
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRedox-active polymer-grafted particles as redox mediators for enhanced charge transport in solution-state electrochemical systems†
Mohd
Avais‡
a,
Ratul Mitra
Thakur‡
c,
Evan
Fox
 b,
Jodie L.
Lutkenhaus
*ac and
Emily B.
Pentzer
*ab
b,
Jodie L.
Lutkenhaus
*ac and
Emily B.
Pentzer
*ab
aDepartment of Materials Science and Engineering, Texas A&M University, College Station, Texas 77840, USA. E-mail: emilypentzer@tamu.edu; jodie.lutkenhaus@tamu.edu
bDepartment of Chemistry, Texas A&M University, College Station, Texas 77840, USA
cArtie McFerrin Department of Chemical Engineering, Texas A&M University, College Station, Texas 77840, USA
First published on 27th March 2025
Abstract
Efficient charge transport pathways in solutions of redox-active polymers are essential for advancing next-generation energy storage systems. Herein, we report the grafting of (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) and poly(2,2,6,6-tetramethyl-1-piperidinyloxy-4-yl methacrylate) (PTMA) polymer brushes onto silica particles with different molecular weights and grafting densities, and the impact of these composite particles in solutions of PTMA. The polymer-grafted particles are characterized using Fourier-transform infrared (FTIR) spectroscopy, X-ray photoelectron spectroscopy (XPS), electron paramagnetic resonance (EPR) spectroscopy, field emission scanning electron microscopy (FESEM), transmission electron microscopy (TEM), and dynamic light scattering (DLS) techniques. The grafted polymers have molecular weights of 2.5 kDa and 5.0 kDa, with corresponding grafting densities of 0.688 and 0.378 chains nm−2 for SiO2-PTMA-2.5k and SiO2-PTMA-5k, respectively, with the grafting density decreasing with increasing graft length. To investigate the effect of these composite particles on charge transport in solutions of PTMA, different concentrations of the grafted particles were added to solutions of PTMA of different concentrations (near overlap concentration, C*) in 0.1 M LiTFSI in acetonitrile. Electrochemical analysis reveals that below C* the addition of SiO2-PTMA-5k increases the apparent diffusion coefficient (Dapp) 15.2% to 1.041 × 10−6 cm2 s−1, the exchange rate constant (kex,app) by 9.5% to 1.546 × 1011 L mol−1 s−1, and the heterogeneous electron transfer rate constant (k0) by 24.6%, to 5.526 × 10−4 cm s−1. These results indicate that the synergistic interactions between unbound PTMA polymer chains in solution and PTMA-grafted particles facilitate interchain charge transfer kinetics. This highlights that grafted redox-active particles can enhance charge transport without the limitations of polymer-only solutions (e.g., chain entanglement) and presents a promising design strategy for high-performance electrochemical applications, such as redox flow batteries (RFBs).
Introduction
Non-conjugated redox-active polymers (NC-RAPs) are promising electroactive materials with potential applications in energy storage and organic electronics, offering a sustainable alternative to current energy storage devices that rely on critical resources like nickel and cobalt.1–5 These polymers are applicable in both solid-state and redox flow batteries (RFBs).6,7 However, in RFBs, achieving high energy density of dissolved redox active material is often limited by poor solubility and high viscosity, hindering kinetics and increasing pumping costs.4,8 Therefore, approaches that could improve energy density and kinetics without significantly raising viscosity are needed.Charge transport in solutions of NC-RAPs varies with concentration, depending on whether the chains overlap or not. The overlap concentration (C*) marks the transition from the dilute to semi-dilute region, where isolated polymer chains (in dilute solution) start to overlap and interpenetrate (in semi-dilute solution) which increases polymer solution viscosity due to entanglements.9 Bello et al.10 identified several modes of charge transport: in the dilute regime, intra-molecular charge transport and segmental motion dominate; in the semi-dilute region, interchain charge transfer becomes important. Walker et al.11 further studied the effect of hydrodynamic interactions on charge transport in solutions of RAPs using Brownian dynamics (BD) simulations; the authors reported that extensional flow enhances the charge transport in the dilute region where segmental motion