growing body of aluminium complexes with non-innocent ligands that can access several ligand oxidation states, all with charged N- or O-ligands, many of which are catalytically active.76–79 These complexes typically affect proton-coupled (PC)-ET or proton transfer (PT) reactions rather than ‘pure’ ET reactions, of which the few examples are rather diverse.77,80–82 We have demonstrated that carboranyl aluminium derivatives can act as reversible electron acceptors and that with further study have potential in catalytic ET reactions.
Trapping of low valent aluminium with K(C10H8) 3
Thereafter, we wondered if further reduction of 3 would lead to a reduced aluminium center. With 3 in hand, we probed its reduction with excess KC8, also in the presence of 1,3-dienes, PCy3 or AlCl3. This only gave complicated mixtures resulting from significant decomposition. Therefore, we tested the reaction of 0.5 equivs of dimer 3 with two molar equivs K(C10H8) in THF in an NMR tube. The reagents reacted instantaneously on solvation forming a dark red solution and colourless precipitate. The 1H NMR spectrum showed the formation of one equiv of free C10H8 and one major species 5a (Scheme 4), with no decomposition observed after 3 weeks. In the scaled-up reaction, 5a was isolated as orange crystals from toluene in 22% yield. The 1H NMR spectrum of 5a strongly suggested that it was a 1,4-naphthalene derivative, with a characteristic set of multiplets (dd) at δH = 3.05, 6.38, 6.80 and 6.93 ppm. We attributed the low isolated yield of 5a to its very poor solubility in inert solvents (except THF). Therefore, we undertook a one-pot reaction with the potentially more soluble analogue 2c. One equiv of 2c was reacted with four molar equivs of K(C10H8) in THF at low temperature (Scheme 4). This furnished the expected product 5b, which was isolated as orange crystals from Et2O in 61% yield. 5a and 5b were characterized by multinuclear NMR spectroscopy in THF and benzene, respectively. | ||
| Scheme 4 Formation of 1,4-naphthalene derivatives 5a and 5b from 3 or 2c and K(C10H8) respectively. | ||
Akin to 3, 5a did not react with [2.2.2]-crypt, which might have aided the crystallization of single crystals of 5a. Eventually, after many crystallization attempts, suitable crystals of 5b from Et2O solution were obtained and measured by scXRD analysis (Fig. 4). The angles and distances measured for 5b are consistent with 3. The carborane C⋯C distance is maintained at approximately 2.60 Å as well as the Si–Al–Si bite angle at 100.95(5)°. The Al(C10H8) unit manifests as two Al–C bonds of lengths of 2.056(4) and 2.066(4) Å, with a C–Al–C angle of 77.49(17)°. The 29Si NMR spectrum of 5b revealed two resonances at δSi = 29.5 and 39.1 ppm. The non-equivalence of the 29Si nuclei can be explained by the orientation of the naphthalene group. 5a and 5b show consistent NMR and structural data with other Al(C10H8)-complexes resulting from AlI activation of aromatics.83–86 This contrasts with the cooperative silylene-aluminylene reactivity of the pincer aluminylene III, which activates aromatic 2-methylquinoline through a 1,4-addition across the Al–Si bond instead.42
 | ||
| Fig. 4 Molecular structure of [5b(Et2O)4]. Thermal ellipsoids have been set at 50% probability. Hydrogens and solvent molecules have been omitted and selected groups are shown in wireframe for clarity. | ||
