
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransformation of a Pd6 trifacial barrel to a Pd8 tetrafacial barrel by C70 as guest and oxidative photolysis of alkenes using the C70 encapsulated barrel under red light†
Ranit
Banerjee‡
a,
Monojit
Roy‡
b,
Medha
Aggarwal
a,
Shyamali
Maji
b,
Debashis
Adhikari
 *b and
Partha Sarathi
Mukherjee
*a
*b and
Partha Sarathi
Mukherjee
*a
aDepartment of Inorganic and Physical Chemistry, Indian Institute of Science, Bangalore-560012, India. E-mail: psm@iisc.ac.in
bDepartment of Chemical Science, Indian Institute of Science Education and Research Mohali, SAS Nagar, Mohali-140306, India. E-mail: adhikari@iisermohali.ac.in
First published on 5th June 2025
Abstract
Fabricating discrete molecular cages as fullerene receptors has been a compelling task. The main objective of confining fullerene molecules is to utilize their physico-chemical properties in commonly used media in which they are insoluble. Herein, a fluorenone-based large trigonal Pd6 molecular barrel (M1) was synthesized by coordination self-assembly to act as a potential fullerene trap. M1, in the presence of fullerene C70, converted to a larger Pd8 tetrafacial barrel (M2) forming stable host–guest adduct, (C70)3@M2. The PF6− analogues of both M1 and (C70)3@M2 were acetonitrile soluble, generated reactive oxygen species due to the presence of photosensitizing fluorenone moieties in their building blocks and catalyzed the oxidative transformation of alkenes into carbonyl compounds under 390 nm irradiation. The presence of encapsulated C70 molecules in (C70)3@M2 enabled its photosensitizing wavelength to be tuned to 650 nm (red light). Subsequently, (C70)3@M2, at low catalyst loadings and under red light irradiation, catalyzed olefin oxidations in acetonitrile wherein free C70 was completely inefficient due to insolubility. In summary, (C70)3@M2 was employed as a photocatalyst to mimic the ozonolysis of olefins without the use of ozone or other metal-oxide oxidants that produce over-oxidized products and generate toxic waste, under innocuous red light irradiation and environment-friendly reaction conditions.
Introduction
Confining guest molecules within supramolecular architectures via non-covalent interactions, for carrying out specific functions or for exploiting their modulated properties upon encapsulation to mimic natural processes, is gaining popularity.1–6 Today, a plethora of metal–organic discrete cages of distinct shapes are available at our disposal or can be easily designed to carry out these applications. They are suitable for encapsulating specific guest molecules, thanks to the synthetic simplicity of coordination-driven self-assembly approach.7–14 The main applications that have emerged as a result of such confinement include catalysis,15–18 selective molecular recognition19,20 and separation,21–23 and stabilization of reactive intermediates or unstable molecules.24–26 In this regard, encapsulation of fullerene molecules or their derivatives that possess phenomenal photophysical and opto-electronic properties is highly desirable. Several groups have reported the synthesis of coordination cages that can act as strong fullerene binders over the last decade.27–30 The stabilization of electron-deficient fullerene molecules within such cavities is achieved mainly through π–π interactions with electron-rich aromatic walls of the host cages.31 In some cases, the hosts have been found to transform into another architecture to maximize non-covalent interactions with the guests to form stable host–guest adducts.32–34 Such receptors have been widely used for the purification of fullerene mixtures35,36 and for their regioselective functionalization.37,38 Another important aspect of fullerene encapsulation is to utilize their visible light photosensitizing ability in commonly used organic media where they are otherwise insoluble and hence cannot catalyze oxidative transformations. Recently, there have been only a few reports on cage-fullerene adducts that were employed for sulfoxidation of sulphides and for endo-peroxide formation.33,39,40 Herein, we investigate whether fullerene-encapsulated cages could carry out demanding oxidations, such as the oxidative transformation of olefins into the corresponding carbonyl compounds.Ozonolysis is one of the most common methods used for the oxidative cleavage of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C of alkenes.41 However, ozone is toxic, and its handling requires a lot of safety measures.42 The other methods for olefin oxidative cleavage involve the use of oxidants such as OsO4 or RuO4. These metal oxide catalysts generate toxic waste and often cause over-oxidation of olefin substrates.43,44 Thus, development of a green and sustainable approach for efficient oxidative transformation of alkenes into the corresponding carbonyl compounds has high demand. Photocatalytic pathways to promote oxidative olefinic cleavage mostly use blue or purple light, which has a problem with scalability, owing to the requirement of low temperatures during the reaction.45a A major limitation of these photochemical reactions is poor light penetrability in the reaction flask, alongside undesirable absorption by intermediates that can prematurely activate or deactivate the reaction. To circumvent these issues, light with higher wavelengths such as red light with superior penetration power is preferable.45b
C of alkenes.41 However, ozone is toxic, and its handling requires a lot of safety measures.42 The other methods for olefin oxidative cleavage involve the use of oxidants such as OsO4 or RuO4. These metal oxide catalysts generate toxic waste and often cause over-oxidation of olefin substrates.43,44 Thus, development of a green and sustainable approach for efficient oxidative transformation of alkenes into the corresponding carbonyl compounds has high demand. Photocatalytic pathways to promote oxidative olefinic cleavage mostly use blue or purple light, which has a problem with scalability, owing to the requirement of low temperatures during the reaction.45a A major limitation of these photochemical reactions is poor light penetrability in the reaction flask, alongside undesirable absorption by intermediates that can prematurely activate or deactivate the reaction. To circumvent these issues, light with higher wavelengths such as red light with superior penetration power is preferable.45b
In the context of olefinic oxidative cleavage, we report the synthesis of a large trigonal Pd6 molecular barrel (M1) as a potential receptor for fullerene molecules. M1 was obtained by coordination-driven self-assembly of a Pd(II) acceptor (A) with a “fluorenone” incorporated tetradentate ligand (L). The presence of fullerene C70 molecules acted as a guest stimulus to trigger the transformation of the trifacial barrel into a larger tetrafacial Pd8 molecular barrel (M2) leading to the formation of a stable host–guest complex, (C70)3@M2 (comprising three C70 molecules bound within the cavity of a tetrafacial barrel M2) (Scheme 1). The acetonitrile soluble PF6− analogues of both M1 and (C70)3@M2 generated reactive oxygen species (ROS), both singlet oxygen (1O2) and superoxide radicals (O2˙−), upon irradiation with a light source of 390 nm that could be utilized for catalyzing the oxidative cleavage of alkenes to generate the corresponding carbonyl compounds in the same solvent (Scheme 2). The ROS generating ability of M1 and (C70)3@M2 may be attributed to the presence of photosensitizing fluorenone46 within their building units and additionally to the presence of encapsulated C70 molecules in the latter. Interestingly, the host–guest complex, (C70)3@M2 could also be photosensitized using red light due to the presence of encapsulated C70 molecules that absorb in the red region of visible light (Scheme 2). The use of red light is advantageous as the associated low energy does not trigger an undesirable, detrimental bond cleavage, directly reflecting on the functional group tolerance of substrates. Subsequently, the PF6− analogue of (C70)3@M2 could photo-catalyze the ROS-mediated oxidative transformation of olefins into the corresponding carbonyl compounds for a wide variety of substrates starting from terminal to sterically hindered alkenes under red-light irradiation in acetonitrile at low catalyst loadings. Thus, the encapsulation of C70 molecules within the host cavity played a vital role in solubilizing C70 fullerene molecules in acetonitrile to utilize their red-light sensitizing activity for photocatalysis in environmentally safe and commonly used organic solvents. Such an approach which involves the utilization of photosensitizing properties of entrapped guest molecules for the oxidative cleavage of alkenes (which otherwise requires oxidants that are hazardous and produce over-oxidized products) under low-energy red light is unknown to the best of our knowledge and opens up new avenues in the direction of green synthesis.
 | ||
| Scheme 1 Schematic representation of the synthesis of trifacial barrel M1, its structural conversion in the presence of C70 to multiple C70 encapsulated tetragonal barrel (C70)3@M2, and its photo-catalytic ability to carry out the oxidative transformation of olefins into the corresponding carbonyl compounds under red light (650 nm) irradiation. | ||
 | ||
| Scheme 2 Schematic representation of alkene oxidative transformation into carbonyl compounds in acetonitrile in the presence of trifacial barrel M1 or (C70)3@M2 under 390 nm irradiation (blue-violet light). The encapsulation of multiple C70 molecules enables tuning of photosensitizing wavelength of host–guest complex (C70)3@M2 to 650 nm (red light) and the oxidative transformation of alkenes proceeds under red light. | ||
Results and discussion
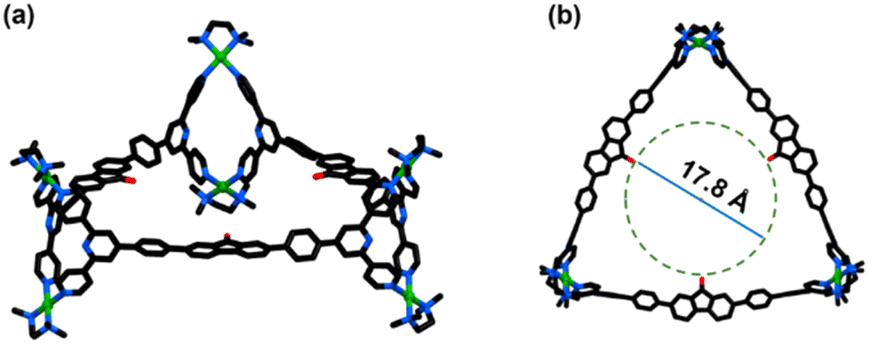
The bent tetra-pyridyl ligand (L) incorporated with a “fluorenone” core was synthesized from the precursor aldehyde, 4,4′-(9-oxo-9H-fluorene-2,7-diyl)dibenzaldehyde (P), following standard procedures for terpyridine synthesis (Scheme S1†).47 The aldehyde (P) was synthesized via a Suzuki coupling reaction between 2,7-dibromo-9H-fluoren-9-one and (4-formylphenyl)boronic acid following a reported procedure (Fig. S1 and Scheme S1†).48 Ligand L was characterized using 1H and 13C NMR spectroscopy and mass spectrometry (Fig. S2–S4†). Self-assembly of L was carried out with a cis-blocked Pd(II) acceptor [(tmeda)Pd(ONO2)2] (A), in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio in dimethyl sulfoxide [tmeda = N,N,N′,N′-tetramethylethane-1,2-diamine]. Although the ligand (L) has poor solubility in DMSO, its reaction with the acceptor (A) for 24 h at 70 °C resulted in the formation of a clear red solution. The self-assembled product (M1) was isolated by treating the DMSO solution with an excess of ethyl acetate which afforded a canary yellow precipitate. The 1H NMR recorded in DMSO-d6 displayed sharp and distinct peaks (Fig. 1a and S5†) indicating the formation of a self-assembled architecture, and the formation of a single product was subsequently confirmed from 1H DOSY NMR (Fig. S6†). The peaks in the 1H NMR spectrum were assigned with the help of 2D 1H-1H COSY and 1H-1H NOESY NMR (Fig. S7 and S8†). A two-fold splitting was observed for the α-pyridyl (a1 and a2) and β-pyridyl (b1 and b2) protons at room temperature (25 °C) due to the rigidification of the pyridyl rings of the ligands upon coordination with the metal centres. Such coordination prevents free rotation in the NMR timescale, which leads to two different chemical environments for the pyridyl rings in the self-assembled product (M1). We carried out the variable temperature (VT) 1H NMR which showed that these split α-pyridyl (a1 and a2) and β-pyridyl (b1 and b2) protons coalesce into single peaks, a and b, respectively at temperatures higher than 85 °C (Fig. S34†). At higher temperatures, the faster rotation of the coordinated pyridyl rings becomes unresolvable in the NMR timescale resulting in coalescence of the peaks corresponding to the pyridyl protons. Finally, to determine the composition of M1, the NO3− analogue was converted to the PF6− analogue for better ionization in ESI-MS. The m/z peaks obtained correspond to an A6L3 composition. The isotopic patterns obtained at 1219.9336, 946.9544, and 764.9654 correspond to the fragments [M1-4PF6]4+, [M1-5PF6]5+, and [M1-6PF6]6+ respectively, which matched well with the theoretically calculated isotopic patterns, confirming the formation of a trigonal barrel (M1) (Fig. 1b–e, S19 and S20†).
:2 molar ratio in dimethyl sulfoxide [tmeda = N,N,N′,N′-tetramethylethane-1,2-diamine]. Although the ligand (L) has poor solubility in DMSO, its reaction with the acceptor (A) for 24 h at 70 °C resulted in the formation of a clear red solution. The self-assembled product (M1) was isolated by treating the DMSO solution with an excess of ethyl acetate which afforded a canary yellow precipitate. The 1H NMR recorded in DMSO-d6 displayed sharp and distinct peaks (Fig. 1a and S5†) indicating the formation of a self-assembled architecture, and the formation of a single product was subsequently confirmed from 1H DOSY NMR (Fig. S6†). The peaks in the 1H NMR spectrum were assigned with the help of 2D 1H-1H COSY and 1H-1H NOESY NMR (Fig. S7 and S8†). A two-fold splitting was observed for the α-pyridyl (a1 and a2) and β-pyridyl (b1 and b2) protons at room temperature (25 °C) due to the rigidification of the pyridyl rings of the ligands upon coordination with the metal centres. Such coordination prevents free rotation in the NMR timescale, which leads to two different chemical environments for the pyridyl rings in the self-assembled product (M1). We carried out the variable temperature (VT) 1H NMR which showed that these split α-pyridyl (a1 and a2) and β-pyridyl (b1 and b2) protons coalesce into single peaks, a and b, respectively at temperatures higher than 85 °C (Fig. S34†). At higher temperatures, the faster rotation of the coordinated pyridyl rings becomes unresolvable in the NMR timescale resulting in coalescence of the peaks corresponding to the pyridyl protons. Finally, to determine the composition of M1, the NO3− analogue was converted to the PF6− analogue for better ionization in ESI-MS. The m/z peaks obtained correspond to an A6L3 composition. The isotopic patterns obtained at 1219.9336, 946.9544, and 764.9654 correspond to the fragments [M1-4PF6]4+, [M1-5PF6]5+, and [M1-6PF6]6+ respectively, which matched well with the theoretically calculated isotopic patterns, confirming the formation of a trigonal barrel (M1) (Fig. 1b–e, S19 and S20†).
 | ||
| Fig. 1 (a) 1H NMR spectrum of trifacial barrel M1 in DMSO-d6 at 298 K. (b) Experimental (blue) and (d) theoretical (red) isotopic patterns for the charged fragment [M1-4PF6]4+. (c) Experimental (blue) and (e) theoretical (red) isotopic patterns for the charged fragment [M1-5PF6]5+ in ESI-MS recorded in acetonitrile. | ||
Unfortunately, all attempts to crystallize M1 were unsuccessful. Thus, the geometry of M1 was optimized by the Density Functional Theory (DFT) method. There are two possible isomers of M1: one in which all ligands assume horizontal orientation (Fig. 2a and b) and are clipped to metal acceptors at the corners, forming a trifacial barrel or vertical orientation of all ligands with more steric requirements, producing a trifacial tube-like architecture (Fig. S37†). Expectedly, the former architecture was found to be more stable by ca. 130 kcal mol−1, ruling out the presence of tube-like architecture.
 | ||
| Fig. 2 DFT optimized structure of trifacial barrel M1 (isomer of M1, where the three ligands are oriented horizontally and clipped to six “Pd” acceptors): (a) side view and (b) top view. Color codes: carbon (black), nitrogen (blue), oxygen (red) and palladium (green). Hydrogen atoms are omitted for clarity. | ||
Moreover, the solvo-dynamic radius (r = 20.4 Å) obtained from 1H DOSY NMR (Fig. S6†) matched reasonably well with the non-solvated radius (the distance of the centroid of the optimized structure to the farthest atom) of the trifacial barrel (r = 21.6 Å) [Fig. S47 and Table S3, Section 6 of the ESI†] confirming it to be the architecture of M1 (A6L3) cage.
The trifacial barrel (M1) with an internal cavity diameter of ca. 17.8 Å and a pair of “terpyridine” moieties at its corners was expected to be a potential host for large fullerene C70 molecules. Accordingly, the DMSO solution of M1 was treated with an excess of C70 (4 equivalents with respect to M1) and the reaction mixture was heated at 70 °C for 12 h. After completion of the reaction, the excess C70 was removed by centrifugation and a clear dark-brown solution was decanted. The host–guest complex was isolated as a dark-brown precipitate by treating the solution with a large excess of ethyl acetate. The 1H NMR recorded in DMSO-d6 showed a completely different pattern, with shifts in peaks compared to the trifacial barrel (M1) indicating the formation of a host–guest complex (Fig. 3a and S10†). All peaks in the 1H NMR had the same diffusion coefficient as confirmed from the 1H DOSY experiment suggesting the presence of only one species in solution (Fig. S11†). Finally, the nitrate analogue was converted to the hexafluorophosphate analogue and subjected to ESI-MS analyses. The peaks and isotopic patterns observed in the mass spectrum correspond to a composition of three C70 molecules bound to a higher analogue, tetrafacial barrel, (C70)3@M2. The isotopic patterns observed at m/z = 1815.3732, 1488.6732, 1255.2926, and 1080.2664 correspond to the charged fragments [(C70)3@M2-5PF6]5+, [(C70)3@M2-6PF6]6+, [(C70)3@M2-7PF6]7+, and [(C70)3@M2-8PF6]8+ respectively, which confirmed the structural transformation of the trifacial barrel (M1) into the tetrafacial barrel (M2) driven by the encapsulation of three C70 molecules (Fig. 3c–f, S21–S23†).
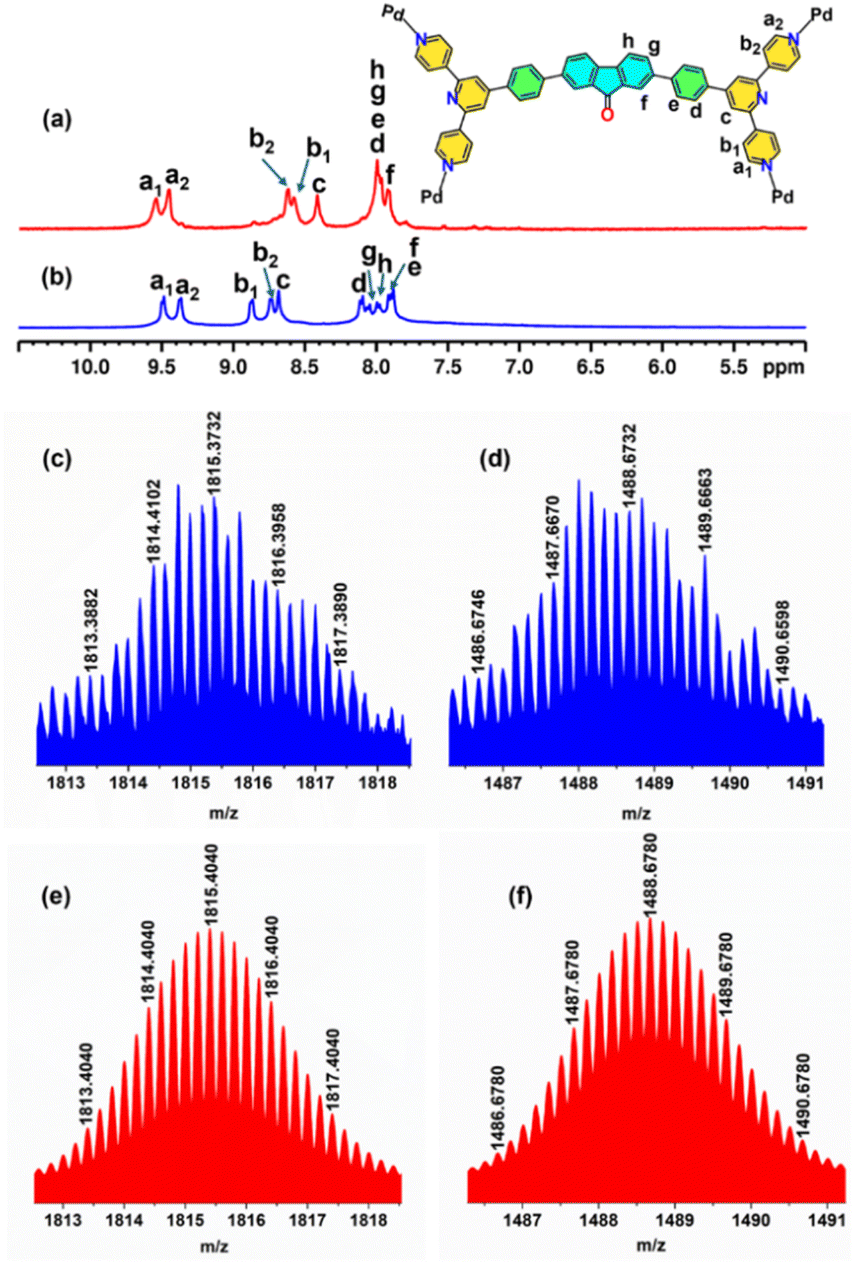 | ||
| Fig. 3 Stacked 1H NMR spectra of (a) host–guest complex (C70)3@M2 and (b) trifacial barrel M1 in DMSO-d6 at 298 K. (c) Experimental (blue) and (e) theoretical (red) isotopic patterns for the charged fragment [(C70)3@M2-5PF6]5+. (d) Experimental (blue) and (f) theoretical (red) isotopic patterns for the charged fragment [(C70)3@M2-6PF6]6+ in ESI-MS recorded in acetonitrile. | ||
The optimized structure depicted that the ellipsoidal C70 molecules are strongly stabilized at the three corners of the tetrafacial barrel via π–π interactions with the “terpyridine” units present at the corners (Fig. 4a). The other possibility of M2 reassembling with three C70 molecules with a vertical orientation for all the ligands is not possible since there is not enough space within such a tube-like structure for stabilizing three C70 molecules.
 | ||
| Fig. 4 PM6 optimized structure of (a) (C70)3@M2 (side view) and (b) (C60)3@M2 (side view). Color codes: carbons of the host (black), nitrogen (blue), oxygen (red) and palladium (green), carbons of guest C70 (brown), and carbons of guest C60 (purple). Hydrogen atoms are omitted for clarity. | ||
All attempts to optimize tube-like architecture with three C70 molecules led to the expulsion of one C70 molecule (Fig. S40†). Moreover, such a conformation was less stable by ca. 365 kcal mol−1 than the conformation with a horizontal arrangement of ligands. Furthermore, the solvo-dynamic radius obtained from 1H-DOSY (r = 25.1 Å) matches closely with the non-solvated radius of the optimized structure of (C70)3@M2 with horizontal orientation of ligands (r = 25.8 Å) [Fig. S49 and Table S3, Section 6 in the ESI†]. These results further rule out the existence of tube-like architecture for the C70 complex.
The 1H NMR (Fig. 3a and S10†) of the complex, (C70)3@M2, at room temperature (25 °C), shows a simple two-fold splitting for the α-pyridyl (a1 and a2) and β-pyridyl (b1 and b2) protons similar to the free cage M1, whereas a higher order splitting was expected for a tetrafacial barrel stabilizing three C70 molecules asymmetrically within its cavity. We further carried out variable temperature (VT) NMR experiments to get additional insights. At higher temperatures, the doubly split α-pyridyl (a1 and a2) and β-pyridyl (b1 and b2) protons coalesce into single peaks, a and b, respectively, like the free cage, M1 (Fig. S35†). However, the coalescence of the protons occurs at a lower temperature (65 °C) compared to the free cage (85 °C).
This observation clearly indicates that the conformational flexibility of the complex, (C70)3@M2, is much higher than that of M1 due to the formation of a large tetragonal architecture resulting in greater molecular tumbling and dynamic motion of the three bound fullerenes among the four potential fullerene stabilization sites. We hypothesize that the three C70 molecules dynamically shuttle among the four corners of the barrel (M2) which is much faster compared to the NMR timescale and thus a time-averaged simple 1H NMR spectrum is obtained for (C70)3@M2 (Fig. 3a and S10†) even at room temperature (25 °C). Since DMSO freezes at 19 °C, we recorded the 1H NMR of the acetonitrile soluble PF6− analogue in CD3CN and carried out the low temperature VT NMR experiments (acetonitrile freezes at −45 °C). The 1H NMR of (C70)3@M2 in CD3CN also shows a two-fold splitting for the α-pyridyl (a) protons at room temperature (25 °C). However, at a lower temperature of −40 °C, the α-pyridyl (a) protons split into multiple peaks (merged and appears broad) due to freezing of molecular tumbling motion and free movement of bound C70 molecules within the cavity of M2. As a result, at lower temperatures, the protons remain in different chemical environments and become resolvable in the NMR timescale (Fig. S36†). The other protons also further split at lower temperatures due to such effects.
Moreover, the kinetics of this C70 induced transformation from M1 into (C70)3@M2 was explored. Accordingly, a solution of M1 in DMSO-d6 was treated with an excess of C70 (4 equivalents with respect to M1), and the reaction mixture was stirred at 70 °C and monitored using 1H NMR at different time intervals. Within 10 min, the peaks corresponding to (C70)3@M2 started appearing and the conversion of M1 to (C70)3@M2 was nearly complete within 2 h (Fig. S30†). This experiment proves that M1 instantly converts to (C70)3@M2 in the presence of C70 under heat treatment without the involvement of any other detectable intermediates. Notably, when the solution of M1 in DMSO-d6 was stirred with an excess of C70 at room temperature, no changes were observed in the 1H NMR indicating no structural transformation or encapsulation at room temperature (Fig. S30b†). Thus, M1 in the presence of C70 and only under heat treatment directly converts to a stable product, (C70)3@M2. To get further insights into the structural transformation and C70 encapsulation, host–guest studies were carried out using lower equivalents of C70 with respect to M1 than that required for the formation of (C70)3@M2. Two solutions of M1 in DMSO-d6 were separately treated with 0.75 equivalents (1 equivalent with respect to transformed M2) and 1.5 equivalents (2 equivalents with respect to transformed M2) of C70, respectively. The reaction mixtures were stirred at 70 °C and monitored using 1H NMR at different time intervals. In both cases, peaks corresponding to (C70)3@M2 started to appear within 20 min and conversion was nearly complete within 3 h (Fig. S31 and S32†). To further validate the compositions, samples of M1 in DMSO, treated with 0.75 equivalents and 1.5 equivalents of C70, respectively at 70 °C for 1 h were converted to their PF6− analogues and subjected to ESI-MS analyses in acetonitrile. The mass spectra displayed only peaks and isotopic patterns corresponding to (C70)3@M2 (Fig. S33c and d†). Even the mass spectrum of the PF6− analogue obtained after treating M1 with 0.75 equivalents of C70 for just 10 min at 70 °C displayed intense peaks and isotopic patterns corresponding to (C70)3@M2 and very less intense peaks and isotopic patterns corresponding to free M1 (Fig. S33b†). No other isotopic distributions were observed corresponding to M2 or other compositions of M1/M2 with C70. These experiments further prove that the conversion of M1 to (C70)3@M2 is concerted and (C70)3@M2 is both the kinetic and thermodynamic product. M1 upon treatment with C70 at higher temperatures instantly degrades and converts to (C70)3@M2 without the involvement of any detectable intermediates.
The direct formation of (C70)3@M2 from M1 was unambiguously confirmed from the time- and temperature-dependent NMR and mass analyses. However, we were curious to investigate why the fourth C70 molecule was not encapsulated into the remaining vacant site of M2. We optimized the structures of the tetrafacial barrel, M2 with one, two, three and four C70 molecules encapsulated (Fig. S41†) and normalized their energies with respect to M2 to calculate the host–guest stabilization energies (pages S30–S32 in the ESI†). The calculations clearly indicate that the addition of each C70 molecule to M2 leads to an increase in non-covalent interactions, enthalpically favouring (C70)4@M2 (Fig. S42†) On the other hand, entropically (C70)4@M2 formation is most disfavoured. Thus, the opposing effects of enthalpy and entropy seem to guide the formation of (C70)3@M2 as the thermodynamically stable product. While fullerene induced structural transformation is known,33,34 here for the first time we observed the encapsulation of three large C70 molecules within the cavity of the transformed cage.
The host–guest studies were also carried out with C60 molecules by treating the solution of the trifacial barrel (M1) in DMSO with an excess of C60 (4 equivalents with respect to M1) followed by stirring the solution at 70 °C for 12 h. The brown precipitate was isolated after treating the reaction mixture with an excess of ethyl acetate. NMR and mass analyses revealed the presence of both free trifacial barrel (M1) and tetrafacial barrel containing three C60 molecules (C60)3@M2 (Fig. S15–S18, S24, S25, and S27–S29†). These results indicated that spherically symmetrical C60 molecules could not induce complete transformation of the cage which was very facile with C70. This may be ascribed to the greater extent of the π–π interactions of the host (M2) with ellipsoidal-shaped C70 rather than with spherically symmetrical C60. This also indicates that the transformed tetrafacial barrel (M2) has a much higher selectivity for C70 compared to C60.
Fluorenone is well known for generating ROS like singlet oxygen or superoxide. The main objective of embedding the “fluorenone” core within our building blocks was to endow the self-assembled product with ROS generating ability. Furthermore, the transformed tetrafacial barrel (M2) encapsulated three C70 molecules which is expected to improve the ROS-generating ability particularly under visible light since fullerene-C70 is a known photosensitizer, with it's broad absorption in the green-red spectrum of the visible light. Interestingly, the PF6− analogues of the trifacial barrel (M1) and C70 complex, (C70)3@M2, were highly soluble in acetonitrile and could function as potential catalysts in the aforementioned solvent.
M1 absorbs in the UV and blue regions of visible light (characteristic absorption of fluorenone). (C70)3@M2 also absorbs in the similar region with a slight shift in the absorption profile due to the formation of host–guest complex (Fig. S43†). Additionally, it displays a broad absorption characteristic of C70 fullerene (starting at ca. 450 nm and tailing up to ca. 700 nm) which further corroborates the presence of encapsulated C70 molecules.
Only recently have there been efforts to replace the hazardous ozonolysis-mediated olefinic cleavage to generate respective aldehydes or ketones under milder conditions.49a–c In this respect, recent approaches involve the combination of nitroarenes and alkenes under purple light, leading to the formation of dioxazolidine intermediates that fragment to yield carbonyl products.50a,b We surmise that use of low-energy red light at ambient temperature to sensitize triplet oxygen and consequent oxidative olefinic cleavage will represent a significant improvement. Accordingly, we investigated whether the PF6− analogues of our barrel M1 or C70 complex, (C70)3@M2, could function as potential catalysts for the ROS-mediated oxidative cleavage of CC of alkenes in acetonitrile. M1 and (C70)3@M2 both absorb strongly in the blue-violet region of visible light. Initially, diphenylethylene was selected as a model substrate and treated separately with catalytic amounts (0.2 mol%) of M1 and (C70)3@M2 in acetonitrile and subjected to irradiation with a LED of wavelength 390 nm for 24 h under an O2 atmosphere using a balloon. After completion of the reaction, the product mixture was extracted with a hexane–ethyl acetate mixture (95:5) and analyzed using NMR spectroscopy. In both cases the formation of the oxidized product, benzophenone, was observed in quite high yields. However, the yield (85%) was slightly higher when (C70)3@M2 was used as a catalyst compared to M1 (71%) (Table 1, entries 1 and 2). The slightly higher yield in the presence of (C70)3@M2 may be ascribed to the ease of Förster Resonance Energy Transfer (FRET) from M2 to the encapsulated C70 molecules. The free trifacial barrel M1 and (C70)3@M2 exhibit similar emission profiles having a significant overlap with the C70 absorption band (Fig. S44†). Furthermore, the proximity of M2 and C70 molecules in (C70)3@M2 provides an ideal platform for FRET from M2 to C70 under 390 nm irradiation which probably is responsible for its slightly better photo-catalytic performance under 390 nm irradiation. Importantly, control reactions were carried out using ligand L and free C70 in acetonitrile, while none of them showed any product formation due to their insolubility in acetonitrile (Table 1, entries 5 and 7). Thus, catalytic amounts of the PF6− analogues of both M1 and (C70)3@M2 were effective in mimicking ozonolysis by oxidatively transforming diphenylethylene into benzophenone and formaldehyde under 390 nm irradiation (Table 1 and S5†).
|
|
|||||
|---|---|---|---|---|---|
| Entry no. | Catalyst | Irradiation wavelength (nm) | Solvent | ROS quencher | P1a |
| Yield (%) | |||||
| a Reactions were carried out using 0.2 mol% of the PF6− analogue of M1, (C70)3@M2, or free ligand L as a catalyst in acetonitrile under continuous oxygen supply and irradiation with a light source of 650 nm or 390 nm for 24 h. b Reactions were carried out using 0.6 mol% of the corresponding catalyst. | |||||
| 1 | M1 (PF6− analogue) | 390 | MeCN | — | 71 |
| 2 | (C70)3@M2 (PF6− analogue) | 390 | MeCN | — | 85 |
| 3 | M1 (PF6− analogue) | 650 | MeCN | — | No reaction |
| 4 | (C70)3@M2 (PF6− analogue) | 650 | MeCN | — | 74 |
| 5b | C70 | 390 | MeCN | — | No reaction |
| 6b | C70 | 650 | MeCN | — | No reaction |
| 7 | Ligand L | 390 | MeCN | — | No reaction |
| 8b | C70 | 650 | Toluene | — | 43 |
| 9 | M1 (PF6− analogue) | 390 | MeCN | DABCO (2 equiv.) | 28% |
| 10 | (C70)3@M2 (PF6− analogue) | 390 | MeCN | DABCO (2 equiv.) | 32% |
| 11 | (C70)3@M2 (PF6− analogue) | 650 | MeCN | DABCO (2 equiv.) | 15% |
| 12 | (C70)3@M2 (PF6− analogue) | 650 | MeCN | TEMPO (2 equiv.) | 11% |
| 13 | (C70)3@M2 (PF6− analogue) | 650 | MeCN | p-Benzoquinone (2 equiv.) | 17% |
| 14 | No catalyst | 390 | MeCN | — | No reaction |
| 15 | No catalyst | 650 | MeCN | — | No reaction |

The PF6− analogue of (C70)3@M2 shows broad absorption from ca. 450 nm which tails up to ca. 700 nm. This prompted us to investigate whether it could show photo-catalytic activity under the irradiation of longer wavelength, red light. Subsequently, we carried out the same reaction using diphenylethylene as a model substrate and a catalytic amount (0.2 mol%) of (C70)3@M2 under 650 nm irradiation for 24 h under an O2 atmosphere maintained using a balloon. The oxidized product, benzophenone, was obtained in 74% yield (Table 1, entry 4). Expectedly, no product formation was observed when M1 was used as the catalyst since it doesn't absorb in red light (Table 1, entry 3). Moreover, free C70 is unable to catalyze the reaction due to insolubility in acetonitrile (Table 1, entry 6). Free C70 is photosensitizing in nature but only soluble in non-polar solvents such as toluene. So, we looked at the possibility of free C70 catalyzing the oxidative transformation of diphenylethylene into benzophenone in toluene. The reaction was carried out using C70 as a catalyst in toluene under 650 nm irradiation keeping other reaction conditions the same. Expectedly, the formation of benzophenone was observed. However, the yield obtained using (C70)3@M2 as a catalyst (74%) in acetonitrile was much higher than that using three times free C70 as a catalyst (43%) in toluene (Table 1, entry 8). The superior performance of (C70)3@M2 in acetonitrile compared to free C70 in toluene in spite of better oxygen solubility in toluene51 compared to acetonitrile52 may be ascribed to slight red shifting of absorption upon C70 encapsulation. A comparison of the absorption profiles of (C70)3@M2 in acetonitrile and free C70 in toluene in the red region of visible light reveals that absorption of (C70)3@M2 is slightly red shifted compared to free C70. While free C70 shows absorption maxima at 639 nm, (C70)3@M2 shows stronger absorption at around 650 nm, which tails up to ca. 700 nm (Fig. S45†). Moreover, carrying out oxidative reactions in acetonitrile is far more advantageous compared to toluene since in toluene different types of ROS form deteriorating the product selectivity.53 However, the host–guest complex, (C70)3@M2, at very low catalyst loading, under safe red-light irradiation and in commonly used acetonitrile media selectively cleaved the olefins oxidatively to produce the corresponding carbonyl compounds without formation of any over-oxidized product(s).
To get further insights into the possible mechanism of this ozonolysis mimicking oxidative transformation, the type of ROS generated in the reaction conditions was investigated. DABCO is a well-known singlet oxygen (1O2) quencher.54 The reactions were carried out under similar conditions using (C70)3@M2 as a catalyst and in the presence of 2 equivalents of DABCO (with respect to the substrate diphenylethylene) both under 390 nm and 650 nm light irradiation. The reactions resulted in reduced yield supporting that 1O2 might be the primary ROS generated under photoirradiation (Table 1, entries 9–11). However, the non-negligible product formation (32% under 390 nm irradiation) strongly suggests the presence of another ROS different from 1O2. To interrogate the nature of the second reactive oxygen species, we conducted further control reactions with TEMPO and p-benzoquinone. TEMPO is a well-known radical scavenger55 while p-benzoquinone is profusely used to trap a superoxide radical.56 In both cases, the reaction was significantly suppressed. From these control reactions it can be concluded that the oxidative cleavage reaction also follows a radical pathway and likely involves a superoxide species (Table 1, entries 12–13). To gather a compelling proof, electron paramagnetic resonance (EPR) experiments were conducted to spectroscopically detect the generation of 1O2 and O2˙− in the catalytic cycle. The EPR spectra were recorded after irradiating the oxygen purged acetonitrile solutions of complex (C70)3@M2 in the presence of 1O2 spin trap, 2,2,6,6-tetramethylpiperidine (TEMP). In another experiment O2˙− spin trap, 5,5-dimethyl-1-pyrroline N-oxide (DMPO) was used. The signature signals obtained in the EPR spectra corresponding to the formation of TEMPO and DMPO–OOH spin radicals in the respective cases confirmed the generation of 1O2 and O2˙−, respectively (Section 13, Fig. S52 and S53 in the ESI†).
The plausible reaction mechanism suggests a [2 + 2] cyclo-addition of the alkene with 1O2 to generate the endoperoxide followed by cleavage of the four membered dioxetane to furnish the corresponding carbonyl compounds (path I, Fig. 5). Thus, the PF6− analogue of (C70)3@M2 generated 1O2 under irradiation with both blue-violet and red light which successfully catalysed the oxidative transformation of diphenylethylene into benzophenone in acetonitrile. Alternatively, diphenylethylene can be oxidized by photoexcited (C70)3@M2 to generate the radical cation of the olefin and the reduced form of the putative complex can donate one electron to oxygen forming a superoxide radical. This superoxide radical can further react with the oxidized radical cation of alkene to generate the same dioxetane intermediate (path II, Fig. 5).
 | ||
| Fig. 5 Plausible mechanisms of ROS mediated oxidative transformation of olefins into carbonyl compounds in the presence of catalytic amounts of (C70)3@M2 under red-light (650 nm) irradiation. Both singlet oxygen (1O2) and superoxide radicals (O2˙−) are generated by (C70)3@M2 under light irradiation via energy transfer (path I) or electron transfer (path II) processes, respectively. | ||
Next, the substrate scope for this catalysis was explored under red light irradiation. The reaction with different non-hindered terminal alkenes proceeded with good yields (Table 2, entries 1–5) under similar reaction conditions. In all these reactions, the other oxidized product formaldehyde (HCHO) could not be detected from NMR analyses due to its volatile nature. On the other hand, when bulky, non-terminal substrates were used (Table 2, entries 6–8) both the oxidized carbonyl compounds could be detected. However, either the product yields were much lower for the bulky olefin substrates (Table 2, entries 6–7), or a trace amount of product formation was observed (Table 2, entry 8), possibly due to steric factors resulting in sluggish endoperoxide formation. Gratifyingly, heterocycle-substituted olefin or olefin possessing nitroaromatics afforded the oxidatively cleaved product, albeit via 390 nm irradiation (Table 2, entries 9–10). As described in Table 2, (C70)3@M2 could catalyze the oxidative transformation of a wide range of aromatic alkene substrates; however it fails to cleave aliphatic olefins.
| Entry no. | Alkene substrates | Product/(s) | Yields |
|---|---|---|---|
| a All reactions were carried out using 0.2 mol% of the PF6− analogue of (C70)3@M2 as a catalyst in acetonitrile (for substrates S7 and S8 an acetonitrile–dichloromethane mixture was used) under continuous oxygen supply and irradiated with a light source of 650 nm for 24 h. b 45% isolated yield under 390 nm light irradiation. c 74% isolated yield under 390 nm light irradiation; the control reaction for substrate S10 carried out in the absence of the catalyst, under the same reaction conditions and under 390 nm light irradiation, generated P10a in ca. 5% yield (crude NMR yield).57 | |||
| 1 |

|

|
P1a (74%) |
| 2 |

|

|
P2a (62%) |
| 3 |

|

|
P3a (69%) |
| 4 |

|

|
P4a (63%) |
| 5 |

|

|
P5a (61%) |
| 6 |

|

|
P6a (58%) |
| 7 |

|
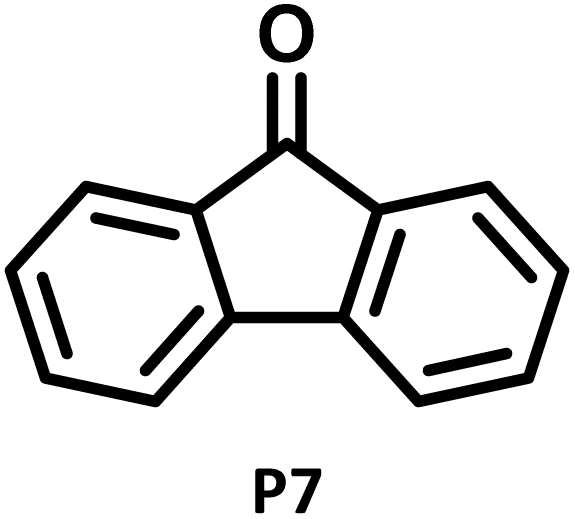
|
P7 (32%) |
| 8 |
|

|
Trace amount of P8a |
| 9 |

|
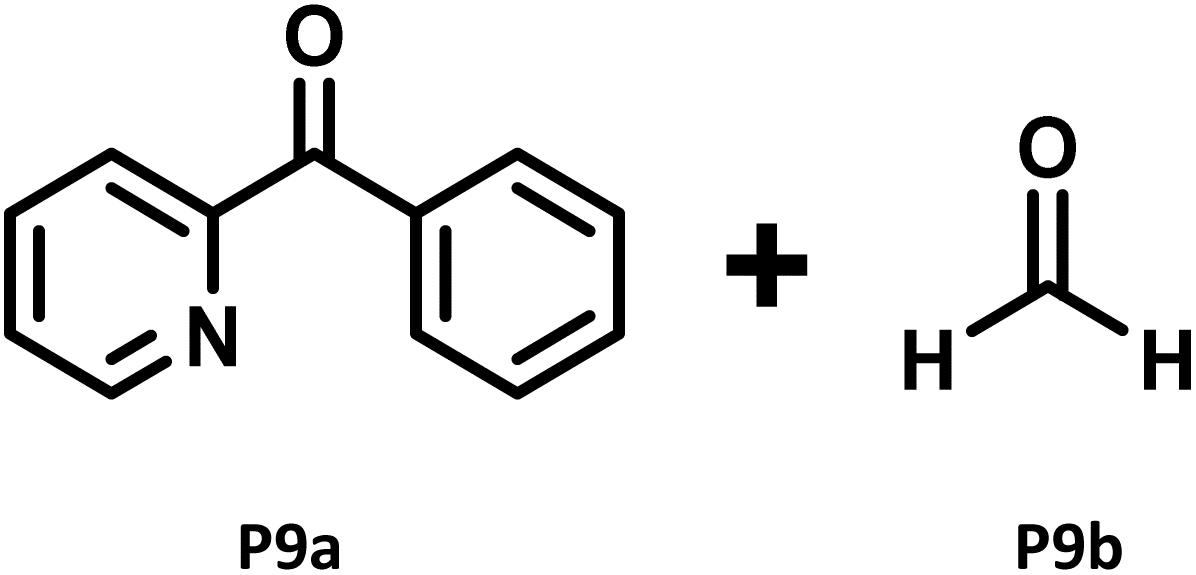
|
Trace amount of P9a (45%)b |
| 10 |

|
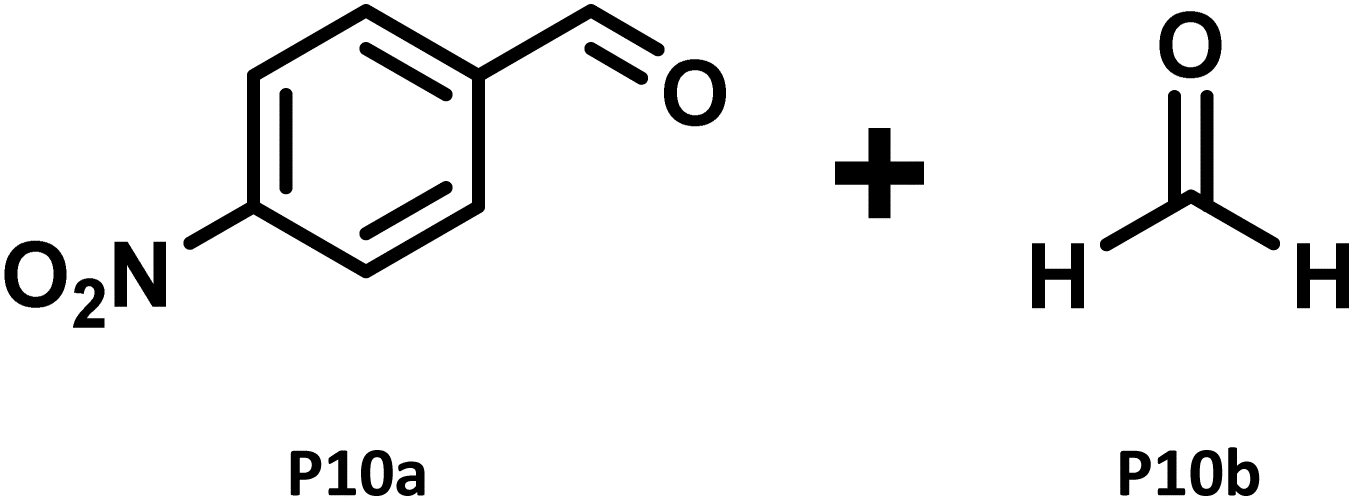
|
P10a 21% (74%)c,57 |
Conclusions
In conclusion, a fluorenone-based Pd6 trifacial barrel (M1) was synthesized by coordination-driven self-assembly of a tetradentate donor (L) with a cis-Pd(II) acceptor (A). The trifacial barrel (M1) displayed a guest-mediated transformation into a Pd8 tetrafacial barrel (M2) in the presence of C70 fullerene molecules leading to the formation of a C70 encapsulated stable host–guest complex, (C70)3@M2. The PF6− analogues of both M1 and (C70)3@M2 were soluble in acetonitrile and on account of the presence of photosensitizing “fluorenone” within their building units along with three encapsulated C70 molecules within (C70)3@M2, their potential to catalyze the ROS-mediated oxidative transformation of alkenes into the corresponding carbonyl compounds was investigated. M1 and (C70)3@M2 both generated ROS, singlet oxygen (1O2) and superoxide radicals (O2˙−) under 390 nm irradiation and could catalyze the oxidative transformation of olefins into carbonyl compounds in acetonitrile. Additionally, (C70)3@M2 absorbs in the red-region of visible light and could photo-catalyze such alkene oxidative transformations under red light irradiation at a low catalyst loading. A wide range of terminal and bulky, sterically hindered alkenes in acetonitrile were successfully cleaved by the cage/complex, while free C70 fails to catalyze due to its poor solubility in the chosen solvent.The formation of host–guest complex, (C70)3@M2 with C70 molecules enabled tuning of the photosensitizing wavelength from hazardous blue-violet (390 nm) to long wavelength red light (650 nm) for carrying out the reactions. Therefore, the encapsulation of C70 molecules was vital for solubilizing C70 and utilizing its photocatalytic activity under red-light irradiation in acetonitrile. Furthermore, (C70)3@M2 in acetonitrile was found to act as a better photocatalyst than free C70 in a toluene owing to the modulated absorption profile of C70 upon encapsulation. The advantage of using (C70)3@M2 as a catalyst is that it can be photosensitized using low-energy red light which not only enables oxidative cleavage of olefins but also curtails chances of substrate over-oxidation. The non-toxic catalyst, (C70)3@M2, thus overcomes the limitations of traditional olefin oxidation by using metal-oxide oxidants or ozonolysis. Such host–guest complex catalyzed ROS-mediated alkene oxidative splitting at a very low catalyst loading, under milder conditions, and under red light irradiation is a promising green approach for simplifying organic synthesis.
Data availability
The data supporting this article have been included as a part of ESI.†Author contributions
R. B. carried out most of the experimental studies: synthesis and characterization and analyzed the data. R. B. carried out the computational studies. M. R. carried out the catalysis. M. A. and S. M. carried out some of the experiments. P. S. M. and D. A. supervised the whole project. All authors contributed to writing of this manuscript and they have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. S. M. thanks the SERB (New Delhi) for a research grant and J. C. Bose Fellowship. R. B. and M. R. gratefully acknowledge PMRF (India) for a research fellowship and contingency grant. M. A. thanks IISc for the research fellowship and S. M. is funded by CSIR, senior research fellowship. D. A. thanks MHRD, India (Grant No. STARS-2/2023/0474), and acknowledges the departmental NMR (SR/FST/CS-II/2019/94, TPN no. 32545) facility. IISER Mohali central NMR facility is also thanked.References
- N. M. A. Speakman, A. W. Heard and J. R. Nitschke, J. Am. Chem. Soc., 2024, 146, 10234–10239 CrossRef CAS PubMed.
- R. Saha, B. Mondal and P. S. Mukherjee, Chem. Rev., 2022, 122, 12244–12307 CrossRef CAS PubMed.
- A. Platzek, S. Juber, C. Yurtseven, S. Hasegawa, L. Schneider, C. Drechsler, K. E. Ebbert, R. Rudolf, Q.-Q. Yan, J. J. Holstein, L. V. Schäfer and G. H. Clever, Angew. Chem., Int. Ed., 2022, 61, e202209305 CrossRef CAS PubMed.
- (a) Y. Hou, C. Mu, Y. Shi, Z. Zhang, H. Liu, Z. Zhou, S. Ling, B. Shi, X. Duan, C. Yang and M. Zhang, Aggregate, 2024, e628 CrossRef CAS; (b) S.-Q. Wang, Y. Wang, X. Yang, Y. Liu, H. Li, Z. Yang, W.-Y. Sun and J. L. Sessler, Angew. Chem., Int. Ed., 2024, 63, e202317775 CrossRef CAS PubMed; (c) L.-X. Cai, S.-H. Li, D.-N. Yan, L.-P. Zhou, F. Guo and Q.-F. Sun, J. Am. Chem. Soc., 2018, 140, 4869–4876 CrossRef CAS PubMed.
- (a) H. Takezawa, K. Iizuka and M. Fujita, Angew. Chem., Int. Ed., 2024, 63, e202319140 CrossRef CAS PubMed; (b) M. R. Crawley, D. Zhang, A. N. Oldacre, C. M. Beavers, A. E. Friedman and T. R. Cook, J. Am. Chem. Soc., 2021, 143, 1098–1106 CrossRef CAS PubMed.
- Q. Li, H. Zhu and F. Huang, Trends Chem., 2020, 2, 850–864 CrossRef CAS.
- S. M. Bierschenk, R. G. Bergman, K. N. Raymond and F. D. Toste, J. Am. Chem. Soc., 2020, 142, 733–737 CrossRef CAS PubMed.
- R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS PubMed.
- S.-D. Guo, J. F. Stoddart and K. Cai, Chem, 2023, 9, 1071–1073 CAS.
- X.-R. Liu, P.-F. Cui, S.-T. Guo, Y.-J. Lin and G.-X. Jin, J. Am. Chem. Soc., 2023, 145, 8569–8575 CrossRef CAS PubMed.
- (a) M. Shuto, R. Sumida, M. Yuasa, T. Sawada and M. Yoshizawa, JACS Au, 2023, 3, 2905–2911 CrossRef CAS PubMed; (b) L. Zhang, R. Das, C.-T. Li, Y.-Y. Wang, F. E. Hahn, L.-Y. Sun and Y.-F. Han, Angew. Chem., Int. Ed., 2019, 58, 13360–13364 CrossRef CAS PubMed; (c) J. Liu, B. Wang, C. Przybylski, O. Bistri-Aslanoff, M. Ménand, Y. Zhang and M. Sollogoub, Angew. Chem., Int. Ed., 2021, 60, 12090–12096 CrossRef CAS PubMed.
- (a) B. Huang, M. Zhou, Q.-Y. Hong, M.-X. Wu, X.-L. Zhao, L. Xu, E.-Q. Gao, H.-B. Yang and X. Shi, Angew. Chem., Int. Ed., 2024, 63, e202407279 CrossRef CAS PubMed; (b) K. Wu, E. Benchimol, A. Bakshi and G. H. Clever, Nat. Chem., 2024, 16, 584–591 CrossRef CAS PubMed.
- (a) C. T. McTernan, J. A. Davies and J. R. Nitschke, Chem. Rev., 2022, 122, 10393–10437 CrossRef CAS PubMed; (b) M. Yoshizawa and J. K. Klosterman, Chem. Soc. Rev., 2014, 43, 1885–1898 RSC; (c) N. Sinha and F. E. Hahn, Acc. Chem. Res., 2017, 50, 2167–2184 CrossRef CAS PubMed; (d) W.-X. Gao, H.-J. Feng, B.-B. Guo, Y. Lu and G.-X. Jin, Chem. Rev., 2020, 120, 6288–6325 CrossRef CAS PubMed.
- R. Banerjee, D. Chakraborty and P. S. Mukherjee, J. Am. Chem. Soc., 2023, 145, 7692–7711 CrossRef CAS PubMed.
- (a) H. Takezawa, K. Shitozawa and M. Fujita, Nat. Chem., 2020, 12, 574–578 CrossRef CAS PubMed; (b) S. Ghosal, A. Das, D. Roy and J. Dasgupta, Nat. Commun., 2024, 15, 1810 CrossRef CAS PubMed.
- V. A. Rinshad, M. Aggarwal, J. K. Clegg and P. S. Mukherjee, JACS Au, 2024, 4, 3238–3247 CrossRef CAS PubMed.
- (a) X. Zhu, G. Xu, L. M. Chamoreau, Y. Zhang, V. Mouriés-Mansuy, L. Fensterbank, O. Bistri-Aslanoff, S. Roland and M. Sollogoub, Chem.–Eur. J., 2020, 26, 15901–15909 CrossRef CAS PubMed; (b) C. Tugny, N. del Rio, M. Koohgard, N. Vanthuyne, D. Lesage, K. Bijouard, P. Zhang, J. M. Suaréz, S. Roland, E. Derat, O. Bistri-Aslanoff, M. Sollogoub, L. Fensterbank and V. Mouriés-Mansuy, ACS Catal., 2020, 10, 5964–5972 CrossRef CAS.
- T. K. Piskorz, V. Martí-Centelles, R. L. Spicer, F. Duarte and P. J. Lusby, Chem. Sci., 2023, 14, 11300–11331 RSC.
- M. Aggarwal, R. Banerjee, N. Hickey and P. S. Mukherjee, Angew. Chem., Int. Ed., 2024, 63, e202411513 CrossRef CAS PubMed.
- X. Fu, G. Ke, F. Peng, X. Hu, J. Li, Y. Shi, G. Kong, X.-B. Zhang and W. Tan, Nat. Commun., 2020, 11, 1518 CrossRef CAS PubMed.
- A. B. Sainaba, M. Venkateswarulu, P. Bhandari, K. S. A. Arachchige, J. K. Clegg and P. S. Mukherjee, J. Am. Chem. Soc., 2022, 144, 7504–7513 CrossRef CAS PubMed.
- D. Chakraborty, R. Saha, J. K. Clegg and P. S. Mukherjee, Chem. Sci., 2022, 13, 11764–11771 RSC.
- J. Martí-Rujas, S. Elli, A. Zanotti, A. Famulari and F. Castiglione, Chem.–Eur. J., 2023, 29, e202302025 CrossRef PubMed.
- D. Samanta, D. Galaktionova, J. Gemen, L. J. W. Shimon, Y. Diskin-Posner, L. Avram, P. Král and R. Klajn, Nat. Commun., 2018, 9, 641 CrossRef PubMed.
- A. Galan and P. Ballester, Chem. Soc. Rev., 2016, 45, 1720–1737 RSC.
- R. Saha, A. Devaraj, S. Bhattacharyya, S. Das, E. Zangrando and P. S. Mukherjee, J. Am. Chem. Soc., 2019, 141, 8638–8645 CrossRef CAS PubMed.
- V. Martínez-Agramunt, D. G. Gusev and E. Peris, Chem.–Eur. J., 2018, 24, 14802–14807 CrossRef PubMed.
- W.-K. Han, H.-X. Zhang, Y. Wang, W. Liu, X. Yan, T. Li and Z.-G. Gu, Chem. Commun., 2018, 54, 12646–12649 RSC.
- S.-Z. Zhan, J.-H. Li, G.-H. Zhang, M.-D. Li, S. Sun, J. Zheng, G.-H. Ning, M. Li, D.-B. Kuang, X.-D. Wang and D. Li, Chem. Commun., 2020, 56, 3325–3328 RSC.
- X. Chang, Y. Xu and M. von Delius, Chem. Soc. Rev., 2024, 53, 47–83 RSC.
- H. Liu, C. Guo, L. Li, Z. Zhang, Y. Hou, C. Mu, G.-l. Hou, Z. Zhang, H. Wang, X. Li and M. Zhang, J. Am. Chem. Soc., 2024, 146, 15787–15795 CrossRef CAS PubMed.
- F. J. Rizzuto and J. R. Nitschke, Nat. Chem., 2017, 9, 903–908 CrossRef CAS PubMed.
- R. Banerjee, D. Chakraborty, W.-T. Jhang, Y.-T. Chan and P. S. Mukherjee, Angew. Chem., Int. Ed., 2023, 62, e202305338 CrossRef CAS PubMed.
- R. Banerjee, S. Bhattacharyya and P. S. Mukherjee, JACS Au, 2023, 3, 1998–2006 CrossRef CAS PubMed.
- C. García-Simón, M. Garcia-Borràs, L. Gómez, T. Parella, S. Osuna, J. Juanhuix, I. Imaz, D. Maspoch, M. Costas and X. Ribas, Nat. Commun., 2014, 5, 5557 CrossRef PubMed.
- J. Kou, Q. Wu, D. Cui, Y. Geng, K. Zhang, M. Zhang, H. Zang, X. Wang, Z. Su and C. Sun, Angew. Chem., Int. Ed., 2023, 62, e202312733 CrossRef CAS PubMed.
- V. Iannace, C. Sabrià, Y. Xu, M. v. Delius, I. Imaz, D. Maspoch, F. Feixas and X. Ribas, J. Am. Chem. Soc., 2024, 146, 5186–5194 CrossRef CAS PubMed.
- Z. Lu, T. K. Ronson, A. W. Heard, S. Feldmann, N. Vanthuyne, A. Martinez and J. R. Nitschke, Nat. Chem., 2023, 15, 405–412 CrossRef CAS PubMed.
- K. Gao, Y. Cheng, Z. Zhang, X. Huo, C. Guo, W. Fu, J. Xu, G.-L. Hou, X. Shang and M. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202319488 CrossRef CAS PubMed.
- E. O. Bobylev, D. A. Poole III, B. de Bruin and J. N. H. Reek, J. Am. Chem. Soc., 2022, 144, 15633–15642 CrossRef CAS PubMed.
- T. J. Fisher and P. H. Dussault, Tetrahedron, 2017, 73, 4233–4258 CrossRef CAS.
- S. G. Van Ornum, R. M. Champeau and R. Pariza, Chem. Rev., 2006, 106, 2990–3001 CrossRef CAS PubMed.
- D. Yang and C. Zhang, J. Org. Chem., 2001, 66, 4814–4818 CrossRef CAS PubMed.
- B. R. Travis, R. S. Narayan and B. Borhan, J. Am. Chem. Soc., 2002, 124, 3824–3825 CrossRef CAS PubMed.
- (a) D. C. Cabanero, S. K. Kariofillis, A. C. Johns, J. Kim, J. Ni, S. Park, D. L. Parker Jr, C. P. Ramil, X. Roy, N. H. Shah and T. Rovis, J. Am. Chem. Soc., 2024, 146, 1337–1345 CrossRef CAS PubMed; (b) D. C. Cabanero and T. Rovis, Nat. Rev. Chem., 2025, 9, 28–45 CrossRef CAS PubMed.
- Y. Wang, X. Feng, J. Cao, X. Zheng, X. Gong, W. Yu, M. Wang and S. Shi, Angew. Chem., Int. Ed., 2024, 63, e202319139 CrossRef CAS PubMed.
- A. Gorczyński, J. M. Harrowfield, V. Patroniak and A. R. Stefankiewicz, Chem. Rev., 2016, 116, 14620–14674 CrossRef PubMed.
- F. Salami and Y. Zhao, New J. Chem., 2020, 44, 9179–9189 RSC.
- (a) C. Wang and J. Xiao, Acc. Chem. Res., 2025, 58, 714–731 CrossRef CAS PubMed; (b) Z. Huang, R. Guan, M. Shanmugam, E. L. Bennett, C. M. Robertson, A. Brookfield, E. J. L. McInnes and J. Xiao, J. Am. Chem. Soc., 2021, 143, 10005–10013 CrossRef CAS PubMed; (c) Y. Liu, D. Xue, C. Li, J. Xiao and C. Wang, Catal. Sci. Technol., 2017, 7, 5510–5514 RSC.
- (a) D. E. Wise, E. S. Gogarnoiu, A. D. Duke, J. M. Paolillo, T. L. Vacala, W. A. Hussain and M. Parasram, J. Am. Chem. Soc., 2022, 144, 15437–15442 CrossRef CAS PubMed; (b) A. Ruffoni, C. Hampton, M. Simonetti and D. Leonori, Nature, 2022, 610, 81–86 CrossRef CAS PubMed.
- A. Li, S. Tang, P. Tan, C. Liu and B. Liang, J. Chem. Eng. Data, 2007, 52, 2339–2344 CrossRef CAS.
- C. Franco and J. Olmsted, Talanta, 1990, 37, 905–909 CrossRef CAS PubMed.
- Y. Ji, J. Zhao, H. Terazono, K. Misawa, N.-P. Levitt, Y. Li, Y. Lin, J. Peng, Y. Wang, L. Duan, B. Pan, F. Zhang, X. Feng, T. An, W. Marrero-Ortiz, J. Secrest, A.-L. Zhang, K. Shibuya, M.-J. Molina and R. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 8169–8174 CrossRef CAS PubMed.
- C. Ouannes and T. Wilson, J. Am. Chem. Soc., 1968, 90, 6527–6528 CrossRef CAS.
- C. R. DeJulius, B. R. Dollinger, T. E. Kavanaugh, E. Dailing, F. Yu, S. Gulati, A. Miskalis, C. Zhang, J. Uddin, S. Dikalov and C. L. Duvall, Bioconjugate Chem., 2021, 32, 928–941 CrossRef CAS PubMed.
- O. Fónagy, E. Szabó-Bardos and O. Horváth, J. Photochem. Photobiol., A, 2021, 407, 113057 CrossRef.
- Footnote to Table 2 (entry 10): the control reaction carried out using S10 as a substrate, in the absence of the catalyst (C70)3@M2 under 390 nm and with all other reaction conditions kept the same, generated P10a in ca. 5% yield (crude NMR yield). The tiny amount of product formation for the substrate S10 under 390 nm irradiation may be ascribed to the ability of nitroarenes and alkenes to undergo a reaction in the absence of any catalyst under near UV irradiation (390 nm) to generate dioxazolidine intermediates that fragment to generate carbonyl compounds. References: 50a and 50b.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section, additional NMR spectra (1H, COSY, NOESY, and DOSY), mass spectra, and optimized structures. See DOI: https://doi.org/10.1039/d5sc01015b |
| ‡ Ranit Banerjee and Monojit Roy contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |