
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWidespread false negatives in DNA-encoded library data: how linker effects impair machine learning-based lead prediction†
Alba L.
Montoya‡
 a,
Adam S.
Hogendorf‡
a,
Steven
Tingey
b,
Aadarsh
Kuberan
c,
Lik Hang
Yuen
a,
Herwig
Schüler
d and
Raphael M.
Franzini
*ae
a,
Adam S.
Hogendorf‡
a,
Steven
Tingey
b,
Aadarsh
Kuberan
c,
Lik Hang
Yuen
a,
Herwig
Schüler
d and
Raphael M.
Franzini
*ae
aDepartment of Medicinal Chemistry, College of Pharmacy, University of Utah, 30 S 2000 E, Salt Lake City, UT 84112, USA. E-mail: raphael.franzini@utah.edu
bWaterford School, 1480 E 9400 S, Sandy, UT 84093, USA
cWest High School, 241 N 300 W, Salt Lake City, UT 84103, USA
dCenter for Molecular Protein Science, Department of Chemistry, Lund University, Lund, 22100, Sweden
eHuntsman Cancer Institute, University of Utah, 2000 Circle of Hope, Salt Lake City, UT 84054, USA
First published on 9th May 2025
Abstract
DNA-encoded chemical libraries (DECLs) have become integral to early-stage drug discovery, yielding active compounds and extensive labeled datasets for machine learning (ML)-based prediction of bioactive molecules. However, the information content of DECL selection data remains scarcely explored. This study systematically investigates for the first time the prevalence of false negatives and the influence of the linker in DECL data. Using a focused DECL targeting the poly-(ADP-ribose) polymerases PARP1/2 and TNKS1/2 as a model system, we found that our DECL selections frequently miss active compounds, with numerous false negatives for each identified hit. The presence of the DNA-conjugation linker emerged as a factor contributing to the underdetection of active molecules. This bias toward false negatives compromises the predictive power of DECL data for prioritizing hits, anticipating target selectivity, and training ML models, as determined by analyzing the effects of undersampling and oversampling techniques in learning the PARP2 data. Conversely, the linker's presence in DECLs offers advantages, such as enabling the identification of target-selective protein engagers, even when the underlying molecules themselves may not be selective. These findings highlight the challenges and opportunities of DECL data, emphasizing the need for best practices in data handling and ML model development in drug discovery.
Introduction
The discovery of biologically active molecules is a central stage in small-molecule drug development. In recent years, DNA-encoded chemical libraries (DECLs) have become an integral part of early lead discovery efforts.1–4 Consisting of synthetic molecules conjugated to DNA sequences that encode their chemical identity, DECLs facilitate the rapid identification of protein binders through a straightforward affinity selection protocol. Routinely employed in pharmaceutical research, DECLs have yielded numerous bioactive molecules across diverse targets.5–11 Building upon this success, an exciting research direction integrates DECLs with computational methods to learn structure–activity relationships from selection data. Successful implementation of this workflow could transform drug discovery, by finding lead molecules at a fraction of the time and costs of conventional approaches. However, the reliability of machine-learning (ML) predictions of active molecules is contingent upon the quality of the input data.12 Despite a recent surge in reports of approaches for computational lead prediction from DECL data,13–23 notably little is known about the fidelity and limitations of DECL data.In this study, we systematically explore the information contained in DECL selections from a chemically focused DECL24 against a set of four poly-(ADP-ribose) polymerase (PARP) targets25 as a model system. Focused DECLs minimize issues related to library heterogeneity26 and undersampling27 and consistently provide enrichment fingerprints suitable for structure–activity analysis. Therefore, focused DECLs have become actively pursued as an alternative to large DECL platforms.24,28–31 Moreover, the selected PARP1/2 and TNKS1/2 targets are structurally and functionally closely related, and their medicinal chemistry is well established,32 with active compounds often inhibiting several of the isoforms.33 The homology among the catalytic domains of the four PARP enzymes34 is ideal for conducting comparative analyses of DECL enrichment patterns and structural features.
This study sheds light on both the potential and limitations of DECL data. While most tested hit molecules exhibited activity, the findings indicate that compounds with enriched sequence reads represent only a fraction of the active chemical space in the present DECL. The presence of the DNA-conjugation linker was identified as one factor contributing to this underdetection. However, the study also provided evidence that DECL data may provide valuable insights into how linker can endow otherwise unselective molecules with target-selectivity, which is relevant for multi-valent drug classes such as proteolysis targeting chimeras (PROTACs).35 Furthermore, we explored how inherent issues in DECL data influence ML-based lead prediction efforts. Overall, this study enhances our understanding of DECL data and offers insights for developing best practices in computational DECL analysis.
Results
Cross-examination of DNA-encoded chemical library data from different PARPs
We investigated the predictive value of DECL affinity selection data using as a model system a NAD+-mimicking DECL (NADEL24Fig. 1c) screened against the four human PARP enzymes PARP1/2 and TNKS1/2 (Fig. 1).24 Advantages of this model system are that NADEL as a focused 2-cycle DECL has a homogeneous composition, low truncation rates, and allows for over-sequencing following selections.36 Despite its modest size of 58![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 302 compounds, NADEL consistently provides chemically diverse hits for PARP enzymes suitable for recognizing molecular trends.21,24 The structural and functional relatedness of the four PARP enzymes,33,34 together with their well-established medicinal chemistry,32 sets an ideal basis for conducting comparative analyses of enrichment patterns and structural features.
302 compounds, NADEL consistently provides chemically diverse hits for PARP enzymes suitable for recognizing molecular trends.21,24 The structural and functional relatedness of the four PARP enzymes,33,34 together with their well-established medicinal chemistry,32 sets an ideal basis for conducting comparative analyses of enrichment patterns and structural features.
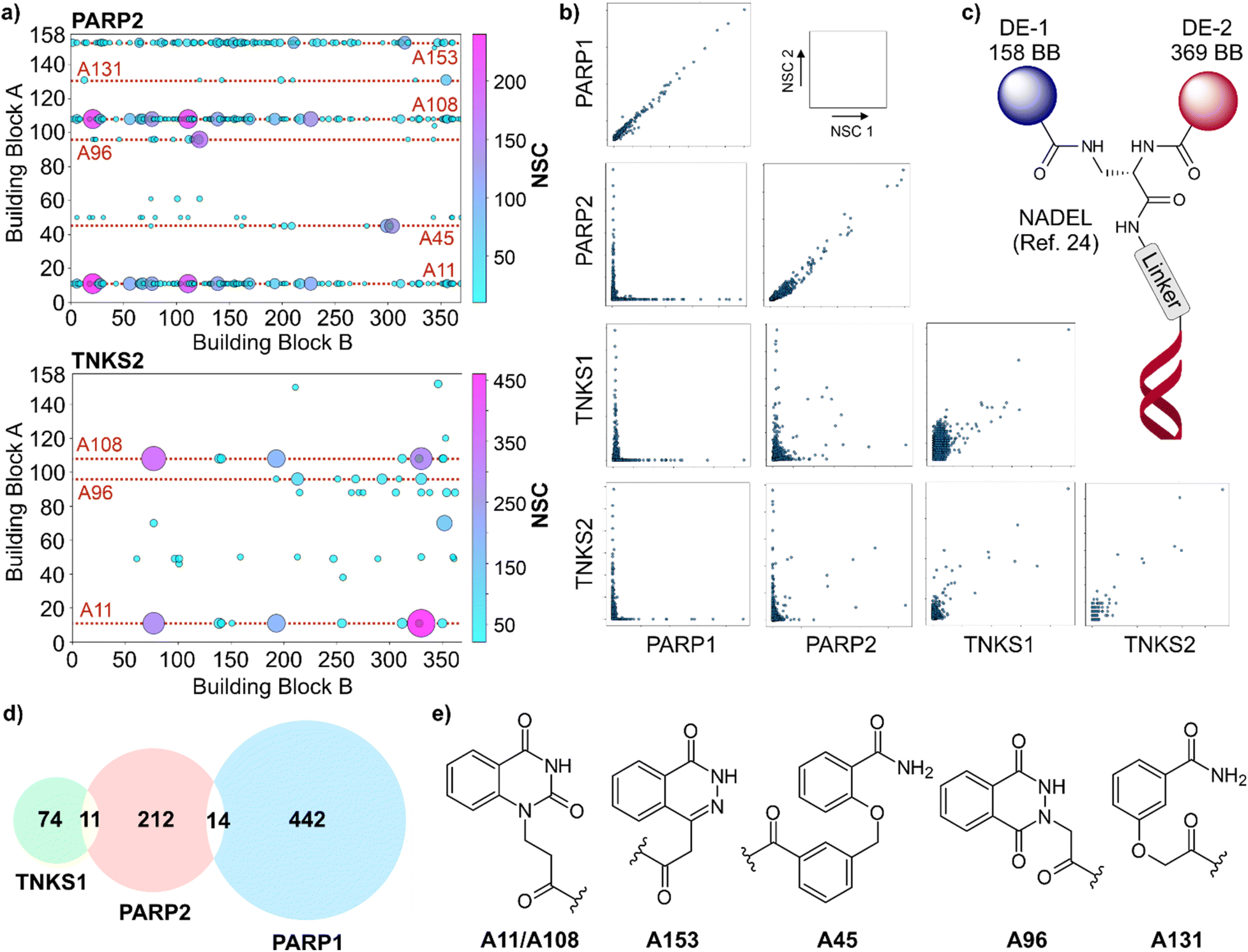 | ||
| Fig. 1 Summary of NADEL-selection results for poly-(ADP-ribose) polymerase (PARP) targets. (a) Two-dimensional scatter representation for NADEL selection results of PARP2 (threshold value = 10) and TNKS2 (threshold value = 20). (b) Correlation of NADEL selection results for PARP1/2 and TNSK1/2 enzymes and reproducibility of selections. (c) Structure of DECL used in this study. DE: diversity element. (d) Venn diagram of overlapping NADEL hits for different PARP enzymes. (e) Representative structures of building blocks at position A of identified NADEL hits for PARP enzymes.§ A11 and A108 refers to the same building block. (NSC: Normalized Sequence Counts). | ||
Affinity selections ofNADEL for the PARP enzymes were performed using a standard protocol37 and resulted in series of highly enriched and chemically diverse compounds. The NADEL library was used at a concentration of 10 nM, containing all 58302 compounds, which translates to 0.2 pM for individual conjugates. Interested readers can refer to the (ESI†) for technical details of the affinity selection procedures. Replicate experiments showed strong correlations, demonstrating reproducibility (Fig. 1b). However, one TNKS1 selection had a lower signal-to-background ratio, and the correlation was weaker for TNKS2 because of lower sequencing depth.
For PARP2, two series of enriched compounds containing the quinazolinedione fragment A11/A108 and the phthalazide A153 were the primary features with several other clusters of enriched structures present (Fig. 1a and e). TNKS2 afforded a pattern of discrete hit molecules based on different heterocycles with A11/A108-containing compounds having the highest enrichment values (Fig. 1a). Selection results for PARP1 and TNKS1 were reported previously.21,24
The overlap of hits for the four targets was compared to assess whether the DECL selections would result in target-specific enrichment patterns or mirror the homology of the enzymes (Fig. 1b). Hits for TNKS1 and TNKS2 showed considerable overlap reflecting the similarity of the targets and the challenge of identifying molecules that are selective to one of the two isoforms.38 In contrast, PARP2 hits remarkably differed from those for TNKS1/2, and there were almost no hits that were found simultaneously for PARP1 and any of the other PARPs (Fig. 1b). Not a single molecule was identified as a hit for PARP1, PARP2, and TNKS1 at the same time (Fig. 1d). This level of divergence contrasts the many medicinal chemistry efforts for these proteins, which established that it is challenging to identify target-specific inhibitors.32 While variable synthesis yields26 and undersampling27 are known to affect data reliability of DECLs, the controlled nature of NADEL makes such technical issues unlikely reasons for this result. Indeed, of 34 hit molecules that we have synthesized and tested for these four targets, 32 (94%) exhibited >50% inhibition of these targets at 10 μM (Table 2 in the ESI†).
Isolated hit compounds mask patterns of active molecules
The lack of correlation between hits across the four targets prompted us to interrogate whether the DECL data accurately reflects the PARP inhibitors, or if it is biased. To address this issue, we investigated isolated hits, focusing on compounds containing the building blocks A45 and A96. Selections for PARP1/2 and TNKS1 all identified A45-and A96-containing compounds as hits; however, only a few such hits were found, and these varied between the targets (Fig. 2a). | ||
| Fig. 2 Analysis of isolated hit compounds across PARP enzymes. (a) Selection results of NADEL for molecules with building blocks A45 or A96 at one position of NADEL and inhibition of PARP1/2 and TNKS1 of synthesized hits at single concentrations (c = 500 nM for A45, and c = 1000 nM for A96) A45–Ac and A96–Bz are control compounds in which the building block at the second position was replaced by a generic acyl group. (b) Chemical similarity of A45-containing compounds including the ethylenediamine linker to the clinically used PARP inhibitor Olaparib. The structures of the building blocks at the B-position are shown in Table 3 in the ESI.† n.d. not determined. | ||
The conventional interpretation of isolated hits is that they require synergistic interactions between fragments at both positions, with these A45/A96-containing compounds expected to be enzyme-specific inhibitors.36 To test this hypothesis, one hit molecule from each target was synthesized and evaluated for inhibitory activity in biochemical assays. Interestingly, all tested compounds exhibited similar levels of inhibition across targets, regardless of the selection they were identified from (Fig. 2a). While correlations between enrichment and activity are generally unreliable and threshold values are somewhat arbitrary, this finding clearly demonstrates that differences in sequence enrichment across targets do not necessarily correspond to true target selectivity.
Near-complete inhibition of PARP1/2 and TNKS1 was observed for A45-containing compounds at c = 500 nM, regardless of what target they were identified for. Similarly, A96-containing compounds exhibited comparable isoform inhibition patterns at c = 1000 nM, despite different enrichments in selections. Even molecules in which the B-building blocks were replaced by generic acetyl or benzoyl groups showed similar inhibitions as the hits.
These findings suggest that many A45-and A96-based compounds within NADEL are PARP inhibitors with their activity being largely independent of the building blocks at the B-position, at least at the tested concentrations. Such findings contradict the interpretation of synergistic binding and target selectivity one would typically infer from the DECL data and suggest the presence of a bias towards false negatives.
A noteworthy feature of A45-containing molecules with the adjoining linker is their structural similarity to the clinically used PARP inhibitor Olaparib (Fig. 2b). Nearly overlooking this chemotype within a small, focused DECL highlights the concern that DECL campaigns may miss many valuable lead opportunities.
False negatives are widespread in DECL selection results
The observation that active molecules appear to be missed in the NADEL selections results led us to investigate the reliability of the data further. We analyzed the inhibition of PARP2 by a total of 33 molecules, categorized based on DECL results. The grouping criteria encompassed molecules identified as PARP2 hits (category 1), those recognized as hits for other PARPs and an A-building block for a PARP2 hit (category 2), those recognized as hits for other PARPs without a shared A-building block for PARP2 hits (category 3), those containing an A-building block present in another PARP2 hit without being a hit for another PARP (category 4), and those unrelated to PARP2 or other PARPs (category 5; Fig. 3a). | ||
| Fig. 3 Analysis of the predictiveness of DECL data on the activity of resynthesized hit compounds. (a) Comparison of PARP2 inhibition among molecules identified in the screen for PARP2, hits for other PARPs or different enzymes, molecules with privileged building blocks, and unrelated molecules. The inhibitory potencies of PARP2 hits are comparable to hits for PARP1, TNKS1, and TNKS2, with a significant contribution observed from the A-building blocks. This finding strongly suggests a high prevalence of false negatives in DECL data. Categories: category 1: PARP2 hits; category 2: not PARP2 hit but A-BB present in another PARP2 hit and hit for PARP1 or TNKS1/2; category 3: unrelated to PARP2 hit but hit for PARP1 or TNKS1/2; category 4: A-BB present in another PARP2 hit but not a hit for PARP1/2 or TNKS1/2; category 5: unenriched for any of the tested enzymes. Structures of compounds and values of percent inhibition of PARP2 are provided in Table 3 in the ESI.† (b) Evaluation of inhibition and selectivity profiles for representative PARP2 hits and hybrid molecules composed of building blocks that confer both potency and selectivity. Values are given in Table 4 in the ESI.† | ||
All but one PARP2 hit (category 1) inhibited PARP2 with >50% at c = 10 μM, while PARP2-unrelated molecules (category 5) exhibited weak activity. Intriguingly, despite not being enriched in the PARP2 selection, molecules in categories 2–4 uniformly inhibited PARP2 at this concentration. Most molecules in categories 1–4 also inhibited PARP2 at c = 500 nM. Molecules with A-building blocks found for only one of the targets, even if it was PARP2, were most susceptible to concentration-dependent loss of activity. Interestingly, the A-building block emerged as a more predictive factor for PARP2 inhibition by molecules than whether a molecule was a PARP2 hit or not.
Collectively, this data indicates a high prevalence of false negatives. While the data lacks the statistical power for quantitative predictions, it appears likely that for every identified PARP2 hit, there exist multiple PARP2 inhibitors that were not identified as hits during the DECL selection. In larger DECLs, reduced synthetic homogeneity and higher selection stringency may worsen the under-identification of active molecules even further.26,27
Potent and selective PARP2 inhibitors were identified
Despite the mentioned challenges, the screening process yielded numerous inhibitors for PARP2, exemplified by A45/B299 with an IC50 = 12 nM in a biochemical assay (Fig. 2 in the ESI†). Notably, the PARP2 data revealed an unexpected structural feature in the form of series of hydrophobic building blocks, including bulky aliphatic ones (e.g., B21), at the B-position. Such structures were absent in selections for all other PARP enzymes as well as in reported PARP2 inhibitors. Indeed, A108/B21 and A108/B111 exhibited favourable selectivity profiles while being potent (Fig. 3b and Table 4 in the ESI†). Subsequent synthesis and testing of hybrid molecules, combining these selectivity-affording fragments B21 and B108 with the A45 fragment resulted in several compounds displaying preference for PARP2 albeit with lower potency compared to A45-En-B299, indicating the necessity for precise geometric positioning to simultaneously achieve potency and selectivity (Table 5 in the ESI†). Nevertheless, these newly identified fragments present potential entry points for developing chemical probes targeting PARP2 in the future.Linkers endow unselective compounds with target selectivity