The naphthalene-derived products 5a and 5b indicate a [1 + 4] cycloaddition reaction involving low oxidation state Al intermediates. Multiple reaction pathways are possible. Since 2c and 3 both generate the same product 5, we propose that 3 is also an intermediate in the reaction of 2c (Scheme 4). One-electron reduction of 3 with K(C10H8) would generate Int1 with an aluminium radical cation center first (Scheme 5). Due to presence of two electrons in the carborane backbone, one electron could be transferred to the Al center to give iodoaluminylene Int2 with an Al-based electron pair. Int1 and Int2 are valence tautomers or electromers. Int1 and Int2 would react with a second equivalent of K(C10H8) to generate aluminylene species Int3 and Int4 respectively. It is additionally possible than Int1 undergoes a radical cycloaddition with [C10H8]˙– directly, while aluminylene species Int2–Int4 would undergo [1 + 4] cycloaddition with C10H8 generated in the reaction (as previously demonstrated by other aluminylene species).84–86 Such transient species as Int1–Int4 are unprecedented for aluminium, but have been proposed as boron intermediates vide infra. In a similar manner, transient boron(I) hydrides have also been captured as [1 + 2] cycloaddition products of naphthalene from reaction with Na(C10H8).87,88 Contrastingly, we did not observe C10H8 incorporation in our previous work with aluminium halide complexes of bidentate donor ligands [SiII(Xant)SiII] or a bis(NHC)-ligand (bis(N-dipp-imidazole-2-ylidene)methylene).41,89 Therefore this cycloaddition reactivity is unique to the Al complexes derived from ligand 1. We expect this difference is related to the stabilizing effects of the dianionic carborane backbone on the low-valent Al center, through countering the positive charge at Al or possibly even reducing the Al center through valence tautomerism.
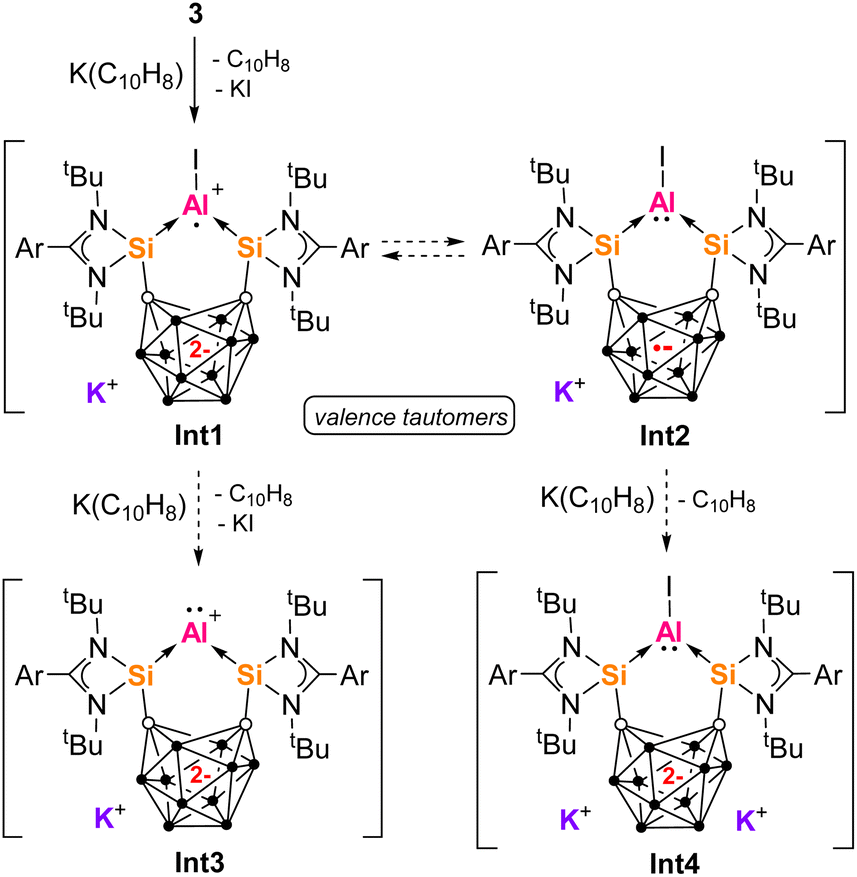 | ||
| Scheme 5 The structures of the fleeting aluminium intermediates Int1–Int4 proposed to form from one- or two-electron reduction. | ||
Donor-stabilized haloaluminylenes have only been isolated as metal carbonyl complexes (e.g.IV, Fig. 1), while no donor-stabilized low valent aluminium cations have been reported.41,89–92 It is noted that a AlI hydride [(cAAC)2AlH], AlI cation [Al(AlCp*)3]+ (I, Fig. 1) and a silyl-substituted AlIII cation [R3Si-Al-SiR3]+ have been isolated and characterized.15,20,93 Radical cations, e.g. [AlH]˙+ and [AlF]˙+, have only been synthesized in matrix conditions.94,95 However, a neutral AlIII radical [(R3Si)3Al]˙ has been synthesized and mononuclear AlII radicals [R2Al]˙ have also been recently detected in situ with EPR.75,96 AlII radicals have also been proposed as intermediates in aluminium-directed reduction of H2, aromatics or alkynes.83,97–99 To our knowledge, electromerism (or valence tautomerism) resulting in the formation of an AlI centre has not been reported.59 For the related boron complexes of ligand 1, species related to Int1–Int3 have been proposed as intermediates or isolated.23 One-electron reduction of bromoborylene II (Fig. 1) with Na(C10H8) gives an isolable BII radical complex of ligand 1a (analogous to Int1), via the initial formation of its valence tautomer (analogous to Int2).61 Two-electron reduction of II yields VI (Fig. 1), with DFT calculations supporting the formation of a transient borylene cation (like Int3) that then reacts with a nearby cage B–H bond. Such a borylene or aluminylene cation like Int3 would be isolobal with the isolable zero-valent beryllium compound [(cAAC)2Be].100
