
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh-capacity organic cathode boosted by coordination chemistry for energy-dense aqueous zinc-organic batteries†
Guanzhong
Ma‡
a,
Zhengyu
Ju‡
b,
Yutong
Chen
a,
Runmo
Wang
a,
Zihao
Yuan
a,
Huiping
Du
a,
Mian
Cai
 a,
Meng
Gao
a,
Yaqun
Wang
*a and
Guihua
Yu
*b
a,
Meng
Gao
a,
Yaqun
Wang
*a and
Guihua
Yu
*b
aCollege of Energy Storage Technology, Shandong University of Science and Technology, Qingdao 266590, China. E-mail: yqwang@sdust.edu.cn
bMaterials Science and Engineering Program, Walker Department of Mechanical Engineering, The University of Texas at Austin, TX 78712, USA. E-mail: ghyu@austin.utexas.edu
First published on 26th February 2025
Abstract
N-type organic cathode materials containing carbonyl and imine groups have emerged as promising candidates for zinc-ion batteries due to their excellent charge storage capability, which arise from the synergic storage of both Zn2+ and H+. However, an increase in active sites also complicates the synthesis, introduces complex multi-electron reactions, and hinders comprehensive understanding of the charge storage mechanism and the evolution of molecular configuration during the electrochemical process. Herein, a 10-electron transfer organic cathode material, featuring imine and quinone groups that are spaced apart, was synthesized in one-step. Its highly conjugated molecular structure promotes electron delocalization, thereby enhancing the stability. The competitive storage mechanism of Zn2+ and H+ was unveiled through multiple quasi in situ spectroscopy techniques and calculations, revealing that Zn2+ are initially coordinated to form O–Zn–N, followed by the co-insertion of H+/Zn2+ during the reduction of the carbonyl groups. Thanks to the Zn2+/H+ co-insertion and coordination stabilization, an ultra-high capacity of 445 mA h g−1 at a current density of 0.2 A g−1 and a retained capacity of 200 mA h g−1 (>80% capacity retention) at 10 A g−1 after 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles can be achieved. The molecular structure-related charge storage mechanism revealed in this study can provide useful design considerations for realizing high-capacity, fast-charging and long-duration organic cathodes for various energy storage systems.
000 cycles can be achieved. The molecular structure-related charge storage mechanism revealed in this study can provide useful design considerations for realizing high-capacity, fast-charging and long-duration organic cathodes for various energy storage systems.
Introduction
With the growing research on aqueous zinc-ion batteries (AZIBs), the design of cathode materials has emerged as a key factor determining the high performance of these batteries.1–3 Significant improvements in charging and discharging performance have been observed when organic materials, such as stable organic radicals,4 conductive polymers,5 triphenylamine,6 dinitrobenzene,7 and phenothiazine,8 are used as cathodes.9–11 Among various organic cathode materials for AZIBs, imine (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) and carbonyl (CO) organics have garnered significant interest owing to their unique molecular structure characterized by stability and designability.12–14 Carbonyl compounds are widely used in AZIBs due to their high theoretical specific capacity and ease with which they can be modified by chemical and physical methods.15,16 However, some carbonyl compounds are unstable in aqueous environments and tend to dissolve, resulting in instability.17 Imine compounds offer notable advantages as cathodes in AZIBs, including higher activity during charging and discharging, which suggests faster reaction kinetics.18,19 However, unmodified imine compounds often exhibit lower discharge voltages when employed as cathodes in AZIBs.20,21 Moreover, repeated charging and discharging cycles can lead to structure changes, highlighting the need for improved electrochemical stability.22,23
N) and carbonyl (CO) organics have garnered significant interest owing to their unique molecular structure characterized by stability and designability.12–14 Carbonyl compounds are widely used in AZIBs due to their high theoretical specific capacity and ease with which they can be modified by chemical and physical methods.15,16 However, some carbonyl compounds are unstable in aqueous environments and tend to dissolve, resulting in instability.17 Imine compounds offer notable advantages as cathodes in AZIBs, including higher activity during charging and discharging, which suggests faster reaction kinetics.18,19 However, unmodified imine compounds often exhibit lower discharge voltages when employed as cathodes in AZIBs.20,21 Moreover, repeated charging and discharging cycles can lead to structure changes, highlighting the need for improved electrochemical stability.22,23