plays a critical role in the charge transfer process. Elsewhere, Blauch et al.12 used Monte-Carlo simulations to predict that upon exceeding the critical percolation concentration of polymer (where there is no physical motion of redox polymers), charge propagation is dominated by electron hopping. On the contrary, rapid molecular motion rearranges the successive electron hopping distribution, thereby eliminating the electron's memory in its previous environment. Recently, Perez Sirkin et al.13 examined effects of the concentration of the redox molecules and the strength of the intermolecular interactions on the charge-transport mechanism. They reported that an increase in intermolecular interactions favours the formation of clusters where electron-hopping is the dominant charge transfer process within the cluster; however, electron hopping is inefficient between the polymer clusters. These prior works highlight that determining the charge transport mechanism is complex due to many simultaneous processes such as physical diffusion, charge hopping, and changing dynamics with varying polymer concentrations.13–15
In previous reports, the concentration of redox species directly influenced solution conductivity, capacity, and energy density.16–18 Kosswattaarachchi et al.16 showed that the cycling behaviour for non-aqueous RFBs is dependent on the concentration of the redox-active species. They reported that as the concentration of a neutral redox active species, like (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) or N-methylphthalimide, increased, conductivity and electrochemical performance decreased due to aggregation. Elsewhere, Burgess et al.19 studied the electrochemical reactivity of poly(benzyl ethyl viologen) (VioRAP), varying the polymer and concentration of the electrolyte tetrabutylammonium hexafluorophosphate, TBAPF6, in acetonitrile. Here, the authors reported that ionic strength influenced chain conformation and that concentration of the RAP affects interchain charge transfer. Nagarjuna et al.20 studied the impact of molecular weight of the RAP poly(vinylbenzyl chloride) (PVBC) on electrochemical properties in 0.1 M LiBF4 in acetonitrile. By varying the molecular weight from 21 to 318 kDa, the authors observed that increasing molecular weight led to increased solution viscosity and decreased solubility and apparent diffusion coefficient.
Poly(2,2,6,6-tetramethyl-1-piperidinyloxy-4-yl methacrylate) (PTMA) is one of the most studied NC-RAPs due to its electrochemical reversibility in both organic and aqueous electrolytes. Electrochemical performance and kinetics of PTMA can be affected by concentration, radical density, chain conformation, and synthesis method.21–24 Our group24,25 used casting solvents that gave favourable PTMA interactions to show that electrochemical performance in the solid state is improved in compact chain conformations due to enhanced inter- and intrachain charge transfer. Martin et al.22 studied the impact of radical content on polymer conformation; increased radical density led to inflexible PTMA chains, which aggregated with neighbouring chains. Recently, Hatakeyama-Sato et al.26 examined the electrochemical charge transfer kinetics in solution for methacrylic copolymers containing TEMPO and the zwitterion [2-(methacryloxy)ethyl]dimethyl-(3-sulfopropyl)ammonium hydroxide (P(TMA-r-SBMA)); a diffusion-based kinetic model predicted that the Dapp was comparable to the physical diffusion (Dphys).
Redox mediation may alter the kinetics of electron transfer between neighbouring polymer chains in solution, where electron transfer may be promoted by pathways formed by the mediator, rather than solely by the physical diffusion of the polymer. Indeed, mediators have improved performance for polymer-based RFBs. Schröter et al.27 utilized N,N,N-2,2,6,6-heptamethylpiperidinyloxy-4-ammonium chloride (TMATEMPO) as a redox mediator for PTMA-based RFBs in 1 M NaCl aqueous electrolyte; an increase in the absolute discharge capacity from 3.7 mA h to 11.4 mA h was observed. The same group28 further reported the use of poly(2,2,6,6-tetramethylpiperdinyloxy-4-yl-methacrylamide) (PTMAm) as the active redox species and N,N,N-trimethyl-2-oxo-2-[(2,2,6,6-tetramethylpiperidin-4-yloxyl)amino]ethan-1-ammoniumchloride (TEMPO amide) as the redox mediator gave improved capacity by 228% (i.e., compared to without the redox mediator).
