
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxidation-induced nucleophilic substitution at the electron-rich B(12) vertex in [CB11H12]− under catalyst-free conditions†
Wanqi
Sun‡
a,
Yujie
Jin‡
ab,
Yongtao
Wang
 *ab,
Zeyu
Wen
a,
Jizeng
Sun
a,
Jia
Yao
ab,
Simon
Duttwyler
a and
Haoran
Li
*abc
*ab,
Zeyu
Wen
a,
Jizeng
Sun
a,
Jia
Yao
ab,
Simon
Duttwyler
a and
Haoran
Li
*abc
aDepartment of Chemistry, Zhejiang University, 866 Yuhangtang Rd, Hangzhou 310058, China. E-mail: wyongtao@zju.edu.cn; lihr@zju.edu.cn
bCenter of Chemistry for Frontier Technologies, ZJU-NHU United R&D Center, Zhejiang University, 866 Yuhangtang Rd, Hangzhou 310058, China
cState Key Laboratory of Chemical Engineering, College of Chemical and Biological Engineering, Zhejiang University, 866 Yuhangtang Rd, Hangzhou 310058, China
First published on 26th February 2025
Abstract
Highly regioselective B(12) substitutions of the monocarborane anion [CB11H12]− has been a challenge. Here, we synthesized a stable B–O–N zwitterionic compound with an impressive yield (isolated yield up to 98%) and excellent regioselectivity at the B(12) position under catalyst-free conditions. The kinetics, substituent effect, and capture experiments are paired with theoretical calculations, showing that the reaction mechanism is oxidation-induced nucleophilic substitution. The hydride anion at the B(12) position is abstracted by an oxoammonium oxidant with lower cleavage energy of 4.2 kcal mol−1 than B(7–11) positions, thereby changing the electronegativity upon the conversion of [CB11H12]− to neutral [CB11H11], in turn giving very high regioselectivity for nucleophilic substitution. This work presents an effective method for synthesizing B(12) oxygen derivatives of the [CB11H12]− anion.
Introduction
12-Vertex boron clusters, characterized by their distinctive icosahedral geometry and exceptional stability,1–4 have garnered significant attention across diverse fields.5–14 Monocarborane anion [CB11H12]−, with a single negative charge, has received particular attention.15–18 Highly regioselective B(12) substitutions are a challenge due to the minimal polarization and high symmetry of the B–H bond.19–23 Currently, there are two mechanisms for achieving monosubstitution at the B(12) position of [CB11H12]−. (1) Electrophilic substitution, the electrophilic halogenation is most commonly used.24–26 However, 7-isomers were always obtained as by-products due to the similar electrostatic potential at B(7–11) to that at B(12) (Fig. 1a). The less regioselective electrophilic substitutions commonly cause lower yields (Fig. 1b). (2) Nucleophilic substitution, boron-centered cations were generally proposed to be key intermediates in the chemistry of polyhedral boron hydrides,27–29 however, limited studies are reported on the functionalization of [CB11H12]−. Hydroxylation at the B(12) position using 80% sulfuric acid yields 12-hydroxy anions under elevated temperatures.30 Electrophile-induced nucleophilic substitution (EINS)31 has been proposed for this reaction.32,33 Recently, Kaszyński and co-workers found that [CB11H12]− reacts with iodobenzene diacetate, yielding zwitterionic products Ar-I+-[CB11H11]− at the B(12) position, which undergo nucleophilic substitution to generate a series of functionalized derivatives of the [CB11H12]− anion (Fig. 1c).34,35 | ||
| Fig. 1 (a) The monocarborane anion [CB11H12]− and electrostatic potential surface; (b) previously reported electrophilic substitution; (c) previously reported nucleophilic substitution; (d) overview and summary of the key finding in this work. | ||
Oxoammonium salts are commonly utilized as oxidants in organic synthesis for their easy access and recyclability,36–41 and they are commonly used as hydride abstracting reagents.42,43 For example, they catalyze the oxidation of alcohols or ether to yield the corresponding aldehydes, ketones, or carboxylic acids via a hydrogen anion abstraction mechanism.44 Recently, Lin and co-workers achieved the oxidation of ethers to lactones with the oxoammonium cation, where the hydride abstraction from C–H was also suggested.45
Inspired by these literature studies, we hypothesized that oxoammonium salts might also abstract hydride from B–H bonds in [CB11H12]−. As expected, we found a novel coupling reaction of oxoammonium salts and [CB11H12]− to form stable B–O–N compounds under catalyst-free conditions. This approach exhibited excellent regioselectivity with high B(12) substituted products. The reaction was found to undergo an oxoammonium oxidation-induced nucleophilic substitution (OINS) by various mechanistic studies, such as kinetic studies, substituent effects, EPR spectroscopy, trapping experiments, and theoretical calculations.
Results and discussion
At the very beginning, optimization of the reaction conditions was conducted (Table S1†). The conditions outlined in entry 12 of Table S1† were identified as optimal. The optimized reaction conditions were [CB11H12]− with 1.25 equivalents of the commercial agent [AcNH-TEMPO]+ [BF4]− in 1,4-dioxane at 80 °C for 180 min.Under optimal conditions, a scale-up synthesis of 2 was carried out with a high isolated yield of 98%. The new compound was characterized through spectroscopic techniques, including 1H, 1H{11B}, 11B, 11B{1H}, and 13C{1H} NMR, as well as high resolution mass spectrometry (HRMS). The 11B NMR spectra displayed a similar pattern of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5:5 with the chemical shifts of B(12) vertices changed from δ = −7.93 ppm to δ = 11.8 ppm. Moreover, single crystals of the composition were obtained from an acetone solution via slow evaporation. And the single-crystal X-ray analysis of 2 confirms the molecular structure (Fig. 2b). For compound 2, the observed distances are B12–O1 1.459 Å, O1–N1 1.423 Å, with an angle of B12–O1–N1 at 123.33°. The distances and angles are comparable to those reported in the previous literature.46 In addition, the B–O–N compound is a zwitterionic compound with high stability.47,48 The theoretically calculated dipole moment is 11.75.
:5:5 with the chemical shifts of B(12) vertices changed from δ = −7.93 ppm to δ = 11.8 ppm. Moreover, single crystals of the composition were obtained from an acetone solution via slow evaporation. And the single-crystal X-ray analysis of 2 confirms the molecular structure (Fig. 2b). For compound 2, the observed distances are B12–O1 1.459 Å, O1–N1 1.423 Å, with an angle of B12–O1–N1 at 123.33°. The distances and angles are comparable to those reported in the previous literature.46 In addition, the B–O–N compound is a zwitterionic compound with high stability.47,48 The theoretically calculated dipole moment is 11.75.
 | ||
| Fig. 2 (a) Gram-scale synthesis and (b) X-ray crystal structures of 2. Cations and H atoms are omitted for clarity; 30% displacement ellipsoids. | ||
In an effort to investigate the reaction mechanism behind such high regioselectivity, we performed density functional theory (DFT) calculations for the heterolytic cleavage of various B–H bonds in [CB11H12]−. Due to the influence of H+ or H− acceptors on the value of Gibbs free energy change (ΔG), the relative Gibbs free energy change (ΔΔG) was calculated in this work by subtracting ΔG with ΔG(B(12)–H) in [CB11H12]−. Δ–HFig. 3a presents the ΔΔG, indicating that the B(12) position is the most favorable for H− loss, followed by B(7) and, ultimately, B(2). In contrast, the ΔΔG trend of losing H+ is exactly opposite as the proton at B(2) was most activated. Given that the regioselectivity was favored at the B(12) site, it was more likely to undergo a hydride abstraction pathway. Simultaneously, we calculated the ΔΔG for various B–H bonds in [CB11H12]− analogues, including [B12H12]2− and m-C2B10H12. By comparison, H− loss was easier in [B12H12]2−, which lacks a carbon vertex, as the ΔΔG is −12.2 kcal mol−1. However, it is more difficult for meta-carborane 1,7-C2B10H12 to lose H− with ΔΔG of B–H in any position exceeding 20 kcal mol−1. To check these calculation results, we carried out the experimental work, and found that neither ortho-C2B10, nor meta-C2B10, nor para-C2B10 reacted with [AcNH-TEMPO]+[BF4]−, and the single-substituted product could not be obtained in [B12H12]2−. These verified that the reaction process involved the loss of H− rather than the loss of H+.
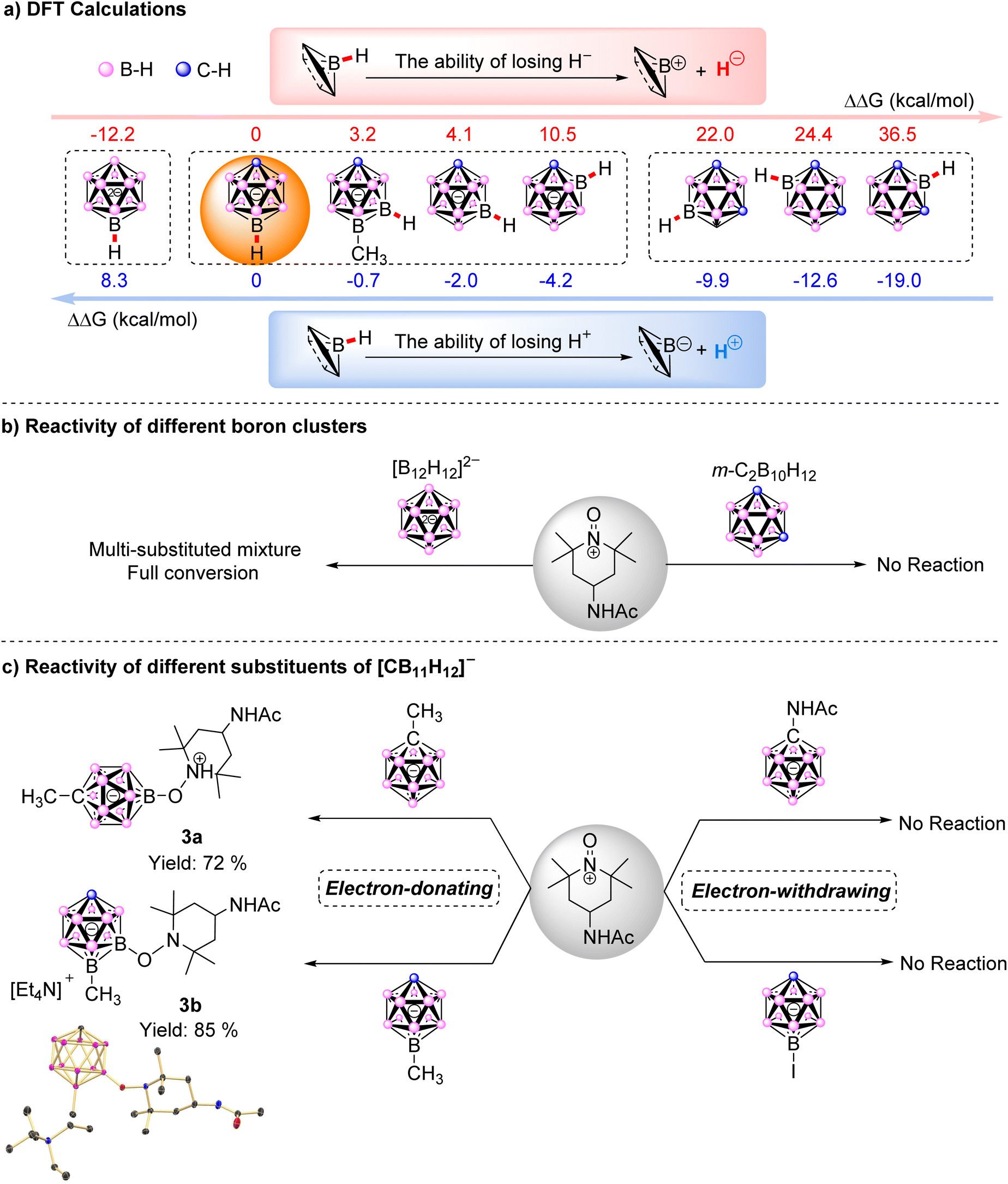 | ||
| Fig. 3 (a) Relative Gibbs free energy change (ΔΔG) for the heterolytic cleavage of B–H bonds, showing the loss of H− (red) and H+ (blue); (b) reactivity of different boron clusters and (c) reactivity of different substituents of the [CB11H12]− anion. | ||
Further evidence came from the fact that the compound did not react with [AcNH-TEMPO]+[BF4]− when the electron-withdrawing iodine substituent occupied position B(12). In contrast, the coupling product at position 7 (the second most likely position to lose H−) was obtained with an isolated yield of 85% when the electron-donating effect [12-CH3-CB11H11]− was used. Additionally, theoretical calculations indicated that the ΔΔG for H− loss at this position was reduced by 0.9 kcal mol−1 compared to [CB11H12]−, which further proved that the reaction involved a hydride anion abstraction (H− loss) process. In addition, when electron-donating –CH3 and electron-withdrawing –NHAc substituents were attached to the C vertex of [CB11H12]−, the results were consistent with our expectations: the desired product was obtained with an isolated yield of 72% when [1-CH3-CB11H11]− was used, whereas [1-AcNH-CB11H11]− did not undergo conversion (Fig. 3c).
Subsequently, the transition state for B(12)–H heterolysis was investigated (Fig. 4a–c). Initially, we tried to find a 4-member ring type transition state to mediate concerted H- abstraction and B–O bond formation, but failed due to the large steric hindrance between the oxoammonium cation and the [CB11H12]− cage. However, a reasonable transition state was found for the transfer of H− to O atom at the oxoammonium cation, and the intrinsic reaction coordinate (IRC) analysis suggested the subsequent O–B bond formation after the formation of the [CB11H11][hydroxylamine] pair. The corresponding activation Gibbs free energy (ΔG‡) was 21.1 kcal mol−1, owing a favored energy barrier of 4.2 kcal mol−1 to that at the B(7–11) positions (ΔG‡ = 25.3 kcal mol−1). The intrinsic bond orbitals (IBOs) along the reaction coordinate supported the hydride anion abstraction by the oxoammonium cation. As illustrated in Fig. 4b, apparent electron flow was observed from the [CB11H12]− σ orbital to the NO π orbital and the transient intermediate CB11H11 and hydroxylamine were formed, indicating the apparent hydride anion transfer. Besides, the IRC plot indicated that this reaction proceeded in two steps and involved a high-energy transition state (Fig. 4c). In the first step, the H atom of [CB11H12]− moves toward the oxoammonium salts, resulting in B–H bond cleavage and O–H bond formation (blank balls). The B–O distance decreases from 2.95 to 2.60 Å. In the next step, starting from the red balls, hydroxylamine attacks the electrophilic intermediate, forming the final product. The B–O distance further decreases from 2.60 to 1.87 Å. The formation of the B–O–N coupling product was exergonic by 56.3 kcal mol−1. Based on the above calculations, we propose the following mechanism: firstly, the [CB11H12]− is oxidized by [AcNH-TEMPO]+[BF4]− to form the paired intermediate, [CB11H11][hydroxylamine]. Subsequently, hydroxylamine, acting as a nucleophilic reagent, attacked the intermediate CB11H11 to form the B–O–N compound as the final product.