Carbenes, specifically cAACs, have been used to tame or trap aluminium radical complexes in situ, where TEMPO is unsuitable.16,19,75 To find out more about the low-valent intermediates that form in the reaction to give 5a and 5b, we attempted reductions of 2c and 3 in the presence of MecAAC-5 with dropwise addition of THF solutions of crystalline [K(THF)(C10H8)] (of varying equivalents) at −78 °C (see ESI†).17,101 Formation of 5 was prevented but no cAAC-containing products were identified. To remove the option of C10H8 addition entirely, we carried out reduction reactions of 2 and 3 with K/KI (5% w/w) powder. Most promising was the reduction of the BPh4 salt 2d with one equiv MecAAC-5 and five equivs K/KI powder in Et2O (see ESI†).102 We observed color changes from colorless, via red, to dark blue within a few hours (Fig. S1 and S2†). 1H NMR aliquots of the reaction mixture in benzene showed no notable products, despite the maintaining the intense color for at least 2 weeks, and unfortunately, no viable single crystals for scXRD were isolated. Despite these experimental challenges, there is still more to uncover about the nature of such low valent Al species supported by 1. Further theoretical study could shine light on this and support the development of new ligand systems.
Conclusions
Building on our previous work to access low oxidation state aluminium–silicon compounds, we employed the bis-silylenyl o-carborane ligands 1a and 1b to synthesize the respective bis-silylene AlIII complexes 2a–2d. Using KC8, the two-electron reduction of 2a could be achieved leading to the bis-silylenyl AlIIInido-C2B10 dianion complex 3, whilst one-electron reduction affords the corresponding AlIII carborane radical anion complex 4a. Thus, we establish a redox series of aluminium complexes with three interconvertible oxidation states characterized. The analogous gallium radical complex 4b was also isolated from the reaction of 1a with ‘GaI2’. Using K(C10H8), the AlI2+ center of 3 or 2c was reduced furnishing the 1,4-naphthalene complexes 5a and 5b, respectively. Such products indicate the formation of transient donor-stabilized low-valent aluminium intermediates. These include the aluminium radical cation Int1 which could react further to form iodo- or cationic aluminylene intermediates Int2–Int4. Overall, it is evident that tunable chelating silylene ligands and redox non-innocent ligands have important roles to play in the continued development of donor-stabilized low oxidation state aluminium complexes towards zero valent aluminium.Data availability
All experimental data associated with this work are available in the ESI.†Author contributions
A. S. carried out the synthetic experiments, analyzed the experimental data and wrote the original manuscript. S. Y. carried out the scXRD refinement of the compounds and edited the manuscript. C. L. collected and analyzed the EPR data. M. D. supervised the work and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by DFG (German Research Foundation) under Germany's Excellence Strategy – EXC 2008-390540038-UniSysCat and DR-226/25-1. We thank Paula Nixdorf for assistance in scXRD measurements.Notes and references
- K. Ota and R. Kinjo, Chem, 2022, 8, 340–350 CAS.