The integration of these two functional groups into a single compound appears to effectively address these challenges.24,25 In recent years, cathode materials featuring CO and CN bonds as dual active sites for energy storage in AZIBs have been widely reported. For example, Shi et al.26 reported a four-electron organic cathode material, benzo[b]phenazine-6,11-dione (BPD), which exhibited a discharge specific capacity of up to 429 mA h g−1 in a 2 M ZnSO4 aqueous electrolyte (at a current density of 0.05 A g−1). Song's team introduced a six-electron cathode material, 5,7,12,14-tetraaza-6,13-pentacenequinone (TAPQ), which demonstrated a discharge specific capacity of up to 443 mA h g−1.27 Chen et al.28 carefully considered the binding effects of CO and CN on zinc ions and designed a twelve-electron material with a triangular planar structure, hexaazatrinaphthalene-quinone (HATNQ), which exhibited strong electrochemical stability. To improve material stability and ion transport capability, Peng et al.29 designed a cathode material with dual active units, dipyrido[3′,2′:5,6;2′′,3′′:7,8]quinoxalino[2,3-i]dipyrido[3,2-a:2′,3′-c]phenazine-10,21-dione (DQDPD), through a strategy of expanding the π-conjugation of N-heterocycles. The synergistic effect of these two functional groups enhances the discharge specific capacity of ZIBs and improves their interfacial chemical stability. Consequently, designing and synthesizing electrode materials with a high density of active units is a crucial approach to improving the electrochemical performance of ZIBs.30,31 One key strategy involves maximizing the number of active sites per molecule to enhance the theoretical specific capacity of the electrode material. However, as the number of active sites increase, it becomes increasingly challenging to synthesize the materials in a straightforward manner. Furthermore, while many reports have partially explored the molecular design of CO- and CN-based compounds, a clear understanding of the insertion mechanisms of Zn2+ and H+ when both functional groups coexist in a single molecule remains elusive.32
In this work, the reaction of 2,3-diaminonaphthalene-1,4-dione (DANQ) and 2,3,5,6-tetrachlorocyclohexa-2,5-diene-1,4-dione (TCBQ) was conducted via a facile one-pot method, synthesizing a 10-electron organic compound named 6,8,15,17-tetrahydrobenzo[b]benzo[6,7] quinoxalino[2,3-i]phenazine-5,7,9,14,16,18-hexaone (TBQPH). The synthetic compound consists of six p-benzoquinone structures connected by four imine groups, supplying sufficient coordination sites for H+/Zn2+ in the electrochemical reaction. As the positive electrode material for aqueous ZIBs in 1 M zinc sulphate (ZnSO4) electrolyte, the TBQPH@GO electrode demonstrated an extraordinary capacity of 445 mA h g−1 at a current density of 0.2 A g−1. Even under a high current density of 10 A g−1, a high capacity of 200 mA h g−1 could be retained. Moreover, a remarkable capacity retention of ∼80% after extensive cycling for 15000 cycles could be achieved. Such high-capacity retention could be attributed to the insoluble property of the molecule, as validated by in situ ultraviolet (UV) spectroscopy. Furthermore, the charge storage mechanism of TBQPH was systematically investigated using quasi in situ X-ray photoelectron spectroscopy (XPS) and Fourier transform infrared spectroscopy (FTIR) techniques. A H+/Zn2+ co-insertion mechanism was revealed for TBQPH in 1 M ZnSO4 electrolyte. In addition, the reaction order of H+/Zn2+ ions was unveiled via electrochemical analysis and theoretical calculations. To sustain the charge storage mechanism of TBQPH, Fukui function (FUKUI) calculations were performed, showing the preferential central carbonyl group as the reactive site in the molecule. Density functional theory (DFT) calculations were also conducted to calculate Gibbs free energy (ΔG) for the redox reaction, verifying that the first reduction platform is contributed by Zn2+ coordination, while subsequent platforms are attributed to the co-insertion of Zn2+ and H+ which leads to a 10-electron transfer reaction in TBQPH@GO. This organic electrode based on TBQPH@GO offers enhanced capacity and stability for ZIBs towards increased performance and practicality.
Results and discussion
Herein, we have designed a straightforward synthetic procedure for TBQPH, a planar organic molecule with conjugated carbonyl and imine groups, via the ammonolysis reaction between DANQ and TCBQ (Fig. 1A). By comparison, it was found that fully conjugated TBQPH with a super-electronic off-domain has a smaller energy gap (Eg) (1.186 eV) between the LUMO and the HOMO, which gives it higher conductivity for fast electron transfer and rapid redox kinetics (Fig. 1B). The decrease in the energy gap can be correlated with the high degree of electron conjugation and electron delocalization in TBQPH, which is further supported by its localized orbital locator profiles (Fig. 1E).33 Through 1H and 13C nuclear magnetic resonance (NMR) spectroscopy, the successful synthesis of TBQPH was verified (Fig. 1F, ESI Appendix, Fig. S1†). The chemical structure of TBQPH was characterized by FTIR (ESI Appendix, Fig. S2†). The prominent peaks located at the wavelengths of ∼1700 and ∼1250 cm−1 can be attributed to the bending vibration of CO and C–N bonds, respectively, in the units of TBQPH. The presence of CO and C–N functional groups was further identified by XPS (Fig. 1C and D). In the N 1s spectra, the peaks located at 399 and 402 eV correspond to the N–H and C–N bonding, respectively (Fig. 1C). Notably, in the C 1s spectra, besides the apparent C–C, CC, C–N and CO peaks from the molecular structure of TBQPH, a distinct π–π stacked structure at 289 eV can also be observed (Fig. 1D). Such π–π interaction can be attributed to the stacking of planar TBQPH molecules, as characterized by transmission electron microscopy (TEM) (Fig. 1G and H). Layered features can be observed in the TEM image (Fig. 1G), and the interval between the molecular planes can be measured to be ∼0.32 nm (Fig. 1H). Irregular powdery structure with a micrometer particle size can be seen from the scanning electron microscopy (SEM) image due to the severe π–π stacking of the TBQPH molecule (ESI Appendix, Fig. S3†). The uniform elemental distribution of C, N and O can be clearly seen via EDS surface mapping (Fig. 1I), and the atomic ratio (C–O–N) was calculated to be 13:3:2, which is close to the ideal elemental ratio of TBQPH (ESI Appendix, Fig. S4†).
