
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLigase-catalyzed transcription and reverse-transcription of XNA-containing nucleic acid polymers using T3 DNA ligase†
Natalie
Khamissi
 ,
Christopher
Korfmann
,
Areeba
Chaudhry
and
Ryan
Hili
*
,
Christopher
Korfmann
,
Areeba
Chaudhry
and
Ryan
Hili
*
Department of Chemistry, Centre for Research on Biomolecular Interactions, York University, 4700 Keele Street, Toronto, ON M3J 1P3, Canada. E-mail: rhili@yorku.ca; Web: www.yorku.ca/rhili
First published on 21st March 2025
Abstract
A method to enable the transliteration between various XNA-containing nucleic acids and canonical DNA is described. Using ligase-catalysed oligonucleotide polymerisation (LOOPER), we show that DNA can be used as a template to generate nucleic acids polymers comprising various levels of 2′-fluoro (2′-F), 2′-fluoro-arabinonucleic acid (FANA), 2′-O-methyl (2′-OMe), and Locked Nucleic Acids (LNA) in moderate yields. The fidelity and biases of the LOOPER process were studied in detail for the 2′-F system by developing a hairpin-based sequencing method, which showed fidelities exceeding 95% along with positional and sequence dependencies within the polymerised XNA-containing anticodons. Lastly, we show the ability of LOOPER to regenerate DNA from 2′-F, FANA, 2′-OMe, and LNA in moderate yield and in fidelities over 95%. Taken together, this study demonstrates the potential of LOOPER to serve as a platform for applications where the transliteration between XNA and DNA is needed, such as the in vitro evolution of XNA-containing nucleic acid polymers.
Introduction
Nature has evolved to use highly specific backbones for its genetically encoded polymers. In proteins, amide-linked α-amino acid are employed, while RNA and DNA use phosphodiester linked ribose and deoxyribose sugars, respectively. These naturally occurring backbones enable efficient and specific root processing of the biopolymers of life, be it synthesis, replication, modification, or degradation.Chemists have long explored the use of unnatural backbones to study and perturb the biology of living systems. For instance, β-peptides can form complex and predictable folding patterns, engage with protein targets, and exhibit profound resistance to proteolysis.1,2 Inspired by their potential, efforts to engineer the protein translation machinery have enabled the translation, evolution, and ultimately the study and application of β-peptides.3,4 Within the domain of genetic polymers, the exploration of alternative phospho-sugar backbones have been investigated for decades5 with the ultimate goal to understand the etiology of DNA and RNA in living systems, possibly from a structurally related progenitor.6 However, more recently, nucleic acid polymers with unnatural backbones have come to the fore as a critical advance in synthetic biology with considerable potential in various therapeutic applications. Such polymers, termed xeno-nucleic acids (XNA), are not recognised by endogenous nucleases, thus slowing, or precluding their degradation in biological samples. Indeed, this property has prompted their emergence in oligonucleotides used in CRISPR guide RNA,7 and various antisense oligonucleotide therapeutics.8
Functional nucleic acids are conventionally discovered through a process called Systematic Evolution of Ligands by EXponential enrichment (SELEX), which involves iterative cycles of a selection pressure against a library of nucleic acids for a desired function (e.g., binding a protein target), and amplifying the survivors. Recent advances in engineered polymerases have enabled the transcription and reverse transcription of several XNA forms,9 including those with nucleobase modifications,10 with high fidelity – a critical step to enable the evolution of functional XNAs using traditional SELEX methods. High-affinity aptamers against protein targets have been isolated from selections involving α-L-threofuranosyl nucleic acid (TNA),11–13 2′-fluoro-arabinonucleic acid (FANA),14–16 and 1′,5′-anhydrohexitol nucleic acid (HNA).17 Despite the considerable advances in polymerase engineering, new conceptual approaches to enzymatically generate XNA are needed to fully explore their potential. In particular, general methods to transcribe and reverse transcribe broad classes of XNA, chimeric XNA, or XNAs bearing nucleobase modifications, are needed to expand the utility of this promising class of biopolymers for therapeutic, diagnostic, and other biotechnology applications.
