
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceL/Z-ligand type amphoterism of an acridinium unit†
Elishua D.
Litle
 ,
Shantabh
Bedajna
and
François P.
Gabbaï
*
,
Shantabh
Bedajna
and
François P.
Gabbaï
*
Department of Chemistry, Texas A&M University, College Station, TX 77843, USA. E-mail: francois@tamu.edu
First published on 5th May 2025
Abstract
As part of our study of ambiphilic carbenium-based ligands, we now report on the coordination chemistry of the known acridinium phosphine ligand [(o-Ph2P(C6H4)Acr)]+ ([Lacr]+, Acr = 9-N-methylacridinium) towards rhodium(I) reagents. While the simple coordination complex [LacrRh(COD)Cl]+ ([1]+) is obtained from [Lacr]+ and [Rh(COD)Cl]2 (COD = 1,5-cyclooctadiene) in CH2Cl2, the use of [Rh(COE)2Cl]2 (COE = cyclooctene) in MeCN affords [(o-Ph2P(C6H4)Acr)Rh(MeCN)2Cl]22+ ([2]22+), a chloride-bridged dimeric complex in which each rhodium atom is involved in a Rh→Ccarb dative bond (Ccarb = acridinium C9 atom), indicating Z-type behavior of the acridinium unit. The same reaction in CH2Cl2 affords [(o-Ph2P(C6H4)Acr)RhCl]22+ ([3]22+), a chloride-bridged dimeric square planar complex in which the acridinium assumes the role of an η2-heteroarene, L-type ligand. All complexes, which have been structurally characterized as their triflate salts, can be interconverted into one another via simple ligand exchange or displacement reactions. These experimental results, supported by computational analyses, show that the acridinium unit of [Lacr]+ displays ligand-type amphoterism as it easily and reversibly switches from L-type to Z-type in response to changes in the coordination sphere of the metal center.
The Covalent Bond Classification (CBC) can be used to define ligands according to their ability to donate or accept electrons when interacting with a metal.1 The extremes of this ligand classification include the most common L-type, for ligands donating two electrons to a metal, and Z-type, for ligands accepting two electrons from a metal center.2,3 Side-on bound arene ligands are examples of L-type ligands that contribute two electrons to the valence of a metal complex.4 The simplest Z-type ligands are boranes capable of accepting two electrons from a metal center.3,5 The diametrically opposed behavior of these two ligands can be used to modulate the electron richness of the metal center, which can be particularly useful in the context of catalysis.2f–h,5 However, ligands that can switch behavior from L-type to Z-type, without ligand-centered compositional changes,6,7 are, to our knowledge, unknown.
Our recent efforts in the chemistry of Z-type ligands has led us to consider cationic phosphines8 featuring a pendent carbenium functionality9–11 positioned to interact with the coordinated transition metal center.12 One of the best-studied examples of such ligands includes the cationic ambiphilic system [Lacr]+ which features an acridinium moiety as the electron-poor unit (Fig. 1).10 Being isoelectronic with anthracene and thus aromatic, the acridinium core could, in principle, behave as an arene-like L-type ligand. While a related behavior has been observed in the case of pyridinium units such as in [I]+,13 the chemistry of acridinium-containing ligands is dominated by the Lewis acidity of the C9 carbon atom, referred to as Ccarb (Fig. 1). This behavior is in full display in complexes of type [II]+,11 where the group 10 metal engages the acridinium unit via a dative M→Ccarb interaction. Building on our interest in the design of atypical and coordination non-innocent ligands, we are now describing a series of rhodium systems in which the acridinium unit of [Lacr]+ displays ligand amphoterism, switching its behavior from L-type to Z-type in response to changes in the coordination environment of the metal center.
 | ||
| Fig. 1 Top: example of L-type and Z-type ligands. Bottom: structure of [Lacr]+ and known complexes illustrating the L-type behavior of a pyridinium unit ([I]+) and the Z-type behavior of an acridnium unit ([II]+). | ||
Results and discussion
Our investigation began with the reaction of [Lacr][OTf] with [Rh(COD)Cl]2 (COD = 1,5-cyclooctadiene) in CH2Cl2 which proceeded smoothly at room temperature to produce a new complex characterized in CDCl3 by a 31P{1H} NMR resonance at 33.95 ppm split into a doublet by a 1JP–Rh coupling constant of 150.7 Hz (Scheme 1). This coupling is analogous to that reported for Ph3PRh(COD)Cl (1JP–Rh = 152.0 Hz),14 suggesting that the product of this reaction is the simple coordination complex [LacrRh(COD)Cl][OTf] ([1][OTf]). Formation of this complex was confirmed by single crystal X-ray diffraction (see ESI†), which also shows that the acridinium ligand is disengaged from the metal and acts as a spectator unit. In agreement with this view, the 13C{1H} NMR spectrum of [1][OTf] in CDCl3 displays a resonance at 162.6 ppm corresponding to the uncompromised carbenium atom of [Lacr]+. Interestingly, dissolving [1][OTf] in CD3CN results in an immediate broadening of the 31P{1H} NMR resonance, suggesting the onset of a reaction. Over time, the resonance assigned to [1][OTf] disappears to give rise to a doublet at 52.95 ppm (1JP–Rh = 128.4 Hz), signaling the formation of a new species referred to as [2][OTf] (Scheme 1). The 1JP–Rh coupling constant measured for [2][OTf] falls within the expected range for phosphine RhIII complex and can be compared to the value of 103 Hz in RhCl3(triphos).15 This new species, which assumes a dark red color in solution, also forms when [Lacr][OTf] is allowed to react with [Rh(COE)2Cl]2 (COE = cyclooctene) in MeCN. The 13C{1H} NMR spectrum of this new complex displays a resonance at 63.02 ppm, assigned to the former carbenium center. This high field resonance indicates coordination of this center to the rhodium atom, a notion supported by the multiplicity of this signal which couples with the rhodium center via a 1JC–Rh coupling constant of 18.3 Hz. This 13C{1H} NMR chemical shift is at a higher field than that measured in the case of [1][OTf] (162.6 ppm), suggesting Rh→Ccarb dative bonding as in complexes of type [II]+.11 | ||
| Scheme 1 Synthesis of the rhodium complexes [1][OTf] and [2][OTf]. | ||
A single crystal X-ray diffraction study afforded the structure shown in Fig. 2. The rhodium atom is coordinated by four primary ligands, including two tightly bound acetonitriles, a chloride anion and the phosphine ligand. The bonds that these ligands form with the rhodium center are quite short, as indicated by the Rh–N (2.025(5) and 2.012(5) Å), Rh–P (2.2301(18) Å), and Rh–Cl (2.4590(16) Å) distances. These four ligands, which define a square plane, are complemented by two additional ligands positioned above and below the rhodium atom. One of these ligands is the carbenium ion of the acridinium moiety which is engaged in a long Rh→Ccarb interaction of 2.229(6) Å with the adjacent acridinium unit. The trans site, which is acidified by the carbenium Z-type ligand,16 is occupied by the bridging chloride ligand Cl′. This bridging chloride ligand forms a Rh–Cl′ distance of 2.5848(16) Å which is significantly longer than the primary Rh–Cl bond (2.4590(16) Å). The Rh→Ccarb bond present in [2][OTf] induces a distinct pyramidalization of the carbenium center as illustrated by the sum of the C–Ccarb–C angle of 336.4°. The polarization of the Rh→Ccarb bond was assessed computationally through a Pipek–Mezey orbital analysis, using an approach previously adopted to probe the polarization of metal–carbon bonds.11,17 This analysis shows that the Rh→Ccarb bond of [2]+ displays a higher parentage from the rhodium (47.1%) than the Ccarb carbon atom (36.8%). Altogether, the data collected for [2][OTf] indicates that the acridinium unit functions as a Z-type ligand through its carbenium ion, as observed in complexes of type [II]+.11
 | ||
| Fig. 2 Top: crystal structure of the dimeric form of [2][OTf] with ellipsoid drawn at the 50% probability level. Hydrogen atoms and triflate anions are omitted for clarity. N atoms shown in blue. Bottom: truncated view of [2]+ showing the Pipek–Mezey orbital defining the Rh→Ccarb bond. The main atomic orbital parentages are also provided. | ||
Elemental analysis of [2][OTf] pointed to the facile dissociation of the MeCN ligand. Intrigued by this possibility, [2][OTf] was dissolved in CH2Cl2 and evacuated to dryness. Repeating this protocol three times produced a green solid product referred to as [3][OTf] (Scheme 2). This complex is poorly soluble in CD2Cl2 which prevented its full characterization in this solvent. Yet, extended acquisition allowed for the detection of a 31P{1H} NMR signal at 64.71 ppm (1JP–Rh = 135.3 Hz), a chemical shift almost identical to that determined by 31P HPDec-MAS NMR spectroscopy (63.24 ppm). An additional clue into the identity of [3][OTf] was derived from the observation that it dissolves in CD3CN to afford [2][OTf], as confirmed by 1H and 31P{1H} NMR spectroscopy. Further information on the nature of [3][OTf], which is also formed by reaction of [Lacr][OTf] with 0.5 equiv. [Rh(COE)2Cl]2 in CH2Cl2 (Scheme 2), were obtained from an X-ray diffraction experiment (Fig. 3) carried out on single crystals obtained from a very dilute CH2Cl2 solution. Like its precursor [2][OTf], complex [3][OTf] exists as a chloride-bridged dimer with the rhodium atom in a distorted square planar geometry (τ4 = 0.20).18 In addition to the two bridging chloride and the phosphine ligand, the rhodium center is also coordinated in a η2-fashion by the flanking acridinium unit which now functions as an L-type ligand. The two carbon atoms involved in this η2-interaction are the carbenium center and the adjacent C10 atom, placing the centroid of the Ccarb–C10 bond at just 2.045(3) Å from the rhodium atom. This bonding description is supported by the moderate pyramidalization of the carbenium atom (Σ(C–Ccarb–C) = 353.7°). The η2-interaction is rather symmetrical, as confirmed by the Rh–Ccarb and Rh–C10 distances of 2.149(4) Å and 2.191(4) Å, respectively. These distances are close to those found in η2-arene rhodium(I) complexes,19 including [Cp*Rh(η2-phenanthrene)(PMe3)] (III, Fig. 4) in which the arene ligand interacts with the rhodium center via bonds of 2.128 (4) and 2.144 (4) Å.20 A comparison can also be made with complex [IV]+ which also features an intramolecular η2-arene coordination motif. In this case, however, the longer Rh–C distance of 2.411(3) and 2.476(3) Å suggest weaker coordination of the arene ligand, possibly because of the hindrance imposed on the metal center by the norbornadiene ligand.21 The structure of [3][OTf] also indicates a noticeable asymmetry in the Rh–Cl distances, with the longest one being formed with the chlorine atom trans to the phosphine ligand (2.4609(13) Å) and the shortest