We propose that TEMPO- and PTMA-functionalized silica particles can serve as mediators for RAPs in solution and enhance the kinetics of electron transfer. Previous literature shows a precedent for the functionalization of silica or carbon surfaces with TEMPO-based species. For example, Rohan et al.29 created a film of PTMA grafts on planar indium tin oxide (ITO) substrates modified with n-octyltrichlorosilane as a capping agent, achieving a discharge capacity of 225 nA h cm−2 at 20 °C with 89% capacity retention after 400 cycles using l.0 M LiTFSI in ethylene carbonate and diethyl carbonate. Elsewhere, researchers modified spherical silica particles with PTMA to effectively enhance the stability of low molecular weight PTMA redox polymers in organic electrolytes.30 Further, Lin and co-workers grafted PTMA brushes onto silica particles, resulting in a discharge capacity of 84.9–111.1 mA h g−1 at 10C with 96.3% retention after 300 cycles.31 In other studies, various NC-RAPs have been grafted onto particles to create redox mediators for both solid state and suspension systems.32–34 Jin et al.34 produced PTMA-grafted reduced graphene oxide nanosheets, achieving an initial discharge capacity of 197 mA h g−1 and retaining a capacity of 101 mA h g−1 after 300 cycles in the solid-state. To the best of our knowledge, no previous study has used TEMPO- or PTMA-grafted particles as redox mediators to improve the charge transport process in PTMA/electrolyte solutions.
Herein, we report the use of TEMPO and PTMA-grafted silica particles as mediators in electrolyte solutions of PTMA. Polymers are grafted from silica particles using surface-initiated atom transfer radical polymerization (SI-ATRP), giving control over molecular weight and radical density in the resulting polymer. The composition of the particles, including molar mass of polymer chains and chain density, are characterized, and TEMPO-modified particles are prepared as a control (e.g., to establish the impact of small molecule versus polymer chain interactions). Different loadings of the modified particles are added to solutions of PTMA, both below and above the C* of PTMA. Importantly, this allows us to probe how the particle mediators might form charge transport pathways in the PTMA solution. This study confirms that the longer PTMA grafts enhance the kinetic rate parameters in PTMA solutions, particularly in dilute solutions at concentrations below PTMA's C*. These findings provide an exciting pathway for achieving faster redox kinetics in polymer solutions, bypassing the limitations of highly concentrated polymer solutions (chain entanglement, high viscosity).
Result and discussion
To create particle-based redox mediators, calcined silica particles (20 nm in diameter) were used as the substrate, and particles were modified in two ways: (i) with TEMPO groups on the surface via direct functionalization (Scheme 1a) and (ii) with PTMA brushes grafted from the particle surface via SI-ATRP (Scheme 1b). To prepare the latter, the particle surface was first modified with an initiator for SI-ATRP (i.e., an alpha-bromo ester), and the monomer 2,2,6,6-tetramethylpiperidin-4-yl-methylacrylate (TMPM) was grafted from the surface. After polymerization, the amine groups of polyTMPM (PTMPM) were oxidized to obtain the nitroxide radical moiety (i.e., grafted PTMA). Polymer grafts of two different molar masses were prepared by altering the monomer-to-initiator ratio. | ||
| Scheme 1 Modification of silica particles with (a) TEMPO units via direct functionalization; and (b) APTES-Br as initiator for SI-ATRP, subsequent grafting-from polymerization of PTMPM, and oxidation to access grafted PTMA. | ||
Modification of silica particles with TEMPO