 | ||
| Fig. 1 Synthesis and structural characterization of TBQPH. (A) One-step synthesis procedure of TBQPH via the ammonolysis reaction. (B) DFT-calculated LUMO/HOMO energy levels of TADQ and TBQPH. (C and D) XPS spectra of N 1s (C) and C 1s (D) of TBQPH, respectively. (E) Colour-filled diagram of the molecular fixed-domain orbitals of TBQPH. (F) NMR images of TBQPH material: C atomic spectrum. (G and H) TEM images of TBQPH. (I) TEM image and the corresponding EDS mapping of C, N, and O in TBQPH. | ||
It is crucial for the ideal cathode materials to possess exceptional electrical conductivity, high reactivity, and remarkable stability.34,35 The reactivity and stability of the material can be directly related to its particle size and stacked structure. To fully exploit the material's reactivity and enhance its stability, graphene oxide (GO) was incorporated into the synthetic process through an in situ method, resulting in the formation of TBQPH@GO (Fig. 2A). The TBQPH nanoparticles grown in situ on GO exhibit no discernible structural changes induced by the substrate, as evidenced by a clear comparison of their FTIR and Raman spectra. Notably, TBQPH and TBQPH@GO exhibit nearly identical FTIR signals (Fig. 2B), while the presence of the D-peak and G-peak of GO weakens the intensity of Raman peaks for these materials (Fig. 2C). The XPS spectra demonstrate the consistency of the active unit material (Fig. 2D and E). It is noteworthy that TBQPH@GO exhibits a stronger π–π structure, attributed to the properties of GO rather than materials stacking or crystallization (Fig. 2D). This can be distinctly observed in SEM images (Fig. 2F), where smaller TBQPH particles grow on the surface of GO, resulting in improved dispersion and a larger contact area for ionic interaction, thereby facilitating the activity of TBQPH. The XRD analysis reveals that the dispersion of the sample on GO enhances the π–π stacking interaction (ESI Appendix, Fig. S5†). Additionally, the exceptional electrochemical stability of GO significantly enhances the stability of TBQPH in electrolytes.
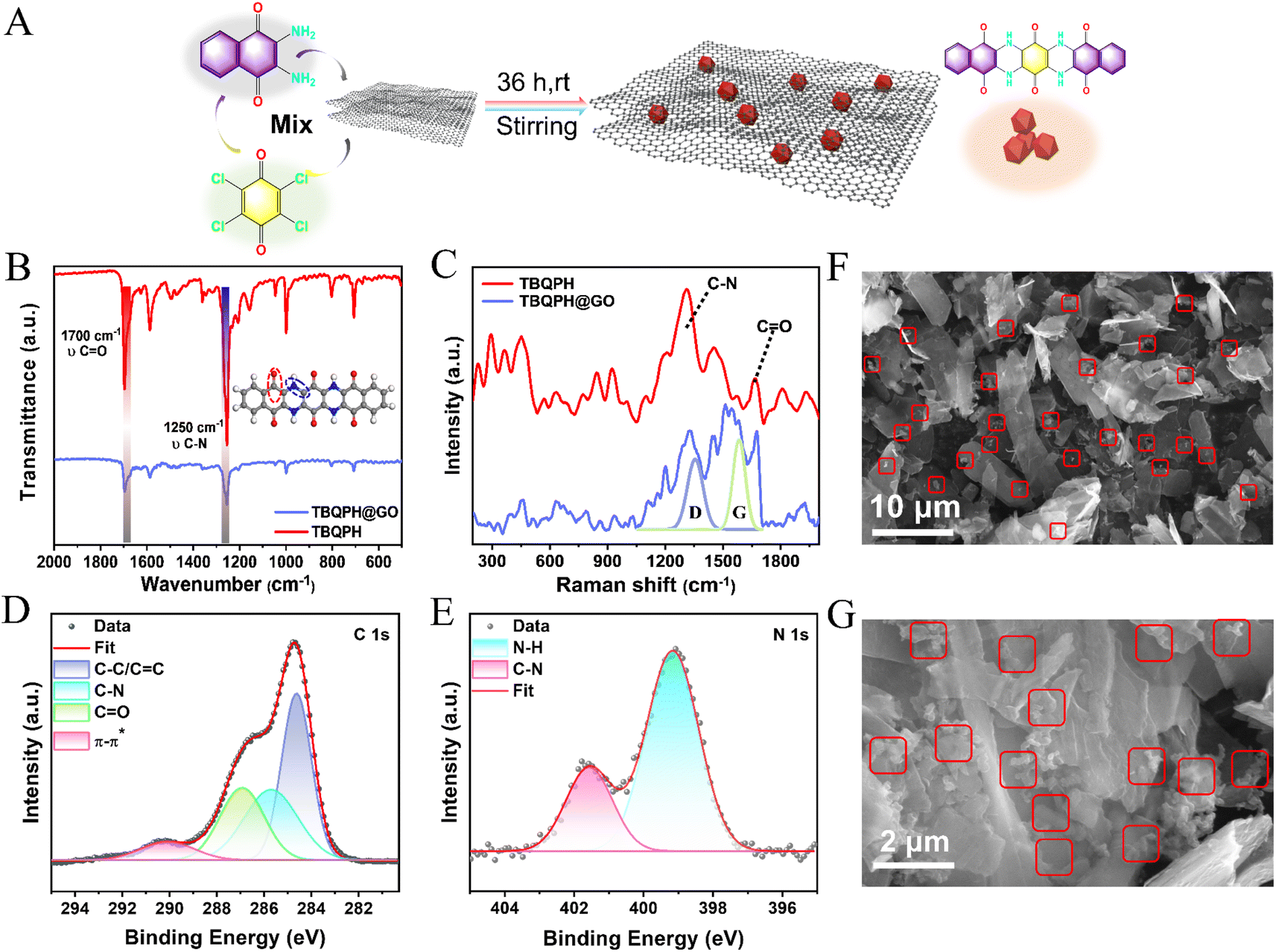 | ||
| Fig. 2 Synthesis and structural characterization of TBQPH@GO. (A) In situ synthetic procedure of TBQPH@GO. (B) FTIR spectra of TBQPH and TBQPH@GO. (C) Raman spectra of TBQPH and TBQPH@GO. (D and E) XPS spectra of C 1s (D) and N 1s (E) of TBQPH@GO. (F and G) SEM images of TBQPH@GO at different magnifications. | ||
The reaction mechanism of conjugated structures containing both carbonyl and imine groups has not been well understood in previous studies.36,37 Due to the polyionic nature of the aqueous electrolyte (containing both H+ and Zn2+), competition between H+ and Zn2+ occurs at the electrodes, resulting in a distinct order of charge storage in n-type organic materials. It is often hypothesized that the smaller H+ ions are more likely to approach the reaction active sites, while the coordinated N–Zn–O structure with enhanced stability leads to reduced energy levels upon electron acquisition. To elucidate the reaction mechanism of TBQPH, combined electrochemical characterization and DFT calculations were conducted. The electrostatic potential (ESP) method was employed to speculate the electrophilic and nucleophilic reaction-active sites to get insights into the active sites.38 In Fig. 3A, the red region represents the positive ESP value (electrophilic center) and the blue region stands for the negative ESP value (nucleophilic center). The sites with more negative ESP areas prefer electrophilic reactions, which are considered highly reactive sites. During the initial discharge, all the capacity can be attributed to the carbonyl group due to the presence of the hydrogen atom in the imine group (Fig. 3A). Fig. 3B shows a typical CV curve of the TBQPH@GO electrode in the first cycle. During the first discharge process, two reduction peaks at 0.55 and 0.80 V can be observed, indicating the reaction between carbonyl groups and Zn2+/H+ ions. Subsequently, during the charging process, in addition to the two oxidation peaks at 0.78 and 1.15 V, which correspond to the two reduction peaks in the discharge process, a third oxidation peak appears at 1.19 V (Fig. 3B), which represents the removal of the hydrogen atoms from the imine groups in TBQPH. Consequently, the electrostatic configuration surrounding the N atoms within the material undergoes molecular transitions to a lower state, indicating its oxidized state (Fig. 3A).