with the chlorine atom trans to the η2-bonded acridinium heteroarene moiety (2.3422(11) Å). A similar asymmetry is seen in [RhCl(C2H4)(PiPr3)]2 which features Rh–Cl distances of 2.376(1) and 2.441(1) Å for the chlorine atom trans to the ethylene and phosphine ligand, respectively.22 These structural results support the description of [3]+ as a square planar rhodium(I) complex featuring an η2-heteroarene unit as one of the four ligands. A Pipek–Mezey orbital analysis suggests that this interaction benefits both from donation from the heteroarene π system, with significant back donation from the metal to the carbenium center (Fig. 3). An NBO calculation, carried out with full atomic disconnection, offers a consistent picture, with the forward-donation from the C10 carbon center to the rhodium center and the backdonation from the rhodium to the carbenium center, respectively associated with second-order energy corrections of 29.9 and 99.1 kcal mol−1. These NBO results show that the backdonation dominates the η2-heteroarene–rhodium interaction present in this complex. The large back donation observed in this complex is typical of η2-arene complexes involving electron-rich transition metals.23 Such bonding features are also seen in simple olefin complexes such as Zeise's salt for which a recent computational investigation indicates the dominant influence of the backbonding interaction.24 Finally, it is worth mentioning that these interactions are asymmetrical, with the donation originating mostly from the C10 carbon atom while the back donation occurs between the rhodium atom and the Ccarb atom. Such asymmetrical π-bonding and backbonding interactions are typical of polar olefin or olefin-like fragments such as borataalkenes.25
 | ||
| Scheme 2 Synthesis of the rhodium complex [3][OTf]. | ||
 | ||
| Fig. 3 Top: crystal structure of the dimeric form of [3][OTf] with ellipsoid draw at the 50% probability level. Hydrogen atoms and triflate anions are omitted for clarity. Bottom: truncated view of [3]+ showing the Pipek–Mezey orbitals defining the Rh–acridinium interactions (orbital parentage: PMO1: 17.0%, Ccarb 12.3%, C10 37.7%; PMO2: Rh 63.7%, Ccarb 20.5%). The corresponding NBOs associated to the Rh–acridinium donor–acceptor bonding interactions are also shown. | ||
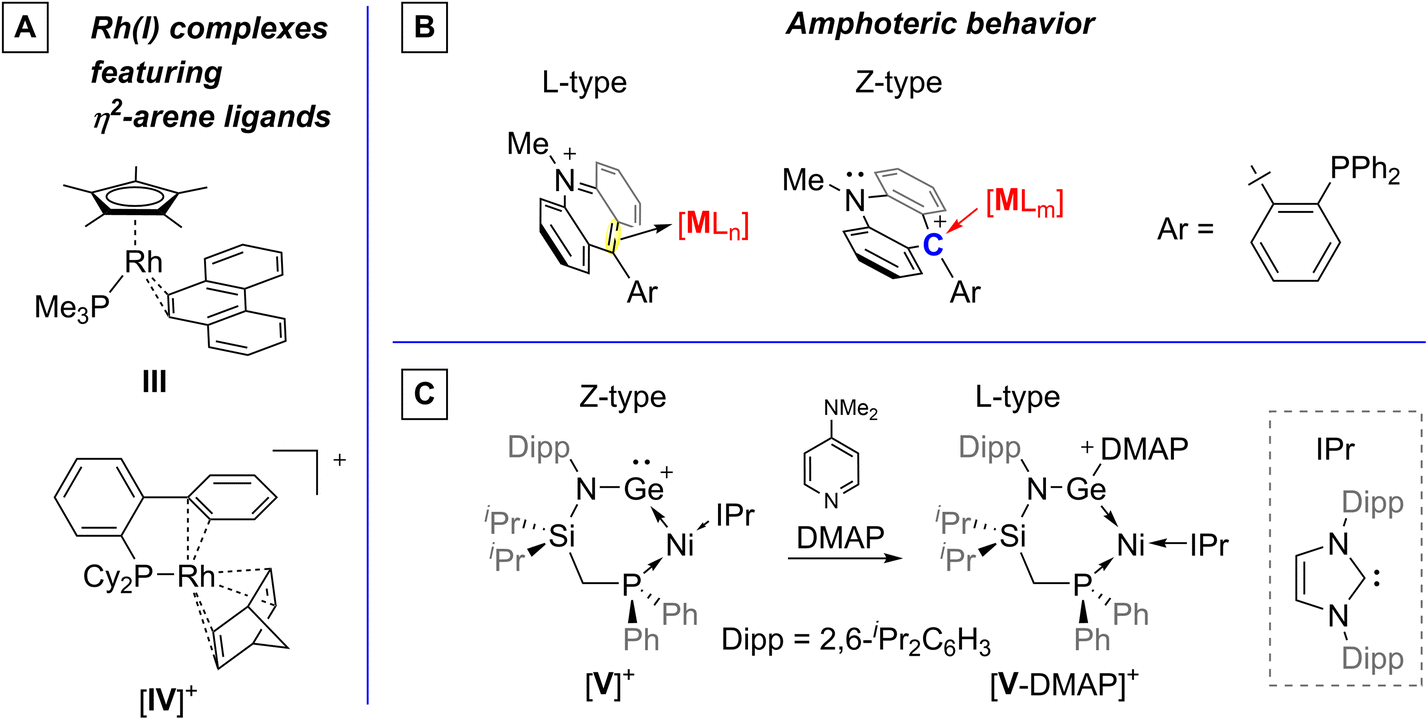 | ||
| Fig. 4 (A) Examples of complexes with η2-arene ligands.19,20 (B) Amphoteric behavior of the acridinium unit of [Lacr]+. (C) A recently documented example of ligand amphoteric behavior triggered by a ligand-centered coordination event.7 | ||
The results discussed above indicate that [Lacr]+ is amphoteric (Fig. 4) in that it is able to switch its behavior from Z-type in [2]+ to L-type in [3]+, simply in response to changes in the coordination environment of the metal center. Moreover, our results show that this behavior is reversible, illustrating the versatility of this system. While ligand-type switching has been previously observed, it has, to our knowledge, always been associated with a change in the composition of the ligand.6 A close example of such behavior arises in the chemistry of the phosphinogermylene complex [V]+ which switches from Z-type to L-type (Fig. 4).7 This switch occurs upon coordination of a base to the Z-type germanium ligand, altering its composition and electronic profile. The switching behavior documented here for [Lacr]+ is thus different since it does not involve any changes in the composition of the ligand.