 | ||
| Fig. 4 (a) Proposed mechanism; (b) the intrinsic bond orbitals (IBOs) illustrate the electron flow during the reaction between [CB11H12]− and oxoammonium cation and proposed mechanism; (c) the change of electronic energies along the intrinsic reaction coordinate for the sequentially occurring step 1 (blank balls) and step 2 (red balls). The dotted lines indicate the change of B–H (black) and B–O (red) bond distances along the intrinsic reaction coordinate; (d) determination of KIE for B–H; (e) capture experiments. | ||
To support the proposed mechanism involving hydride abstraction and subsequently nuleophilic substitution, hydrogen kinetic isotope effect (KIE) experiments were conducted (Fig. 4d). [H-CB11D11]− was synthesized from [CB11H12]−via microwave reaction in D2O containing 20 wt% DCl. A 1:1 reaction between [CB11H12]− and [H-CB11D11]− was conducted under standard reaction conditions. At 95% conversion, an apparent KIE of 1.83 was calculated using the equation R/R0 = (1 − F)(1/KIE)−1 (Fig. 4d).49 In general, primary KIEs typically range from 2 to 8, while secondary KIEs fall within 1.0 to 1.2.50 The observed KIE value of 1.83 is closer to the range of primary KIEs, suggesting that the cleavage of the B–H/D bond was the rate-determining step.
In addition, the radical pathway was excluded by the control experiment and electron paramagnetic resonance spectroscopy (EPR). The reaction could run the same way in the dark. The [AcNH-TEMPO] radical could not take the place of [AcNH-TEMPO]+[BF4]− to oxidize [CB11H12]− under standard conditions. No significant signals were detected during the reaction by EPR (Fig. S2†).
Next, in order to prove the nucleophilic step, 4-methylpyridine, a nucleophile, was introduced into the reaction and found to be attached to the [CB11H12]− anion in the presence of trifluoromethanesulfonic acid (Fig. S3†), which supported an SN1 mechanism. Besides, the oxide form of 4-methylpyridine-N-oxide did not appear, excluding the 4-methylpyridine oxidation route. Furthermore, when 4-methylpyridine was replaced with 4-methoxypyridine, the product with 4-methoxypyridine attached to the [CB11H12]− anion was also observed in HRMS (Fig. S4†). Water, acting as a nucleophile, was also employed in varying amounts to perform the capturing reaction. Anhydrous 1,4-dioxane, untreated commercial 1,4-dioxane, and 1,4-dioxane containing 10 v% water were used as solvents. HRMS data revealed an increasing ratio of hydroxyl-substituted [CB11H12]− anion to the hydroxylamine-substituted [CB11H12]− anion (Fig. S5–S7†). However, the yields of these captured compounds were relatively low, making isolation and precise determination of the ratio challenging. This could be attributed to the preferential reaction within the in situ-formed [CB11H11] [hydroxylamine] pair, which suppresses competition from other nucleophiles. In brief, these experimental results demonstrated that the reaction proceeded in two steps, likely following an SN1-type nucleophilic substitution mechanism rather than an SN2. The oxidation of the B–H bond in [CB11H12]− by oxoammonium salts leads to hydride abstraction, which then facilitates nucleophilic attack by hydroxylamine. This results in the highly selective formation of B(12) oxygen derivatives of the [CB11H12]− anion. Based on these observations, we propose a novel mechanism: oxoammonium oxidation-induced nucleophilic substitution (OINS).
Finally, we employed various N–O compounds for the reaction to verify the universality of the method. All reactions were conveniently set up and conducted under ambient air conditions. Considering that oxoammonium cations can be generated by the disproportionation of nitroxide radicals in acidic media, we attempted to conduct a one-pot reaction by directly employing nitroxide radicals. As shown in Fig. 5, common piperidine nitroxide radicals also worked. Additionally, for some substrates with active groups, moderate to high yields were achieved. Moreover, during the reaction with free radicals, we detected the structure of the hydroxy-oxoammonium adduct, which further proved that the mechanism of the reaction involved free radicals first generating hydroxy-oxoammonium adducts and oxoammonium cations via disproportionation. This method enables the construction of similar B–O–N structures with various nitroxide radicals, providing a reference for potential future applications.
 | ||
| Fig. 5 Substrate scope of B(12) derivatives. | ||
Conclusions
In conclusion, we have developed a new strategy to achieve high selectivity B–H functionalization of [CB11H12]−, and an OINS mechanism was proposed. With this approach, a series of novel and stable B–O–N derivatives were synthesized. SN1 type nucleophilic substitution reactions on aromatic rings have long been recognized as challenging.51–53 Our research indicates that the successful nucleophilic substitution on the [CB11H12]− anion is attributed to three primary factors: (1) the B–H bond is more easily cleaved compared to the C–H bond in conventional aromatic systems; (2) oxoammonium cations can abstract the hydride (H−) due to their unique oxidizing abilities; and (3) the resulting product is a zwitterionic compound, which contributes to its stability. This work establishes a novel regioselective derivatization method and advances the mechanistic understanding of derivatization in aromatic compounds.Data availability
The data supporting this article have been included as part of the ESI.† All data can be obtained from the authors. Crystallographic data for compounds 2, 3b, 4b and 4c have been deposited with the Cambridge Crystallographic Data Centre (CCDC) under accession numbers 2407119–247122. The data can be obtained free of charge viahttps://www.ccdc.cam.ac.uk/data_request/cif.Author contributions
H. L. conceived the project and supervised the work with Y. W. and J. Y. W. S. conducted the experiments and drafted the original manuscript. Y. J. contributed to the development of the methodology. Y. W. performed theoretical calculations. Z. W. participated in the experimental design. J. S. provided critical feedback and assisted in data analysis. S. D. offered constructive comments. All authors discussed the results, reviewed, and contributed to the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Key R&D Program of China (2022YFA1503200) and the National Natural Science Foundation of China (No. 22303079 and 22073081). We gratefully thank Dr Xinyu Wang, Prof. Dr Qiaohong He, Mr Jiyong Liu, Dr Yaqin Liu and Dr Lina Gao (Chemistry Instrumentation Center Zhejiang University) for EPR/ESI-HRMS/single-crystal X-ray diffraction/NMR technical support, respectively.Notes and references
- A. J. Baublis and A. M. Spokoyny, Chem, 2024, 10, 29–32 CAS.
- R. J. Grams, W. L. Santos, I. R. Scorei, A. Abad-Garcia, C. A. Rosenblum, A. Bita, H. Cerecetto, C. Vinas and M. A. Soriano-Ursua, Chem. Rev., 2024, 124, 2441–2511 CrossRef CAS PubMed.
- A. D. Ready, Y. A. Nelson, D. F. Torres Pomares and A. M. Spokoyny, Acc. Chem. Res., 2024, 57, 1310–1324 CrossRef CAS PubMed.
- W.-B. Yu, P.-F. Cui, W.-X. Gao and G.-X. Jin, Coord. Chem. Rev., 2017, 350, 300–319 CrossRef CAS.
- Z. Qiu and Z. Xie, Chem. Soc. Rev., 2022, 51, 3164–3180 RSC.
- H. A. Mills, J. L. Martin, A. L. Rheingold and A. M. Spokoyny, J. Am. Chem. Soc., 2020, 142, 4586–4591 CrossRef CAS PubMed.
- J. Wang, L. Xiang, X. Liu, A. Matler, Z. Lin and Q. Ye, Chem. Sci., 2024, 15, 4839–4845 RSC.
- M. Chen, J. Xu, D. Zhao, F. Sun, S. Tian, D. Tu, C. Lu and H. Yan, Angew. Chem., Int. Ed., 2022, 61, e202205672 CrossRef CAS PubMed.
- A. Marfavi, P. Kavianpour and L. M. Rendina, Nat. Rev. Chem, 2022, 6, 486–504 CrossRef PubMed.
- P. Stockmann, M. Gozzi, R. Kuhnert, M. B. Sárosi and E. Hey-Hawkins, Chem. Soc. Rev., 2019, 48, 3497–3512 RSC.
- A. W. Tomich, J. Chen, V. Carta, J. Guo and V. Lavallo, ACS Cent. Sci., 2024, 10, 264–271 CrossRef CAS PubMed.
- P. F. Cui, X. R. Liu and G. X. Jin, J. Am. Chem. Soc., 2023, 145, 19440–19457 CrossRef CAS PubMed.
- S. O. Gunther, Q. Lai, T. Senecal, R. Huacuja, S. Bremer, D. M. Pearson, J. C. DeMott, N. Bhuvanesh, O. V. Ozerov and J. Klosin, ACS Catal., 2021, 11, 3335–3342 CrossRef CAS.
- L. Wang, S. Wu, J. Hu, Y. Jiang, J. Li, Y. Hu, Y. Han, T. Ben, B. Chen and Y. Zhang, Chem. Sci., 2024, 15, 5653–5659 RSC.
- K. Zhang, K. Cao, J. Sun, Y. Jin, J. Liu and S. Duttwyler, Inorg. Chem., 2024, 63, 16595–16599 CrossRef CAS PubMed.
- P. Ragué Schleyer and K. Najafian, Inorg. Chem., 1998, 37, 3454–3470 CrossRef PubMed.
- R. Dontha, T. C. Zhu, Y. Shen, M. Wörle, X. Hong and S. Duttwyler, Angew. Chem., Int. Ed., 2019, 58, 19007–19013 CrossRef CAS PubMed.
- M. Nava, I. V. Stoyanova, S. Cummings, E. S. Stoyanov and C. A. Reed, Angew. Chem., Int. Ed., 2013, 53, 1131–1134 CrossRef PubMed.
- D. Olid, R. Núñez, C. Viñas and F. Teixidor, Chem. Soc. Rev., 2013, 42, 3318–3336 RSC.
- G. E. Dwulet and M. A. Juhasz, Inorg. Chem. Commun., 2015, 51, 26–28 CrossRef CAS.
- J. Pecyna, B. Ringstrand, S. Domagała, P. Kaszyński and K. Woźniak, Inorg. Chem., 2014, 53, 12617–12626 CrossRef CAS PubMed.
- A. Himmelspach, G. J. Reiss and M. Finze, Inorg. Chem., 2012, 51, 2679–2688 CrossRef CAS PubMed.
- Y. Shen, Y. Pan, K. Zhang, X. Liang, J. Liu, B. Spingler and S. Duttwyler, Dalton Trans., 2017, 46, 3135–3140 RSC.
- S. V. Ivanov, A. J. Lupinetti, S. M. Miller, O. P. Anderson, K. A. Solntsev and S. H. Strauss, Inorg. Chem., 1995, 34, 6419–6420 CrossRef CAS.
- I. Krossing and I. Raabe, Angew. Chem., Int. Ed., 2004, 43, 2066–2090 CrossRef CAS PubMed.
- C. Douvris and J. Michl, Chem. Rev., 2013, 113, 179–233 CrossRef PubMed.
- S. Kim, J. W. Treacy, Y. A. Nelson, J. A. M. Gonzalez, M. Gembicky, K. N. Houk and A. M. Spokoyny, Nat. Commun., 2023, 14, 1671 CrossRef CAS PubMed.
- V. I. Bregadze, I. D. Kosenko, I. A. Lobanova, Z. A. Starikova, I. A. Godovikov and I. B. Sivaev, Organometallics, 2010, 29, 5366–5372 CrossRef CAS.
- D. Zhao and Z. Xie, Chem. Sci., 2016, 7, 5635–5639 RSC.
- B. Grüner, I. Císařová, J. Čáslavský, B. Bonnetot and D. Cornu, Collect. Czech. Chem. Commun., 2002, 67, 953–964 CrossRef.
- T. Jelínek, B. Stíbr, F. Mares, J. Plesek and S. Hermanek, Polyhedron, 1987, 6, 1737 CrossRef.
- S. Kőrbe, P. J. Schreiber and J. Michl, Chem. Rev., 2006, 106, 5208–5249 CrossRef PubMed.
- L. Wang, Y. Jiang, S. Duttwyler, F. Lin and Y. Zhang, Coord. Chem. Rev., 2024, 516, 215974 CrossRef CAS.
- P. Kaszynski and B. Ringstrand, Angew. Chem., Int. Ed., 2015, 54, 6576–6581 CrossRef CAS PubMed.
- J. Pecyna, P. Kaszyński, B. Ringstrand, D. Pociecha, S. Pakhomov, A. G. Douglass and V. G. Young, Inorg. Chem., 2016, 55, 4016–4025 CrossRef CAS PubMed.
- T. Sahana, A. Mondal, B. S. Anju and S. Kundu, Angew. Chem., Int. Ed., 2021, 60, 20661–20665 CrossRef CAS PubMed.
- Y. Wang, J. Liu, W. Sun, Y. Zhou, X. Wang, Q. Hu, Z. Wen, J. Yao and H. Li, J. Org. Chem., 2024, 89, 2440–2447 CrossRef CAS PubMed.
- Z. Lu, M. Ju, Y. Wang, J. M. Meinhardt, J. I. Martinez Alvarado, E. Villemure, J. A. Terrett and S. Lin, Nature, 2023, 619, 514–520 CrossRef CAS PubMed.
- C. Gerleve and A. Studer, Angew. Chem., Int. Ed., 2020, 59, 15468–15473 CrossRef CAS PubMed.
- W. Wang, X. Li, X. Yang, L. Ai, Z. Gong, N. Jiao and S. Song, Nat. Commun., 2021, 12, 3873 CrossRef CAS PubMed.
- J. M. Bobbitt, A. L. Bartelson, W. F. Bailey, T. A. Hamlin and C. B. Kelly, J. Org. Chem., 2014, 79, 1055–1067 CrossRef CAS PubMed.
- D. Leifert and A. Studer, Chem. Rev., 2023, 123, 10302–10380 CrossRef CAS PubMed.
- J. M. Bray, S. M. Stephens, S. M. Weierbach, K. Vargas and K. M. Lambert, Chem. Commun., 2023, 59, 14063–14092 RSC.
- Y. Wang, Z. Wen, J. Liu, W. Sun, W. Sun, Q. Hu and H. Li, Eur. J. Org Chem., 2024, 27, e202400867 CrossRef CAS.
- Y. Cheng, J. Rein, N. Le and S. Lin, J. Am. Chem. Soc., 2024, 146, 31420–31432 CrossRef CAS PubMed.
- H. Ren, P. Zhang, J. Xu, W. Ma, D. Tu, C. S. Lu and H. Yan, J. Am. Chem. Soc., 2023, 145, 7638–7647 CrossRef CAS PubMed.
- S. Liu, J. Tang, F. Ji, W. Lin and S. Chen, Gels, 2022, 8, 46 CrossRef CAS PubMed.
- J. Ding, X. Ding and J. Sun, Materials, 2022, 15, 4498 CrossRef CAS PubMed.
- D. A. Singleton and A. A. Thomas, J. Am. Chem. Soc., 1995, 117, 9357–9358 CrossRef CAS.
- K. B. Wiberg and L. H. Slaugh, J. Am. Chem. Soc., 1958, 80, 3033–3039 CrossRef CAS.
- R. Glaser, C. J. Horan, E. D. Nelson and M. Kirk Hall, J. Org. Chem., 1991, 57, 215–228 CrossRef.
- C. Tang, J. Zhang and Z. Xie, Angew. Chem., Int. Ed., 2017, 56, 8642–8646 CrossRef CAS PubMed.
- C. Tang, J. Zhang, J. Zhang and Z. Xie, J. Am. Chem. Soc., 2018, 140, 16423–16427 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2407119–2407122. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc00234f |
| ‡ These authors contributed equally: Wanqi Sun and Yujie Jin. |
| This journal is © The Royal Society of Chemistry 2025 |