- G. Frenking, M. Hermann, D. M. Andrada and N. Holzmann, Chem. Soc. Rev., 2016, 45, 1129–1144 RSC.
- Y. Wang and G. H. Robinson, J. Am. Chem. Soc., 2023, 145, 5592–5612 CrossRef CAS PubMed.
- R. Tonner, F. Öxler, B. Neumüller, W. Petz and G. Frenking, Angew. Chem., Int. Ed., 2006, 45, 8038–8042 CrossRef CAS PubMed.
- S. Yao, Y. Xiong, A. Saddington and M. Driess, Chem. Commun., 2021, 57, 10139–10153 RSC.
- S. Yao, Y. Xiong and M. Driess, Acc. Chem. Res., 2017, 50, 2026–2037 CrossRef CAS PubMed.
- C. Panda, P. W. Menezes and M. Driess, Angew. Chem., Int. Ed., 2018, 57, 11130–11139 CrossRef CAS PubMed.
- M. A. Malik, M. Afzaal and P. O'Brien, Chem. Rev., 2010, 110, 4417–4446 CrossRef CAS PubMed.
- P. W. Menezes, S. Yao, R. Beltrán-Suito, J. N. Hausmann, P. V. Menezes and M. Driess, Angew. Chem., Int. Ed., 2021, 60, 4640–4647 CrossRef CAS PubMed.
- H. Braunschweig, R. D. Dewhurst, K. Hammond, J. Mies, K. Radacki and A. Vargas, Science, 2012, 336, 1420–1422 CrossRef CAS PubMed.
- J. Böhnke, H. Braunschweig, W. C. Ewing, C. Hörl, T. Kramer, I. Krummenacher, J. Mies and A. Vargas, Angew. Chem., Int. Ed., 2014, 53, 9082–9085 CrossRef PubMed.
- J. Böhnke, M. Arrowsmith and H. Braunschweig, J. Am. Chem. Soc., 2018, 140, 10368–10373 CrossRef PubMed.
- W. Lu, Y. Li and R. Kinjo, J. Am. Chem. Soc., 2019, 141, 5164–5168 CrossRef CAS PubMed.
- W. Kennedy, V. Pattathil, Y. Wei, F. Fantuzzi and C. Pranckevicius, J. Am. Chem. Soc., 2025, 3500–3506 CrossRef CAS PubMed.
- S. K. Mellerup, Y. Cui, F. Fantuzzi, P. Schmid, J. T. Goettel, G. Bélanger-Chabot, M. Arrowsmith, I. Krummenacher, Q. Ye, V. Engel, B. Engels and H. Braunschweig, J. Am. Chem. Soc., 2019, 141, 16954–16960 CrossRef CAS PubMed.
- B. Li, S. Kundu, A. C. Stückl, H. Zhu, H. Keil, R. Herbst-Irmer, D. Stalke, B. Schwederski, W. Kaim, D. M. Andrada, G. Frenking and H. W. Roesky, Angew. Chem., Int. Ed., 2017, 56, 397–400 CrossRef CAS PubMed.
- M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2015, 48, 256–266 CrossRef CAS PubMed.
- L. Werner and U. Radius, Dalton Trans., 2024, 53, 16436–16454 RSC.
- D. Dhara, L. Endres, I. Krummenacher, M. Arrowsmith, R. D. Dewhurst, B. Engels, R. Bertermann, M. Finze, S. Demeshko, F. Meyer, F. Fantuzzi and H. Braunschweig, Angew. Chem., Int. Ed., 2024, 63, e202401052 CrossRef CAS PubMed.
- P. Dabringhaus, J. Willrett and I. Krossing, Nat. Chem., 2022, 14, 1151–1157 CrossRef CAS PubMed.
- J. M. Slattery, A. Higelin, T. Bayer and I. Krossing, Angew. Chem., Int. Ed., 2010, 49(18), 3228–3231 CrossRef CAS PubMed.
- A. Higelin, S. Keller, C. Göhringer, C. Jones and I. Krossing, Angew. Chem., Int. Ed., 2013, 52(18), 4941–4944 CrossRef CAS PubMed.
- H. Wang, L. Wu, Z. Lin and Z. Xie, J. Am. Chem. Soc., 2017, 139, 13680–13683 CrossRef CAS PubMed.
- S. Yao, A. Saddington, Y. Xiong and M. Driess, Acc. Chem. Res., 2023, 56, 475–488 CrossRef CAS PubMed.
- J. Xu, C. Dai, S. Yao, J. Zhu and M. Driess, Angew. Chem., Int. Ed., 2022, 61, e202114073 CrossRef CAS PubMed.
- J. Xu, S. Pan, S. Yao, G. Frenking and M. Driess, Angew. Chem., Int. Ed., 2022, 61, e202209442 CrossRef CAS PubMed.
- Y. Wang, T. Szilvási, S. Yao and M. Driess, Nat. Chem., 2020, 12, 801–807 CrossRef CAS PubMed.
- Y. Wang, M. Karni, S. Yao, A. Kaushansky, Y. Apeloig and M. Driess, J. Am. Chem. Soc., 2019, 141, 12916–12927 CrossRef CAS PubMed.
- Y. Wang, M. Karni, S. Yao, Y. Apeloig and M. Driess, J. Am. Chem. Soc., 2019, 141, 1655–1664 CrossRef CAS PubMed.
- C. Shan, S. Yao and M. Driess, Chem. Soc. Rev., 2020, 49, 6733–6754 RSC.
- M. Denk, R. Lennon, R. Hayashi, R. West, A. V. Belyakov, H. P. Verne, A. Haaland, M. Wagner and N. Metzler, J. Am. Chem. Soc., 1994, 116, 2691–2692 CrossRef CAS.
- L. Wang, Y. Li, Z. Li and M. Kira, Coord. Chem. Rev., 2022, 457, 214413 CrossRef CAS.
- C.-W. So, H. W. Roesky, J. Magull and R. B. Oswald, Angew. Chem., Int. Ed., 2006, 45, 3948–3950 CrossRef CAS PubMed.
- W. Wang, S. Inoue, S. Yao and M. Driess, J. Am. Chem. Soc., 2010, 132, 15890–15892 CrossRef CAS PubMed.
- A. Saddington, S. Yao and M. Driess, in Advances in Inorganic Chemistry, 2023, pp. 119–156 Search PubMed.
- Y. Zhang, L. Wu and H. Wang, Coord. Chem. Rev., 2023, 477, 214942 CrossRef CAS.
- P. Dabringhaus, S. Zedlitz, L. Giarrana, D. Scheschkewitz and I. Krossing, Angew. Chem., Int. Ed., 2023, 62, e202215170 CrossRef CAS PubMed.
- M. Ludwig, D. Franz, A. Espinosa Ferao, M. Bolte, F. Hanusch and S. Inoue, Nat. Chem., 2023, 15, 1452–1460 CrossRef CAS PubMed.
- L. Guo, J. Zhang and C. Cui, J. Am. Chem. Soc., 2023, 145, 27911–27915 CrossRef CAS PubMed.
- J. Schoening, A. Gehlhaar, C. Wölper and S. Schulz, Chem.–Eur. J., 2022, 28, e202201031 CrossRef CAS PubMed.
- A. Saddington, S. Dong, S. Yao, J. Zhu and M. Driess, Angew. Chem., Int. Ed., 2024, e202410790 CAS.
- X. Chen, D. Yang, F. Cao and Z. Mo, J. Am. Chem. Soc., 2024, 146, 29278–29284 CrossRef CAS PubMed.
- Y. Wang, A. Kostenko, S. Yao and M. Driess, J. Am. Chem. Soc., 2017, 139, 13499–13506 CrossRef CAS PubMed.
- Y.-P. Zhou, S. Raoufmoghaddam, T. Szilvási and M. Driess, Angew. Chem., Int. Ed., 2016, 55, 12868–12872 CrossRef CAS PubMed.
- H. Wang, Chin. Chem. Lett., 2022, 33, 3672–3680 CrossRef CAS.
- Y. Wang, A. Kostenko, T. J. Hadlington, M.-P. Luecke, S. Yao and M. Driess, J. Am. Chem. Soc., 2019, 141, 626–634 CrossRef CAS PubMed.
- Y. Xiong, S. Yao, T. Szilvási, A. Ruzicka and M. Driess, Chem. Commun., 2020, 56, 747–750 RSC.
- Y. Xiong, S. Dong, S. Yao, J. Zhu and M. Driess, Angew. Chem., Int. Ed., 2022, 61, e202205358 CrossRef CAS PubMed.
- M. F. Hawthorne, T. E. Berry and P. A. Wegner, J. Am. Chem. Soc., 1965, 87, 4746–4750 CrossRef CAS PubMed.
- J. H. Morris, H. J. Gysling and D. Reed, Chem. Rev., 1985, 85, 51–76 CrossRef CAS.
- R. Núñez, M. Tarrés, A. Ferrer-Ugalde, F. F. De Biani and F. Teixidor, Chem. Rev., 2016, 116, 14307–14378 CrossRef PubMed.
- S. Yao, A. Kostenko, Y. Xiong, A. Ruzicka and M. Driess, J. Am. Chem. Soc., 2020, 142, 12608–12612 CrossRef CAS PubMed.
- S. Yao, T. Szilvási, Y. Xiong, C. Lorent, A. Ruzicka and M. Driess, Angew. Chem., Int. Ed., 2020, 59, 22043–22047 CrossRef CAS PubMed.
- S. Yao, A. Kostenko, Y. Xiong, C. Lorent, A. Ruzicka and M. Driess, Angew. Chem., Int. Ed., 2021, 60, 14864–14868 CrossRef CAS PubMed.
- H. Wang, Chin. Chem. Lett., 2022, 33, 3672–3680 CrossRef CAS.
- B. Lei, F. Cao, M. Chen, X. Wang and Z. Mo, J. Am. Chem. Soc., 2024, 146, 17817–17826 CrossRef CAS PubMed.
- Y. Xiong, D. Chen, S. Yao, J. Zhu, A. Ruzicka and M. Driess, J. Am. Chem. Soc., 2021, 143, 6229–6237 CrossRef CAS PubMed.
- M. Chen, B. Lei, X. Wang, H. Rong, H. Song and Z. Mo, Angew. Chem., Int. Ed., 2022, 61, e202204495 CrossRef CAS PubMed.
- L. Greb, Eur. J. Inorg. Chem., 2022, 2022, e202100871 CrossRef CAS PubMed.
- S. Suthar and K. C. Mondal, Chem. Eur. J., 2024, 30, e202303355 CrossRef CAS PubMed.
- H. Wang, J. Zhang, H. K. Lee and Z. Xie, J. Am. Chem. Soc., 2018, 140, 3888–3891 CrossRef CAS PubMed.
- D. A. T. Young, R. J. Wiersema and M. F. Hawthorne, J. Am. Chem. Soc., 1971, 93, 5687–5694 CrossRef CAS.
- K.-C. Son, Y.-J. Lee, M. Cheong, J. Ko and S. O. Kang, J. Am. Chem. Soc., 2006, 128, 12086–12087 CrossRef CAS PubMed.
- H. Sohn and J.-D. Lee, Crystals, 2023, 13, 877 CrossRef CAS.
- H.-Y. Liu, K. G. Pearce, M. S. Hill and M. F. Mahon, Inorganics, 2024, 12, 309 CrossRef CAS.
- Z.-J. Yao and G.-X. Jin, Coord. Chem. Rev., 2013, 257, 2522–2535 CrossRef CAS.
- Y. Kang, B. Wang, R. Nan, Y. Li, Z. Zhu and X.-Q. Xiao, Inorg. Chem., 2022, 61, 8806–8814 CrossRef CAS PubMed.
- C. Shan, S. Dong, S. Yao, J. Zhu and M. Driess, J. Am. Chem. Soc., 2023, 145, 7084–7089 CrossRef CAS PubMed.
- D. F. Moser, T. Bosse, J. Olson, J. L. Moser, I. A. Guzei and R. West, J. Am. Chem. Soc., 2002, 124, 4186–4187 CrossRef CAS PubMed.
- J. C. Beamish, M. Wilkinson and I. J. Worrall, Inorg. Chem., 1978, 17, 2026–2027 CrossRef CAS.
- J. C. Beamish, A. Boardman and I. J. Worrall, Polyhedron, 1991, 10, 95–99 CrossRef CAS.
- I. L. Fedushkin, M. V. Moskalev, A. N. Lukoyanov, A. N. Tishkina, E. V. Baranov and G. A. Abakumov, Chem. Eur. J., 2012, 18, 11264–11276 CrossRef CAS PubMed.
- J. Scott, S. Gambarotta, I. Korobkov, Q. Knijnenburg, B. De Bruin and P. H. M. Budzelaar, J. Am. Chem. Soc., 2005, 127, 17204–17206 CrossRef CAS PubMed.
- J. Li, K. Zhang, H. Huang, A. Yu, H. Hu, H. Cui and C. Cui, Organometallics, 2013, 32, 1630–1635 CrossRef CAS.
- D. Mandal, T. I. Demirer, T. Sergeieva, B. Morgenstern, H. T. A. Wiedemann, C. W. M. Kay and D. M. Andrada, Angew. Chem., Int. Ed., 2023, 62, e202217184 CrossRef CAS PubMed.
- P. S. Gahlaut, K. Yadav, D. Gautam and B. Jana, J. Organomet. Chem., 2022, 963, 122298 CrossRef CAS.
- L. W. T. Parsons and L. A. Berben, Acc. Chem. Res., 2024, 57, 1087–1097 CrossRef CAS PubMed.
- G. Szigethy and A. F. Heyduk, Dalton Trans., 2012, 41, 8144 RSC.
- T. W. Myers, N. Kazem, S. Stoll, R. D. Britt, M. Shanmugam and L. A. Berben, J. Am. Chem. Soc., 2011, 133, 8662–8672 CrossRef CAS PubMed.
- I. L. Fedushkin, A. N. Lukoyanov, G. K. Fukin, M. Hummert and H. Schumann, Russ. Chem. Bull., 2006, 55, 1177–1183 CrossRef CAS.
- T. W. Myers, A. L. Holmes and L. A. Berben, Inorg. Chem., 2012, 51, 8997–9004 CrossRef CAS PubMed.
- T. J. Sherbow, L. W. T. Parsons, N. A. Phan, J. C. Fettinger and L. A. Berben, Inorg. Chem., 2020, 59, 17614–17619 CrossRef CAS PubMed.
- C. Cui, S. Ko, M. Noltemeyer, H.-G. Schmidt and B. Wrackmeyer, J. Am. Chem. Soc., 2001, 123, 9091–9098 CrossRef CAS PubMed.
- X. Zhang and L. L. Liu, Angew. Chem., Int. Ed., 2022, 61, e202116658 CrossRef CAS PubMed.