To conclude this study, we have also explored how changes in the ligand type would affect the reactivity of the metal center. A simple test using COD shows that [3]+ reacts almost immediately to form the corresponding COD complex [1]+ in CH2Cl2 (Scheme 3). To test the reactivity of [2]+ toward COD, we carried out an analogous experiment in which a solution of [3]+ in CH2Cl2 was first combined with MeCN (15% vol.) to ensure formation of [2]+ and subsequently COD, which left [2]+ unchanged. This lack of reactivity illustrates the ability of [Lacr]+ to alter the coordination environment and the reactivity of the metal center when switching into a Z-type ligand. The differing reactivity of [2]+ and [3]+ was further tested by investigating the behavior of these two compounds towards O2 and H2O, which reacted with [3]+ immediately to give the known phosphine oxide [(o-Ph2P(O)(C6H4)Acr)]+ in the case of O2.26 As in the reaction involving COD, [2]+, generated in situ by the addition of MeCN, shows much greater robustness and remains unaffected when evaluated within 1 hour after exposure.
 | ||
| Scheme 3 Reactions of [2][OTf] and [3][OTf] with COD. | ||
Conclusions
In summary, the interaction of the acridinium–phosphine ligand [Lacr]+ with [Rh(COE)2Cl]2 leads to different complexes, the nature of which is influenced by the conditions of the reaction. When carried out in MeCN, this reaction affords a chloride-bridged octahedral 18-electron complex ([2]+) featuring two acetonitrile ligands as well as a Rh→Ccarb donor–acceptor motif indicating that the acridinium moiety acts as Z-type ligand. The same reaction in CH2Cl2 follows a different course and affords a square planar complex ([3]+) in which the acridinium functionality behaves as a η2-heteroarene L-type ligand, providing two electrons to the 16-electron square planar metal center. Isolation and characterization of these two complexes highlight the amphoteric ligand behavior of the acridinium unit which can switch from L-type to Z-type based on the coordination of auxiliary ligands to the metal center. These results document a unique features of aromatic carbenium functionalities which can assume a different role in response to the electronic environment of the transition metal center. We are currently aiming to exploit these attributes in the context of catalysis.Data availability
The data supporting this article have been included as part of the ESI.† CCDC 4 ([1][OTf]), 2421485 ([2][OTf]) and 2421486 ([3][OTf]) contains the supplementary crystallographic data for this paper.Author contributions
Elishua D. Litle: conceptualization, data curation, investigation, methodology, writing – original draft. Shantabh Bedajna: data curation, validation, writing – original draft. François P. Gabbaï: conceptualization, data curation, funding acquisition, methodology, visualization, writing – original draft, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge support from the National Science Foundation (CHE-2154972) and the Welch Foundation (A-1423). All calculations were conducted with the advanced computing resources provided by Texas A&M High Performance Research Computing.Notes and references
- M. L. H. Green, J. Organomet. Chem., 1995, 500, 127–148 CrossRef CAS.