To functionalize the calcined silica particles with TEMPO, the surface silanol groups of the particles were reacted with 3-(4-oxy-TEMPO)propylsilane, which was prepared from 4-hydroxy-TEMPO (Scheme 1a and Fig. S1†); see ESI† for more details.35 Briefly, the hydroxyl group of 4-hydroxy TEMPO was allylated, and, then, the alkene underwent hydrosilylation with trimethoxysilane in the presence of chloroplatinic acid to produce 3-(4-oxy-TEMPO)propylsilane; this was next used to directly modify the surface of the silica particles.35 Successful modification of the silica particles was confirmed using Fourier transform infrared (FTIR) spectroscopy and X-ray photoelectron spectroscopy (XPS), as shown in Fig. 1. The FTIR spectrum of SiO2 particles (Fig. 1a) shows a stretching frequency at 3462 cm−1, attributed to the O–H bond. The stretching frequencies at 1644 cm−1 and 1087 cm−1 correspond to H–O–H bending of absorbed water and Si–O–Si asymmetric stretching, respectively. The FTIR spectrum for SiO2-TEMPO reveals a methylene C–H stretching vibration at 2950 cm−1, an N–O˙ stretching vibration at 1365 cm−1, and a signal at ∼1280 cm−1, which is assigned to the C–O stretching of the ether, all indicative of modification. The XPS spectrum of the modified particles revealed the presence of C 1s, N 1s, and O 1s binding energies at 286.5 eV, 399.7 eV, and 531.4 eV, respectively (Fig. 1b). These results support the successful functionalization of the silica particles with TEMPO units. | ||
| Fig. 1 Characterization of SiO2, SiO2-TEMPO, SiO2-PTMA-2.5k and SiO2-PTMA-5k by: (a) FTIR, (b) XPS, and (c) TGA. | ||
Modification of silica particles with TEMPO-containing polymer
Modification of calcined silica particles with PTMA polymer brushes was accomplished using a multi-step approach, as depicted in Scheme 1b. First, 2-bromo-2-methyl-N-(3-(triethoxysilyl)propyl) propanamide (APTES-Br) was prepared as previously reported (Fig. S2 and S3†)36 and then covalently attached to the surface of the silica particles via silanization.37 The FTIR spectrum for the APTES-Br-modified silica particles shows peaks at 2962 cm−1 and 1643 cm−1, which are assigned to the methylene C–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of the amide, respectively (Fig. S4†). The alpha-bromo amide groups served as initiator for SI-ATRP of TMPM, using different monomer
O of the amide, respectively (Fig. S4†). The alpha-bromo amide groups served as initiator for SI-ATRP of TMPM, using different monomer![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :initiator ratios (M:I) to target two molar masses of grafted polymer (Table 1, Fig. S5 and S6†); the ESI† provides additional details. Sample nomenclature describes the polymer and the molar mass of the graft: SiO2-PTMPM-5k and SiO2-PTMPM-2.5k have polymer grafts of 5 kg mol−1vs. 2.5 kg mol−1, respectively. The molar mass and dispersity of the grafted polymer were determined by dissolving the silica core using aqueous HF, isolating the polymer by precipitation, and characterization using size exclusion chromatography (SEC). As shown in Fig. S7,† the SEC analysis of the isolated polymers revealed a Mn of ∼2.5 kDa (Ð = 1.16) and ∼5 kDa (Ð = 1.29), for SiO2-PTMPM-2.5k and SiO2-PTMPM-5k, respectively.
:initiator ratios (M:I) to target two molar masses of grafted polymer (Table 1, Fig. S5 and S6†); the ESI† provides additional details. Sample nomenclature describes the polymer and the molar mass of the graft: SiO2-PTMPM-5k and SiO2-PTMPM-2.5k have polymer grafts of 5 kg mol−1vs. 2.5 kg mol−1, respectively. The molar mass and dispersity of the grafted polymer were determined by dissolving the silica core using aqueous HF, isolating the polymer by precipitation, and characterization using size exclusion chromatography (SEC). As shown in Fig. S7,† the SEC analysis of the isolated polymers revealed a Mn of ∼2.5 kDa (Ð = 1.16) and ∼5 kDa (Ð = 1.29), for SiO2-PTMPM-2.5k and SiO2-PTMPM-5k, respectively.
| Sample | [M]0/[I]0 | M n, SEC (kDa) | Đ | DPb | Grafting density (chain nm−2) | D h (nm) | h dry (nm) | h wet (nm) |
|---|---|---|---|---|---|---|---|---|
| a Determined from SEC relative to polystyrene standards. b Calculated from Mn. c Hydrodynamic diameter (Dh) from DLS in acetone. d Height of grafted polymer brushes from FESEM of dried particles. e Height of grafted polymer brushes from DLS in the solution. | ||||||||
| SiO2-TEMPO | — | — | — | — | 2.88 | ∼60 | 3.0 | 20 |
| SiO2-PTMPM-2.5k | 15:1 |
∼2.5 | 1.14 | 10 | 0.688 | ∼367 | 4.5 | 173.5 |
| SiO2-PTMPM-5k | 60:1 |
∼5.0 | 1.29 | 20 | 0.378 | ∼415 | 7.5 | 197.5 |
The PTMPM polymer grafts were converted to redox-active PTMA grafts by oxidation of the secondary amine of the pendant groups using meta-chloroperoxybenzoic acid (mCPBA). The production of SiO2-PTMA was confirmed using FTIR spectroscopy and XPS, as well as EPR spectroscopy (see below). The thermal stability was established using thermogravimetric analysis (TGA). A comparison of the FTIR spectra of SiO2 and SiO2-PTMA-5k confirms successful oxidation of the amines by the observation of the N–O˙ stretching vibration at 1365 cm−1 (Fig. 1a). Additionally, XPS also supported functionalization: the binding energy of the N 1s of PTMPM was observed at 398.3 eV for SiO2-PTMPM-5k, which corresponds to a secondary amine, whereas the binding energy of the N 1s shifted to 399.7 eV for SiO2-PTMA-5k, which corresponds to nitroxide radicals (inset of Fig. 1b).38 Similar results were obtained for SiO2-PTMA-2.5k, demonstrating oxidation. The thermal weight loss profiles of SiO2-PTMA-5k, SiO2-PTMA-2.5k, and SiO2-TEMPO are shown in Fig. 1c. The major mass loss events for SiO2-PTMA-5k occurred from ∼150 to 300 °C, attributed to thermolysis of the TEMPO side group, and ∼300 to 600 °C, attributed to oxidation of the polymer backbone.31 The mass loss profile for SiO2-PTMA-2.5k was similar to that for the 5k sample. For SiO2-TEMPO, the mass started to decrease steadily from about 200 °C. Notably, varying compositions exhibited different mass loss percentages, with SiO2-PTMA-5k displaying the highest mass loss (∼33%), followed by SiO2-PTMA-2.5k (∼31%) and SiO2-TEMPO (∼19%), as expected. Notably, the theoretical organic mass fractions were calculated based on the grafted polymer composition: ∼53% for SiO2-TEMPO, ∼62% for SiO2-PTMA-2.5k, and ∼63% for SiO2-PTMA-5k. These values closely match the results obtained from TGA (∼49%, ∼59%, and ∼61%, respectively), demonstrating good agreement between expected and observed organic fractions in the modified particles.
Grafting density ‘σ’ (chains nm−2)
Grafting density of PTMA on the surface of the silica particles was determined using eqn (1), where Wgrafted is the weight of the grafted polymer brush (determined by TGA), Mgrafted is molar mass (Mn) of the grafted polymer brush (determined by SEC), NA is Avogadro's number, Wsilica is the weight loss of the silica particles without modification (Fig. S8†), and S is the surface area of the silica particles. | (1) |
To estimate the brush height (h) in the dry state, we examined the morphology of as-received silica particles and the three modified SiO2 particles using field emission scanning electron microscopy (FESEM). The micrographs of as received calcined silica particles reveal a spherical morphology with an approximate average diameter of ∼20 nm (Fig. 2a). FESEM micrographs of SiO2-TEMPO show a slightly larger average particles diameter of ∼26 nm, which indicates that the 20 nm silica core was enveloped by a ∼3 nm TEMPO corona (Fig. 2b). In comparison, SiO2-PTMA-2.5k particles displayed an average diameter of 29 nm and that of SiO2-PTMA-5k was much larger at ∼35 nm (Fig. 2c and d). This increase in size is attributed to the increase in the molar mass of the polymer grafts. The micrographs of all modified nanoparticles reveal particle–particle aggregation, attributed to favourable inter-particle interactions during sample preparation (i.e., drying). TEM images further confirmed the size of the SiO2-PTMA-5k particles is ∼35 nm (Fig. 2e). Thus, accounting for a SiO2 core diameter of 20 nm, the brush heights are ∼4.5 nm and 7.5 nm for SiO2-PTMA-2.5k and SiO2-PTMA-5k, respectively (Table 1). Based on grafting density, degree of polymerization (N), and brush height (h), the conformation of the polymer graft can be classified as a mushroom, semi-dilute brush, or concentrated brush with increasing grafting density.39–41 As described in the ESI,† we estimate that both polymer grafts have a semi-dilute brush conformation in the dry state.