To this end, various ligases have been explored to enzymatically ligate XNA fragments on a DNA template using ligases such as T3, T4 and T7 DNA ligase18–20 and RNA ligase.21 While this approach enables rapid synthesis of discrete XNAs sequences, the generation and reverse transcription of XNA libraries using ligases has not been investigated. The ligase-catalyzed oligonucleotide polymerisation (LOOPER) provides a platform for analogous transcription of DNA into synthetic biopolymers (Fig. 1).22–29 LOOPER involves the nucleic acid-templated ligation of very short oligonucleotide fragments, termed anticodons, in a sequence-defined manner and proceeds with high fidelity when using large combinatorial libraries of templates. Importantly, LOOPER has been shown to accommodate nucleobase-modified anticodons. Indeed, complex libraries can be generated with up to 16 different chemical modifications25 or with peptide fragments up to 8-residues in length.23 LOOPER has also been implemented in SELEX to identify high-affinity aptamers against protein targets, including human α-thrombin,28 PCSK9 and IL-6.27 Due to the potential flexibility of LOOPER, we reasoned that DNA-templated XNA library synthesis and XNA-templated DNA library synthesis may be within the scope of the process, thus enabling a potentially general approach to analogous transcription and reverse transcription of XNAs that can be ported into SELEX or other applications, including memory storage (Fig. 2).30
 | ||
| Fig. 1 General process for ligase-catalyzed oligonucleotide polymerisation (LOOPER) for the generation of modified DNA. | ||
 | ||
| Fig. 2 General schematic for the ligase-mediated “transcription” of DNA into XNA using a DNA ligase and 5′-phosphorylated XNA trinucleotides and the ligase-mediated “reverse transcription” of XNA into DNA using a DNA ligase and 5′-phosphorylated DNA trinucleotides. | ||
The ligase used during LOOPER can have profound influence over the outcome of the process. T4 DNA ligase, which was the original enzyme used in LOOPER, can most efficiently polymerise anticodons that are at least pentanucleotides in length.31 Importantly, the anticodon must be chemically modified to enable high fidelity. When polymerising anticodons comprising the ANNNN sequence, whereby N = A, C, G, T, the incorporation of a C8 modification at the 5′-end adenine increases fidelity from 87% to 95%, presumably through an anti to syn conformational switch along the adenine glycosidic bond, resulting in a more stringent annealing process.31 With fully degenerate pentanucleotide anticodons, NNNNN, which would be required for the proposed XNA system, fidelities are decreased to 81% which are likely too low for most applications. T3 DNA ligase on the other hand has been shown to accommodate anticodons as short as three nucleotides in length.27,31 Importantly, while fully degenerate unmodified anticodons such as NNNNN result in very low fidelity (67%), shorter anticodons, such as NNNN and NNN are incorporated with much high fidelities of 89% and 97%, respectively.31 For this reason, we pursued T3 as a potential ligase for XNA-based libraries in LOOPER.
Results and discussion
Polymerisation efficiency of XNA anticodons in LOOPER
Using degenerate trinucleotide libraries, we tested commonly used and commercially available XNA modifications including 2′-F, 2′-OMe, FANA, and LNA (Fig. 2). For each XNA type, four NNN anticodon libraries were synthesised: one with the XNA modification at every position within the anticodon, and then three corresponding libraries where the XNA modification was either in the 5′, middle, or 3′ position of the anticodon. This allowed us to evaluate the efficiency and positional preferences of polymerisation, which has been an important parameter in earlier developments.32 Each library was subjected to LOOPER along a template comprising a 39-nt (13 codon) reading frame using T3 DNA ligase and the yield was evaluated by polyacrylamide electrophoresis (PAGE) and densitometry (Fig. 3). While both 2′-OMe and FANA at any position failed to yield full-length products, T3 DNA ligase was able to generate full-length product using any of the four 2′-F libraries. Surprisingly, LNA was also well-tolerated by T3 DNA ligase, specifically when the modification was on the middle ribose sugar of the trinucleotide. For both LNA and 2′-F, the greatest yields were found with modifications at the middle position, suggesting that the modifications may be interfering with the enzymatic synthesis of the phosphodiester linkages between the nucleotides at each terminus. | ||
| Fig. 3 LOOPER of XNAs 2′F and LNA. Denaturing PAGE analysis of LOOPER process containing a DNA template comprising a 39-nucleotide random region and degenerate trinucleotides (NNN) bearing XNA modifications at noted trinucleotide positions. | ||
Although there was no full-length product observed when using 2′-OMe or FANA-modified trinucleotides in LOOPER, partially polymerised products were detected depending on the position of the modification on the anticodon (Fig. S1 and S2†); further optimisation may enable these classes of XNA to be more efficiently polymerised. 2′-OMe modifications at the middle position of the trinucleotide were tolerated the best, incorporating up to four trinucleotides. It is important to note that LOOPER with 2′-OMe-modified trinucleotides of a discrete sequence perform much better than in library contexts. For instance, TTT trinucleotides with 2′-OMe modifications at the middle position were well tolerated, resulting in full-length product (Fig. S2a†). While the reason for this have not been fully explored, our lab has previously observed that yield typically decreases as the LOOPER anticodon library size increases.25 This may be due to the decrease in relative concentration of the cognate anticodon at each ligation site. Furthermore, certain anticodon sequences may have superior annealing and ligation kinetics with T3 DNA ligase, which creates challenges in library-based polymerisations. For FANA-modified trinucleotide libraries, the 3′-modified library was the most tolerated by T3 DNA ligase resulting in three trinucleotide incorporations (Fig. S1b†). This is consistent with what was observed with FANA-modified TTT sequences, where the 3′-end modification exhibited the most efficient polymerisation (Fig. S1†).
Fidelity and bias analysis of XNA LOOPER
With a degenerate trinucleotide anticodon set, there are 43 = 64 trinucleotide sequences used for polymerisation during LOOPER. This gives rise to a combinatorially challenging system, where errors can occur due to the ligation of a misannealed anticodon across from a codon within the reading frame. Assessing the fidelity of a LOOPER system is necessary to evaluate its potential in downstream applications. Examining the fidelity of polymerases is more straightforward as they typically use just four monomers during transcription and reverse transcription, enabling the use of sequencing methods in a non-library format (e.g., a long discrete template). For LOOPER, in order to obtain sequencing coverage of each anticodon in different sequence contexts, and to establish codon incorporation bias for each anticodon, we previously developed a duplex sequencing method to allow for the direct comparison of template and polymerised product in a library format using high-throughput sequencing.24 Since LNA was not amplifiable using commercial polymerases, we focused on 2′-F XNA as a model system. Unfortunately, while duplex sequencing could accurately assess the fidelities of nucleobase-modified DNA anticodons,25 the method failed to yield the sequencing coverage needed for accurate fidelity characterisations with 2′-F XNA. When analyzing duplex sequencing data, 28 nt barcodes on the sense and antisense strands are paired together during the fidelity analysis. We found that very few pairs could be matched from the sequencing data. We attempted reducing the amount of input into the sequencing runs to maximise the probability of finding pairs, but this did not result in significant improvements. Q5U polymerase was then used instead of standard Q5 since this polymerase is reported to better tolerate uridine bases present on the 2′-F and 2′-OMe modified nucleotides.33 Unfortunately, Q5U polymerase did not improve matching rates. Encouraged by the high fidelities from the limited number of matched sequences (see Table S1†), we sought to redevelop the duplex sequencing into a more robust method for XNA.We hypothesised that the lack of pairing was resulting from the disparity of PCR efficiency between the template (DNA) and products (XNA) strands. Even with more permissive polymerases, it was difficult to control this bias. To overcome this issue, we chose to evaluate the fidelity using a hairpin architecture (Fig. 4). Using this approach, full-length amplicons that go into sequencing would necessarily have read through both the template and product strand, allowing for straightforward pairing for fidelity analysis.
 | ||
| Fig. 4 General pipeline for hairpin sequencing (HP-seq) to assess LOOPER fidelity with poorly amplifying XNA templates. Hairpin library template (reading frame = N39) containing primer-binding site and sequencing adapters is subjected to LOOPER. PCR amplification of the product results in an amplicon comprising the original template read and the LOOPER strand read. High-throughput sequencing an post-sequencing analysis is used to compare the template and LOOPER reads to identify errors in LOOPER within a library context. | ||
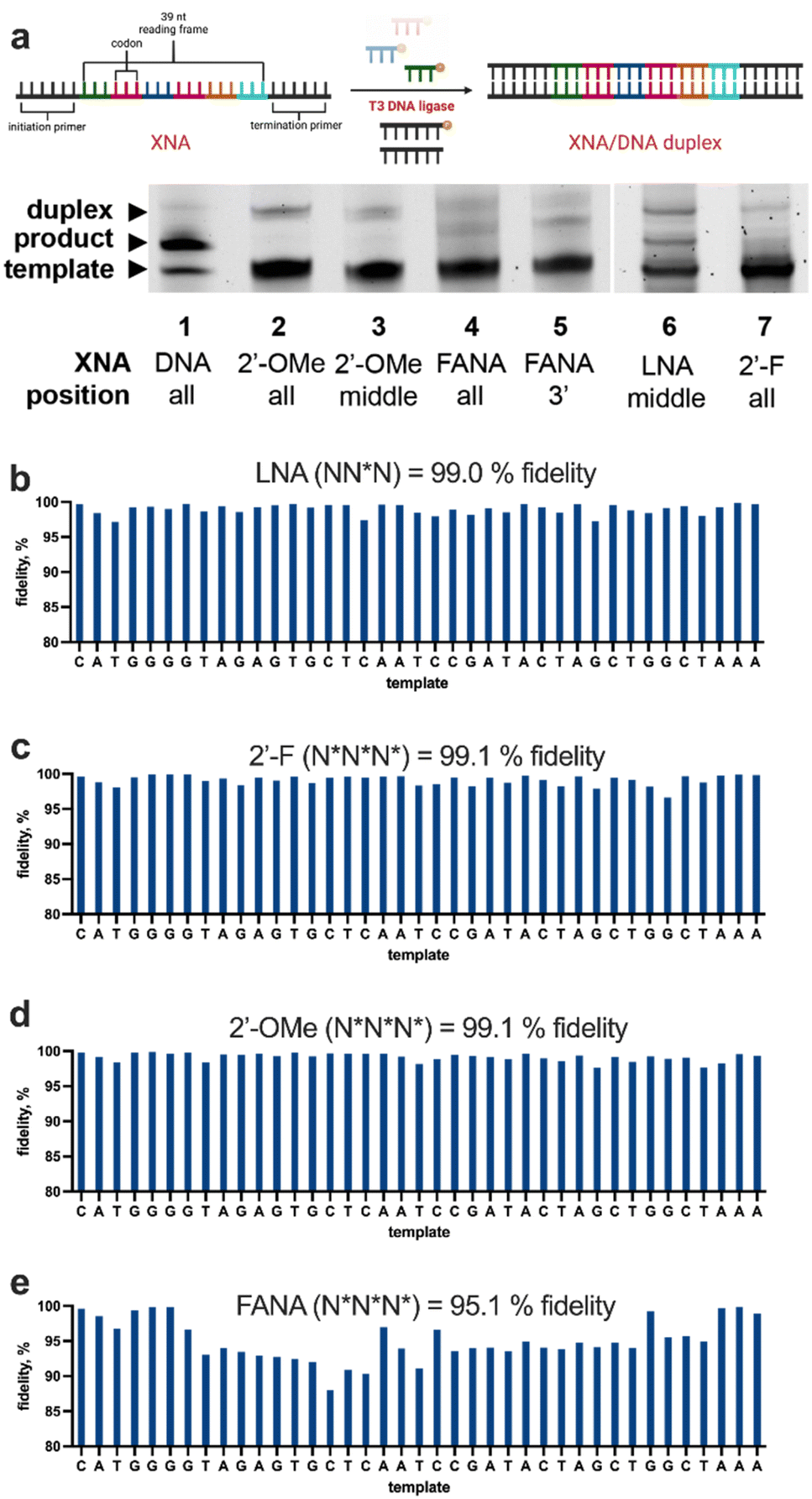
LOOPER was performed along a hairpin template library and PCR amplified before high-throughput sequencing. An advantage to purifying the hairpin product is that the fully extended product is well resolved during PAGE from the partial products, allowing for efficient purification. The product was validated by attaching a fluorophore to the 5′-end of the primer. This allowed for confirmation of the full-length product band for extraction. The correct amplicon length prior to sequencing further validated the fully extended product. All modified hairpin products demonstrated identical PAGE gels before purification, except for the completely modified 2′-F product. Although the modification was successful during LOOPER, PCR amplification using Q5 DNA polymerase did not result in any product. This could be due to the highly structured hairpin in conjunction with the densely modified strand that makes the polymerisation difficult. BST 3.0 polymerase, a polymerase with a larger, more accommodating active site, did not improve PCR efficiency. KOD DNA polymerase had a faint product band that was not well resolved; however, sequencing confirmed that this band was not the product. Therefore, we were unable to determine the fidelity of the completely modified 2′-F product using this current approach. In addition, the LNA product could not be PCR amplified by Q5 DNA polymerase, so we were not able to perform sequencing on these products.
After fidelity sequencing of the hairpin products, we noticed that the sequencer had difficulties sequencing through the hairpin structure. We determined this using a control hairpin that was extended using Q5 DNA polymerase and sequenced alongside the modified products. This resulted in a single nucleotide fidelity of 97.0% and a trinucleotide fidelity of 91.7%; we normalised the modified fidelities to this Q5 DNA control (Table 1). Analysis of the LOOPER control (unmodified NNN) resulted in a 95.9% normalised fidelity, which is comparable to the fidelity obtained from duplex sequencing (97.0%).31 The data shows that the middle-modified 2′-F trinucleotide system, while having the highest polymerisation yield, resulted in the lowest fidelity, which is consistent with our preliminary data using duplex sequencing (Table S1†). This may indicate that this anticodon set has a stronger polymerisation bias for subsets of anticodons. While the single nucleotide fidelities using anticodons with the 2′-F at either the 3′-end or the 5′-end were equivalent, at the trinucleotide level, the trinucleotide library with the 2′-F at the 5′-end resulted in the highest fidelities. On the whole, these levels of fidelities are consistent with those used for LOOPER-based aptamer selections,28 which highlights the utility of this method.
| Anticodonb | Fidelityc (1N) | N Fidelity (1N) | Fidelitye (3N) | N Fidelity (3N) |
|---|---|---|---|---|
| a Fidelities were determined by Ion Torrent sequencing on an Ion GeneStudio S5 Plus. All LOOPER experiments were conducted on a template comprising a 13 codon (39 mer) reading frame. b Italicised letters indicate the location of the XNA nucleotide. c Fidelity was calculated at the single nucleotide (1N) level. d Fidelity at the single nucleotide (1N) level normalised to Q5 DNA polymerase standard, which was benchmarked at 100%. e Fidelity at the trinucleotide (3N) codon level. f Fidelity at the trinucleotide level (3N) normalised to Q5 DNA polymerase standard, which was benchmarked at 100%. g Q5 DNA polymerase control. | ||||
| NNN | 94.5% | 97.4% | 87.9% | 95.9% |
| N NN | 95.5% | 98.5% | 86.3% | 94.0% |
| NNN | 92.6% | 95.5% | 77.3% | 84.3% |
| NNN | 95.5% | 98.5% | 84.3% | 92.0% |
| NNN | N/A | N/A | N/A | N/A |
| Q5 ctrlg | 97% | 100% | 91.7% | 100% |
We sought to explore trends in the sequencing data to better understand the strengths and shortcomings of the XNA library synthesis method (Fig. 5). We first evaluated the effect of GC content in each trinucleotide library. We observed in the unmodified DNA control that increasing GC content resulted in decreasing fidelity; this can be rationalised by higher GC-content facilitating the ligation of stable misannealed anticodons (Fig. 5a). However, with the 2′-F modified libraries the effect was surprisingly reversed. Upon further analysis, we noticed that there are a select few trinucleotides with 0% GC-content in each of the 2′-F libraries that have unusually poor fidelity. For example, the average fidelity of all 0% GC-containing anticodons in the 5′-modified 2′-F library is just 77.3%. However, when the ATT anticodon is removed from the average, the 0%-GC fidelity increases to 86.0%. Indeed, analysis of sublibraries in each trinucleotide library (Fig. 5b) show that the A and T sublibraries have significantly lower fidelities at all positions within the trinucleotide anticodon. It is known that the incorporation of 2′-F modifications have considerable effects on DNA thermodynamics, including the increase in thermal melting,34 and changes in conformational equilibrium,35 both of which may be at play here.
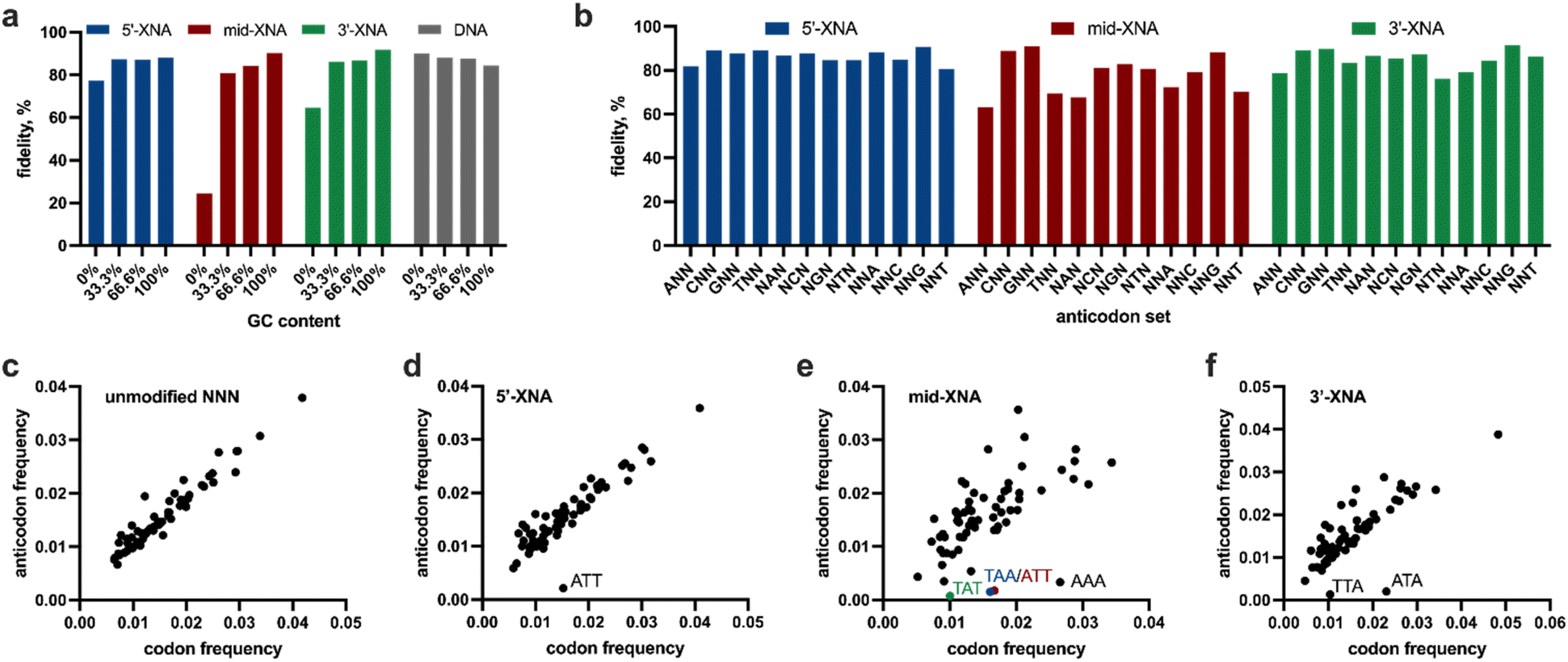 | ||
| Fig. 5 Analysis of codon sets used during LOOPER of 2′-F XNA trinucleotide anticodons. (a) GC-content dependence on LOOPER fidelity for 2′-F XNA at various positions within the NNN anticodon; (b) LOOPER fidelity of anticodon sublibraries for 2′-F XNA at various positions within the NNN anticodon; (c) codon bias observed for LOOPER involving the control unmodified NNN anticodon; (d) codon bias observed for LOOPER involving the NNN anticodon with the 2′-F XNA at the 5′-position; (e) codon bias observed for LOOPER involving the NNN anticodon with the 2′-F XNA at the middle position; (f) codon bias observed for LOOPER involving the NNN anticodon with the 2′-F XNA at the 3′-position. The anticodon sequence of significant outliers are denoted in each graph. | ||
While fidelity plays a large role in oligonucleotide polymerisations, so does polymerisation bias. Biases can occur when anticodons are incorporated into the product strand at different frequencies than they occur in the starting template. This can happen in low fidelity systems but can also happen to some degree in high fidelity systems when polymerisation yields are lower. High bias can convolute analysis in downstream applications, such as in vitro selections of functional nucleic acids. Here, bias analysis has revealed several trends (Fig. 5c–f). While the unmodified DNA control library displayed little bias (as indicated by data distributed along the diagonal), the XNA-based systems exhibit some outliers (Fig. 5d–f, noted in plots). Amongst the XNA libraries, having the 2′F-XNA at the 5′-end of the anticodon resulted in the lowest level of library bias, with just one significant outlier, namely ATT, which had a considerable negative bias. Across all libraries we observed that trinucleotide sequences with a 5′-G base tend to have positive biases, where T3 DNA ligase prefers incorporating these trinucleotide sequences over others. This positive bias has in part resulted in several 0% GC sequences having low fidelities. For example, the ATT trinucleotide sequence found in the 5′-modified 2′-F library resulted in a fidelity of only 11.0%. Upon analysis, we observed that GTT has 4-fold more incorporations across from the TAA codon in the template compared to the cognate ATT anticodon.
LOOPER of unmodified DNA anticodons along XNA templates
The faithful conversion of XNA into DNA is an essential process in the development of XNA-based technologies. This is largely due to traditional amplification and sequencing methods requiring DNA as the genetic medium. While LOOPER has been extensively explored to generate nucleobase-modified oligonucleotides, the ability to use LOOPER to effectively reverse transcribe XNA back into DNA would be of considerable value to the field.During our fidelity analysis of XNA synthesis using LOOPER, we experienced difficulty with the reverse transcription of the completely modified 2′-F products and LNA products, and thus we first explored the reverse transcription ability of LOOPER within this context. In this method, polymerisation occurs using an unmodified trinucleotide DNA library along a defined XNA-modified template (Fig. 6). We attempted an LNA-modified template modified at every third base, preserving the modification structure of the successful middle-modified LNA transcription product; this template could not be PCR amplified by Q5 DNA polymerase (Fig. S6†). LOOPER was able to successfully reverse transcribe the LNA-modified template into DNA (Fig. 6a, lane 6). A considerable amount of duplex remains intact during the denaturing PAGE conditions, likely due to the known increase in thermal melting stability of LNA-containing duplexes.36 We also tested completely modified XNA templates including 2′-F, 2′-OMe, and FANA, the latter two of which could not be directly amplified by Q5 (Fig. S6†); all were found to be within the scope of the LOOPER method. Similar to LNA, they also exhibited varying levels of duplex:single strand ratios on denaturing PAGE, which can be attributed to their known increases in thermal stability relative to canonical DNA.34,37,38 Unfortunately, the completely-modified LNA synthesis could not be included in this study due to difficulties in its synthesis and lack of available commercial availability for oligos with >20 LNA incorporations.
 | ||
| Fig. 6 Evaluation of LOOPER of unmodified DNA anticodons along XNA-containing templates. (a) Denaturing PAGE analysis of LOOPER process of degenerate DNA trinucleotides (NNN) along a corresponding XNA template comprising a discrete 39-nucleotide region. LOOPER fidelities at each position for templates containing (b) LNA at middle position of codon, (c) 2′-F at every position in codon, (d) 2′-OMe at every position in codon, and (e) FANA at every position in codon. | ||
The fidelities of the LOOPER-mediated reverse transcriptions were purified and assessed by Illumina sequencing and were found to be between 95–99% average at the single-nucleotide level (Fig. 6b–e). Interesting, FANA exhibited considerably lower fidelity during the reverse transcription, particularly within the middle of the reading frame. Further investigation into this is ongoing as it is unclear if this effect is dependent on sequence or length of the hybrid duplex. Nonetheless, these results provide a platform for the evolution of XNA-containing nucleic acid polymers.
Conclusions
T3 DNA ligase has been shown to enable LOOPER-mediated synthesis of XNA from DNA templates and DNA from XNA templates by using trinucleotide anticodon libraries. Both LNA and 2′-F libraries were successfully synthesised from corresponding DNA template libraries in moderate yields. A hairpin-based sequencing method was developed to calculate the fidelity and bias of trinucleotide incorporation, which were consistent with those used in LOOPER-mediated in vitro selections, highlighting the strength of this approach. While LOOPER with 2′-OMe and FANA trinucleotides were only partially polymerised, further investigation and optimisation may yield improved conditions to make these XNAs products tractable. Lastly, the reverse transcription of XNAs, including LNA, FANA, 2′-OMe, and 2′-F were achieved using LOOPER in moderate yields. A discrete template was used as a model to assess the fidelity of the method, which showed average fidelities over 95% for all XNA types, which is within the range of LOOPER methods that have been used to evolve aptamers. Taken together, we have shown that LOOPER is capable of the transliteration of various genetic polymers and can potentially serve as a mediator for their use in various biotechnology applications.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
R. H. and N. K. conceived of the idea for this study. R. H. and N. K. designed the methods and experiments. N. K. and A. C. conducted the experiments. C. K. developed the bioinformatic analysis. R. H and N. K analysed the data. R. H. and N. K. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The R. H. laboratory is supported by the Natural Sciences and Engineering Research Council of Canada (RGPIN-2023-05070), Ontario Ministry of Research and Innovation, Canada Foundation for Innovation, and York University.Notes and references
- R. P. Cheng, S. H. Gellman and W. F. DeGrado, Chem. Rev., 2001, 101, 3219–3232 CAS.
- D. Seebach and J. Gardiner, Acc. Chem. Res., 2008, 41, 1366–1375 CrossRef CAS PubMed.
- T. Katoh and H. Suga, J. Am. Chem. Soc., 2018, 140, 12159–12167 CrossRef CAS PubMed.
- T. Katoh, T. Sengoku, K. Hirata, K. Ogata and H. Suga, Nat. Chem., 2020, 12, 1081–1088 CrossRef CAS PubMed.
- A. Eschenmoser, Science, 1999, 284, 2118–2124 CrossRef CAS PubMed.
- J. C. Chaput, Acc. Chem. Res., 2021, 54, 1056–1065 CrossRef CAS PubMed.
- C. R. Cromwell, K. Sung, J. Park, A. R. Krysler, J. Jovel, S. K. Kim and B. P. Hubbard, Nat. Commun., 2018, 9, 1448 CrossRef PubMed.
- W. B. Wan and P. P. Seth, J. Med. Chem., 2016, 59, 9645–9667 CAS.
- C. Blackstock, C. Walters-Freke, N. Richards and A. Williamson, Biochem. J., 2025, 482, 39–56 CrossRef PubMed.
- B. Majumdar, D. Sarma, Y. Yu, A. Lozoya-Colinas and J. C. Chaput, RSC Chem. Biol., 2024, 5, 41–48 RSC.
- M. R. Dunn, C. M. McCloskey, P. Buckley, K. Rhea and J. C. Chaput, J. Am. Chem. Soc., 2020, 142, 7721–7724 CAS.
- X. Li, Z. Li and H. Yu, Chem. Commun., 2020, 56, 14653–14656 CAS.
- H. Mei, J. Y. Liao, R. M. Jimenez, Y. Wang, S. Bala, C. McCloskey, C. Switzer and J. C. Chaput, J. Am. Chem. Soc., 2018, 140, 5706–5713 CrossRef CAS PubMed.
- I. A. Ferreira-Bravo, C. Cozens, P. Holliger and J. J. DeStefano, Nucleic Acids Res., 2015, 43, 9587–9599 CAS.
- I. A. Ferreira-Bravo and J. J. DeStefano, Viruses, 2021, 13, 1983 Search PubMed.
- K. M. Rose, I. A. Ferreira-Bravo, M. Li, R. Craigie, M. A. Ditzler, P. Holliger and J. J. DeStefano, ACS Chem. Biol., 2019, 14, 2166–2175 CAS.
- E. Eremeeva, A. Fikatas, L. Margamuljana, M. Abramov, D. Schols, E. Groaz and P. Herdewijn, Nucleic Acids Res., 2019, 47, 4927–4939 CrossRef CAS PubMed.
- D. Kestemont, M. Renders, P. Leonczak, M. Abramov, G. Schepers, V. B. Pinheiro, J. Rozenski and P. Herdewijn, Chem. Commun., 2018, 54, 6408–6411 RSC.
- C. M. McCloskey, J.-Y. Liao, S. Bala and J. C. Chaput, ACS Synth. Biol., 2019, 8, 282–286 CrossRef CAS PubMed.
- N. Sabat, A. Stämpfli, S. Hanlon, S. Basagni, F. Sladojevich, K. Püntener and M. Hollenstein, Nat. Commun., 2024, 15, 8009 CrossRef CAS PubMed.
- S. Paul, D. Gray, J. Caswell, J. Brooks, W. Ye, T. S. Moody, R. Radinov and L. Nechev, ACS Chem. Biol., 2023, 18, 2183–2187 CrossRef CAS PubMed.
- R. Hili, J. Niu and D. R. Liu, J. Am. Chem. Soc., 2013, 135, 98–101 CrossRef CAS PubMed.
- C. Guo, C. P. Watkins and R. Hili, J. Am. Chem. Soc., 2015, 137, 11191–11196 CrossRef CAS PubMed.
- Y. Lei, D. Kong and R. Hili, ACS Comb. Sci., 2015, 17, 716–721 CrossRef CAS PubMed.
- D. Kong, Y. Lei, W. Yeung and R. Hili, Angew. Chem., Int. Ed., 2016, 55, 13164–13168 CrossRef CAS PubMed.
- C. Guo and R. Hili, Bioconjugate Chem., 2017, 28, 314–318 CrossRef CAS PubMed.
- Z. Chen, P. A. Lichtor, A. P. Berliner, J. C. Chen and D. R. Liu, Nat. Chem., 2018, 10, 420–427 CrossRef CAS PubMed.
- D. Kong, W. Yeung and R. Hili, J. Am. Chem. Soc., 2017, 139, 13977–13980 CrossRef CAS PubMed.
- D. Kong, M. Movahedi, Y. Mahdavi-Amiri, W. Yeung, T. R. Tiburcio, D. Chen and R. Hili, ACS Synth. Biol., 2020, 9, 43–52 CrossRef CAS PubMed.
- L. Ceze, J. Nivala and K. Strauss, Nat. Rev. Genet., 2019, 20, 456–466 CrossRef CAS PubMed.
- Y. Lei, J. Washington and R. Hili, Org. Biomol. Chem., 2019, 17, 1962–1965 RSC.
- M. Movahedi, N. Khamissi and R. Hili, Aptamers, 2021, 5, 22–30 Search PubMed.
- L. Liu, Y. Huang and H. H. Wang, Nat. Methods, 2023, 20, 841–848 CrossRef CAS PubMed.
- P. S. Pallan, E. M. Greene, P. A. Jicman, R. K. Pandey, M. Manoharan, E. Rozners and M. Egli, Nucleic Acids Res., 2011, 39, 3482–3495 CrossRef CAS PubMed.
- W. Guschlbauer and K. Jankowski, Nucleic Acids Res., 1980, 8, 1421–1433 CrossRef CAS PubMed.
- S. K. Singh, A. A. Koshkin, J. Wengel and P. Nielsen, Chem. Commun., 1998, 455–456 RSC.
- Y. Masaki, R. Miyasaka, A. Ohkubo, K. Seio and M. Sekine, J. Phys. Chem. B, 2010, 114, 2517–2524 CrossRef CAS PubMed.
- R. El-Khoury and M. J. Damha, Acc. Chem. Res., 2021, 54, 2287–2297 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc00834d |
| This journal is © The Royal Society of Chemistry 2025 |