- (a) D. F. Shriver, Acc. Chem. Res., 1970, 3, 231–238 CrossRef CAS; (b) F.-G. Fontaine, J. Boudreau and M.-H. Thibault, Eur. J. Inorg. Chem., 2008, 2008, 5439–5454 CrossRef; (c) J. Bauer, H. Braunschweig and R. D. Dewhurst, Chem. Rev., 2012, 112, 4329–4346 CrossRef CAS PubMed; (d) R. J. Eisenhart, L. J. Clouston and C. C. Lu, Acc. Chem. Res., 2015, 48, 2885–2894 CrossRef CAS PubMed; (e) J. S. Jones and F. P. Gabbaï, Acc. Chem. Res., 2016, 49, 857–867 CrossRef CAS PubMed; (f) R. C. Cammarota, L. J. Clouston and C. C. Lu, Coord. Chem. Rev., 2017, 334, 100–111 CrossRef CAS; (g) J. Takaya, Chem. Sci., 2021, 12, 1964–1981 RSC; (h) M. T. Whited, Dalton Trans., 2021, 50, 16443–16450 RSC.
- A. Amgoune and D. Bourissou, Chem. Commun., 2011, 47, 859–871 RSC.
- (a) E. L. Muetterties, J. R. Bleeke, E. J. Wucherer and T. Albright, Chem. Rev., 1982, 82, 499–525 CrossRef CAS; (b) J. R. Sweet and W. A. G. Graham, J. Am. Chem. Soc., 1983, 105, 305–306 CrossRef CAS; (c) S. M. Hubig, S. V. Lindeman and J. K. Kochi, Coord. Chem. Rev., 2000, 200–202, 831–873 CrossRef CAS; (d) G. Pampaloni, Coord. Chem. Rev., 2010, 254, 402–419 CrossRef CAS.
- (a) M. Devillard, G. Bouhadir and D. Bourissou, Angew. Chem., Int. Ed., 2015, 54, 730–732 CrossRef CAS PubMed; (b) G. Bouhadir and D. Bourissou, Chem. Soc. Rev., 2016, 45, 1065–1079 RSC; (c) M. J. Chalkley, M. W. Drover and J. C. Peters, Chem. Rev., 2020, 120, 5582–5636 CrossRef CAS.
- (a) I.-S. Ke, J. S. Jones and F. P. Gabbaï, Angew. Chem., Int. Ed., 2014, 53, 2633–2637 CrossRef CAS; (b) J. S. Jones, C. R. Wade and F. P. Gabbaï, Angew. Chem., Int. Ed., 2014, 53, 8876–8879 CrossRef CAS; (c) J. Schwarzmann, T. Eskelinen, S. Reith, J. Ramler, A. J. Karttunen, J. Poater and C. Lichtenberg, Angew. Chem., Int. Ed., 2024, 63, e202410291 CrossRef CAS; (d) N. Ansmann, M. Kerscher and L. Greb, Angew. Chem., Int. Ed., 2025, 64, e202417581 CrossRef CAS PubMed.
- A. Schulz, T. L. Kalkuhl, P. M. Keil and T. J. Hadlington, Angew. Chem., Int. Ed., 2023, 62, e202305996 CrossRef CAS PubMed.
- (a) J. Petuškova, H. Bruns and M. Alcarazo, Angew. Chem., Int. Ed., 2011, 50, 3799–3802 CrossRef; (b) Y. Canac, C. Maaliki, I. Abdellah and R. Chauvin, New J. Chem., 2012, 36, 17–27 RSC; (c) C. Maaliki, C. Lepetit, Y. Canac, C. Bijani, C. Duhayon and R. Chauvin, Chem.–Eur. J., 2012, 18, 7705–7714 CrossRef CAS; (d) M. Alcarazo, Chem.–Eur. J., 2014, 20, 7868–7877 CrossRef CAS; (e) M. Alcarazo, Acc. Chem. Res., 2016, 49, 1797–1805 CrossRef CAS PubMed; (f) K. Schwedtmann, G. Zanoni and J. J. Weigand, Chem.–Asian J., 2018, 13, 1388–1405 CrossRef CAS.