 | ||
| Fig. 3 Charge storage mechanism of TBQPH during stabilization by quasi in situ electrochemical and structural characterization. (A) Electrochemical redox reaction of TBQPH in the first cycle, with DFT-calculated electrostatic distributions of the pristine and stabilized states of TBQPH. (B) CV profile of TBQPH@GO. (C) GCD profile of TBQPH@GO. (D–F) Quasi in situ XPS spectra of O 1s (D), N 1s (E), and Zn 2p (F) of TBQPH@GO in the first charge–discharge cycle. | ||
The charge storage mechanism of TBQPH can be unveiled via quasi in situ XPS measurements, wherein the data points collected at different voltages during the first charge–discharge cycle are shown in Fig. 3C. It is worth noting that the shift of the XPS peaks arises from the electron gain and loss and the change of electron cloud density surrounding the redox material during the reaction. During the discharge of TBQPH, electrons accumulate in the material, increasing the electron cloud density and shifting the XPS peak towards a lower binding energy. Conversely, when TBQPH is charged, electrons are depleted, reducing the electron cloud density and shifting the XPS peak towards a higher binding energy. As seen from the O 1s spectra, the binding of O atoms on the carbonyl groups with Zn2+ and H+ can be clearly observed as the formation of C–O–Zn and C–O–H bonds in the initial discharge (Fig. 3D). However, there is no significant change as N atoms do not participate in this process (Fig. 3E). During the charging process of TBQPH, C–O–Zn and C–O–H bonds disappear, while CO bonds reform. Additionally, –NH– groups are oxidized to –N in the fully charged state (Fig. 3D and E). Furthermore, the change of Zn2+ states during the charge and discharge process was investigated (Fig. 3F). The intensity of Zn 2p peaks is enhanced upon discharge and weakened upon charge, indicating the insertion and removal of Zn2+ within the material. The slight peaks observed in the initial state originate from the electrolyte. In addition, the C 1s spectra clearly reveal the evolution of the active functional groups in different charge and discharge states (ESI Appendix, Fig. S6†). As the discharge proceeds, the CO bond proportion decreases and disappears, which is then restored upon charging. Moreover, the CN bond does not form in the discharge process but gradually appears during the charging process, which is well aligned with the conclusion from the N 1s spectra. Through the electrochemical and structural analysis of TBQPH during the first charge–discharge cycle, the charge storage mechanism, that is, the 6-electron discharge to the reduced state and 10-electron charge process to the stabilized oxidized state, can be clearly presented.
The charge storage mechanism of TBQPH was further thoroughly investigated to gain a comprehensive understanding of the subsequent redox processes after its stabilization. The Fukui function (FUKUI) calculations (ESI Appendix, Fig. S7 and Table S1†) were conducted on the oxidized state of TBQPH to analyze the specific reactive sites where nucleophilic attack first took place in the reduction process. The FUKUI calculation results reveal that the carbonyl oxygen atoms at the central position have the maximum f+ value, which have the highest reactivity with the electron acquisition preference. The significant difference between the first and second cycles can be observed in the CV curves (Fig. 4A and 3B). It is noted that three pairs of redox peaks can be observed at 0.50/0.78 V, 0.62/1.15 V and 0.85/1.19 V in the 2nd to 4th cycles (ESI Appendix, Fig. S8†). In addition, the GCD profiles of these cycles demonstrate that TBQPH was oxidized to a stable molecular configuration with three redox plateaus after the first cycle. Changes in the ion concentration in the ZnSO4 electrolyte affect the magnitude of the response current but do not influence the material's primary reaction (ESI Appendix, Fig. S9†). To minimize the impact of excessively high or low electrolyte concentrations on battery performance (such as the formation of basic zinc sulfate at high zinc ion concentrations), 1 M ZnSO4 electrolyte was chosen as the primary test concentration.
 | ||
| Fig. 4 Charge storage mechanism of TBQPH after stabilization with quasi in situ electrochemical and structural characterization. (A) CV profile of TBQPH@GO during the second cycle. (B) Galvanostatic discharge curves of TBQPH@GO//Zn cells in different electrolytes. (C) GCD profile of TBQPH@GO. (D and E) Quasi in situ XPS spectra of O 1s (D) and N 1s (E) of TBQPH@GO during the second charge–discharge cycle. (F) Quasi in situ FTIR spectra of TBQPH@GO at different voltages. (G) DFT calculations of Gibbs free energy of TBQPH for the insertion of Zn2+ and H+. (H) Images showing pH change with the indicator during charge–discharge cycles in a TBQPH@GO//Zn cell. | ||
To figure out whether the first reduction peak R1 is attributed to Zn2+ insertion, we conducted CV tests on the TBQPH@GO/Zn cell using a proton-free deep eutectic electrolyte of butanedinitrile and zinc perchlorate (ESI Appendix, Fig. S10†). The reduction peak at ∼0.8 V agrees with the insertion behavior observed in 1 M ZnSO4 electrolyte, implying that the R1 is related to Zn2+ insertion. Furthermore, charge–discharge tests were conducted on the TBQPH@GO/Zn cells in different electrolyte systems to investigate the insertion order of Zn2+ and H+ (Fig. 4B). It is evident that a consistent discharge plateau (approximately 0.95–1 V) can be observed in the 1 M ZnSO4 electrolyte and the deep eutectic electrolyte of nitrile butanedioic acid and zinc perchlorate, further validating the Zn2+ insertion mechanism in this reduction process. However, in the H2SO4 electrolyte (pH = 1 M ZnSO4), the plateau of R1 disappears, and obvious plateaus of R2 and R3 at low potentials can be observed, indicating that H+ insertion is not related to the R1 process but involves the R2 and R3 processes. The GITT profile of the TBQPH@GO//Zn cell (ESI Appendix, Fig. S11†) reveals a large IR compensation for the plateau at high potentials, which is probably because the embedding of Zn2+ is more difficult compared to H+, verifying that the first R1 plateau of discharge belongs to the embedding of Zn2+.
The quasi in situ XPS measurements of the stabilized TBQPH structure provide further understanding of the insertion mechanisms of Zn2+ and H+. The XPS spectra were collected at different voltages during the second charge–discharge cycle (Fig. 4C). As seen in the O 1s spectra, it is evident that Zn2+ initially binds to the carbonyl group by the formation of C–O–Zn, followed by the H+ insertion forming C–O–H with the carbonyl group as the discharge process proceeds (Fig. 4D). Notably, the insertion potential of H+ is generally lower than that of Zn2+, consistent with the distinct plateaus observed using different electrolytes. For the N 1s spectra in Fig. 4E, when discharged to 0.8 V, the peak generated at the lower binding energy can be attributed to the formation of N–Zn as H+ will not participate in the reduction process above this potential, as seen in the discharge voltage profiles with the H2SO4 electrolyte (Fig. 4C). In the further discharge process to 0.35 V, the increase of the peak can be attributed to the insertion of H+, as the peaks of N–Zn and N–H cannot be clearly separated here. The C 1s spectra exhibit decreased CN and CO peaks in the discharge process, which agrees with the molecular evolution from the stabilized state to the reduced state (ESI Appendix, Fig. S12†). The Zn 2p spectra also show an increased Zn peak during discharge as the Zn2+ insertion reaction proceeds. In the quasi in situ FTIR spectra, as the cell undergoes discharge, a decrease of CO and CN peaks can be observed, attributed to their reduction into C–O and C–N species (Fig. 4F). Intriguingly, a decline in the C–N peak is also observed with decreasing potentials; however, the peak position shifts to a higher wavenumber. Such peak shift is probably due to the N–Zn coordination along with Zn2+ insertion which serves to stabilize the structure and dampen C–N vibrations.
Theoretical calculations were further conducted to reveal the charge storage mechanism during the redox process. To analyze the Gibbs free energies associated with different ions inserted at each stage, we conducted DFT calculations on the potential insertion order of Zn2+ and H+ in TBQPH. As shown in Fig. 4G, consistent with the FUKUI function prediction, the carbonyl groups at the center prefer to coordinate two Zn2+ along with the adjacent imine groups. The Gibbs free energy associated with binding of Zn2+ to the neighboring carbonyl group was also calculated to see if more Zn2+ can be inserted. However, insertion of another two Zn2+ is not possible as such molecular configuration exhibits a higher free energy of −2.05 eV compared to that for the insertion of four H+ (−4.46 eV). In addition, we considered the co-storage of Zn between two TBQPH molecules and showed that CO and CN co-bound a Zn2+ with the lowest energy (−4.56 eV), suggesting a high synergistic effect of CO and CN on Zn2+ (ESI Appendix, Fig. S13†). The subsequent insertion of H+ appears to enhance the molecular stability, which is consistent with the experimental results where H+ insertion occurs at low potentials.
With the integration of experimental characterization and calculations, we can draw the conclusion that TBQPH preferentially induces the insertion of Zn2+, resulting in the formation of a stabilized N–Zn–O coordinated structure, followed by the co-insertion of Zn2+ and H+. The pH changes in the electrolyte caused by H+ insertion into TBQPH@GO were further visualized by introducing a bromocresol green indicator into the electrolyte (Fig. 4H). The indicator exhibited a pale green color as the pH of 1 M ZnSO4 was ∼4.0. As the discharge reached the second plateau, the color of the electrolyte near the TBQPH@GO electrode turned blue, indicating an increase in pH. This color alteration can be attributed to H+ insertion and manifested as a blue-to-green transition near the positive electrode during the charging process. However, no discernible change in color occurred near the negative electrode due to the inevitable hydrogen precipitation reaction on the Zn metal negative electrode, resulting in an elevation of the surrounding electrolyte pH. The change in zinc sulfate base can be monitored from the in situ XRD data (ESI Appendix, Fig. S14†). This change is more pronounced when discharged to low potentials, indicating that the intercalation of H+ predominantly occurs at low potentials, which is consistent with the previous test results.
The reaction kinetics of TBQPH@GO was investigated using CV at various sweep rates ranging from 0.2 to 1.0 mV s−1. As shown in Fig. 5A, the three pairs of redox peaks of TBAPH@GO exhibit slight shifts with increasing sweep rates, indicating the excellent charge transfer kinetics of TBAPH@GO. According to the relationship between the peak current (i) and the sweep rate (ν) in the formula i = aνb, b values (O1-3 and R1-3) are 0.804, 0.929, 1.015, 1.013, 0.866 and 0.766 for the six redox peaks, respectively (Fig. 5B). The value of b equals 1, indicating a surface-controlled process, while it equals 0.5, suggesting a diffusion-controlled process for the charge storage. The high b values of TBAPH@GO indicate that the redox processes are more capacitive with rapid charge storage kinetics. The proportions of each process are further evaluated via the equation i = k1ν + k2ν1/2, wherein the surface-controlled capacity accounts for 64.7% at 0.2 mV s−1 and increases to 80.2% when the sweep rate increases to 1.0 mV s−1, indicating that the surface-controlled process dominates throughout the reaction process (Fig. 5C and ESI Appendix, Fig. S15†). The impedance spectroscopy measurements of the TBQPH@GO//Zn cell under different voltages reveal that the charge transfer resistance decreases during discharge, indicating the enhanced off-domain transport property and mobility of electrons inside the highly conjugated molecular structure (ESI Appendix, Fig. S16†).
 | ||
| Fig. 5 Electrochemical properties of TBQPH@GO//Zn batteries. (A) CV profiles of TBQPH@GO at varying sweep rates. (B) Plot of ln(i) versus ln(v) for redox peaks. (C) Capacity contribution of TBQPH@GO at different sweep rates. (D) Rate capabilities of the TBQPH@GO//Zn battery at current densities from 0.2 to 10 A g−1. (E and F) Long-term cycling performance (E) and the corresponding charge–discharge voltage profiles (F) of the TBQPH@GO//Zn battery at a current density of 10 A g−1. (G) Performance comparison of the TBQPH@GO//Zn battery with other state-of-the-art zinc-organic batteries. (H) Comparison of TBQPH with other state-of-the-art organic cathode materials in terms of specific capacity, operating voltage, and energy density. | ||
The electrochemical performance of the TBAPH@GO//Zn batteries was further investigated under different current densities. An ultra-high discharge capacity of 445 mA h g−1 can be achieved at a current density of 0.2 A g−1, and a retained capacity of 230 mA h g−1 can be delivered at a high current density of 10 A g−1 (Fig. 5D). It is noted that the high capacity and fast kinetics of TBAPH@GO are also related to the decreased particle size and conductive substrate for efficient charge transport and good electrolyte penetration, as the TBAPH//Zn cell only exhibits limited capacity (ESI Appendix, Fig. S17†). When cycled at a current density of 10 A g−1, an impressive capacity retention of ∼80% can be achieved even after 15000 cycles with a coulombic efficiency of ∼99.6% (Fig. 5E). The excellent cycling stability of TBAPH@GO can be attributed to the unique stabilization process in the first cycle, as the charge–discharge voltage profiles change slowly with the cycle number (Fig. 5F). It can be further evidenced by the high material stability through in situ UV tests using the electrolyte containing 1 M ZnSO4 during the 1000th cycle (ESI Appendix, Fig. S18†). Notably, there are no peaks in the UV spectra representing material dissolution observed at different voltages, suggesting that the TBQPH@GO composite possesses exceptional stability after extended cycling in aqueous environments. Moreover, at a low current density of 0.3 A g−1, a high capacity retention of ∼88.2% can be realized after 1000 cycles due to the conjugated molecular structure which facilitates electron delocalization and structural stability (ESI Appendix, Fig. S19†). However, the TBQPH//Zn cell exhibited poorer cycling stability and weaker rate performance at 0.3 A g−1 over long cycles, clearly demonstrating that the addition of GO enhances the exposure of the material's active surface area and improves both electronic and ionic transport rates (ESI Appendix, Fig. S20 and S21†). We also analyzed the SEM images and XRD patterns of the zinc anode after 1000 cycles. The results show no significant dendrite formation on the electrode before and after cycling; however, basic zinc sulfate was observed on the surface, which may play a critical role in influencing the battery's cycling performance (ESI Appendix, Fig. S22 and S23†). Compared with the state-of-the-art zinc-organic batteries, the TBQPH@GO//Zn battery exhibits simultaneously high rate capability, coulombic efficiency and cycling stability (Fig. 5G).39–48 We conducted cycling tests on TBQPH@GO material at a high loading (20 mg cm−2) and found that its capacity contribution approached 70 mA h g−1 at 0.5 A g−1 (ESI Appendix, Fig. S24†). In terms of specific capacity, working voltage and energy density of organic cathode materials, TBQPH@GO exhibits an ultrahigh capacity (∼445 mA h g−1) as well as a high working voltage (∼1.0 V vs. Zn2+/Zn), resulting in a high energy density of ∼300 W h kg−1, which is among the best values reported in the literature.
Conclusions
In conclusion, a new organic cathode TBQPH with a spacer composition of imine groups (CN) and carbonyl groups (CO) was designed and synthesized for aqueous zinc ion batteries with high theoretical specific capacity. Theoretical calculations and experimental data demonstrate the relationship between the conjugated structure, energy gap reduction, electron delocalization enhancement and material stability. Importantly, the combination of TBQPH with GO material enhances its electrochemical performance and stability, exhibiting ultra-high capacity contributions of 445 mA h g−1 at a current density of 0.2 A g−1 and 200 mA h g−1 at a current density of 10 A g−1, while maintaining 80% capacity retention after 15000 cycles. Calculations and electrochemical tests reveal distinct mechanisms for the first and subsequent cycle reactions in TBQPH, with the imine groups not participating in the discharge reaction during the initial cycle. The stabilized TBQPH exhibits a stabilization mode of synergistic storage involving carbonyl and imine groups, where the first discharge plateau is attributed to Zn2+ insertion, while the second and third discharge plateaus are triggered by the co-insertion of Zn2+ and H+. Our work proposes an extended conjugation and “group synergy” approach, which enhances the conductivity, stability, and electrochemical activity of the molecule. Furthermore, this synergistic cation storage mechanism can be extrapolated to the design and application of other n-type molecules. This strategy offers novel insights for designing and developing organic cathode materials for aqueous zinc ion batteries.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Y. W. and G. Y. directed the project. G. M. and Y. C. carried out material preparation, characterization and testing. R. W., Z. Y. and H. Y. performed XPS, XRD, FTIR and Raman tests. Y. W., H. D., and M. C. provided some constructive suggestions for the experiments. G. M. and J. Z. wrote the first draft. All authors discussed the experimental results and prepared the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Y. W. acknowledges the support from National Natural Science Foundation of China (61904097). G. Y. acknowledges the support from the Welch Foundation F-1861.Notes and references
- Z. Fan, J. Wang, Y. Wu, X. Yan, D. Dai and X.-L. Wu, J. Energy Chem., 2024, 97, 237–264 CrossRef CAS.
- Y. Fan, Q. Wang, Y. Xie, N. Zhou, Y. Yang, Y. Ding, Y. Wei and G. Qu, Prog. Mater. Sci., 2025, 149, 101393 CrossRef CAS.
- J. Chen, W. Zhao, J. Jiang, X. Zhao, S. Zheng, Z. Pan and X. Yang, Energy Storage Mater., 2023, 59, 102767 CrossRef.
- Y. Ou, Y. Xiong, Z. Hu, Y. Zhang and L. Dong, J. Mater. Chem. A, 2022, 10, 10373–10382 RSC.
- F. Hua, Y. Zhang, S. Cao, M. Li, L. Jing and J. Cui, J. Energy Storage, 2023, 68, 107779 CrossRef.
- K. Yamamoto, D. Suemasa, K. Masuda, K. Aita and T. Endo, ACS Appl. Mater. Interfaces, 2018, 10, 6346–6353 CrossRef CAS PubMed.
- Z. Song, L. Miao, H. Duan, L. Ruhlmann, Y. Lv, D. Zhu, L. Li, L. Gan and M. Liu, Angew. Chem., Int. Ed., 2022, 61, e202208821 CrossRef CAS PubMed.
- N. Wang, Z. Guo, Z. Ni, J. Xu, X. Qiu, J. Ma, P. Wei and Y. Wang, Angew. Chem., Int. Ed., 2021, 60, 20826–20832 CrossRef CAS PubMed.
- D. Li, Y. Guo, C. Zhang, X. Chen, W. Zhang, S. Mei and C.-J. Yao, Nano-Micro Lett., 2024, 16, 194 CrossRef CAS PubMed.
- H.-g. Wang, Q. Wu, L. Cheng and G. Zhu, Coord. Chem. Rev., 2022, 472, 214772 CrossRef CAS.
- Z. Li, J. Tan, Y. Wang, C. Gao, Y. Wang, M. Ye and J. Shen, Energy Environ. Sci., 2023, 16, 2398–2431 RSC.
- Y. Gao, J. Yin, X. Xu and Y. Cheng, J. Mater. Chem. A, 2022, 10, 9773–9787 RSC.
- F. Ye, Q. Liu, H. Dong, K. Guan, Z. Chen, N. Ju and L. Hu, Angew. Chem., Int. Ed., 2022, 61, e202214244 CrossRef CAS PubMed.
- W. Wang, V. S. Kale, Z. Cao, Y. Lei, S. Kandambeth, G. Zou, Y. Zhu, E. Abouhamad, O. Shekhah, L. Cavallo, M. Eddaoudi and H. N. Alshareef, Adv. Mater., 2021, 33, 2103617 CrossRef CAS PubMed.
- X. Geng, H. Ma, F. Lv, K. Yang, J. Ma, Y. Jiang, Q. Liu, D. Chen, Y. Jiang and N. Zhu, Chem. Eng. J., 2022, 446, 137289 CrossRef CAS.
- J. Kumankuma-Sarpong, S. Tang, W. Guo and Y. Fu, ACS Appl. Mater. Interfaces, 2021, 13, 4084–4092 CrossRef CAS PubMed.
- D. Ma, H. Zhao, F. Cao, H. Zhao, J. Li, L. Wang and K. Liu, Chem. Sci., 2022, 13, 2385–2390 RSC.
- M. Li, M. Liu, Y. Lu, G. Zhang, Y. Zhang, Z. Li, Q. Xu, H. Liu and Y. Wang, Adv. Funct. Mater., 2024, 34, 2312789 CrossRef CAS.
- J. Yang, H. Hua, H. Yang, P. Lai, M. Zhang, Z. Lv, Z. Wen, C. C. Li, J. Zhao and Y. Yang, Adv. Energy Mater., 2023, 13, 2204005 CrossRef CAS.