 | ||
| Fig. 2 FESEM micrographs of (a) SiO2, (b) SiO2-TEMPO, (c) SiO2-PTMA-2.5k, (d) SiO2-PTMA-5k, (e) TEM micrograms of SiO2-PTMA-5k, and (f) DLS of modified particles. | ||
Although the polymer chains are assumed to be collapsed in the solid state, they are expected to be solvated and expanded when in a good solvent. Dynamic light scattering (DLS) analysis provided insights into the swelling of the brushes in acetone and the particle size distribution. Contin plots revealed narrow, approximately monomodal size distributions for SiO2-PTMA-2.5k and SiO2-PTMA-5k, indicating uniform particle distributions. Hydrodynamic diameters (Dh) of 60 nm, 367 nm, and 415 nm were observed for SiO2-TEMPO, SiO2-PTMA-2.5k, and SiO2-PTMA-5k, respectively (Fig. 2f). Notably, the particle diameters obtained from DLS in solution were significantly larger than those observed from FESEM and TEM images, which can be attributed to the swelling (e.g., solvation) of the grafted polymers. Assuming a SiO2 core diameter of 20 nm, the solvated brush heights are 20 nm, 173.5 nm and 197.5 nm for SiO2-TEMPO, SiO2-PTMA-2.5k and SiO2-PTMA-5k, respectively (Table 1). The conformation of the polymer grafts was further validated by estimating the radius of gyration (Rg) for the grafted PTMA chains. Using the established equation for grafted polymers (see ESI†), the Rg values were calculated as 3.16 nm for SiO2-PTMA-2.5k and 4.47 nm for SiO2-PTMA-5k. In both cases, the brush height (h) in solution was observed to be significantly greater than twice the Rg, further supporting that the polymer chains are in a semi-dilute brush regime (see ESI†).
Electron paramagnetic resonance (EPR) spectroscopy was used to understand how functionalization affects the density of radicals on the silica particles (e.g., TEMPO units or polymers with pendant TEMPO units). As shown in Fig. 3, The EPR spectrum of homopolymer PTMA shows a singlet, which indicates spin–spin interactions among closely spaced radicals.42 For the modified particles, the intensity of the quantitative EPR signal increases with increased radical concentrations, with SiO2-PTMA-5k giving a significantly higher signal compared to SiO2-TEMPO and SiO2-PTMA-2.5k. Analysis of the EPR spectra multiplicity provides valuable insights into the distribution and interactions of radical units within the sample. SiO2-PTMA-5k displayed broad Lorentzian singlets, whereas the sample with the lowest loading of radicals, SiO2-TEMPO, exhibited triplet multiplicity spins; this is consistent with the splitting formula of N = 2I + 1, with nitrogen having a spin quantum number of I = +1, suggesting comparatively isolated radical electrons, as observed for the small molecule TEMPO.43 SiO2-PTMA-2.5k, of intermediate radical loading, showed an EPR signal that could be classified as a broad doublet, indicative of increased spin–spin interactions compared to SiO2-TEMPO, leading to band broadening that results in a decreased ability to resolve hyperfine splitting. Notably, no discernible EPR peaks were observed from as received silica particles, confirming the specificity of the functionalization process (Fig. S9†).
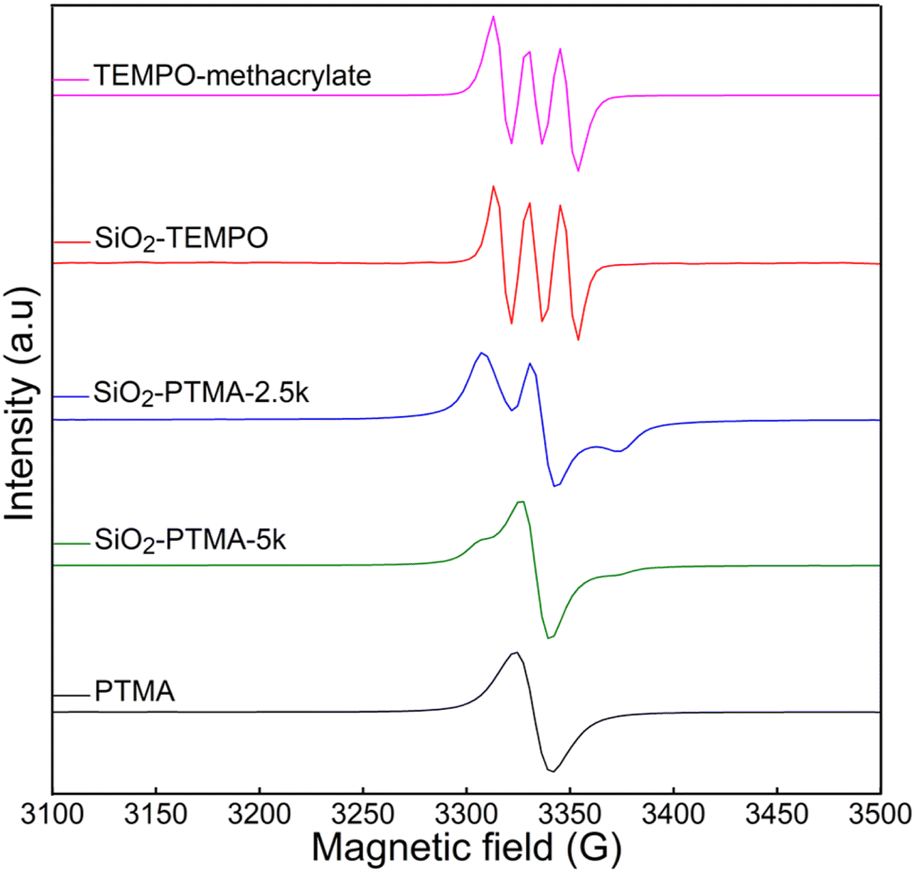 | ||
| Fig. 3 EPR spectra of (from top to bottom) TEMPO methacrylate, SiO2-TEMPO, SiO2-PTMA-2.5k, SiO2-PTMA-5k, and PTMA. | ||
Overlap concentration (C*)