The finding that DECL selections miss many active molecules raises the question for the underlying cause of this effect. We hypothesized that the structural constraints imposed by the DNA-attachment linker might hinder target engagement of molecules that would otherwise exhibit activity. To test the impact of the linker, we synthesized hexa–thymidine conjugates of A45/B299 (PARP2 hit) and A45/B145 (TNKS1 hit), using the same linker employed in NADEL and compared inhibition of PARP1, PARP2, and TNKS1 in enzyme-activity assays to that of the molecules with ethylenediamine linkers. In accordance with our hypothesis, the presence of attached oligonucleotides diminished the activity of the molecules, and this effect was more pronounced for off-target protein-ligand combinations than for proteins to which the molecule was identified as a hit (Fig. 4). These outcomes suggest that the linker contributes to the discrepancy between DECL results and off-DNA activities, as well as the widespread false negatives in DECL data, although other factors likely play a role.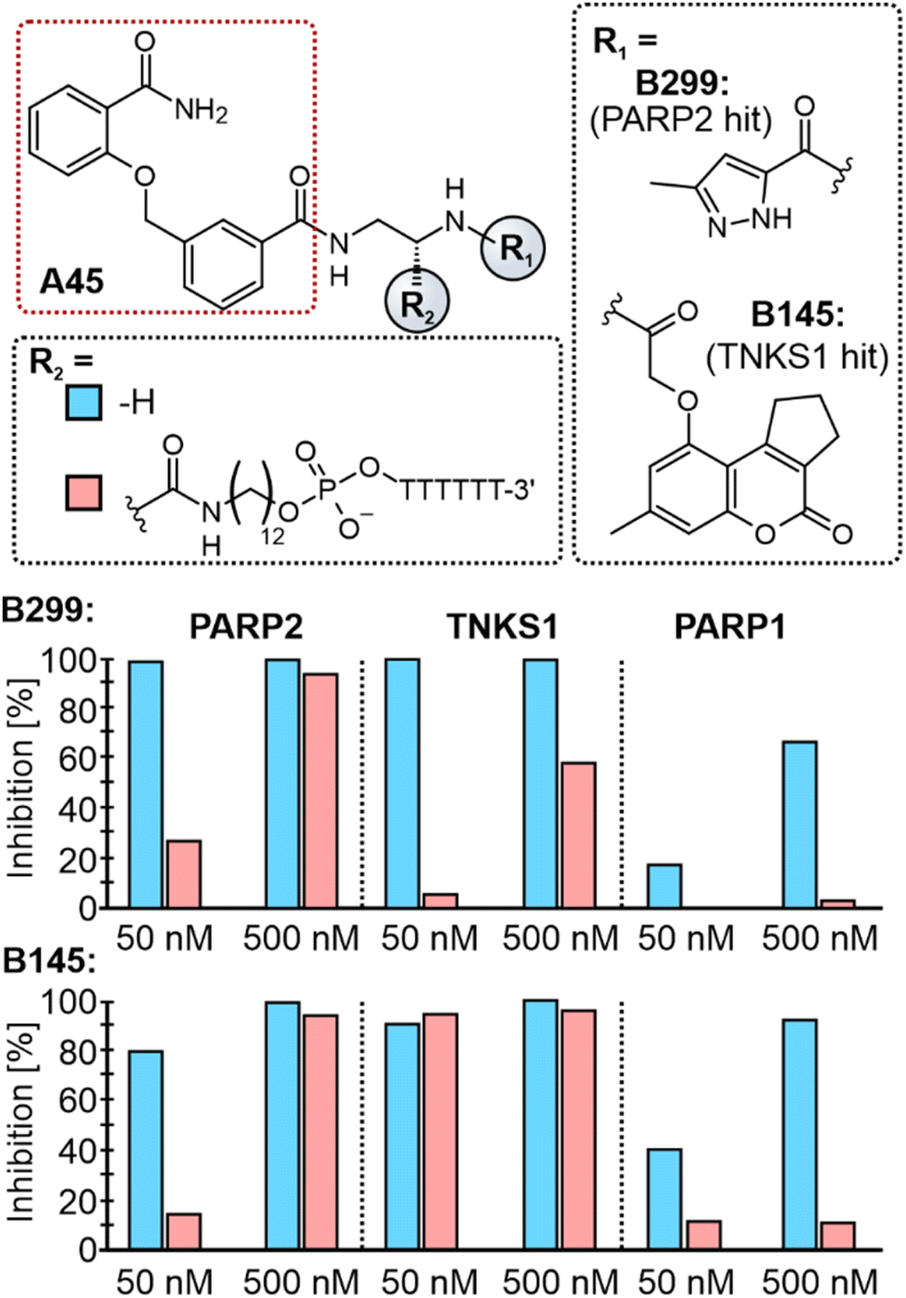 | ||
| Fig. 4 Influence of DNA-attachment linker on inhibitory potency of PARP hits. Structures of tested molecules are shown on top. Bar plots indicate inhibition of PARP2, TNKS1, and PARP1 for two pairs of tested molecules at c = 50 nM and 500 nM. | ||
While the utility of DECL data to predict target selectivity of inhibitors without a linker may be limited, DECL data might guide the design of molecules that retain a linker component. This capability may be valuable for the discovery of protein-engaging molecules such as PROTACs,35 where DECLs already play a significant role.39–41 To test this possibility, we synthesized two sets of five PROTAC-like molecules each derived from the PARP2 hit A45/B299 and the TNKS1 hit A45/B145. These molecules featured a cereblon binding pomalidomide fragment conjugated via representative linkers to the respective PARP inhibitors (Fig. 5). In biochemical assays, the A45/B299-based PROTAC-like molecules showed a preference for inhibiting PARP2 over TNKS1, whereas the A45/B145-based molecules were more potent for TNKS1. Although the structure of the linker affects selectivity and potency, the trend is mostly determined on the building blocks and the presence and absence of a linker. While these findings align with the DECL results and suggest accurate prediction of PROTAC-like molecule selectivity, it is difficult to parse out the individual contributions of the building blocks, linkers, and DNA. No enzyme degradation was observed in cell-based experiments (Fig. 3 in the ESI†), likely because of limited cell permeability or suboptimal positioning of the E3 ligase relative to the target. Nevertheless, the results underscore the potential of DECL data to guide the development of molecules that include a linker component.
 | ||
| Fig. 5 PROTAC-like molecules based on PARP2 and TNKS1 hits A45/B299 and A45/B145. Increased selectivity can be observed when comparing the putative degraders to their parent molecules, which indicates a linker effect. The selectivity patterns are similar to that of DNA-conjugates. The structures can be found in Table 6 in the ESI.† %I: percent inhibition of enzyme activity at c = 50 nM for 1a–5a and c = 10 nM for 1b–5b. | ||
Data bias and processing affect prediction of lead compounds through machine learning of DNA-encoded library data
These studies indicate that DECL data provides a potentially misleading view of molecule/protein interactions, with a bias toward false negatives. Such irregularities in DECL data may negatively affect ML models.36 Therefore, using this model system, we wanted to explore how limitations of DECL data influence the predictiveness of such models.For this study, we used logistic regression (LR), a binary classification method commonly used because of its interpretability, ability to output class probabilities, and relative resistance to overfitting. NADEL compounds were classified as PARP2 hits or non-hits and encoded as extended-connectivity fingerprints. In a leave-one-out cross-validation analysis of the LR model, many hits were misclassified as inactive, especially isolated hits such as A45-or A96-compounds (Fig. 6a). This outcome underscores the limited predictiveness of the DECL-LR model and highlights the challenge that ML models may inadvertently discard valuable chemotypes.
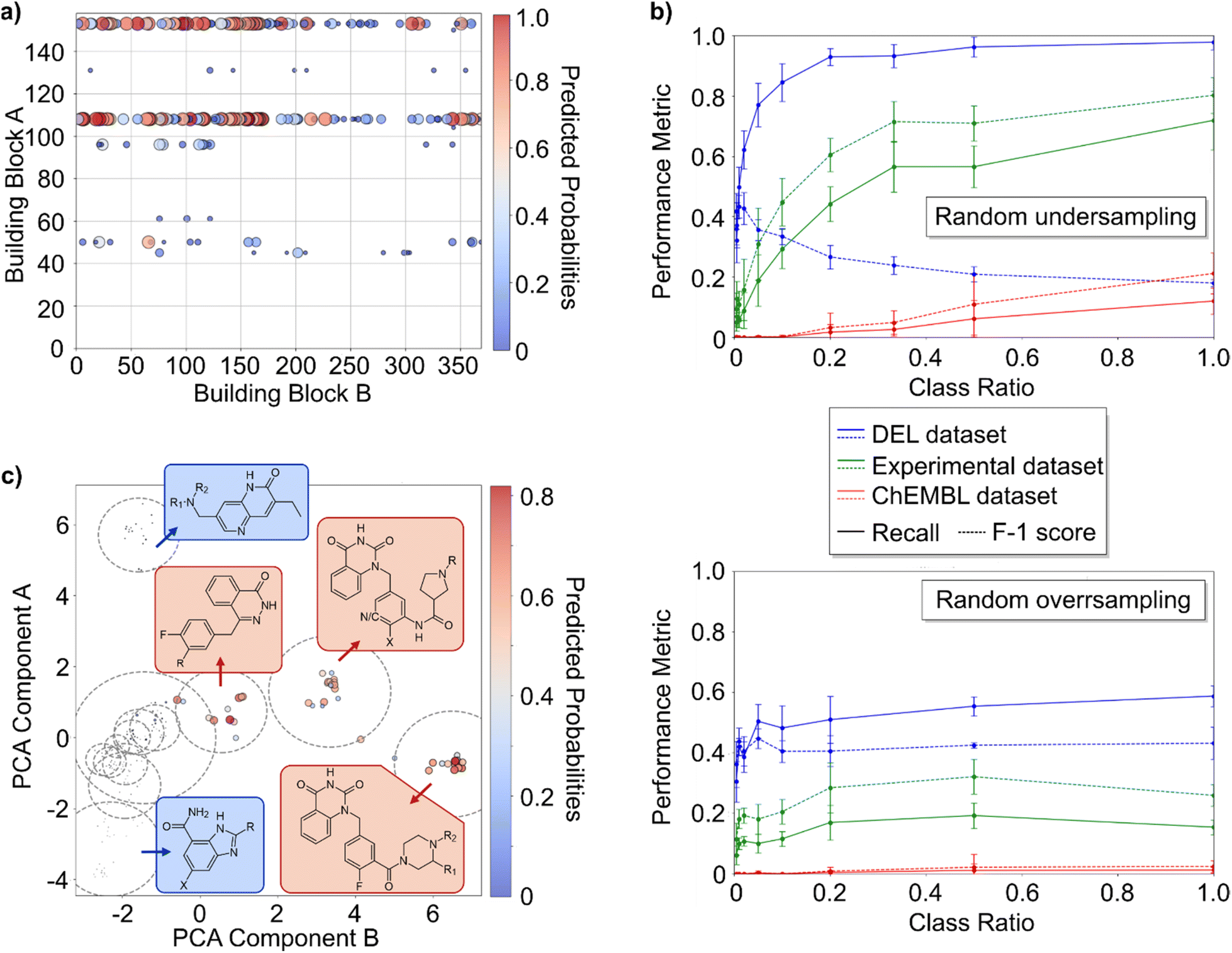 | ||
| Fig. 6 Analysis of how limitations of DECL data affect activity prediction by machine learning (ML) models. (a) Prediction of hits by logistic regression (LR) model without class balancing. Hits of PARP2 selection with sequence count indicated by circle size (threshold: NSC > 10). The colour predicts the predicted probability from a leave-one-out logarithmic regression analysis. A11 compounds were removed to avoid biasing of the model. For the corresponding selection data see Fig. 1a (b) effect of random undersampling of the majority class (upper graph) and random oversampling of the minority class (lower figure) in the learning set on recall (solid) and F1-score (dashed) of the internal validation set (blue), validation set of experimentally validated NADEL compounds (green), and ChEMBL validation set (red). Each datapoint is the average of five replicates of logistic regression analyses. (c) PARP2 inhibitors from ChEMBL databased are clustered by k-means clustering (14 clusters) displayed on principal component analysis (PCA) coordinates with marker size and colour scale indicating the predicted probability of a PARP2 inhibitor in the ChEMBL database to be a PARP2 inhibitor according to the logistic regression model balanced by random undersampling. | ||
Several factors could contribute to the inadequate ML performance. Besides bias, class imbalance, which means that there are many more non-hits than hits, is a recognized challenge with DECL data.23 To test for the relative importance of the two effects, we investigated the effect of class balancing by either removing non-hits (undersampling) or amplifying the number hits (oversampling). Three validation datasets were used for testing the DECL-LR model: (1) the test set of library compounds (internal validation set; all NADEL compounds with 80/20 training-test split), (2) experimentally validated compounds classified according to PARP2 inhibition (experimental validation set; 41 molecules), and (3) 228 PARP2 inhibitors from the ChEMBL database (IC50 or Kd < 500 nM) combined with 1000 random inactive ChEMBL compounds (ChEMBL validation set). Datasets are provided as ESI.†
The effects of undersampling and oversampling on the DECL-LR model were distinct (Fig. 6b). Undersampling greatly increased recall, which specifies the model's ability to identify actives, for all three datasets. For the internal dataset, higher recall came with lower F1 scores, which balances recognizing actives with false positives, likely because the model correctly identified actives that were not DECL hits. Importantly, the recall and F1 scores increase substantially with undersampling for the experimental and ChEMBL datasets. Balancing yielded near-perfect predictions for the experimental dataset and predicted ∼20% of the actives ChEMBL with no misclassification of inactives. This performance is impressive for a DECL that covers only a fraction of the PARP inhibitor chemical space. The LR model's performance aligns with what one might expect from a medicinal chemist analyzing the same data, which is to recognize recurring patterns but ultimately constrained by the biases and limitations inherent in the dataset.
In contrast, the effect of oversampling either by random oversampling (Fig. 6b) or SMOTEN42 (Fig. 5 in the ESI†) on the performance metrics was low. Therefore, correcting for bias rather than imbalance appears to improve the performance for undersampling.
To ensure the observed effects were not specific to the LR model, we repeated the analysis using alternative ML approaches, including random forest, support vector machine, multilayer perceptron, naive Bayes, elastic net, and histogram gradient tree. Similar or worse recall values and F1-scores were observed across these models relative to LR (Fig. 4 in the ESI†). These findings confirm that the observed effects stem from limitations of the DECL data rather than the chosen ML model.
While the performance metrics for data balanced by undersampling suggest that the LR model learned the PARP2 data, it is equally possible that it memorized structures of specific building blocks. Clustering the ChEMBL compounds revealed that all predicted actives belonged to three families of PARP2 inhibitors, each containing features resembling the recurrent building blocks A108 and A153 (Fig. 6c). This result provides strong evidence for memorization of these building blocks rather than ability to generalize the learning set to larger datasets. This outcome can be explained by eliminating non-hits that contain these building blocks during undersampling, which leads the model to associate such fragments exclusively with hits (Fig. 9, ESI†). Class balancing is a standard processing step in ML activities, and because of the combinatorial nature of DECLs undersampling biases the model toward classifying molecules with related substructures as active. Furthermore, the study shows that good performance metrics are no proof that an ML model can generalize DECL data.
Discussions
This study provides valuable insights into the strengths and limitations of DECLs for drug discovery, using PARP enzymes as model targets. The findings deepen the understanding of the chemical information embedded in DECL data, particularly highlighting the prevalence of false negatives and the impact of linker presence. These findings also underscore constraints regarding DECL data for use as learning data for ML-based lead prediction.36The DECL selections for PARP2 and TNKS2 yielded nanomolar hits. Some of the nanomolar PARP2 inhibitors show selectivity over related enzymes. Notably, certain fragments appear to confer selectivity for PARP2 over related targets, offering a foundation for developing selective chemical probes or drug leads.
This study assessed the activity of non-hits in DECLs for the first time, uncovering a concerningly high frequency of false negatives. Numerous compounds not enriched in the DECL selection were found to potently inhibit PARP2 indicating that many actives may be overlooked. The near-miss of Olaparib-related A45-containing compounds (Fig. 2b) exemplifies this issue. Given the experimental setting, incomplete synthesis or undersampling cannot account for the frequency of false negatives. Instead, experimental results confirmed linker positioning as one contributor to false negatives, because the structural constraints of DNA-conjugation linkers may hinder target engagement. Other factors besides the linker likely contribute to the discrepancy between selection data and experimental validation, and it is noteworthy that the experimental conditions differ substantially between the selection protocol and the inhibition assays used for validation.
These findings have important implications for interpreting DECL data. They challenge the assumption that isolated hits or singletons necessarily indicate synergistic interactions between BBs. Instead, isolated hits may serve as indicators of otherwise overlooked clusters of active molecules. Emphasizing feature-based structure–activity relationships during hit triaging can exacerbate this issue, and isolated hits hold great value for validation efforts, provided that the sampling depth is sufficient to distinguish them from background noise.36 The results also caution that denoising algorithms may inadvertently discard structurally significant compounds. Importantly, on-DNA methods often used for early hit validation43 may also be prone to linker-related false negatives.
The study highlights challenges in using DECL data for predicting target selectivity. Although some DECL hits appeared selective for individual PARPs, validation experiments showed that most of the tested molecules lacked selectivity. While there are reports of successful DECL-based selectivity predictions,44 systematic studies are needed to establish the general applicability of such approaches. However, the results also suggests that DECL data predicts selectivity more effectively when a linker is present, highlighting the potential of DECLs to access selectivity for chimeric molecules such as PROTACs.
While integrating DECL data with ML holds considerable promise for accelerating drug discovery, our findings suggest that several inherent characteristics of DECL data complicate the development of predictive models. Beyond its noisiness, DECL data is often biased toward false negatives, exhibits severe class imbalance (there are less hits than non-hits), is heteroscedastic (error is unequally distributed), and is multicollinear (features are correlated because of the combinatorial structure of DECLs). These limitations can significantly impact the ability of supervised ML models to generalize from DECL data to a broad chemical space. Furthermore, our analysis indicates that successful predictions of active molecules should not automatically be taken as proof that an ML model has genuinely learned meaningful relationships within the data, as such predictions could result from inherent data biases or processing artifacts.
Identifying the linker as a source of false negatives in DECL selections raises the question of how to mitigate this issue. One option is using DECLs where compounds are released from DNA, which, while incompatible with standard affinity selections, could work in phenotypic assays.45 Smaller libraries in such setups may yield results comparable to larger DECLs by reducing false negatives. Another approach is attaching compounds in multiple orientations, such as through late-stage functionalization.46 Alternatively, using a panel of DECLs with varied geometries should ensure finding relevant chemical space even if it is hidden for certain libraries. Along this line, cross-dataset learning could help refine ML models by integrating data from different DECLs. Additionally, clustering and aligning hit compounds may provide an alternative approach, mitigating uncertainties regarding non-hits in the dataset.21
While the study reinforces that linkers impact DECL results, the atomic-level contributions remain unclear. This is a case study, and computational and structural studies are needed to clarify their effects on hit activity.and further research is needed to assess the universality of its findings. The branched structure of NADEL, where two fragments engage deep binding pockets on PARPs, may impose unique constraints that differ from other target proteins and DECL designs. Additionally, the relatively small size of NADEL leaves opens the possibility that larger, more diverse DECL platforms might mitigate some of the limitations we observed in ML model building. However, making DECLs larger also introduces additional noise because of synthetic and sampling challenges. Similar considerations apply to hit triaging. Whether to follow up on singletons is debated,36 but our study supports their inclusion, showing they can indicate families of active compounds. If linker effects drive widespread false negatives, pursuing singletons should also be valuable for large DECLs. However, larger DECLs may also have higher false positive rates, so practitioners must balance expanding hit space with the risk of artifacts from library limitations. Therefore, further studies are essential to establish best practices for managing DECL data and to develop ML methods that fully harness the potential of DECLs in lead discovery.
Conclusions
In conclusion, this study highlights both the potential and challenges of using DECLs in drug discovery. While we identified potent PARP2 inhibitors, our findings expose significant issues, including a high rate of false negatives likely caused by DNA linker constraints. These limitations raise concerns about DECL selections excluding valuable chemical series and hinder the reliability of DECL data for ML-based bioactivity predictions. This case study emphasizes the need for further optimization of DECL methodologies and data processing to enhance ML-driven lead discovery.Data availability
Detailed experimental protocols, in vitro assay data, and in silico methodology are provided in the ESI.† DEL dataset and validation sets are available at available at https://github.com/SnowyTheWestie/DEL_PARP_ML.Author contributions
A. L. M., A. S. H., L. H. Y., S. T., A. K. synthesized the compounds. A. L. M. and A. S. H. coordinated the inhibition assays. H. S. provided the PARP enzymes for DECL selections. L. H. Y. and R. M. F. synthesized the NADEL DECL. RMF performed the ML analyses. R. M. F., A. S. H., and A. L. M. prepared the manuscript. All co-authors carefully reviewed the manuscript and approved of the content.Conflicts of interest
RMF serves as an independent consultant and a member of the scientific advisory board for companies involved in DNA-encoded chemical library (DECL) technologies.Acknowledgements
RMF gratefully acknowledges financial support from the National Institutes of Health (R35GM138335). A. K. received support from the National Science Foundation (CHE2004010). HS was supported by the Swedish Research Council (2019-04871). NMR spectroscopy was performed using the Health Sciences Centre Cores facility at the University of Utah. We thank Dr K. Diehl and Faheem for testing the PROTAC-like molecules in cells. We thank Brayden Halverson from Leash Bio for help with DNA sequencing.Notes and references
- D. Neri and R. A. Lerner, DNA-Encoded Chemical Libraries: A Selection System Based on Endowing Organic Compounds with Amplifiable Information, Annu. Rev. Biochem., 2018, 87, 479–502, DOI:10.1146/annurev-biochem-062917-012550
.
- A. Gironda-Martinez, E. J. Donckele, F. Samain and D. Neri, DNA-Encoded Chemical Libraries: A Comprehensive Review with Successful Stories and Future Challenges, ACS Pharmacol. Transl. Sci., 2021, 4(4), 1265–1279, DOI:10.1021/acsptsci.1c00118
- A. A. Peterson and D. R. Liu, Small-molecule discovery through DNA-encoded libraries, Nat. Rev. Drug Discov., 2023, 22(9), 699–722, DOI:10.1038/s41573-023-00713-6
- R. M. Franzini and C. Randolph, Chemical Space of DNA-Encoded Libraries, J. Med. Chem., 2016, 59(14), 6629–6644, DOI:10.1021/acs.jmedchem.5b01874
- A. F. Ku, K. L. Sharma, H. M. Ta, C. M. Sutton, K. M. Bohren, Y. Wang, S. Chamakuri, R. Chen, J. M. Hakenjos and R. Jimmidi,
et al., Reversible male contraception by targeted inhibition of serine/threonine kinase 33, Science, 2024, 384(6698), 885–890, DOI:10.1126/science.adl2688
- E. S. O’Brien, V. A. Rangari, A. El Daibani, S. O. Eans, H. R. Hammond, E. White, H. Wang, Y. Shiimura, K. Krishna Kumar and Q. Jiang,
et al., A μ-opioid receptor modulator that works cooperatively with naloxone, Nature, 2024, 631(8021), 686–693, DOI:10.1038/s41586-024-07587-7
- J. Liang, M. J. Lambrecht, T. L. Arenzana, S. Aubert-Nicol, L. Bao, F. Broccatelli, J. Cai, C. Eidenschenk, C. Everett and T. Garner,
et al., Optimization of a Novel DEL Hit That Binds in the Cbl-b SH2 Domain and Blocks Substrate Binding, ACS Med. Chem. Lett., 2024, 15(6), 864–872, DOI:10.1021/acsmedchemlett.4c00068
- W. You, A. L. Montoya, S. Dana, R. M. Franzini and C. Steegborn, Elucidating the Unconventional Binding Mode of a DNA-Encoded Library Hit Provides a Blueprint for Sirtuin 6 Inhibitor Development, ChemMedChem, 2024, 19(20), e202400273, DOI:10.1002/cmdc.202400273
- Y. Suo, K. Li, X. Ling, K. Yan, W. Lu, J. Yue, X. Chen, Z. Duan and X. Lu, Discovery Small-Molecule p300 Inhibitors Derived from a Newly Developed Indazolone-Focused DNA-Encoded Library, Bioconjug. Chem., 2024, 35(8), 1251–1257, DOI:10.1021/acs.bioconjchem.4c00307
- K. Li, M. W. Krone, A. Butrin, M. J. Bond, B. M. Linhares and C. M. Crews, Development of Ligands and Degraders Targeting MAGE-A3, J. Am. Chem. Soc., 2024, 146(36), 24884–24891, DOI:10.1021/jacs.4c05393
- W. G. Cochrane, M. L. Malone, V. Q. Dang, V. Cavett, A. L. Satz and B. M. Paegel, Activity-Based DNA-Encoded Library Screening, ACS Comb. Sci., 2019, 21(5), 425–435, DOI:10.1021/acscombsci.9b00037
- W. P. Walters and M. A. Murcko, Prediction of ‘drug-likeness’, Adv. Drug Deliv. Rev., 2002, 54(3), 255–271, DOI:10.1016/s0169-409x(02)00003-0
- K. McCloskey, E. A. Sigel, S. Kearnes, L. Xue, X. Tian, D. Moccia, D. Gikunju, S. Bazzaz, B. Chan and M. A. Clark,
et al., Machine Learning on DNA-Encoded Libraries: A New Paradigm for Hit Finding, J. Med. Chem., 2020, 63(16), 8857–8866, DOI:10.1021/acs.jmedchem.0c00452
- P. Komar and M. Kalinic, Denoising DNA Encoded Library Screens with Sparse Learning, ACS Comb. Sci., 2020, 22(8), 410–421, DOI:10.1021/acscombsci.0c00007
- M. Lemke, H. Ravenscroft, N. J. Rueb, D. Kireev, D. Ferraris and R. M. Franzini, Integrating DNA-encoded chemical libraries with virtual combinatorial library screening: Optimizing a PARP10 inhibitor, Bioorg. Med. Chem. Lett., 2020, 30(19), 127464, DOI:10.1016/j.bmcl.2020.127464
- L. Guasch, M. Reutlinger, D. Stoffler and M. Wichert, Augmenting Chemical Space with DNA-encoded Library Technology and Machine Learning, Chimia, 2021, 75(1), 105–107, DOI:10.2533/chimia.2021.105
- K. S. Lim, A. G. Reidenbach, B. K. Hua, J. W. Mason, C. J. Gerry, P. A. Clemons and C. W. Coley, Machine Learning on DNA-Encoded Library Count Data Using an Uncertainty-Aware Probabilistic Loss Function, J. Chem. Inf. Model., 2022, 62(10), 2316–2331, DOI:10.1021/acs.jcim.2c00041
- C. Zhang, M. Pitman, A. Dixit, S. Leelananda, H. Palacci, M. Lawler, S. Belyanskaya, L. Grady, J. Franklin and N. Tilmans,
et al., Building Block-Based Binding Predictions for DNA-Encoded Libraries, J. Chem. Inf. Model., 2023, 63(16), 5120–5132, DOI:10.1021/acs.jcim.3c00588
- R. Hou, C. Xie, Y. Gui, G. Li and X. Li, Machine-Learning-Based Data Analysis Method for Cell-Based Selection of DNA-Encoded Libraries, ACS Omega, 2023, 8(21), 19057–19071, DOI:10.1021/acsomega.3c02152
- K. Shmilovich, B. Chen, T. Karaletsos and M. M. Sultan, DEL-Dock: Molecular Docking-Enabled Modeling of DNA-Encoded Libraries, J. Chem. Inf. Model., 2023, 63(9), 2719–2727, DOI:10.1021/acs.jcim.2c01608
- A. L. Montoya, M. Glavatskikh, B. J. Halverson, L. H. Yuen, H. Schuler, D. Kireev and R. M. Franzini, Combining pharmacophore models derived from DNA-encoded chemical libraries with structure-based exploration to predict Tankyrase 1 inhibitors, Eur. J. Med. Chem., 2023, 246, 114980, DOI:10.1016/j.ejmech.2022.114980
- W. Torng, I. Biancofiore, S. Oehler, J. Xu, J. Xu, I. Watson, B. Masina, L. Prati, N. Favalli and G. Bassi,
et al., Deep Learning Approach for the Discovery of Tumor-Targeting Small Organic Ligands from DNA-Encoded Chemical Libraries, ACS Omega, 2023, 8(28), 25090–25100, DOI:10.1021/acsomega.3c01775
- S. Han, X. Guo, M. Wang, H. Liu, Y. Song, Y. He, K. L. Hsueh, W. Cui, W. Su and L. Kuai,
et al., Highly Selective Novel Heme Oxygenase-1 Hits Found by DNA-Encoded Library Machine Learning beyond the DEL Chemical Space, ACS Med. Chem. Lett., 2024, 15(9), 1456–1466, DOI:10.1021/acsmedchemlett.4c00121
- L. H. Yuen, S. Dana, Y. Liu, S. I. Bloom, A. G. Thorsell, D. Neri, A. J. Donato, D. Kireev, H. Schuler and R. M. Franzini, A Focused DNA-Encoded Chemical Library for the Discovery of Inhibitors of NAD(+)-Dependent Enzymes, J. Am. Chem. Soc., 2019, 141(13), 5169–5181, DOI:10.1021/jacs.8b08039
- M. F. Langelier, T. Eisemann, A. A. Riccio and J. M. Pascal, PARP family enzymes: regulation and catalysis of the poly(ADP-ribose) posttranslational modification, Curr. Opin. Struct. Biol., 2018, 53, 187–198, DOI:10.1016/j.sbi.2018.11.002
- A. L. Satz, DNA Encoded Library Selections and Insights Provided by Computational Simulations, ACS Chem. Biol., 2015, 10(10), 2237–2245, DOI:10.1021/acschembio.5b00378
- A. L. Satz, R. Hochstrasser and A. C. Petersen, Analysis of Current DNA Encoded Library Screening Data Indicates Higher False Negative Rates for Numerically Larger Libraries, ACS Comb. Sci., 2017, 19(4), 234–238, DOI:10.1021/acscombsci.7b00023
- S. Dawadi, N. Simmons, G. Miklossy, K. M. Bohren, J. C. Faver, M. N. Ucisik, P. Nyshadham, Z. Yu and M. M. Matzuk, Discovery of potent thrombin inhibitors from a protease-focused DNA-encoded chemical library, Proc. Natl. Acad. Sci. U. S. A., 2020, 117(29), 16782–16789, DOI:10.1073/pnas.2005447117
- R. Jimmidi, S. Chamakuri, S. Lu, M. N. Ucisik, P. J. Chen, K. M. Bohren, S. A. Moghadasi, L. Versteeg, C. Nnabuife and J. Y. Li,
et al., DNA-encoded chemical libraries yield non-covalent and non-peptidic SARS-CoV-2 main protease inhibitors, Commun. Chem., 2023, 6(1), 164, DOI:10.1038/s42004-023-00961-y
- J. M. Rectenwald, S. K. R. Guduru, Z. Dang, L. B. Collins, Y. E. Liao, J. L. Norris-Drouin, S. H. Cholensky, K. W. Kaufmann, S. M. Hammond and D. B. Kireev,
et al., Design and Construction of a Focused DNA-Encoded Library for Multivalent Chromatin Reader Proteins, Molecules, 2020, 25(4), 979–990, DOI:10.3390/molecules25040979
- S. Wang, K. E. Denton, K. F. Hobbs, T. Weaver, J. M. B. McFarlane, K. E. Connelly, M. C. Gignac, N. Milosevich, F. Hof and I. Paci,
et al., Optimization of Ligands Using Focused DNA-Encoded Libraries To Develop a Selective, Cell-Permeable CBX8 Chromodomain Inhibitor, ACS Chem. Biol., 2020, 15(1), 112–131, DOI:10.1021/acschembio.9b00654
- N. J. Curtin and C. Szabo, Poly(ADP-ribose) polymerase inhibition: past, present and future, Nat. Rev. Drug Discov., 2020, 19(10), 711–736, DOI:10.1038/s41573-020-0076-6
- A. G. Thorsell, T. Ekblad, T. Karlberg, M. Low, A. F. Pinto, L. Tresaugues, M. Moche, M. S. Cohen and H. Schuler, Structural Basis for Potency and Promiscuity in Poly(ADP-ribose) Polymerase (PARP) and Tankyrase Inhibitors, J. Med. Chem., 2017, 60(4), 1262–1271, DOI:10.1021/acs.jmedchem.6b00990
- E. Wahlberg, T. Karlberg, E. Kouznetsova, N. Markova, A. Macchiarulo, A. G. Thorsell, E. Pol, A. Frostell, T. Ekblad and D. Oncu,
et al., Family-wide chemical profiling and structural analysis of PARP and tankyrase inhibitors, Nat. Biotechnol., 2012, 30(3), 283–288, DOI:10.1038/nbt.2121
- M. Bekes, D. R. Langley and C. M. Crews, PROTAC targeted protein degraders: the past is prologue, Nat. Rev. Drug Discov., 2022, 21(3), 181–200, DOI:10.1038/s41573-021-00371-6
- M. Wichert, L. Guasch and R. M. Franzini, Challenges and Prospects of DNA-Encoded Library Data Interpretation, Chem. Rev., 2024, 124(22), 12551–12572 CrossRef CAS
- W. Decurtins, M. Wichert, R. M. Franzini, F. Buller, M. A. Stravs, Y. Zhang, D. Neri and J. Scheuermann, Automated screening for small organic ligands using DNA-encoded chemical libraries, Nat. Protoc., 2016, 11(4), 764–780, DOI:10.1038/nprot.2016.039
- C. C. Mehta and H. G. Bhatt, Tankyrase inhibitors as antitumor agents: a patent update (2013–2020), Expert Opin. Ther. Pat., 2021, 31(7), 645–661, DOI:10.1080/13543776.2021.1888929
- Q. Chen, C. Liu, W. Wang, X. Meng, X. Cheng, X. Li, L. Cai, L. Luo, X. He and H. Qu,
et al., Optimization of PROTAC Ternary Complex Using DNA Encoded Library Approach, ACS Chem. Biol., 2023, 18(1), 25–33, DOI:10.1021/acschembio.2c00797
- C. Huang, X. Liu, X. Wu, L. Meng, X. Lu and X. Li, Selection of DNA-encoded Chemical Libraries for Compounds that can Induce Protein Ubiquitination, ChemRxiv, 2024 DOI:10.26434/chemrxiv-2024-rmgtv
- S. Liu, B. Tong, J. W. Mason, J. M. Ostrem, A. Tutter, B. K. Hua, S. A. Tang, S. Bonazzi, K. Briner and F. Berst,
et al., Rational Screening for Cooperativity in Small-Molecule Inducers of Protein-Protein Associations, J. Am. Chem. Soc., 2023, 145(42), 23281–23291, DOI:10.1021/jacs.3c08307
- N. V. Chawla, K. W. Bowyer, L. O. Hall and W. P. Kegelmeyer, SMOTE: synthetic minority over-sampling technique, J. Artif. Intell. Res., 2002, 16(1), 321–357 CrossRef
- L. Prati, M. Bigatti, E. J. Donckele, D. Neri and F. Samain, On-DNA hit validation methodologies for ligands identified from DNA-encoded chemical libraries, Biochem. Biophys. Res. Commun., 2020, 533(2), 235–240, DOI:10.1016/j.bbrc.2020.04.030
- R. M. Franzini, A. Nauer, J. Scheuermann and D. Neri, Interrogating target-specificity by parallel screening of a DNA-encoded chemical library against closely related proteins, Chem. Commun., 2015, 51(38), 8014–8016, 10.1039/c5cc01230a
- H. Barhoosh, A. Dixit, W. G. Cochrane, V. Cavett, R. N. Prince, B. O. Blair, F. R. Ward, K. F. McClure, P. A. Patten and M. G. Paulick,
et al., Activity-Based DNA-Encoded Library Screening for Selective Inhibitors of Eukaryotic Translation, ACS Cent. Sci., 2024, 10(10), 1960–1968, DOI:10.1021/acscentsci.4c01218
- P. Ma, H. Xu, J. Li, F. Lu, F. Ma, S. Wang, H. Xiong, W. Wang, D. Buratto and F. Zonta,
et al., Functionality-Independent DNA Encoding of Complex Natural Products, Angew Chem. Int. Ed. Engl., 2019, 58(27), 9254–9261, DOI:10.1002/anie.201901485
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc00844a |
| ‡ These authors contributed equally. |
| § A11 and A108 correspond to the same building block.24 |
| This journal is © The Royal Society of Chemistry 2025 |