- C. Bakewell, M. Garçon, R. Y. Kong, L. O'Hare, A. J. P. White and M. R. Crimmin, Inorg. Chem., 2020, 59, 4608–4616 CrossRef CAS PubMed.
- D. Dhara, A. Jayaraman, M. Härterich, R. D. Dewhurst and H. Braunschweig, Chem. Sci., 2022, 13, 5631–5638 RSC.
- P. Bissinger, H. Braunschweig, K. Kraft and T. Kupfer, Angew. Chem., Int. Ed., 2011, 50, 4704–4707 CrossRef CAS PubMed.
- D. P. Curran, A. Boussonnière, S. J. Geib and E. Lacôte, Angew. Chem., Int. Ed., 2012, 51, 1602–1605 CrossRef CAS PubMed.
- G. Tan, T. Szilvási, S. Inoue, B. Blom and M. Driess, J. Am. Chem. Soc., 2014, 136, 9732–9742 CrossRef CAS PubMed.
- R. A. Fischer, M. M. Schulte, J. Weiss, L. Zsolnai, A. Jacobi, G. Huttner, G. Frenking, C. Boehme and S. F. Vyboishchikov, J. Am. Chem. Soc., 1998, 120, 1237–1248 CrossRef CAS.
- H. Fölsing, O. Segnitz, U. Bossek, K. Merz, M. Winter and R. A. Fischer, J. Organomet. Chem., 2000, 606, 132–140 CrossRef.
- D. Franz and S. Inoue, Chem. Eur. J., 2019, 25, 2898–2926 CrossRef CAS PubMed.
- M. Nakamoto, K. Shimizu and A. Sekiguchi, Chem. Lett., 2007, 36, 984–985 CrossRef CAS.
- L. B. Knight, S. T. Cobranchi, B. W. Gregory and E. Earl, J. Chem. Phys., 1987, 86, 3143–3150 CrossRef CAS.
- L. B. Knight, E. Earl, A. R. Ligon, D. P. Cobranchi, J. R. Woodward, J. M. Bostick, E. R. Davidson and D. Feller, J. Am. Chem. Soc., 1986, 108, 5065–5071 CrossRef CAS.
- M. Nakamoto, T. Yamasaki and A. Sekiguchi, J. Am. Chem. Soc., 2005, 127, 6954–6955 CrossRef CAS PubMed.
- N. Wiberg, K. Amelunxen, T. Blank, H. Nöth and J. Knizek, Organometallics, 1998, 17, 5431–5433 CrossRef CAS.
- D. Dhara, F. Fantuzzi, M. Härterich, R. D. Dewhurst, I. Krummenacher, M. Arrowsmith, C. Pranckevicius and H. Braunschweig, Chem. Sci., 2022, 13, 9693–9700 RSC.
- J. O. Wenzel, J. Werner, A. Allgaier, J. Van Slageren, I. Fernández, A. Unterreiner and F. Breher, Angew. Chem., Int. Ed., 2024, 63, e202402885 CrossRef CAS PubMed.
- M. Arrowsmith, H. Braunschweig, M. A. Celik, T. Dellermann, R. D. Dewhurst, W. C. Ewing, K. Hammond, T. Kramer, I. Krummenacher, J. Mies, K. Radacki and J. K. Schuster, Nat. Chem., 2016, 8, 890–894 CrossRef CAS PubMed.
- T. A. Scott, B. A. Ooro, D. J. Collins, M. Shatruk, A. Yakovenko, K. R. Dunbar and H.-C. Zhou, Chem. Commun., 2009, 65–67 RSC.
- J. Hicks, M. Juckel, A. Paparo, D. Dange and C. Jones, Organometallics, 2018, 37, 4810–4813 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2418300 (compound 2b), 2418301 (compound 3), 2418298 (compound 4a), 2418297 (compound 4b), 2418302 (compound 5b), For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc01104c |
| This journal is © The Royal Society of Chemistry 2025 |