- D. Kundu, P. Oberholzer, C. Glaros, A. Bouzid, E. Tervoort, A. Pasquarello and M. Niederberger, Chem. Mater., 2018, 30, 3874–3881 CrossRef CAS.
- Y. Zhang, Q. Huang, Z. Song, L. Miao, Y. Lv, L. Gan and M. Liu, Adv. Funct. Mater., 2024, 2416415 CrossRef.
- N. T. H. Luu, A. S. Ivanov, T.-H. Chen, I. Popovs, J.-C. Lee and W. Kaveevivitchai, J. Mater. Chem. A, 2022, 10, 12371–12377 RSC.
- Z. Song, L. Miao, H. Duan, Y. Lv, L. Gan and M. Liu, Angew. Chem., Int. Ed., 2024, 63, e202401049 CrossRef CAS PubMed.
- X. Deng, J. K. Sarpong, G. Zhang, J. Hao, X. Zhao, L. Li, H. Li, C. Han and B. Li, InfoMat, 2023, 5, e12382 CrossRef CAS.
- Q. Wang, Y. Liu, C. Wang, X. Xu, W. Zhao, Y. Li and H. Dong, Chem. Eng. J., 2023, 451, 138776 CrossRef CAS.
- Y. Shi, P. Wang, H. Gao, W. Jin, Y. Chen, Y. Huang, T.-R. Wu, D.-Y. Wu, J. Xu and J. Cao, Chem. Eng. J., 2023, 461, 141850 CrossRef CAS.
- Y. Gao, G. Li, F. Wang, J. Chu, P. Yu, B. Wang, H. Zhan and Z. Song, Energy Storage Mater., 2021, 40, 31–40 CrossRef.
- Y. Chen, J. Li, Q. Zhu, K. Fan, Y. Cao, G. Zhang, C. Zhang, Y. Gao, J. Zou, T. Zhai and C. Wang, Angew. Chem., Int. Ed., 2022, 61, e202116289 CrossRef CAS PubMed.
- H. Peng, J. Xiao, Z. Wu, L. Zhang, Y. Geng, W. Xin, J. Li, Z. Yan, K. Zhang and Z. Zhu, CCS Chem., 2022, 5, 1789–1801 CrossRef.
- W. Ji, D. Du, J. Liang, G. Li, G. Feng, Z. Yin, J. Zhou, J. Zhao, Y. Shen, H. Huang and S. Pang, Battery Energy, 2023, 2, 20230020 CrossRef CAS.
- Z. Song, Q. Huang, Y. Lv, L. Gan and M. Liu, Angew. Chem., Int. Ed., 2025, 64, e202418237 CrossRef CAS PubMed.
- W. Sun, M. Ma, M. Zhu, K. Xu, T. Xu, Y. Zhu and Y. Qian, Small, 2022, 18, 2106604 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- T. Zhou, L. Zhu, L. Xie, Q. Han, X. Yang, L. Chen, G. Wang and X. Cao, J. Colloid Interface Sci., 2022, 605, 828–850 CrossRef CAS PubMed.
- G. Li, L. Sun, S. Zhang, C. Zhang, H. Jin, K. Davey, G. Liang, S. Liu, J. Mao and Z. Guo, Adv. Funct. Mater., 2024, 34, 2301291 CrossRef CAS.
- Y. Wang, C. Wang, Z. Ni, Y. Gu, B. Wang, Z. Guo, Z. Wang, D. Bin, J. Ma and Y. Wang, Adv. Mater., 2020, 32, 2000338 CrossRef CAS PubMed.
- L. Liu, L. Miao, L. Li, F. Li, Y. Lu, Z. Shang, J. Chen and T. J. Phy, Chem. Lett., 2018, 9, 3573–3579 CAS.
- J. Murillo-Gelvez, K. P. Hickey, D. M. Di Toro, H. E. Allen, R. F. Carbonaro and P. C. Chiu, Environ. Sci. Technol., 2019, 53, 5816–5827 CrossRef CAS PubMed.
- Q. Zhao, W. Huang, Z. Luo, L. Liu, Y. Lu, Y. Li, L. Li, J. Hu, H. Ma and J. Chen, Sci. Adv., 2018, 4, eaao1761 CrossRef PubMed.
- Z. Tie, L. Liu, S. Deng, D. Zhao and Z. Niu, Angew. Chem., Int. Ed., 2020, 59, 4920–4924 CrossRef CAS PubMed.
- T. Sun, W. Zhang, Q. Nian and Z. Tao, Chem. Eng. J., 2023, 452, 139324 CrossRef CAS.
- Z. Lin, H.-Y. Shi, L. Lin, X. Yang, W. Wu and X. Sun, Nat. Commun., 2021, 12, 4424 CrossRef CAS PubMed.
- K. W. Nam, H. Kim, Y. Beldjoudi, T.-w. Kwon, D. J. Kim and J. F. Stoddart, J. Am. Chem. Soc., 2020, 142, 2541–2548 CrossRef CAS PubMed.
- Q. Wang, Y. Liu and P. Chen, J. Power Sources, 2020, 468, 228401 CrossRef CAS.
- Q. Wang, X. Xu, G. Yang, Y. Liu and X. Yao, Chem. Commun., 2020, 56, 11859–11862 RSC.
- G. Dawut, Y. Lu, L. Miao and J. Chen, Inorg. Chem. Front., 2018, 5, 1391–1396 RSC.
- T. Sun, Z.-J. Li, Y.-F. Zhi, Y.-J. Huang, H. J. Fan and Q. Zhang, Adv. Funct. Mater., 2021, 31, 2010049 CrossRef CAS.
- X. Wang, L. Chen, F. Lu, J. Liu, X. Chen and G. Shao, ChemElectroChem, 2019, 6, 3644–3647 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc00311c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |