
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAtroposelective [4+1] annulation for the synthesis of isotopic isoindolinones bearing both central and axial chirality†
Jun
Gu
,
Li-Hong
Zhang
,
Hong-Feng
Zhuang
and
Ying
He
 *
*
School of Chemistry and Chemical Engineering, Nanjing University of Science and Technology, Nanjing 210094, China. E-mail: yhe@njust.edu.cn
First published on 26th February 2025
Abstract
Isotopically chiral molecules have drawn much attention due to their practical applications in drug discovery. However, existing studies in this area are mainly limited to centrally chiral molecules and H/D exchange. Herein, we report a chiral phosphoric acid-catalyzed atroposelective [4+1] annulation of ketoaldehydes and 1H-indol-1-amines. By means of this strategy, a series of D- and 18O-labeled atropisomers featuring both central and axial chiralities are synthesized with high enantioselectivities and diastereoselectivities and good to excellent isotopic incorporation. Experimental and density functional theory studies suggest that the reaction involves a sequential condensation, cyclization and isomerization cascade, in which the second step is the enantio-determining process.
Introduction
The isotope labeling strategy has been widely used in many fields such as quantitative proteomics, organic reaction mechanisms, and new drug discovery.1 In this context, deuterium (D), as a nonradioactive isotope of hydrogen, was intentionally introduced into bioactive molecules since D-incorporation may not only improve the pharmacokinetic (PK) profile but also provide a solution when bioactive molecules face metabolism-mediated toxicity, drug interactions and low bioactivation.2 In 2017, the U.S. Food and Drug Administration (FDA) granted marketing approval for the first deuterated drug molecule deutetrabenazine, which is used to treat chorea associated with Huntington's disease and tardive dyskinesia (Fig. 1a, left).3 In addition, deucravacitinib, a tyrosine kinase 2 (TYK2) inhibitor approved for the treatment of psoriasis, represents another example of a deuterated FDA-approved drug in 2022 (Fig. 1a, right).4 Therefore, much attention has been paid to synthesizing deuterated bioactive molecules.5 Despite these advances, the synthesis of optically active isotopic molecules is mainly limited to H/D exchange based on either centrally or axially chiral compounds.6 The efficient construction of deuterated molecules featuring multiple chiral elements is far more challenging and yet to be explored. More importantly, beyond the H/D exchange,7 the incorporation of other nonradioactive isotope (e.g.18O) into chiral molecules bearing multiple chiral elements is still undeveloped, despite the fact that these kinds of molecules may also have superior comprehensive performance compared to non-isotopic molecules.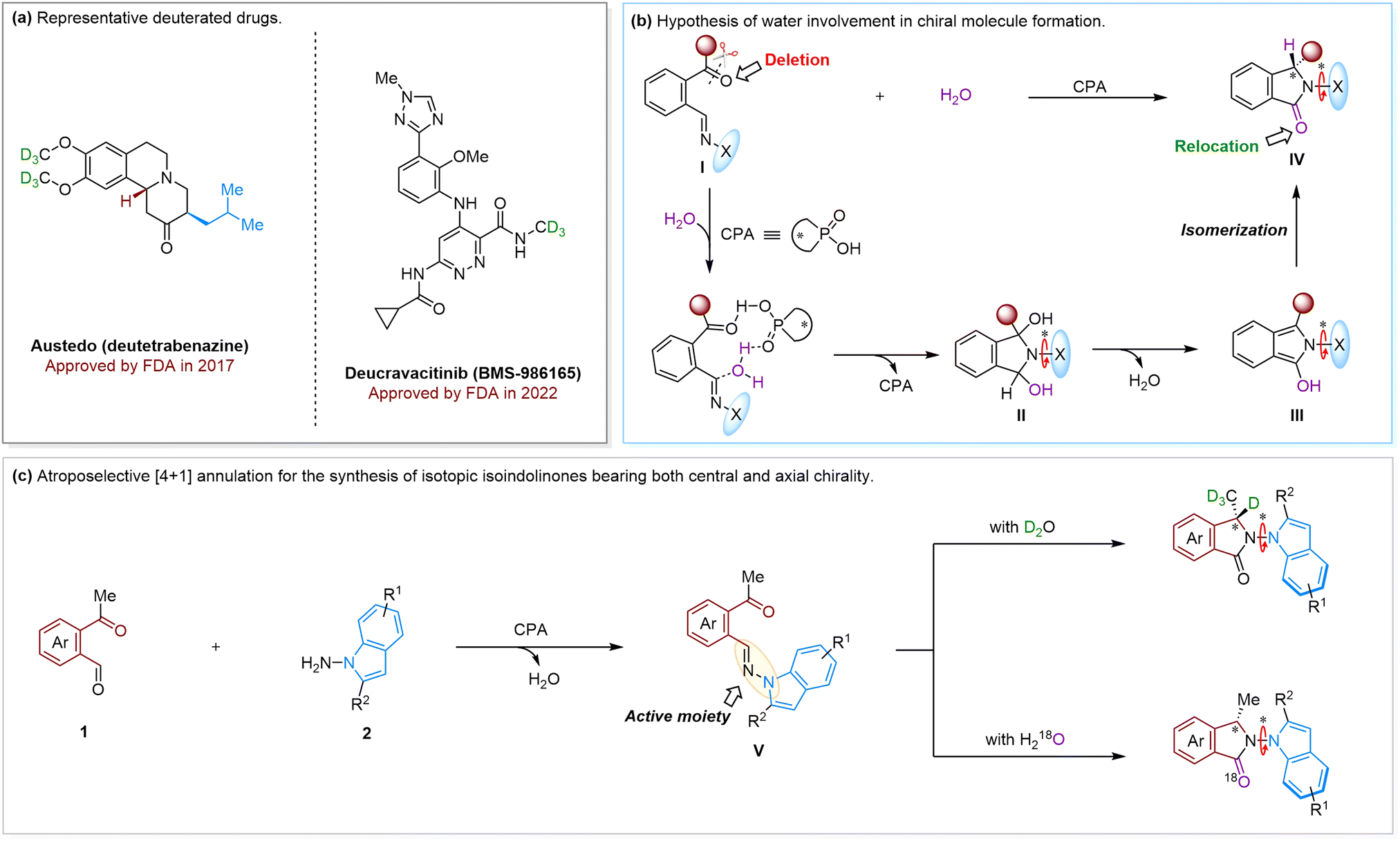 | ||
| Fig. 1 State of the art for the synthesis of isotopically chiral molecules. (a) Representative deuterated drugs. (b) Hypothesis of water involvement in chiral molecule formation. (c) Atroposelective [4+1] annulation for the synthesis of isotopic isoindolinones bearing both central and axial chirality. | ||
Isotopic water (D2O and H218O) is one of the most desirable reagents for the synthesis of isotopically chiral molecules. In this regard, water-involved reactions offer an efficient approach for introducing D or 18O without the need for tedious synthetic procedures of isotopic starting materials. However, the asymmetric introduction of isotopes (D and 18O) by using isotopic water is still in its early stages.8 To this end, we envision the strategy of asymmetric water-nucleophilic attack that could be used for the construction of chiral molecules featuring multiple chiral elements. As shown in Fig. 1b, by using compound I as the substrate, we posited that H2O as the nucleophile could be introduced into compound I to initiate asymmetric cyclization via chiral phosphoric acid (CPA) catalysis. This may cause the carbonyl moiety to first convert to the corresponding hydroxyl group, which affords the chiral intermediate II. The sequential elimination of one molecule of water would then give intermediate III, thus achieving the deletion of the carbonyl group of I. It should be noted that, if the X group is bulky enough, the axially chiral intermediate II can be afforded. Finally, the intramolecular isomerization would occur to generate the isoindolinones IV bearing both axial and central chirality, which accomplishes carbonyl relocation. More importantly, since H2O is a key reactant of the reaction, the isotopically chiral IV may be easily afforded by simply adding D2O or H218O into the reaction system.
Inspired by the reports on practical synthesis of isoindolinones,9 we propose that the reaction of acetaldehyde 1 with 1H-indol-1-amine 2 by CPA catalysis could generate intermediate (V), featuring the active N-amine moiety10 with the release of one molecule of H2O (Fig. 1c). Herein, extra isotopic D2O could be added to the reaction system to replace the H2O and then react with V. On the other hand, due to the keto–enol tautomerism of V (or 1), D-incorporation at the benzylic site would also be expected. Similarly, the 18O-labeled products could also be afforded by subjecting H218O to the reaction mixture. In this regard, isotopically chiral isoindolinones bearing both carbon central and N–N axial chirality11,12 would be produced that may possess potential applications in medicinal chemistry.13
Results and discussion
We commenced our study by investigating the CPA-catalyzed reaction of 2-acetylbenzaldehyde 1a with 2-(phenylsulfonyl)-1H-indol-1-amine 2a, envisioning that the molecular H2O released during the amine generation would participate in the asymmetric annulation to 3a (Table 1). Gratifyingly, the reaction proceeded smoothly at 35 °C in toluene by CPA1 catalysis, affording 3a in 73% enantiomeric excess (ee) with 89% yield, albeit with a low diastereomeric ratio (dr) of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Table 1, entry 1). We then screened a variety of CPA catalysts for the reaction and identified CPA4 as the optimal catalyst that gave 3a in 94% ee and 91% yield with a dr of over 20:1 (entries 2–7). The absolute configuration of 3a was determined to be (P, S) by single-crystal X-ray analysis. The use of other solvents such as CH2Cl2, CHCl3, CCl4, chlorobenzene or EtOAc gave no better results than toluene (entries 8–12). A lower concentration of the reaction mixture resulted in comparable ee of 3a but with a relatively lower yield of 68% and 17:1 dr (entry 13). Moreover, as shown in entries 14–16, decreasing the reaction temperature and catalyst loading both led to 3a with lower ee (77–91%). When the SO2Ph group was replaced with the H group, 68% ee of isoindolinone 3a was obtained. In order to examine the axial configurational stability of 3a, a density functional theory (DFT) study was performed. As a result, the rotational barrier of 3a around the N–N axis in toluene at room temperature is 31.5 kcal mol−1, suggesting the high configurational stability of the N–N moiety of 3a.
:1 (Table 1, entry 1). We then screened a variety of CPA catalysts for the reaction and identified CPA4 as the optimal catalyst that gave 3a in 94% ee and 91% yield with a dr of over 20:1 (entries 2–7). The absolute configuration of 3a was determined to be (P, S) by single-crystal X-ray analysis. The use of other solvents such as CH2Cl2, CHCl3, CCl4, chlorobenzene or EtOAc gave no better results than toluene (entries 8–12). A lower concentration of the reaction mixture resulted in comparable ee of 3a but with a relatively lower yield of 68% and 17:1 dr (entry 13). Moreover, as shown in entries 14–16, decreasing the reaction temperature and catalyst loading both led to 3a with lower ee (77–91%). When the SO2Ph group was replaced with the H group, 68% ee of isoindolinone 3a was obtained. In order to examine the axial configurational stability of 3a, a density functional theory (DFT) study was performed. As a result, the rotational barrier of 3a around the N–N axis in toluene at room temperature is 31.5 kcal mol−1, suggesting the high configurational stability of the N–N moiety of 3a.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | ee (%) | Yield (%) | dr |
| a Reaction conditions: 1a (0.2 mmol), 2a (0.1 mmol), CPA catalyst (10 mol%), solvent (1.0 mL), 35 °C and 24 h. Isolated yield, ee values were determined by high performance liquid chromatography (HPLC), and dr values were determined by 1H-NMR analysis of the crude reaction mixture. b Toluene (2.0 mL). c The reaction was carried out at 25 °C. d 5 mol% of CPA4 was used. e PhSO2 group was replaced with H. | |||||
| 1 | CPA1 | Toluene | 73 | 89 | 3:1 |
| 2 | CPA2 | Toluene | 27 | 71 | 4:1 |
| 3 | CPA3 | Toluene | −75 | 76 | 12:1 |
| 4 | CPA4 | Toluene | 94 | 91 | >20:1 |
| 5 | CPA5 | Toluene | 94 | 91 | 17:1 |
| 6 | CPA6 | Toluene | −51 | 88 | 15:1 |
| 7 | CPA7 | Toluene | 5 | 80 | 11:1 |
| 8 | CPA4 | CH2Cl2 | 91 | 91 | >20:1 |
| 9 | CPA4 | CHCl3 | 90 | 90 | >20:1 |
| 10 | CPA4 | CCl4 | 71 | 91 | 20:1 |
| 11 | CPA4 | Cl-Ph | 80 | 88 | 18:1 |
| 12 | CPA4 | EtOAc | 80 | 44 | 10:1 |
| 13b | CPA4 | Toluene | 93 | 68 | 17:1 |
| 14c | CPA4 | Toluene | 91 | 80 | 15:1 |
| 15d | CPA4 | Toluene | 83 | 66 | 16:1 |
| 16e | CPA4 | Toluene | 68 | 36 | — |
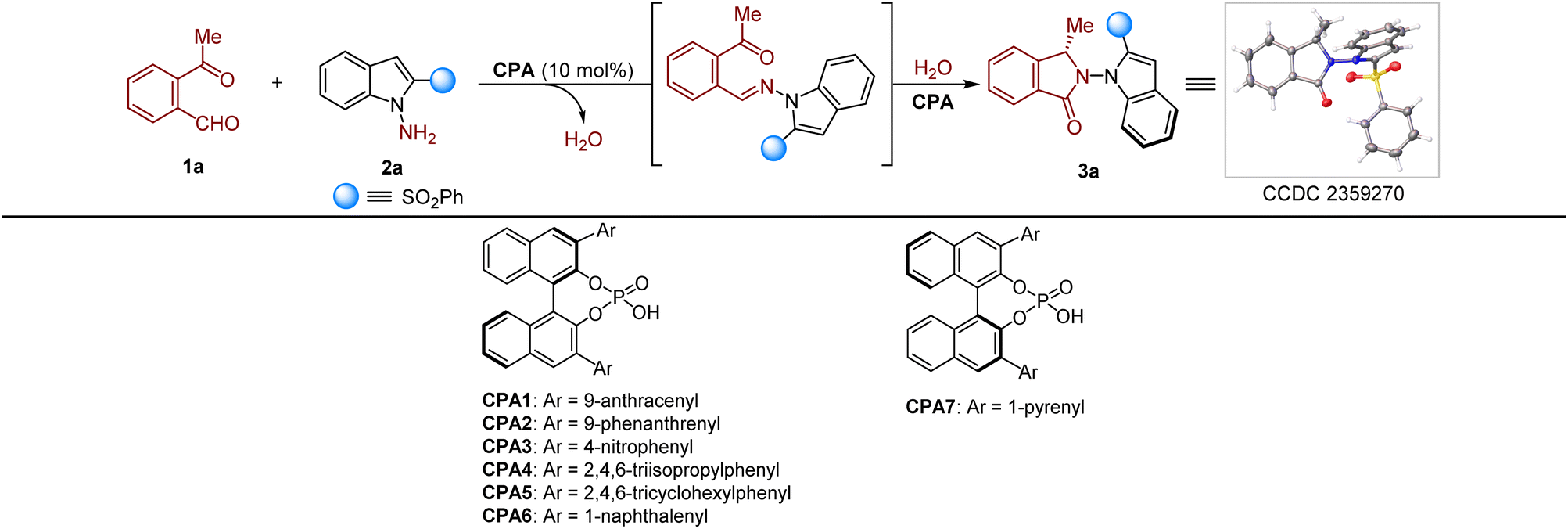
With the optimal conditions in hand, the scope of the reaction was investigated. As shown in Table 2, the reaction proceeded smoothly when substrates 2 with substituents at the C3 or C4 position, affording 3b–3e in 62–80% yields and 86–94% ee with >20:1 dr in all cases. Substrates 2 bearing electron withdrawing and donating groups at the C5 position were compatible with the reaction, providing the desired products (3f–3k) in good to excellent yields and high enantioselectivities (92–96% ee), with excellent to high levels of diastereomeric control (10:1 to >20:1 dr). However, relatively lower ee and dr were obtained when substrates 2 possessing C6 substituents (3l and 3m) were used. On the other hand, the phenylsulfonyl group could be verified by other groups including Ts, Fs and different ester groups, delivering products 3n–3r in high ee and excellent to high dr. Moreover, the reaction also occurred when 2-(trifluoromethyl)-1H-benzo[d]imidazole was used as the substrate, leading to 3s in 42% yield and 82% ee, but only with 2:1 dr. Finally, different ketoaldehydes 1 were examined for the reaction. To our delight, isoindolinones (3t–3a′) were readily isolated in high ee (88–98%) with 10:1 to 20:1 dr, but only 37% yield and 60% ee of 3b′ were obtained when 2-acetyl-6-chlorobenzaldehyde was used as the substrate. Notably, long-chain ketones also tolerated the reaction, delivering 3c′ and 3d′ in 94% ee and 92% ee with 18:1 and 14:1 dr, respectively. However, no desired product 3e′ was generated when 2-benzoylbenzaldehyde was used for the reaction under the optimal conditions.
| a Reaction conditions: 1a (0.2 mmol), 2a (0.1 mmol), CPA4 (10 mol%), toluene (1.0 mL), 35 °C and 24 h. Isolated yield, ee values were determined by HPLC, and dr values were determined by 1H-NMR analysis of the crude reaction mixture. Ts = tosyl and Fs = p-fluorobenzenesulfonyl. b Reaction conditions: 1a (0.2 mmol), 2a (0.1 mmol), CPA catalyst (10 mol%), solvent: chlorobenzene (1.0 mL), −10 °C and 12 h. |
|---|

|
Having established the strategy for the synthesis of chiral isoindolinones 3, we next explored the potential isotopic incorporation of 3 by adding D2O or H18O into the reaction mixture. As shown in Table 3, the use of D2O led to D-incorporation at two different sites, affording D-labeled isoindolinones 4 featuring both central and axial chirality. Substrates 2 bearing substituents with electron-neutral, -withdrawing and -donating groups at the C3, C4 or C5 position were well tolerated to produce the corresponding products in high yields with good to high dr (4a–4e). In addition, ketoaldehydes 1 bearing different groups at the phenyl ring were tolerated under the reaction conditions, affording 4f–4h in good yield, high ee and excellent dr. It should be noted that, in all cases, the isoindolinones 4 were obtained with high D-incorporation at the methyl group (89–91%) and moderate D-incorporation at the benzylic position (63–70%). The moderate D-incorporation at the benzylic position may be attributed to the fact that the H/D exchange of keto–enol tautomerism of 1 would generate extra H2O rebounded into the reaction system, thus decreasing the D-incorporation at the benzylic position. On the other hand, replacing the Me with the C7H15 group gave 4i in high ee and dr with moderate D-incorporation.
| a Reaction conditions: 1a (0.2 mmol), 2a (0.1 mmol), CPA4 (10 mol%), toluene (1.0 mL), D2O (50 μL), 35 °C and 24 h. Isolated yield and ee values were determined by HPLC. The dr values and D-incorporation were determined by 1H-NMR analysis of the crude reaction mixture. |
|---|

|
Subsequently, reactions of representative ketoaldehydes 1 and 2-(phenylsulfonyl)-1H-indol-1-amines 2 with H218O were studied (Table 4). As a result, substituents attached at different positions of the indolyl ring were well-accommodated, affording 18O-labeled isoindolinones 5a–5e in high ee and dr with an excellent level of 18O incorporation (70–86%). The 82–91% 18O incorporation of 5 was also obtained in good yield, high ee and good to high dr (5f–5i). It should be noted that this reaction represents a very rare example of the synthesis of 18O-labeled atropisomers bearing both central and axial chirality.
| a Reaction conditions: 1a (0.2 mmol), 2a (0.1 mmol), CPA4 (10 mol%), toluene (1.0 mL), H218O (50 μL), 35 °C and 24 h. Isolated yield, ee values were determined by HPLC, and dr values were determined by 1H-NMR analysis of the crude reaction mixture. The 18O-incorporation was determined by high-resolution mass spectrometry (HRMS). |
|---|

|
To gain some insight into the details of the reaction, we performed a series of experiments to probe the mechanism. As shown in Fig. 2a, we probed the reaction intermediates by performing the reaction of 1a with 2a at low temperature. As a result, compound Int0 was readily afforded and isolated in E-configuration under either CPA4 or ent-CPA4 catalysis. The configuration of Int0 was unambiguously determined by single crystal X-ray diffraction analysis. Moreover, CPA4 catalyzed the reaction of Int0 with H2O converted to (P, S)-3a in 71% yield with 93% ee. The use of ent-CPA4 for the reaction led to (M, R)-3a in 90% ee with >20:1 dr. It should be noted that lower yield and ee would be obtained when the reactions proceeded in the absence of extra H2O in the system. All these results indicated that Int0 is the key intermediate of the annulation, and the enantioselectivity and diastereoselectivity are derived from the transformation of Int0 with H2O to 3a.
 | ||
| Fig. 2 Mechanistic studies. (a) Control experiments. (b) Deuterium- and 18O-labeling experiments. (c) Proposed reaction pathway. (d) Enantio-determining-step investigation. | ||
To probe the isotope introduction step of the reaction, we then performed D- and 18O-labeling experiments (Fig. 2b). The reaction of D-1a with 2a resulted in 3a in 90% yield and 94% ee, but with no D-incorporation observed in 3a. In addition, the reaction of Int0 with D2O or H218O yielded 4a or 5a with excellent D- or 18O-incorporation, respectively. These results suggested that H2O participated in the reaction and is responsible for the isotope introduction of isoindolinones. Finally, we explored the origin of enantioselectivity. Based on these results, we proposed that the reaction may proceed via concerted or stepwise annulation (Fig. 2c). However, currently, the isolation of this intermediate ii or iii has failed. Nevertheless, we explored the reaction of 1a with 9H-carbazol-9-amine 6 since achiral iii-6a would be generated in this case (Fig. 2d). As a result, compound 7 was obtained in 77% yield but only with 7% ee. This result indicated that the generated axial chirality is a key factor for the efficient synthesis of isoindolinones 3 in high ee. Thus, the origin of enantioselectivity for the reaction of 1 and 2 is mainly attributed to the axially chiral generation, derived from the chiral intermediate ii (Fig. 2c). Our calculation suggested that the rotational barrier of ii at room temperature is 30.7 kcal mol−1, indicating that the axial chirality is generated in this step. Furthermore, the high diastereoselectivity is attributed to the stereospecific intramolecular isomerization induced by axial information rather than the chiral induction catalyzed by CPA.
DFT studies were then performed to gain deep insight into the reaction mechanism. We first examined whether the annulation of Int0 and H2O occurred stepwise or concertedly to afford the intermediate ii (Fig. 2c). The energy profiles of the stepwise process are shown in Fig. 3, and the coordination of Int0 with CPA4 was selected as the reference point. With H2O participating in the reaction, the dual H-bonding effect was generated with an energy barrier of 16.0 kcal mol−1 (Int0 + H2O). The Re-face attack of the amine by water formed Int1Rvia transition state TS1Re with an energy barrier of 24.5 kcal mol−1, whereas the Si-face attack formed Int6Svia transition state TS6Si with a higher energy barrier of 28.4 kcal mol−1. Thus, Int1R is the more favorable intermediate, which undergoes sequential N-nucleophilic addition to the carbonyl group. In this case, four intermediates Int2–5 may be generated accordingly, with Int31S-Sa-3R was superior to others because of the relatively lower energy barrier of TS31Re-Sa-3R (24.3 kcal mol−1).
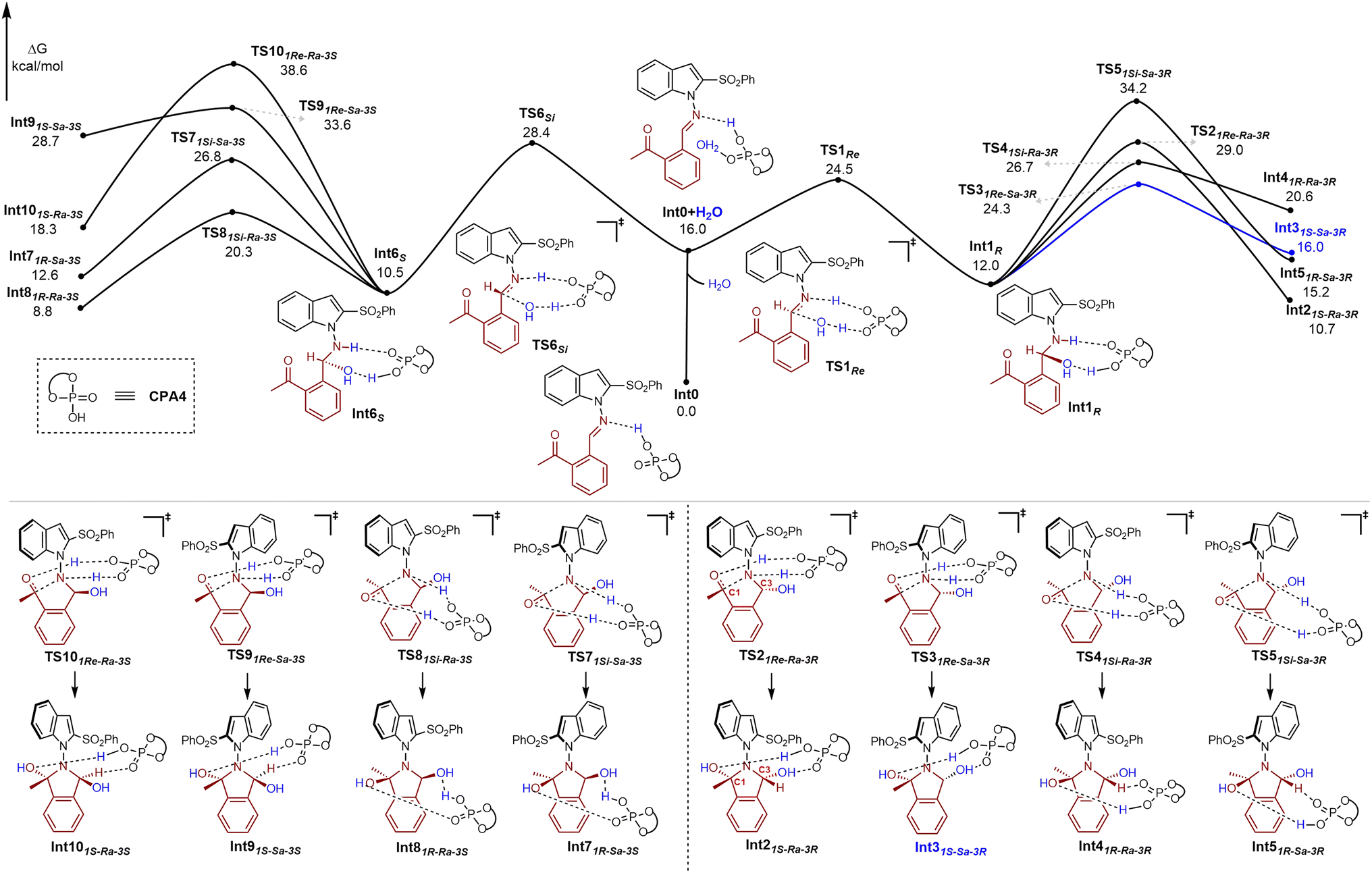 | ||
| Fig. 3 DFT studies of stepwise annulation. Free energy diagrams of the chiral phosphoric acid-catalyzed stepwise mechanism in asymmetric annulation. Gibbs free energy obtained at the M06-2X/def2-TZVPP(SMD, Toluene)//M06-2X/def2-SVP level. The meaning of the corner mark in the structure name: Re: the rectus face; Si: the sinister face; Ra: the (potential) R-configuration for the N–N axis; Sa: the (potential) S-configuration for the N–N axis; R: the R-configuration for the central chirality; S: the S-configuration for the central chirality. | ||
Alternatively, if concerted annulation occurred, only four pathways might be experienced via transition states TS111Re-Ra-3Re, TS121Si-Ra-3Si, TS171Re-Sa-3Re or TS181Si-Sa-3Si (Fig. 4). This is mainly because the bulky indole moiety existing in Int0 induced exclusive suprafacial activation by CPA4. In this regard, the annulation that proceeded viaTS181Si-Sa-3Si has an overcome energy barrier of 30.5 kcal mol−1, a finding that is not possible. On the other hand, although Int31S-Sa-3R could be easily afforded viaTS31Re-Sa-3R, the sequential dehydration step has an extremely high energy barrier of 38.8 kcal mol−1viaTS191S-Sa-3R, which is not possible either. This result also ruled out the only possible stepwise pathway that occurred from Int31S-Sa-3R (Fig. 3, blue line). In contrast, our calculation revealed that Int21S-Ra-3R and Int81R-Ra-3S are more favorable intermediates, which are generated viaTS111Re-Ra-3Re and TS121Si-Ra-3Si with energy barriers of 23.4 and 24.3 kcal mol−1, respectively. However, the dehydration from Int21S-Ra-3R required overcoming a relatively higher energy barrier than that of Int81R-Ra-3S. Therefore, the reaction preferred to undergo dehydration viaTS141R-Ra-3S to afford Int11P. The intramolecular isomerization then occurred viaTS15P-Re to afford (P, S)-3a with an energy barrier of 26.9 kcal mol−1. This result is consistent with our experimental observation that isoindolinone 3a was obtained in preferential (P, S) configuration.
 | ||
| Fig. 4 DFT studies of concerted annulation. Free energy diagrams of the chiral phosphoric acid-catalyzed concerted mechanism for asymmetric annulation, and the investigation of enantioselectivity. M and P were used to replace the R and S configurations of axial chirality, respectively. The structures of Int2, Int3, Int7 and Int8 are shown in Fig. 3. | ||
Finally, synthetic transformations of isoindolinones 3a were performed (Fig. 5). First, the large-scale experiment of 3a was carried out, which afforded 3a in 80% yield with 95% ee. Interestingly, treatment of 3a with LiAlH4 resulted in semi-reduction, affording centrally chiral product 8 in 95% ee with > 20:1 dr. When the reduction was performed with diisobutylaluminium hydride (DIBAL-H) in THF, complete reduction of the carbonyl moiety was observed, producing compound 9 in 85% yield with 90% ee. Additionally, the bromo group was easily introduced via the bromination of 3a with NBS, affording 10 in 76% yield with 91% ee. The stereospecific addition of 3a could be achieved by using MeMgCl, which furnished 11 in 53% yield with 95% ee and >20:1 dr.
 | ||
| Fig. 5 Synthetic transformations. | ||
Conclusions
In conclusion, we have reported the atroposelective [4+1] annulation of ketoaldehydes with 1H-indol-1-amines for the synthesis of centrally and axially chiral isoindolinones bearing N–N axes. By subjecting D2O or H218O to the reaction system, a series of isotopically chiral isoindolinones could be easily obtained with D- or 18O-incorporation in good to excellent levels, respectively. Control experiments and DFT studies indicated that the reaction proceeded through condensation to E-imine intermediates, followed by nucleophilic addition-driven CPA-catalyzed enantioselective cyclization and isomerization. We anticipate that the strategy herein could inspire more studies on the synthesis of isotopically chiral molecules with multiple chiral elements.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
H. Y. conceived and designed the experiment. G. J., Z. L.-H. and Z. H.-F. performed experiments and collected the data. G. J. provided the DFT calculations. H. Y. wrote the manuscript with the revision of all authors.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (22201131) and the Natural Science Foundation of Jiangsu Province (BK20220137).Notes and references
- (a) C. S. McCaughey, J. A. van Santen, J. J. J. van der Hooft, M. H. Medema and R. G. Linington, Nat. Chem. Biol., 2022, 18, 295–304 CrossRef CAS PubMed; (b) M. Miyagi and K. C. S. Rao, Mass Spectrom. Rev., 2007, 26, 121–136 CrossRef CAS PubMed; (c) X. Tian, H. P. Permentier and R. Bischoff, Mass Spectrom. Rev., 2023, 42, 546–576 CrossRef CAS PubMed; (d) X. Chen, Y. Sun, T. Zhang, L. Shu, P. Roepstorff and F. Yang, Genomics, Proteomics Bioinf., 2021, 19, 689–706 CrossRef PubMed; (e) J. Liu, Y. Shan, Y. Zhou, Z. Liang, L. Zhang and Y. Zhang, Trac. Trends Anal. Chem., 2020, 124, 115815 CrossRef CAS; (f) S. Kopf, F. Bourriquen, W. Li, H. Neumann, K. Junge and M. Beller, Chem. Rev., 2022, 122, 6634–6718 CrossRef CAS PubMed; (g) J. Atzrodt, V. Derdau, W. J. Kerr and M. Reid, Angew. Chem., Int. Ed., 2018, 57, 1758–1784 CrossRef CAS PubMed; (h) A. Labiche, A. Malandain, M. Molins, F. Taran and D. Audisio, Angew. Chem., Int. Ed., 2023, 62, e202303535 CrossRef CAS PubMed.
- (a) R. M. C. Di Martino, B. D. Maxwell and T. Pirali, Nat. Rev. Drug Discovery, 2023, 22, 562–584 CrossRef CAS PubMed; (b) T. Pirali, M. Serafini, S. Cargnin and A. A. Genazzani, J. Med. Chem., 2019, 62, 5276–5297 CrossRef CAS PubMed; (c) T. M. Belete, Drug Des., Dev. Ther., 2022, 16, 3465–3472 CrossRef CAS PubMed; (d) T. G. Gant, J. Med. Chem., 2014, 57, 3595–3611 CrossRef CAS PubMed.
- S. H. DeWitt and B. E. Maryanoff, Biochemistry, 2018, 57, 472–473 CrossRef CAS PubMed.
- (a) S. M. Hoy, Drugs, 2022, 82, 1671–1679 CrossRef CAS PubMed; (b) T. M. Truong, G. N. Pathak, A. Singal, V. Taranto and B. K. Rao, Ann. Pharmacother., 2024, 58, 416–427 CrossRef CAS PubMed.
- For selected examples on the synthesis of deuterated drugs, see: (a) H. M. C. Mouli, A. Vinod, S. Kumari, A. K. Tiwari, M. K. Kathiravan, V. Ravichandiran and R. Peraman, Bioorg. Chem., 2023, 135, 10649 Search PubMed; (b) F. Liu, B. Wang, Y. Liu, W. Shi, X. Tang, X. Wang, Z. Hu, Y. Zhang, Y. Guo, X. Chang, X. He, H. Xu and Y. He, ACS Med. Chem. Lett., 2022, 13, 1730–1738 CrossRef CAS PubMed; (c) F. Liu, B. Wang, Y. Liu, W. Shi, Z. Hu, X. Chang, X. Tang, Y. Zhang, H. Xu and Y. He, Bioorg. Med. Chem. Lett., 2023, 86, 129235 Search PubMed; (d) Z. Zhang, Y. Lv, W. Renee Ong, X. Zhao, Z. Jia and T.-P. Loh, Angew. Chem., Int. Ed., 2024, 63, e202408509 CrossRef CAS PubMed; (e) P. Ma, T. Guo and H. Lu, Nat. Commun., 2024, 15, 10190 CrossRef CAS PubMed.
- (a) S. De Witt, A. W. Czarnik and V. Jacques, ACS Med. Chem. Lett., 2020, 11, 1789–1792 CrossRef CAS PubMed; (b) B. Barabás, L. Caglioti, K. Micskei, C. Zucchi and G. Pályi, Origins Life Evol. Biospheres, 2008, 38, 317–327 CrossRef PubMed; (c) X. Chang, X. Cheng and C.-J. Wang, Chem. Sci., 2022, 13, 4041–4049 RSC; (d) J. S. Rowbotham, M. A. Ramirez, O. Lenz, H. A. Reeve and K. A. Vincent, Nat. Commun., 2020, 11, 1454 CrossRef CAS PubMed; (e) A. Li, X. Song, Q. Ren, P. Bao, X. Long, F. Huang, L. Yuan, J. Steve Zhou and X. Qin, Angew. Chem., Int. Ed., 2023, 62, e202301091 CrossRef CAS PubMed; (f) S. Miwa, R. Senda, K. Saito, A. Sato, Y. Nakamura and O. Kitagawa, J. Org. Chem., 2022, 87, 13501–13507 CrossRef CAS PubMed; (g) K. Saito, S. Miwa, A. Iida, Y. Fujimoto, E. Caytan, C. Roussel and O. Kitagawa, Org. Lett., 2021, 23, 7492–7496 CrossRef CAS PubMed.
- (a) R. Senda, Y. Watanabe, S. Miwa, A. Sato and O. Kitagawa, J. Org. Chem., 2023, 88, 9579–9583 CrossRef CAS PubMed; (b) Z. Zhu, X. Wu, G. T. Bida, H. Deng, X. Ma, S. Qian, Z. Wu, Z. Li and D. A. Nicewicz, Nat. Synth., 2024, 4, 97–105 CrossRef; (c) T. Miura, T. Nakamuro, Y. Nagata, D. Moriyama, S. G. Stewart and M. Murakami, J. Am. Chem. Soc., 2019, 141, 13341–13345 CrossRef CAS PubMed; (d) R. H. Beddoe, D. C. Edwards, L. Goodman and R. M. Denton, Chem. Commun., 2020, 56, 6480–6483 RSC; (e) D. Chen, L. Xu, Z. Wang and C. Liu, Chem, 2023, 9, 3212–3223 CrossRef CAS; (f) Z. A. Tolchin and J. M. Smith, J. Am. Chem. Soc., 2024, 146, 2939–2943 CrossRef CAS PubMed; (g) H. M. H. Nguyen, D. C. Thomas, M. A. Hart, K. R. Steenback, J. N. Levy and A. McNally, J. Am. Chem. Soc., 2024, 146, 2944–2949 CrossRef CAS PubMed; (h) G. L. Bartholomew, S. L. Kraus, L. J. Karas, F. Carpaneto, R. Bennett, M. S. Sigman, C. S. Yeung and R. Sarpong, J. Am. Chem. Soc., 2024, 146, 2950–2958 CrossRef CAS PubMed; (i) P. Xu, H.-Q. Jiang, H. Xu, S. Wang, H.-X. Jiang, S.-L. Zhu, L. Yin, D. Guo and X. Zhu, Chem. Sci., 2024, 15, 13041–13048 RSC; (j) X. Li, J. Zhou, W. Deng, Z. Wang, Y. Wen, Z. Li, Y. Qiu and Y. Huang, Chem. Sci., 2024, 15, 11418–11427 RSC.
- For selected papers by using D2O as the reactant to access D-stereogenic compounds, see: (a) L.-W. Zhan, C.-J. Lu, J. Feng and R.-R. Liu, Angew. Chem., Int. Ed., 2023, 62, e202312930 CrossRef CAS PubMed; (b) Q. Shi, M. Xu, R. Chang, D. Ramanathan, B. Peñin, I. Funes-Ardoiz and J. Ye, Nat. Commun., 2022, 13, 4453 Search PubMed; (c) W. Kong, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2017, 56, 3987–3991 Search PubMed; (d) Y. Yan, J. Yang, L. Wang, D. Xu, Z. Yu, X. Guo, G. P. Horsman, S. Lin, M. Tao and S.-X. Huang, Chem. Sci., 2020, 11, 3959–3964 RSC.
- For selected papers on the synthesis of chiral isoindolinones, see: (a) L. Chen and Y.-X. Zou, Adv. Synth. Catal., 2021, 363, 4159–4176 CrossRef CAS; (b) R. Savela and C. Méndez-Gálvez, Chem.–Eur. J., 2021, 27, 5344–5378 CrossRef CAS PubMed; (c) S. Samanta, S. A. Ali, A. Bera, S. Giri and K. Samanta, New J. Chem., 2022, 46, 7780–7830 RSC; (d) V. Bisai, A. Suneja and V. K. Singh, Angew. Chem., Int. Ed., 2014, 53, 10737–10741 CrossRef CAS PubMed; (e) Y. Zhang, Y.-F. Ao, Z.-T. Huang, D.-X. Wang, M.-X. Wang and J. Zhu, Angew. Chem., Int. Ed., 2016, 55, 5282–5285 CrossRef CAS PubMed; (f) C. Min, Y. Lin and D. Seidel, Angew. Chem., Int. Ed., 2017, 56, 15353–15357 CrossRef CAS PubMed; (g) D. Augner, D. C. Gerbino, N. Slavov, J.-M. Neudörfl and H.-G. Schmalz, Org. Lett., 2011, 13, 5374–5377 CrossRef CAS PubMed; (h) X. Yu, A. Lu, Y. Wang, G. Wu, H. Song, Z. Zhou and C. Tang, Eur. J. Org Chem., 2011, 2011, 892–897 CrossRef; (i) A. Suneja, V. Bisai and V. K. Singh, J. Org. Chem., 2016, 81, 4779–4788 CrossRef CAS PubMed; (j) S. Lebrun, R. Sallio, M. Dubois, F. Agbossou-Niedercorn, E. Deniau and C. Michon, Eur. J. Org Chem., 2015, 2015, 1995–2004 CrossRef CAS; (k) A. Di Mola, M. Tiffner, F. Scorzelli, L. Palombi, R. Filosa, P. D. Caprariis, M. Waser and A. Massa, Beilstein J. Org. Chem., 2015, 11, 2591–2599 CrossRef CAS PubMed; (l) A. Beriša, D. Glavač, C. Zheng, S.-L. You and M. Gredičak, Org. Chem. Front., 2022, 9, 428–435 RSC; (m) P. Mukherjee, A. Sairaman, H. J. Deka, S. Jain, S. K. Mishra, S. Roy, P. Bhaumik and D. Maiti, Nat. Synth., 2024, 3, 835–845 CrossRef CAS; (n) G. Yang, C. Shen and W. Zhang, Angew. Chem., Int. Ed., 2012, 51, 9141–9145 CrossRef CAS PubMed; (o) M.-W. Chen, Q.-A. Chen, Y. Duan, Z.-S. Ye and Y.-G. Zhou, Chem. Commun., 2012, 48, 1698–1700 RSC; (p) W.-J. Cui, Z.-J. Wu, J. Gu and S.-L. You, J. Am. Chem. Soc., 2020, 142, 7379–7385 CrossRef CAS PubMed; (q) K. Fang, W. Huang, C. Shan, J. Qu and Y. Chen, Org. Lett., 2021, 23, 5523–5527 CrossRef CAS PubMed; (r) M.-Y. Teng, Y.-J. Wu, J.-H. Chen, F.-R. Huang, D.-Y. Liu, Q.-J. Yao and B.-F. Shi, Angew. Chem., Int. Ed., 2024, 63, e202318803 CrossRef CAS PubMed.
- R. Appel, S. Chelli, T. Tokuyasu, K. Troshin and H. Mayr, J. Am. Chem. Soc., 2013, 135, 6579–6587 CrossRef CAS PubMed.
- For selected papers on the synthesis of N-N atropisomers bearing two chiral elements, see: (a) J. Feng and R.-R. Liu, Chem.–Eur. J., 2024, 30, e202303165 Search PubMed; (b) X.-F. Bai, Y.-M. Cui, J. Cao and L.-W. Xu, Acc. Chem. Res., 2022, 55, 2545–2561 Search PubMed; (c) H.-H. Zhang, T.-Z. Li, S.-J. Liu and F. Shi, Angew. Chem., Int. Ed., 2024, 63, e202311053 CrossRef CAS PubMed; (d) A. Gaucherand, E. Yen-Pon, A. Domain, A. Bourhis, J. Rodriguez and D. Bonne, Chem. Soc. Rev., 2024, 53, 11165–11206 RSC; (e) S.-J. Wang, X. Wang, X. Xin, S. Zhang, H. Yang, M. W. Wong and S. Lu, Nat. Commun., 2024, 15, 518 CrossRef CAS PubMed; (f) W.-T. Wang, S. Zhang, W. Lin, Z.-H. Luo, D. Hu, F. Huang, R. Bai, Y. Lan, L. Qian and J.-Y. Liao, Org. Chem. Front., 2024, 11, 3308–3319 RSC; (g) T.-T. Wang, J. Cao and X. Li, Org. Lett., 2024, 26, 6179–6184 CrossRef CAS PubMed; (h) S. S. Ranganathappa, B. S. Dehury, G. K. Singh, S. Shee and A. T. Biju, ACS Catal., 2024, 14, 6965–6972 CrossRef CAS; (i) C. Portolani, G. Centonze, S. Luciani, A. Pellegrini, P. Righi, A. Mazzanti, A. Ciogli, A. Sorato and G. Bencivenni, Angew. Chem., Int. Ed., 2022, 61, e202209895 CrossRef CAS PubMed; (j) X. Wang, S.-J. Wang, X. Xin, H. An, Z. Tu, H. Yang, M. W. Wong and S. Lu, Chem. Sci., 2024, 15, 13240–13249 RSC; (k) F.-B. Ge, C.-J. Lu, X. Chen, W. Yao, M. An, Y.-K. Jiang, L.-P. Xu and R.-R. Liu, Angew. Chem., Int. Ed., 2024, 63, e202400441 CrossRef CAS PubMed; (l) T.-J. Han, Q.-L. Yang, J. Hu, M.-C. Wang and G.-J. Mei, JACS Au, 2024, 4, 4445–4454 CrossRef CAS PubMed; (m) Y. Wang, X. Zhu, D. Pan, J. Jing, F. Wang, R. Mi, G. Huang and X. Li, Nat. Commun., 2023, 14, 4661 CrossRef CAS PubMed.
- For selected papers on the synthesis of N–N atropisomers, see: (a) G. Centonze, C. Portolani, P. Righi and G. Bencivenni, Angew. Chem., Int. Ed., 2023, 62, e202303966 CrossRef PubMed; (b) Z.-H. Chen, T.-Z. Li, N.-Y. Wang, X.-F. Ma, S.-F. Ni, Y.-C. Zhang and F. Shi, Angew. Chem., Int. Ed., 2023, 62, e202300419 CrossRef CAS PubMed; (c) K.-W. Chen, Z.-H. Chen, S. Yang, S.-F. Wu, Y.-C. Zhang and F. Shi, Angew. Chem., Int. Ed., 2022, 61, e202116829 CrossRef CAS PubMed; (d) Y. Gao, L.-Y. Wang, T. Zhang, B.-M. Yang and Y. Zhao, Angew. Chem., Int. Ed., 2022, 61, e202200371 CrossRef CAS PubMed; (e) C.-S. Wang, Q. Xiong, H. Xu, H.-R. Yang, Y. Dang, X.-Q. Dong and C.-J. Wang, Chem. Sci., 2023, 14, 12091–12097 RSC; (f) Q. Huang, Y. Li, C. Yang, W. Wu, J. Hai and X. Li, Org. Chem. Front., 2024, 11, 726–734 RSC; (g) T.-Z. Li, S.-F. Wu, N.-Y. Wang, C.-S. Hong, Y.-C. Zhang and F. Shi, J. Org. Chem., 2024, 89, 12559–12575 Search PubMed; (h) Z. Huang, Y. Xu, W. Lin, R. Qian, W. Zhang and X. Li, Org. Chem. Front., 2024, 11, 5437–5442 RSC; (i) J. Wang, D. Pan, F. Wang, S. Yu, G. Huang and X. Li, Sci. Adv., 2024, 10, eado4489 CrossRef CAS PubMed; (j) X.-M. Wang, P. Zhang, Q. Xu, C.-Q. Guo, D.-B. Zhang, C.-J. Lu and R.-R. Liu, J. Am. Chem. Soc., 2021, 143, 15005–15010 CrossRef CAS PubMed; (k) G.-J. Mei, J. J. Wong, W. Zhang, A. A. Nangia, K. N. Houk and Y. Lu, Chem, 2021, 7, 2743–2757 CrossRef CAS; (l) S.-Y. Yin, Q. Zhou, C.-X. Liu, Q. Gu and S.-L. You, Angew. Chem., Int. Ed., 2023, 62, e202305067 CrossRef CAS PubMed; (m) X. Zhu, H. Wu, Y. Wang, G. Huang, F. Wang and X. Li, Chem. Sci., 2023, 14, 8564–8569 RSC.
- (a) J. E. Smyth, N. M. Butler and P. A. Keller, Nat. Prod. Rep., 2015, 32, 1562–1583 RSC; (b) S. R. LaPlante, L. D. Fader, K. R. Fandrick, D. R. Fandrick, O. Hucke, R. Kemper, S. P. F. Miller and P. J. Edwards, J. Med. Chem., 2011, 54, 7005–7022 CrossRef CAS PubMed; (c) M. Basilaia, M. H. Chen, J. Secka and J. L. Gustafson, Acc. Chem. Res., 2022, 55, 2904–2919 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and characterization data for all new compounds, computational methods, and Cartesian coordinates. CCDC 2359270, 2354174, 2362138, and 2393057. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc00594a |
| This journal is © The Royal Society of Chemistry 2025 |