- (a) L. C. Wilkins, Y. Kim, E. D. Litle and F. P. Gabbaï, Angew. Chem., Int. Ed., 2019, 58, 18266–18270 CrossRef CAS; (b) J. Zhou, E. D. Litle and F. P. Gabbaï, Chem. Commun., 2021, 57, 10154–10157 RSC; (c) G. Park, M. Karimi, W.-C. Liu and F. P. Gabbaï, Angew. Chem., Int. Ed., 2022, 61, e202206265 CrossRef CAS; (d) W.-C. Liu and F. P. Gabbaï, Chem. Sci., 2023, 14, 277–283 RSC; (e) E. D. Litle and F. P. Gabbaï, Chem. Commun., 2023, 59, 603–606 RSC.
- E. D. Litle, L. C. Wilkins and F. P. Gabbaï, Chem. Sci., 2021, 12, 3929–3936 RSC.
- E. D. Litle and F. P. Gabbaï, Angew. Chem., Int. Ed., 2022, 61, e202201841 CrossRef CAS PubMed.
- (a) H. Goodman, L. Mei and T. L. Gianetti, Front. Chem., 2019, 7, 365 CrossRef CAS PubMed; (b) L. Mei, J. M. Veleta, J. Bloch, H. J. Goodman, D. Pierce-Navarro, A. Villalobos and T. L. Gianetti, Dalton Trans., 2020, 49, 16095–16105 RSC.
- K. T. Horak, D. G. VanderVelde and T. Agapie, Organometallics, 2015, 34, 4753–4765 CrossRef CAS.
- E. Tomás-Mendivil, R. García-Álvarez, C. Vidal, P. Crochet and V. Cadierno, ACS Catal., 2014, 4, 1901–1910 CrossRef.
- G. G. Johnston and M. C. Baird, Organometallics, 1989, 8, 1894–1903 CrossRef CAS.
- (a) T.-P. Lin, C. R. Wade, L. M. Pérez and F. P. Gabbaï, Angew. Chem., Int. Ed., 2010, 49, 6357–6360 CrossRef CAS PubMed; (b) B. R. Barnett, C. E. Moore, P. Chandrasekaran, S. Sproules, A. L. Rheingold, S. DeBeer and J. S. Figueroa, Chem. Sci., 2015, 6, 7169–7178 RSC.
- S. Yogendra, T. Weyhermüller, A. W. Hahn and S. DeBeer, Inorg. Chem., 2019, 58, 9358–9367 CrossRef CAS.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- (a) L. Cronin, C. L. Higgitt and R. N. Perutz, Organometallics, 2000, 19, 672–683 CrossRef CAS; (b) A. Woolf, A. B. Chaplin, J. E. McGrady, M. A. M. Alibadi, N. Rees, S. Draper, F. Murphy and A. S. Weller, Eur. J. Inorg. Chem., 2011, 2011, 1614–1625 CrossRef.
- W. D. Jones and L. Dong, J. Am. Chem. Soc., 1989, 111, 8722–8723 CrossRef CAS.
- A. R. O'Connor, W. Kaminsky, D. M. Heinekey and K. I. Goldberg, Organometallics, 2011, 30, 2105–2116 CrossRef.
- A. Angelini, M. Aresta, A. Dibenedetto, C. Pastore, E. Quaranta, M. R. Chierotti, R. Gobetto, I. Pápai, C. Graiff and A. Tiripicchio, Dalton Trans., 2009, 7924–7933 RSC.
- L. A. Essex, A. McSkimming, N. B. Thompson, M. L. Kelty, E. A. Hill and W. H. Harman, Organometallics, 2020, 39, 2545–2552 CrossRef CAS.
- T. Yang, Z. Li, X.-B. Wang and G.-L. Hou, ChemPhysChem, 2023, 24, e202200835 Search PubMed.
- (a) Y. Yang, J. Jiang, H. Yu and J. Shi, Chem.–Eur. J., 2018, 24, 178–186 Search PubMed; (b) M. Eaton, Y. Zhang and S.-Y. Liu, Chem. Soc. Rev., 2024, 53, 1915–1935 RSC.
- W.-C. Liu, Y. Kim and F. P. Gabbaï, Chem.–Eur. J., 2021, 27, 6701–6705 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental and computational details and crystallographic data in cif format. CCDC 2421484–2421486. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc00965k |
| This journal is © The Royal Society of Chemistry 2025 |