
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnveiling the molecular basis of selective fluorination of SAM-dependent fluorinases†
Ravi Kumar
Verma‡
 a,
Wan Lin
Yeo‡
b,
Elaine
Tiong‡
c,
Ee Lui
Ang
*be,
Yee Hwee
Lim
*de,
Fong Tian
Wong
*cd and
Hao
Fan
*aef
a,
Wan Lin
Yeo‡
b,
Elaine
Tiong‡
c,
Ee Lui
Ang
*be,
Yee Hwee
Lim
*de,
Fong Tian
Wong
*cd and
Hao
Fan
*aef
aBioinformatics Institute, Agency for Science, Technology and Research, 30 Biopolis Street, #07-01 Matrix Building, Singapore 138671, Republic of Singapore. E-mail: ravikumarv@bii.a-star.edu.sg; hfan2006@gmail.com; fanh@bii.a-star.edu.sg
bSingapore Institute of Food and Biotechnology Innovation, Agency for Science, Technology and Research, 31 Biopolis Way, #04-01 Nanos, Singapore 138669, Republic of Singapore. E-mail: yeo_wan_lin@sifbi.a-star.edu.sg; ang_ee_lui@sifbi.a-star.edu.sg
cInstitute of Molecular and Cell Biology, Agency for Science, Technology and Research, 61 Biopolis Drive, Proteos, Singapore 138673, Republic of Singapore. E-mail: elaine_tiong@imcb.a-star.edu.sg; wongft@imcb.a-star.edu.sg
dInstitute of Sustainability for Chemicals, Energy and Environment, Agency for Science, Technology and Research, 8 Biomedical Grove, #07-01 Neuros Building, Singapore 138665, Republic of Singapore. E-mail: lim_yee_hwee@isce2.a-star.edu.sg
eSynthetic Biology Translational Research Program, Yong Loo Lin School of Medicine, National University of Singapore, 10 Medical Drive, Singapore 117597, Republic of Singapore
fDepartment of Biochemistry, Yong Loo Lin School of Medicine, National University of Singapore, 8 Medical Drive, Singapore 117596, Republic of Singapore
First published on 5th May 2025
Abstract
S-Adenosylmethionine (SAM)-dependent fluorinases have emerged as environmentally friendly enzymatic alternatives for organofluorine chemical synthesis. However, their use remains limited by their rarity; only 16 fluorinases have been found in nature so far. Here we report two new fluorinases, FLASbac from Streptosporangiales bacterium and a modified FLAAdig_Nter from Actinoplanes digitatis. Through molecular dynamics (MD) simulations, we have identified the crucial roles of the SAM-binding site and an ion-egress site (IES) for fluorination reaction, particularly regarding its preference for fluoride ions. We have validated these findings by testing mutants of the two new fluorinases and the known fluorinase from Streptomyces sp. MA37 (FLAMA37). Through these targeted mutations, we identified, for the first time, specific sites in certain variants that significantly enhance the enzyme's specificity for fluorination over chlorination while maintaining its fluorination activity. In these particular variants, this refinement led to a remarkable increase in fluorine preference, improving from approximately 10-fold to over 200-fold. Overall, this research advances our fundamental understanding of enzymatic fluorination, providing a basis for further exploration of fluorinase optimization. In turn, these advancements could open new opportunities for the pharmaceutical industry in the development of organofluorine drugs and other fluorine-reliant biotechnologies.
1. Introduction
In recent years, organofluorines have garnered a lot of attention due to their unique physicochemical properties,1,2 such as high thermal and chemical stability and enhanced pharmacokinetic parameters such as metabolic stability, bioavailability and membrane permeability, when compared to their non-fluorinated counterparts. Presently, organofluorine compounds are widely used in various industries,2,3 including agrochemicals, electronics, and pharmaceutical industries. So far, organofluorines have been synthesized using organic chemistry methods, however, the use of hazardous and noxious reagents used in organofluorine synthesis has compelled researchers to hunt for safer, less toxic alternatives. In this context, fluorinase enzymes, belonging to the “S-Adenosylmethionine (SAM)-Dependent Hydrolases/Halogenases” superfamily, have emerged as an environmentally friendly and sustainable alternative to traditional organic chemistry methods for organofluorine synthesis. SAM-dependent fluorinases are trimeric enzymes (Fig. 1A), with an ability to introduce fluorine atoms at the C-5′ position of their natural substrate SAM,7 demonstrating their unique and valuable enzymatic function (Fig. 1B). While crystal structures demonstrate both trimeric and hexameric forms, a recent study has confirmed that trimers, rather than hexamers, are the functional units responsible for fluorination activity.8 Notably, fluorinases share key structural characteristics with chlorinases9 and hydrolases,6 all members of the “SAM-Dependent Hydrolases/Halogenases” (Fig. 1C). | ||
Fig. 1 (A) Biological unit of “SAM Hydrolases/SAM-Dependent Halogenases” superfamily members (PDB 1RQP4). (B) Diversity of the reaction mechanisms in the “SAM Hydrolases/SAM-Dependent Halogenases” superfamily. (C) Identical structural fold in the “SAM Hydrolases/SAM-Dependent Halogenases” superfamily. The fluorinase (PDB 1RQP4), chlorinase (PDB 6RYZ5), hydrolase (PDB 2WR8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 6) are shown in magenta, yellow, and grey cartoons, respectively, with bound SAM (orange) and the co-crystallized chloride ion (green) in chlorinase. Inset highlights different IBS residues in chlorinase and fluorinase, with the modelled fluoride ion shown as a cyan sphere. (D) Schematic illustration of results from SSN analysis. Four subnetworks containing putative fluorinase sequences are represented in different shapes and colours. The dashed line indicates a sequence identity threshold of 60%, meaning the maximum sequence identity between any two sequences from different subnetworks is below 60%. The subnetwork-1 including 16 known fluorinases and the four newly discovered putative fluorinases (FLASbac, FLAAdig, FLASmor, and FLAGbac) are highlighted in bold font. 6) are shown in magenta, yellow, and grey cartoons, respectively, with bound SAM (orange) and the co-crystallized chloride ion (green) in chlorinase. Inset highlights different IBS residues in chlorinase and fluorinase, with the modelled fluoride ion shown as a cyan sphere. (D) Schematic illustration of results from SSN analysis. Four subnetworks containing putative fluorinase sequences are represented in different shapes and colours. The dashed line indicates a sequence identity threshold of 60%, meaning the maximum sequence identity between any two sequences from different subnetworks is below 60%. The subnetwork-1 including 16 known fluorinases and the four newly discovered putative fluorinases (FLASbac, FLAAdig, FLASmor, and FLAGbac) are highlighted in bold font. | ||
Despite the promising prospects, applications of fluorinases have been restricted by their scarcity in nature, i.e., to date only 16 fluorinases have been identified,10 of which two fluorinases, FLAScat from Streptomyces cattleya4 and FLAMA37 from Streptomyces sp. MA37,11 have crystal structures. Furthermore, these enzymes exhibit a degree of promiscuity, catalysing not only fluorination reaction, but also chlorination reactions.10,12 Previous studies comparing fluorinases and chlorinases have established that the halogen specificity is influenced by the architecture of the ion-binding site (IBS). This site has been previously referred to as various names, such as “halide binding pocket”13 or “anion binding site”.14 Here, we refer to it as IBS to distinguish it from ion-egress site (IES). A key difference in our definition is the treatment of residue T80. While previous studies grouped T80 and S158 together, we classify S158 under IBS and T80 under the IES. IBS is composed of four consecutive and conserved residues: a threonine, a phenylalanine/tryptophan, a tyrosine/phenylalanine, and a serine (Fig. 1C and Table S1†).13 Mutating the fourth serine residue (S158) in the IBS of FLAScat, specifically S158G and S158A,15 led to reduced fluorinase activity, emphasizing the role of this S158's hydroxyl group in fluoride ion binding and catalysis. However, introducing serine mutation in the 4th residue in the IBS of chlorinases did not produce fluorinases,9,16 indicating that other residues are also crucial for the halogenation specificity. Interestingly, despite understanding its significance, previous attempts at IBS modifications often had negative effects, even with increased specificity for fluorination.15,17
In this study, we first expanded the known sequence space of fluorinases by identifying two new fluorinases, FLASbac from Streptosporangiales bacterium and FLAAdig_Nter from Actinoplanes digitatis using Sequence Similarity Network (SSN) analysis and experimental testing. Guided by MD simulations, we analysed FLASbac, FLAAdig_Nter, and a known fluorinase from Streptomyces sp. 37 (FLAMA37).18 This yielded additional insights into the known SAM-binding site and ion-binding site, along with the discovery of an ion-egress site (IES), which serves as a critical gateway to the IBS. Our targeted mutations at the SAM-binding and the IES sites, in contrast to previous studies, enabled us to successfully engineer fluorinase variants for the first time with increased specificity for fluorination over chlorination, while maintaining their fluorination activity. Collectively, these findings provide a comprehensive new perspective on the molecular mechanisms governing fluorinase specificity and ion-binding preferences.
2. Experimental section
2.1 Identification of novel fluorinases using sequence-based approaches
The protein sequence of fluorinase from Streptomyces cattleya, FLAScat (UniProt Q70GK9), was downloaded from the UniProt sequence database.19 Using this sequence, a BLAST search was performed using an E-value threshold of 10−5 against the UniProt sequence database and a set of 7074 sequences from bacteria, archaea, and metagenomic samples was identified. We removed 134 sequences with fewer than 250 amino acid residues and obtained a set of 6940 sequences. To analyse these sequences and decipher the potential functional relationship among the computed protein sequence we employed the sequence similarity network (SSN). In SSN, protein sequences are represented as nodes in a network graph that are connected to other nodes by edges when the pairwise sequence similarity is greater than a predetermined threshold. Enzyme Function Initiative-Enzyme Similarity Tool (EFI-EST),20 with a permissive edge detection threshold equal to 40% sequence identity, was used to compute the SSN. SSN was visualized in Cytoscape 3.9.121 and a cut-off of 60% sequence identity was employed to remove edges and divide SSN into smaller subnetworks. Further, using fluorinase enzyme classification number (EC: 2.5.1.63) as a filtering criterion, we identified four distinct subnetworks (subnetwork 1–4), each containing at least one sequence annotated as a fluorinase.
2.2 Homology modeling and structure preparation
Protein sequences for the newly discovered FLAs, FLASbac and FLAAdig were downloaded from the UniProt database. A mutant of FLAAdig was created by N-terminal addition of beta-strand forming peptide (sequence: MAANGSQRPIIAF) from FLAMA37, yielding FLAAdig_Nter (Fig. 2A). The 3D coordinates for FLA trimers were built using Prime in Schrodinger Suite,22 using Streptomyces cattleya fluorinase, FLAScat (PDB 1RQP4), as template. Since 1RQP was solved without fluoride ion in the IBS, the fluoride ion was introduced in 1RQP prior to homology modeling. The location of the fluoride ion was inferred from the crystal structure of Salinispora tropica CNB-440 chlorinase (PDB 6RYZ5), SaIL, which was solved in the presence of chloride ion at the IBS. The fluorinase crystal structure was superposed on chlorinase crystal structure and a fluoride ion was introduced at the corresponding location of chloride ion in 6RYZ. Throughout the homology model building process, we used the implicit solvent model VSGB223 and the force field OPLS4.24 To elucidate why fluorinases prefer fluorination reactions over chlorination, additional systems with chloride ion in the IBS were also prepared for molecular dynamics simulations.
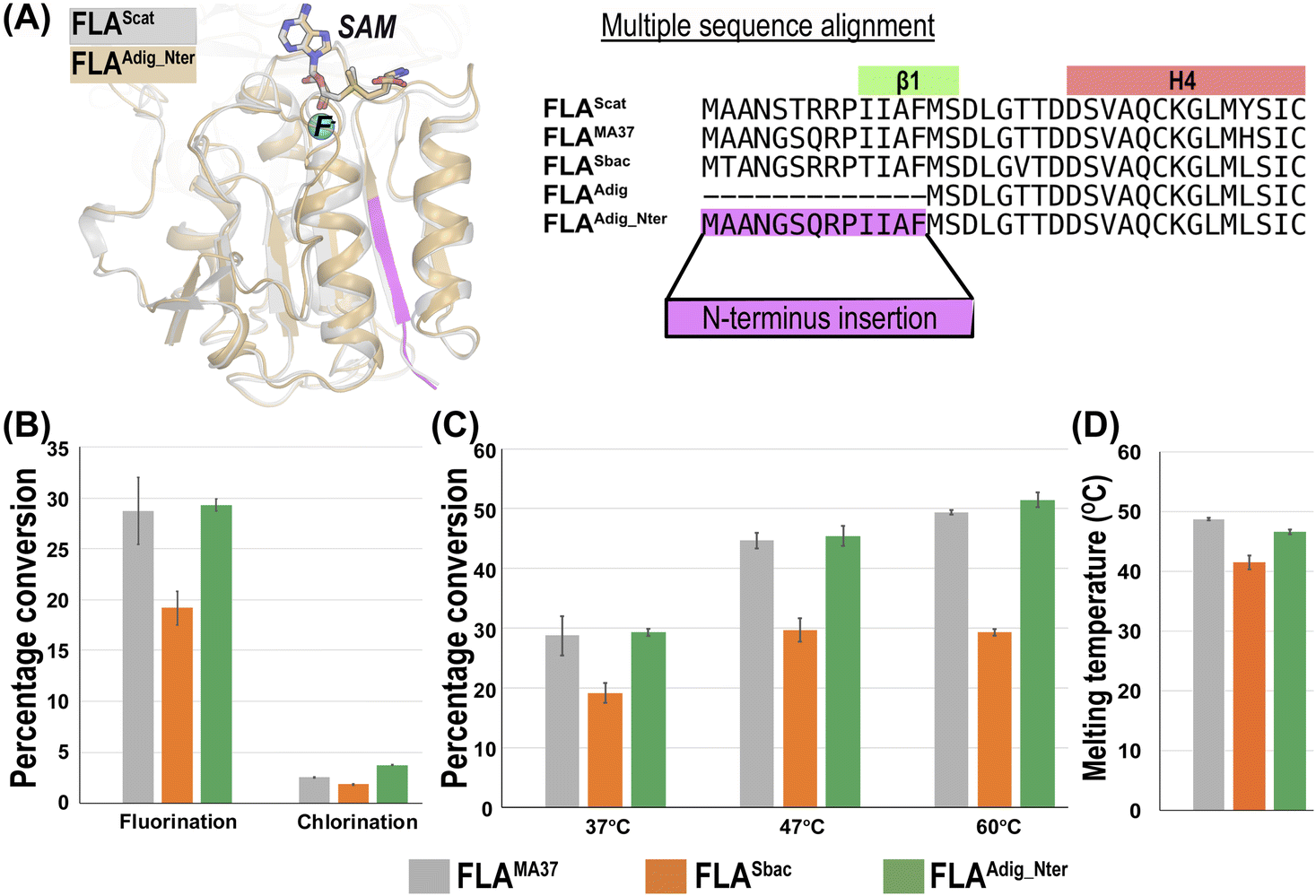 | ||
| Fig. 2 Structural and functional characterization of FLASbac and FLAAdig_Nter in comparison to FLAMA37. (A) Superposition of the FLAAdig_Nter homology model on the FLAScat crystal structure and multiple sequence alignment of the N-terminal region. The crucial beta-strand added to the N-terminus in FLAAdig_Nter is highlighted in magenta. (B) Enzymatic activity comparison of FLASbac and FLAAdig_Nter with FLAMA37 at 37 °C. (C) Temperature effect on fluorination activity. (D) Melting temperatures of FLAMA37, FLASbac and FLAAdig_Nter. | ||
2.3 Molecular dynamics simulation protocol
The comparative MD-simulations of FLASbac, FLAAdig_Nter, FLAMA37 were carried out in Desmond MD System.25 We employed the OPLS4 force field24 to model the entire system, including the protein, ions, and the cofactor S-adenosylmethionine (SAM). The input for MD-simulations was prepared using system builder.26 The systems were solvated with TIP3P explicit water and system charges were neutralized using counterions. In addition, sodium and chloride ions were added for macroscopic concentrations of 150 mM NaCl. Overall, the simulation systems consisted of ∼95000 atoms. The initial MD-simulation systems were relaxed in multiple short MD steps. Briefly, first the systems were relaxed using Brownian dynamics at 10 K with restraints on the solute atoms, followed by another relaxation step at 100 K with restraints on the solute heavy atoms under the NVT ensemble. Next, the system temperature was gradually raised to the desired simulation temperature using simulated annealing with restraints on the solute heavy atoms under the NPT ensemble. This was followed by a relaxation step with restraints applied only to the protein backbone and ligand heavy atoms. The system was then equilibrated for 12 nanoseconds (ns) with restraints only on the ligand heavy atoms. Finally, the equilibrated system was subjected to unrestrained 300 ns production run. For both equilibration and the following unrestrained production runs, we used Langevin constant pressure and temperature dynamical system27 to maintain pressure at 1 atm with a time step of 2 femtoseconds under the NPT ensemble. Long-range electrostatic interactions were calculated using the particle mesh Ewald method.28 A radius of 12.0 Å was used for coulomb interactions. The systems were simulated at 47 °C. To improve conformational sampling, two independent trajectories for each system with different random initial velocities were computed. The data analysis was carried out with the MDTraj program.29
2.4 Chemicals
All chemicals were purchased from Sigma-Aldrich and used as received unless otherwise stated. S-Adenosyl-L-methionine (SAM) was purchased from Santa Cruz Biotechnology. 5′-Fluoro-5′-deoxyadenosine (5′-FDA) was synthesized as previously mentioned.112.5 Construction of expression plasmids for expression of fluorinases in E. coli
DNA sequences optimized for E. coli codons were synthesized by Twist Biosciences, Singapore. Subsequently, these sequences were assembled into the pET28a (+) vector using Golden Gate assembly. The assembled constructs, which carry a C-terminal 6xHis tag for protein purification, were verified by sanger sequencing.2.6 New variants construction
Single amino acid mutant expression plasmids were generated through the QuikChange site-directed mutagenesis protocol (Agilent, USA), utilizing the respective expression plasmids for the wild-type genes as the template. Primers listed in Table S8† were employed for this process. To replace N-terminal regions, NEBuilder HiFi DNA assembly was used to assemble the N-terminal portion of the FLAMA37 with FLAAdig. Sequences of resulting constructs were confirmed through Sanger sequencing.2.7 High throughput (HTP) growth and lysis
The plasmids harbouring the fluorinase genes were transformed into E. coli BL21(DE3) ΔPNP strain.11 84 colonies of the various constructs, 6 colonies of FLAMA37 (as positive control),18 and 6 colonies of pET28a (as negative control) were inoculated into a 96-well plate. Individual colony was inoculated in 180 μL LB medium containing kanamycin (50 μg mL−1) and cultured overnight at 37 °C. 20 μL of cultures were transferred to 380 μL 2xYT medium containing 50 μg mL−1 kanamycin and grown until an absorbance of 0.5 at 600 nm was reached. Overexpression was induced by adding 0.2 mM isopropylthiogalactoside (IPTG) and incubation was continued at 22 °C for 20 h. Cells were harvested by centrifugation (2182 × g for 10 min at 4 °C). The cell pellets were lysed with 250 μL lysis buffer (50 mM sodium phosphate buffer, pH 7.8, 10% glycerol, 0.1% Triton-X) at room temperature for 3 h at speed 6.5 on titer plate shaker (Barnstead). The lysates were obtained by centrifugation (2182 × g for 20 min at 4 °C).2.8 Large scale fluorinase expression in E. coli and purification
The E. coli containing the plasmid was grown in 500 mL 2xYT medium with 50 μg mL−1 kanamycin at 37 °C until an absorbance of 0.5 at 600 nm was reached. Overexpression was induced by adding 0.2 mM IPTG and incubation was continued at 22 °C for 20 h. Cells were harvested by centrifugation (3830 × g for 20 min). The cell pellet was resuspended in 20 mL binding buffer (50 mM sodium phosphate buffer, pH 7.8, 10% glycerol, 300 mM NaCl, 5 mM imidazole) and the cells were lysed using a cell disruptor (Constant Systems) at 40 kpsi. The lysate was then centrifuged, and the clarified supernatant was added to TALON metal affinity resin (Clontech). The supernatant-resin mixture was incubated at 4 °C with rotation for 3 h. The His-tagged protein bound resin was washed with the binding buffer and eluted with the elution buffer (50 mM sodium phosphate buffer, pH 7.8, 10% glycerol, 300 mM NaCl, 250 mM imidazole). The elution was desalted using PD-10 desalting column (GE Healthcare) in storage buffer (50 mM sodium phosphate buffer, pH 7.8, 10% glycerol). The eluted protein was then concentrated using a 30 kDa concentrator (Merck Millipore) with centrifugation at 5000 × g. The protein concentration was measured using a NanoDrop 2000 spectrophotometer (Thermo Scientific). The extinction coefficient was determined using the ExPAsy ProtParam tool.2.9 Enzymatic reaction
HTP enzymatic reaction conditions: SAM (1 mM), NaF (200 mM), and fluorinase lysates (182 μL) in a final volume of 260 μL. HTP enzymatic reaction was carried out in a 96-well deepwell plate at 37 °C for 4 h and reaction was stopped by quenching with 1:1 volume of methanol. The quenched reaction mixtures were centrifuged (2182 × g for 20 min) and 10 μL was used for HPLC-UV analysis.
All reactions using purified proteins were carried out in triplicates in 1.5 mL tubes in shaking incubator at 250 rpm. Reactions were stopped by heating the samples at 95 °C for 1 min (using a PCR machine). The precipitated protein was then removed by centrifugation (20238 × g for 2 min). 10 μL of the reaction mixture was used for HPLC-UV analysis.
Tube enzymatic reaction conditions (activity): SAM (1 mM), NaF or NaCl (200 mM), and fluorinase purified proteins (20 μM) in a final volume of 100 μL at 37 °C for 1.5 h and 24 h.
Tube enzymatic reaction conditions (thermal stability): SAM (1 mM), NaF or NaCl (200 mM), and fluorinase purified proteins (20 μM) in a final volume of 100 μL at 37 °C for 1.5 h at either 47 °C or 60 °C.
Kinetic assay reaction conditions: various concentrations of SAM (10–1000 μM), NaF (200 mM), and fluorinase purified proteins (20 μM) in a final volume of 100 μL at 37 °C. Reactions were carried out at various time points. All reactions were carried out in triplicates, in 200 μL PCR tubes using the PCR machine. Reactions were stopped by heating the samples at 95 °C for 1 min (using a PCR machine). The precipitated protein was then removed by centrifugation (20238 × g for 2 min). 10 μL of the reaction mixture was used for HPLC-UV analysis. Kinetic parameters were obtained by the best-fit model of initial velocity against substrate concentrations based on Michaelis–Menten equation using GraphPad Prism 9 (GraphPad Software).
2.10 HPLC analysis
HPLC analysis was carried out using Agilent HPLC 1200 Infinity series paired with CTC Analytics HTS PAL LC Autosampler.Mobile phase A: 0.1% formic acid in water; B: 0.1% formic acid in methanol, Phenomenex, Kinetex® 2.6 μm biphenyl 100 Å, LC Column 150 × 4.6 mm, 0.6 mL min−1 flow rate, isocratic elution, 23% B for 15 min. The retention time of 5′-FDA is approximately 3.95 min. The retention time of 5′-ClDA is approximately 5.93 min.
2.11 Protein thermal shift assay
Protein thermal shift assay conditions: 0.25 μg μL−1 purified proteins, 1× GloMelt dye, 20 μL total volume, SYBR Green channel, 95 °C for 1 min, melting curve of 10 °C–95 °C with increment of 0.1 °C s−1, 95 °C for 1.5 min. Protein thermal shift assay were carried out using Biorad CFX96™ Real-Time System and melting temperatures were obtained from analysis using its respective software.3. Results and discussion
3.1 Identification of novel fluorinases
We used the sequence of fluorinase from Streptomyces cattleya (FLAScat) to search the UniProt database, resulting in 7074 homologous sequences. After removing 134 partial fragments with fewer than 250 amino acid residues, we constructed a SSN of these sequences and identified four subnetworks that contain at least one sequence with fluorinase EC number (Fig. 1D). Notably, none of the four subnetworks include SalL chlorinase, which belongs to a separate subnetwork.Subnetwork-1 includes 20 unique sequences, 16 of which are known fluorinases, including FLAScat and FLAMA37 (Table S1 and Fig. S1†). Newly identified members FLAGbac, FLASbac, FLAAdig, and FLASmor exhibit significant maximal sequence identities ranging from 68.9%, 78.8%, 93.7%, and 97.9%, respectively, with known fluorinases. FLASmor was not screened due to its high sequence identity to known fluorinases. FLAGbac could not be expressed in any soluble form, hence no further experiment was performed. FLASbac and FLAAdig were expressed and assessed for fluorination and chlorination activity against SAM substrate, as well as thermal stability (Fig. 2). FLASbac exhibited fluorination activity in its native sequence, whilst activity for FLAAdig had to be rescued through a N-terminal addition of beta-strand forming peptide from FLAMA37, yielding FLAAdig_Nter (Fig. 2A and Table S2†). Further discussion on these findings is provided in Section 3.2.
The remaining three subnetworks (subnetworks 2–4) consist of 370, 10, and 2 unique sequences, respectively, with experimentally uncharacterized functions. These highly divergent sequences show low sequence identity to known fluorinases, with maximum identities of 24.2–31.4%, 26.9–29.3%, and 26.7–27.5% for subnetworks 2, 3, and 4. Furthermore, members of these subnetworks show notable differences in the IBS residues (Fig. S2†). We selected 20 sequences from subnetworks 2–4 (Table S3†) for testing, but none of these sequences exhibited detectable fluorination or chlorination activity. Importantly, none of the sequences in these subnetworks, or any other sequences in the SSN, exhibited the key IBS feature of known fluorinases: a threonine at the first position, two aromatic residues at second and third positions, and a serine at the last position. This suggests the significance of these specific residues in fluorinase function.
Further analysis of sequence features revealed critical variations within the β3–β4 loop, located between the β3 and β4 strands, among members of subnetworks 1–4 (Fig. S3†). Subnetwork-1 sequences contain two conserved threonine residues at positions 80 (T80) and 82 (T82). Of these, the T80 is uniquely conserved in subnetwork-1 only, while T82 is also conserved in subnetwork-3 and subnetwork-4. In contrast subnetwork-2 sequences lack threonine at both these positions. Additionally, subnetwork-1 contains a single conserved positively charged arginine at position 85 (R85), whereas subnetworks 2–4 exhibit an overabundance of positively charged residues at multiple positions in the β3–β4 loop. Prior mutagenesis studies on FLAScat highlighted the role of T80 in fluorination activity, with the T80A substitution reducing the catalytic activity to 15% of the wild-type.30 We hypothesize that, in addition to the IBS residues, the distinct sequence features in the β3–β4 loop, particularly the conserved threonine and arginine residues in subnetwork-1 and the divergences in subnetworks 2–4, have functional implications. This hypothesis is explored in detail under Section 3.4, where we experimentally investigate the structural and functional implications of these variations.
3.2 Ion selectivity through IBS residues
FLAMA37, FLASbac, and FLAAdig_Nter exhibit, 11.5-, 10.4-, and 8.7-fold specificity for fluorination over chlorination, respectively (Table S4†). We hypothesized that the IBS dynamics can explain the preference of these proteins for fluorination over chlorination.We compared the F− and Cl− binding free energies for the IBS residues from the initial MD-relaxed initial conformations of FLAMA37, FLASbac, and FLAAdig_Nter using MMGBSA method.22 Fluoride ion showed stabilizing negative binding free energy values (−2.87, −0.33, and −3.21 kcal mol−1) for S158 respectively, which are consistent with the experimental mutational data on S158, where S158G and S158A variants led to reduced fluorinase activity.15 In contrast, the chloride ion showed destabilizing positive values (2.09, 2.79, and 2.19 kcal mol−1) likely due to its larger van der Waals radius and lower electronegativity. Additionally, the other three IBS residues, T155, F156, and Y157, also contributed to the relative preference for binding F− over Cl− by the three enzymes (Table S5†), except for T155 in FLASbac. Although being useful, the MMGBSA method cannot fully capture the breadth of molecular interactions as it relies on approximate models for solvation and often treat entropic contributions at a simplified level.32 Therefore, we further explored the stability of the F− and Cl− ion in the IBS with MD simulations. Specifically, we computed the minimum distance between the fluoride/chloride ion and the polar hydrogen atoms of S158 in the IBS (Fig. S4A†). Our analysis revealed that F− shows much higher occupancy than Cl− in the IBS of all the three fluorinases (Fig. S4B†). In FLAMA37 and newly identified FLASbac, the minimum distance between the F− and the sidechain/backbone polar hydrogen atoms remained less than 2.5 Å throughout the entire MD simulation trajectories, while the engineered FLAAdig_Nter showed 4.77% loss of this critical interaction, with the loss occurring exclusively in one of the two simulation trajectories over a duration of 300 ns. Unlike the fluoride ion, chloride ion demonstrated a tendency to exit the IBS during the MD simulations. Specifically, in every trimer simulation conducted, at least one monomer exhibited a clear egress of the chloride ion from the IBS. This behaviour highlights a key difference in the interaction dynamics of fluoride and chloride ions within the fluorinase enzyme. To rule out potential force field artifacts, we also conducted additional MD simulations of F− and Cl− bound to the related chlorinase, SalL. Interestingly, these MD simulations revealed a opposite trend: F− displayed a greater tendency to dissociate compared to Cl− ions (Fig. S5†). We observed that in three of the six monomers, specifically monomer M1–2 in replicate-1 and M2 in replicate-2, the F− exited the IES (distance > 8 Å) in both F−-bound SalL trajectories. In contrast, no complete event was observed in the Cl−-bound SalL trajectories.
Analysis of minimum distance between the F156 and S158 in the IBS revealed smaller median distances in Cl−-bound trajectories compared to F−-bound fluorinase trajectories. The differences in these distances were 0.5 Å, 0.3 Å, and 0.2 Å, for FLAMA37, FLASbac, and FLAAdig_Nter, respectively (Fig. S6†). The closer proximity observed in Cl−-bound trajectories likely destabilizes Cl− binding by facilitating its displacement from the IBS, consistent with the enzymes' specificity for fluorination. Previously, it was proposed that in FLAScat, F− bind to the active site in solvated form and subsequently exchange the bound water molecules by forming interactions with the polar hydrogens of S158 residue in IBS.15 Building on this model, we hypothesize that the reverse process occurs during the release of the F−/Cl− ion from IBS, wherein water molecules re-enter and reoccupy the IBS as the ion exits. To test this hypothesis, we estimated water penetration events within IBS using a minimum distance criterion of 3.0 Å between the sidechain and backbone polar hydrogens of S158 in the IBS and water molecules, considering only frames where F− or Cl− remained within 8.0 Å of the IBS. Water penetration was minimal in F−-bound trajectories, occurring in 2.5%, 12.6%, and 2.7%, of frames for FLAMA37, FLASbac, and FLAAdig_Nter, respectively, but was significantly higher in Cl−-bound trajectories at 26.5%, 36.9%, and 28.8% (Fig. S4C†). These results further suggest that chloride ions exhibit weaker binding affinity within the IBS compared to fluoride. Furthermore, fluorinases with higher fluorination activity, FLAMA37 (kcat/KM: 13.7 ± 2.2 mM−1 min−1) and FLAAdig_Nter (kcat/KM: 12.9 ± 1.9 mM−1 min−1) showed less water penetration than FLASbac (kcat/KM: 10.0 ± 1.9 mM−1 min−1), which has lower fluorinase activity (Table S6†). MD simulations of wildtype FLAAdig, lacking the N-terminal β-strand (Fig. 2A), showed higher incidences of water penetration in both F−- and Cl−-bound trajectories (22.3% and 34.1%, respectively) compared to FLAAdig_Nter, highlighting the critical role of the N-terminal β-strand on fluorinase function.
3.3 Improving F−/Cl− specificity by mutating SAM-binding site
Since SAM-binding site is located next to the IBS, we hypothesized that mutations affecting SAM-binding could lead to altered fluorination and chlorination activities in fluorinases. With analysis of MD simulations, we identified a set of seven residue positions within the SAM-binding site that form specific interactions with SAM, each occurring with > 60% frequency (FLASbac sequence used as the reference for residue numbering) (Fig. 3A and Table S7†). Among these seven positions, F50 (W50 in FLAMA37 and FLAAdig_Nter), F213, and F254 form pi–pi interactions, while N215 forms a hydrogen bond with the adenosine moiety of SAM. The methionine moiety of SAM is stabilized by R270, D21, and D210 through salt-bridges. Among these interactions, D210 was observed to form the most stable salt-bridge interaction with SAM (Fig. 3A, B and Table S7†). We also compared the SAM binding residues in fluorinases with the SalL chlorinase. Interestingly, these seven residues are identical between FLASbac and SalL. Notably, the aspartate residue at position 210 is highly conserved among known fluorinases (Fig. 3D) and SalL chlorinase (Fig. S1†). As a result, we decided to first investigate D210A mutations in the three fluorinases. The D210A mutation caused notable changes in the interactions involving several of the seven residue positions mentioned above (Table S7†). Experimental testing revealed that, FLASbac_D210A retained reasonable fluorinase activity (0.75-fold of wild-type) but had negligible chlorinase activity (0.04-fold of wild-type), resulting in greater than 200-fold specificity for fluorination over chlorination (Fig. 3E and Table S4†). By contrast, FLAMA37_D210A and FLAAdig_Nter_D211A exhibited almost complete loss of fluorination capabilities (Fig. 3E and Table S4†), similar to prior mutagenesis study aimed at improving specificity.15 | ||
| Fig. 3 Structural and functional analysis of SAM-binding site. (A) Ligplot+31 generated 2D ligand interaction plot illustrating the type of molecular interactions between SAM (depicted in purple) and SAM-binding residues in the FLASbac. FLASbac homology model, was constructed using FLAScat (PDB 1RQP4) as the template. Location of H211, which do not interact directly with SAM, is shown in ellipse. (B) Interaction (magenta) between the SAM-binding site residue D210 (green) and SAM (grey) within FLASbac, with adjacent H211 (pink) and fluoride ion (cyan). (C) Interaction between the R211 and E54 in FLASbac_H211R mutant (green) compared to wild-type FLASbac (grey). (D) Residue conservation at 210–211 position in known 16 fluorinases. (E) Effect of mutations on halogenation activity. | ||
Building on insights from the D210A mutation results, we also targeted the adjacent H211 that is less conserved than D210 in known fluorinases (Fig. 3C, D and Fig. S1†). This residue is hypothesized to play a role in maintaining the structural integrity of the SAM-binding site. Analysis of our wild-type MD simulation trajectories revealed that H211 forms inter-monomer hydrogen bond interactions with the sidechain or backbone of nearby residue T20 (Movie S1†). Mutating H211 to arginine (H211R) could alter these inter-monomer interactions affecting the dimer interface where SAM binds. Our MD simulations of H211R mutant supported this hypothesis. The R211 maintained interactions with the backbone of T20 but showed reduced interactions with the T20 sidechain. Additionally, the extended sidechain of R211 allowed formation of new intra-monomer interactions with E54 (Movie S2†). The E54 is positioned near the F50 (W50 in FLAMA37 and FLAAdig_Nter) which stabilizes the adenosine-moiety of SAM trough pi–pi interactions (Movie S3†). Thus, the R211–E54 interaction could potentially influence intra-monomer SAM binding as well. Experimentally, FLAMA37_H211R, FLASbac_H211R, and FLAAdig_Nter_H212R largely retained fluorination activities, with values of, 0.96-, 0.87-, and 0.88-fold of their respective wild-types (Fig. 3E). The FLASbac_H211R and FLAMA37_H211R exhibit increased F−/Cl− specificities of 28.8- and 16.6-fold, respectively, compared to the wild-type FLASbac (10.4-fold) and FLAMA37 (11.5-fold). In contrast, the FLAAdig_Nter_H212R showed a slightly reduced F−/Cl− specificity of 8.3-fold (Table S4†) compared to 8.65-fold specificity in FLAAdig_Nter.
3.4 Insights into the ion egress pathway
Delving further into F− and Cl− specificities, we compared MD trajectories of F−-bound against Cl−-bound FLAMA37, FLASbac, and FLAAdig_Nter. An important hallmark of our MD trajectories was the release of Cl− from the IBS in at least one monomer of each trimer simulation, a phenomenon not observed in the F−-bound trajectories. However, in one of the FLAAdig_Nter trajectory, the fluoride ion exited the IBS and was observed interacting with both sidechain and backbone polar hydrogens of T82 and T84 residues that are located on the protein surface, outside the IBS (Fig. 4A and B). By extending this FLAAdig_Nter trajectory, we observed complete dissociation of the fluoride ion into the solvent (Movie S4†). Our analysis revealed a common pathway utilized by both fluoride and chloride ions to exit the IBS, involving the β3–β4 loop which we designated as the “ion-egress site” (IES). The β3–β4 loop is enriched with threonine residues T80, T82, and anchored by arginine and serine residues R85, S86 (Fig. 4C). Specifically, after leaving the IBS, the fluoride ion formed stable interactions with T82 and T84, before diffusing into water. The movement of fluoride ion from IBS to IES is facilitated by T80 (Movie S4 and Fig. S7†). Consistent with these findings, we extended one of our FLAMA37 trajectories to 1050 ns and observed a similar trend (Fig. S7†). We hypothesized that the conserved R85 which is distant from IBS, can act as a “hook” to attract negatively charged halide ions and to guide their movement towards threonine residues for subsequent dehydration process. R85's role was confirmed by the FLASbac_R85A mutant that showed no detectable fluorination/chlorination activity (Table S4†).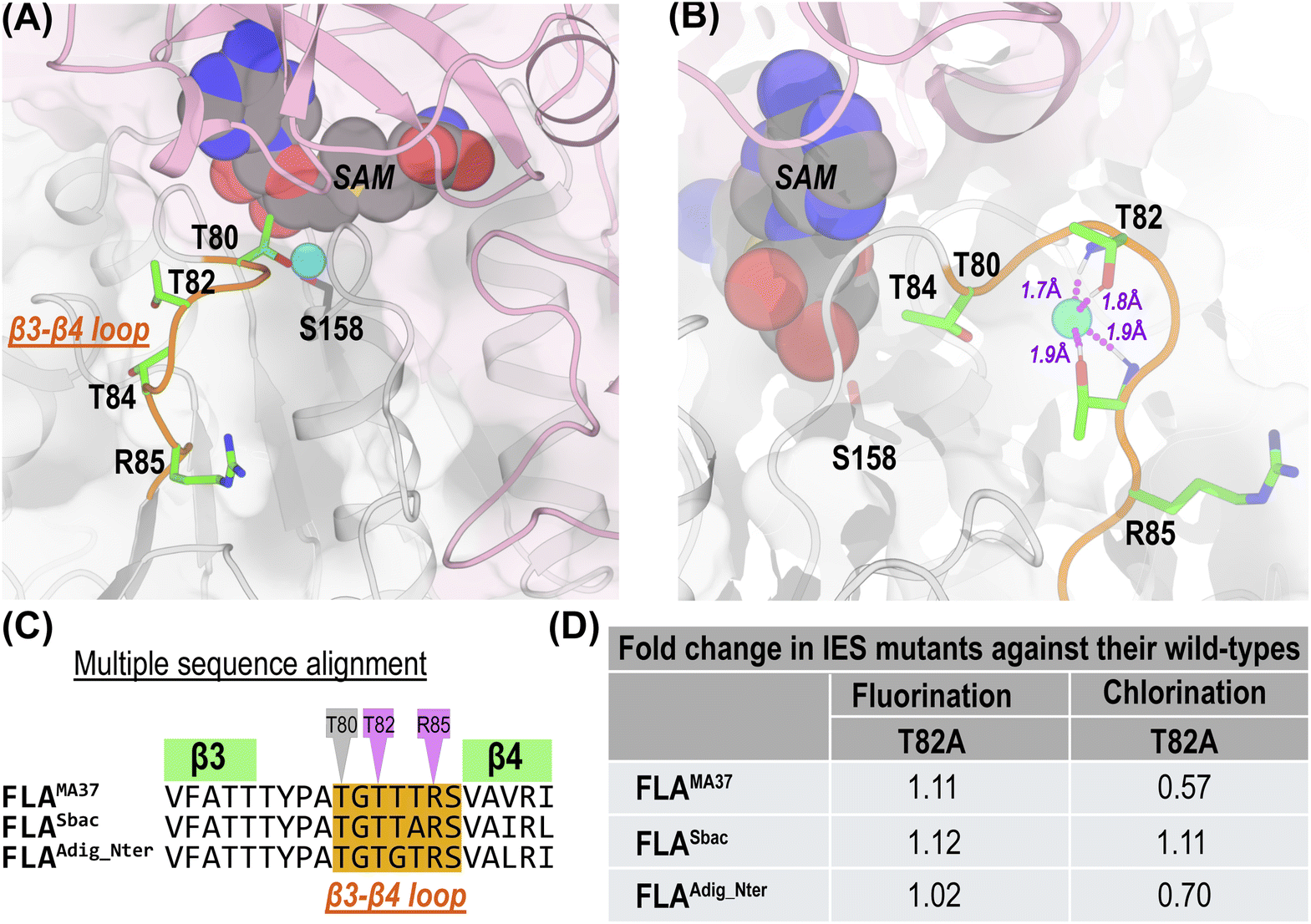 | ||
| Fig. 4 Structural and functional analysis of IES. (A) Relative location of the IES (orange) to the IBS in FLAAdig_Nter. Polar/charged residues in green. Fluoride ion interacting with IBS S158 is shown in cyan sphere. (B) Interactions between the fluoride ion and threonine residues on the IES β3–β4 loop in MD simulations of FLAAdig_Nter. (C) Sequence alignment of β3–β4 containing IES (orange). Mutated T82 and R85 positions in magenta. Conserved T80 is marked in grey. (D) Effect of IES region mutations on halogenation activity. | ||
Within the β3–β4 loop, threonine residues are conserved at positions 80 and 82, and are often observed at positions 83 and 84 in subnetwork-1 members (Fig. S3†). In contrast, the threonine residues are absent in these positions (except for 82) in members of subnetworks 2 to 4 (Fig. S3†). Prior studies on FLAScat suggested that T80 could stabilize the fluoride ion.30 However, our MD simulation results indicate that T80, positioned between the IBS and IES, can also play a crucial role in facilitating ion transport between these regions (Movie S4†). Hence the absence of T80 in subnetworks 2–4 may contribute to the lack of detectable fluorination or chlorination activity in those sequences. In addition, as prior dehydration of F− and Cl− ions is required for their accommodation in IBS,17 we hypothesized that other threonine residues within the β3–β4 loop, although not directly next to the IBS, might also help dehydrate and guide ions into the IBS. Therefore, perturbing IES could also affect enzyme's F−/Cl− specificity. To test this hypothesis, the highly conserved threonine at position 82 was mutated to alanine in FLAMA37, FLASbac, and FLAAdig_Nter. FLASbac_T82A showed 1.12-fold fluorination and 1.11-fold chlorination activity (Fig. 4D). In contrast, FLAMA37_T82A and FLAAdig_Nter_T82A retained their fluorination activity but displayed only 0.6- and 0.7-fold chlorination activity respectively compared to their wildtypes (Fig. 4D). This underscores the potential significance of polar residues within the β3–β4 loop in governing ion egress dynamics and in turn affecting ion specificities.
4. Conclusions
In summary, our research involved computational modeling and experimental validation of three different fluorinases, including two newly identified fluorinases from this study and the previously known FLAMA37. We gained key insights into the role of residues around the SAM-binding site and the newly identified “ion-egress” pathway on enzymatic catalysis and specificities. Most importantly, insights from these mutations have enabled us to take the first critical steps towards being able to fine-tune fluorine specificity whilst still maintaining fluorination activity. Our results also underscore the context-dependent nature of these mutations, as their effects are influenced by the local structure and sequence of each homolog. This highlights the inherent complexity of fine-tuning selectivity in fluorinases. Our finding provides a foundation to facilitate the rational design and engineering of fluorinases for enhanced catalytic efficiency and expanded substrate scope, aiding the development of novel organofluorine compounds for use in agrochemicals, electronics, and pharmaceuticals.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
R. K. V. performed the in silico work and drafted the manuscript. W. L. Y. and E. T. performed the experimental work and contributed to the manuscript writing. Conceptualization, funding acquisition, work supervision, and manuscript finalization were carried out by E. L. A., Y. H. L., F. T. W., and H. F. All authors have approved the final version of the manuscript.Conflicts of interest
The authors have filed an international patent application (PCT/SG2025/050066) for the mutants developed in this study.Acknowledgements
The authors acknowledge funding support from Manufacturing Trade and Connectivity (MTC) Individual Research Grant (IRG) (M22K2c0086), and Agency for Science, Technology, and Research, Singapore, A*STAR (C211917003, C233017006, and C233017010).References
- E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, Applications of Fluorine in Medicinal Chemistry, J. Med. Chem., 2015, 58, 8315–8359 CrossRef CAS.
- M. Inoue, Y. Sumii and N. Shibata, Contribution of Organofluorine Compounds to Pharmaceuticals, ACS Omega, 2020, 5, 10633–10640 CrossRef CAS PubMed.
- T. Okazoe, Overview on the history of organofluorine chemistry from the viewpoint of material industry, Proc. Jpn. Acad., Ser. B, 2009, 85, 276–289 CrossRef CAS.
- C. Dong, F. Huang, H. Deng, C. Schaffrath, J. B. Spencer, D. O'Hagan and J. H. Naismith, Crystal structure and mechanism of a bacterial fluorinating enzyme, Nature, 2004, 427, 561–565 CrossRef CAS PubMed.
- I. J. W. McKean, J. C. Sadler, A. Cuetos, A. Frese, L. D. Humphreys, G. Grogan, P. A. Hoskisson and G. A. Burley, S-Adenosyl Methionine Cofactor Modifications Enhance the Biocatalytic Repertoire of Small Molecule C-Alkylation, Angew Chem. Int. Ed. Engl., 2019, 58, 17583–17588 CrossRef CAS PubMed.
- H. Deng, S. A. McMahon, A. S. Eustaquio, B. S. Moore, J. H. Naismith and D. O'Hagan, Mechanistic insights into water activation in SAM hydroxide adenosyltransferase (duf-62), ChemBioChem, 2009, 10, 2455–2459 CrossRef CAS PubMed.
- S. Thompson, M. Onega, S. Ashworth, I. N. Fleming, J. Passchier and D. O'Hagan, A two-step fluorinase enzyme mediated (18)F labelling of an RGD peptide for positron emission tomography, Chem. Commun., 2015, 51, 13542–13545 RSC.
- T. Kittila, P. Calero, F. Fredslund, P. T. Lowe, D. Teze, M. Nieto-Dominguez, D. O'Hagan, P. I. Nikel and D. H. Welner, Oligomerization engineering of the fluorinase enzyme leads to an active trimer that supports synthesis of fluorometabolites in vitro, Microb. Biotechnol., 2022, 15, 1622–1632 CrossRef CAS PubMed.
- A. S. Eustaquio, F. Pojer, J. P. Noel and B. S. Moore, Discovery and characterization of a marine bacterial SAM-dependent chlorinase, Nat. Chem. Biol., 2008, 4, 69–74 CrossRef CAS.
- I. Pardo, D. Bednar, P. Calero, D. C. Volke, J. Damborsky and P. I. Nikel, A Nonconventional Archaeal Fluorinase Identified by In Silico Mining for Enhanced Fluorine Biocatalysis, ACS Catal., 2022, 12, 6570–6577 CrossRef CAS.
- H. Sun, W. L. Yeo, Y. H. Lim, X. Chew, D. J. Smith, B. Xue, K. P. Chan, R. C. Robinson, E. G. Robins, H. Zhao and E. L. Ang, Directed Evolution of a Fluorinase for Improved Fluorination Efficiency with a Non-native Substrate, Angew Chem. Int. Ed. Engl., 2016, 55, 14277–14280 CrossRef CAS.
- H. Sun, H. Zhao and E. L. Ang, A coupled chlorinase-fluorinase system with a high efficiency of trans-halogenation and a shared substrate tolerance, Chem. Commun., 2018, 54, 9458–9461 RSC.
- L. C. Blasiak and C. L. Drennan, Structural perspective on enzymatic halogenation, Acc. Chem. Res., 2009, 42, 147–155 CrossRef CAS PubMed.
- H. M. Senn, Insights into enzymatic halogenation from computational studies, Front. Chem., 2014, 2, 98 Search PubMed.
- X. Zhu, D. A. Robinson, A. R. McEwan, D. O'Hagan and J. H. Naismith, Mechanism of enzymatic fluorination in Streptomyces cattleya, J. Am. Chem. Soc., 2007, 129, 14597–14604 CrossRef CAS.
- Y. Jiang, M. Yao, H. Niu, W. Wang, J. He, B. Qiao, B. Li, M. Dong, W. Xiao and Y. Yuan, Enzyme Engineering Renders Chlorinase the Activity of Fluorinase, J. Agric. Food Chem., 2024, 72, 1203–1212 CrossRef CAS.
- L. Ma, Y. Li, L. Meng, H. Deng, Y. Li, Q. Zhang and A. Diao, Biological fluorination from the sea: discovery of a SAM-dependent nucleophilic fluorinating enzyme from the marine-derived bacterium Streptomyces xinghaiensis NRRL B24674, RSC Adv., 2016, 6, 91024 RSC.
- H. Deng, L. Ma, N. Bandaranayaka, Z. Qin, G. Mann, K. Kyeremeh, Y. Yu, T. Shepherd, J. H. Naismith and D. O'Hagan, Identification of fluorinases from Streptomyces sp MA37, Norcardia brasiliensis, and Actinoplanes sp N902-109 by genome mining, ChemBioChem, 2014, 15, 364–368 CrossRef CAS PubMed.
- T. U. Consortium, UniProt: the Universal Protein Knowledgebase in 2023, Nucleic Acids Res., 2022, 51, D523–D531 CrossRef.
- R. Zallot, N. Oberg and J. A. Gerlt, The EFI Web Resource for Genomic Enzymology Tools: Leveraging Protein, Genome, and Metagenome Databases to Discover Novel Enzymes and Metabolic Pathways, Biochemistry, 2019, 58, 4169–4182 CrossRef CAS.
- P. Shannon, A. Markiel, O. Ozier, N. S. Baliga, J. T. Wang, D. Ramage, N. Amin, B. Schwikowski and T. Ideker, Cytoscape: a software environment for integrated models of biomolecular interaction networks, Genome Res., 2003, 13, 2498–2504 CrossRef CAS PubMed.
- Schrödinger Release 2021-3: Prime, Schrödinger, LLC, New York, NY Search PubMed.
- J. Li, R. Abel, K. Zhu, Y. Cao, S. Zhao and R. A. Friesner, The VSGB 2.0 model: a next generation energy model for high resolution protein structure modeling, Proteins, 2011, 79, 2794–2812 CrossRef CAS PubMed.
- C. Lu, C. Wu, D. Ghoreishi, W. Chen, L. Wang, W. Damm, G. A. Ross, M. K. Dahlgren, E. Russell, C. D. Von Bargen, R. Abel, R. A. Friesner and E. D. Harder, OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space, J. Chem. Theory Comput., 2021, 17, 4291–4300 CrossRef CAS PubMed.
- Schrödinger Release 2021-3: Desmond Molecular Dynamics System, D. E. Shaw Research, New York, NY Search PubMed.
- Maestro-Desmond Interoperability Tools, Schrödinger, New York, NY Search PubMed.
- S. E. Feller, Y. Zhang, R. W. Pastor and B. R. Brooks, Constant pressure molecular dynamics simulation: the Langevin piston method, J. Chem. Phys., 1995, 103, 4613–4621 CrossRef CAS.
- A. Y. Toukmaji and J. A. Board, Ewald summation techniques in perspective: a survey, Comput. Phys. Commun., 1996, 95, 73–92 CrossRef CAS.
- R. T. McGibbon, K. A. Beauchamp, M. P. Harrigan, C. Klein, J. M. Swails, C. X. Hernandez, C. R. Schwantes, L. P. Wang, T. J. Lane and V. S. Pande, MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories, Biophys. J., 2015, 109, 1528–1532 CrossRef CAS PubMed.
- H. M. Senn, D. O'Hagan and W. Thiel, Insight into enzymatic C-F bond formation from QM and QM/MM calculations, J. Am. Chem. Soc., 2005, 127, 13643–13655 CrossRef CAS PubMed.
- R. A. Laskowski and M. B. Swindells, LigPlot+: multiple ligand-protein interaction diagrams for drug discovery, J. Chem. Inf. Model., 2011, 51, 2778–2786 CrossRef CAS PubMed.
- S. S. Cinaroglu and E. Timucin, Comprehensive evaluation of the MM-GBSA method on bromodomain-inhibitor sets, Briefings Bioinf., 2020, 21, 2112–2125 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc00081e |
| ‡ These authors contributed equally to the manuscript. |
| This journal is © The Royal Society of Chemistry 2025 |