The polymer solution's C* plays a critical role in defining the behaviour of polymer solutions, marking the transition between dilute and semi-dilute regimes where individual polymer chains begin to overlap and interact (Fig. 4a–c). In this study, we examined the influence of C* on charge transport in PTMA solutions, specifically investigating how modified SiO2 particles could promote interchain charge transport in solutions near or below C*. We hypothesized that these modified particles would act as redox mediators, facilitating charge transport between polymer chains even in dilute PTMA solutions. To test this, PTMA homopolymer (Mn ∼50 kDa, Đ = 4.62) was synthesized via free radical polymerization of TMPM then by oxidation.44 The calculated Rg of PTMA was 14.4 nm (see ESI, Fig. S10†). We determined the C* for PTMA both with and without the modified silica particles using a viscometer and 0.1 M LiTFSI in acetonitrile as the solvent, which also serves as the electrolyte for electrochemical characterization. Fig. 4d illustrates the low-shear viscosity of the PTMA solution at a range of polymer concentrations. As evident from the graph, a noticeable change in the viscosity–concentration slope occurs at 7.2 mM, indicative of C*. Phenomenologically, below this concentration the sample is dilute and polymer chains do not interact with each other (low viscosity), and above this concentration the sample is in a semi-dilute state, correlating to formation of entanglement or other cooperative interactions between polymer chains. A rheometer further confirmed C* by revealing a noticeable change in the viscosity slope as a function of polymer concentration at 7.3 mM (Fig. S11†). Upon the addition of SiO2-PTMA-5k to the PTMA solutions, a significant increase in the viscosity and decrease in C* were observed (Fig. S12†). As shown in Fig. 4e, viscometer measurements reveal a change in the slope at 6.1 mM of PTMA polymer, suggesting that the presence of modified silica particles reduces the C*, as expected. Fig. 4f illustrates a schematic representation of the solution of PTMA and modified particles near the C*. | ||
| Fig. 4 Schematics of PTMA in (a) dilute, (b) semi-dilute, and (c) concentrated regions. Determination of the C* of PTMA using different PTMA concentrations with and without modified silica particles (SiO2-PTMA-5k) in 0.1 M lithium bis(trifluoromethane)sulfonimide (LiTFSI) in acetonitrile via viscometry. (d) C* for PTMA without modified silica particles, (e) C* for PTMA with SiO2-PTMA-5k particles, where the grafted particles contribute 10% of the total TEMPO concentration, and (f) schematic of TEMPO/PTMA-modified silica particles added to a PTMA solution (near C*). | ||
Electrochemical characterization
Initially, the charge transport behaviour was studied with the variation of PTMA solution concentration without added particles in 0.1 M LiTFSI in acetonitrile, as shown in Fig. S13.† At a scan rate of 10 mV s−1, the peak-to-peak separation (ΔEp) remained less than 100 mV for all concentrations studied. Fig. S13b† shows peak current density and half-wave (E1/2) potential vs. PTMA concentration. The peak current density increased linearly with concentration, as expected because ip ∼ [TEMPO]. Meanwhile, E1/2 was relatively constant across different concentrations (0.20 to 0.22 V vs. Fc0/Fc+), only increasing slightly above C*.To assess the impact of the addition of particle-based redox mediators, SiO2-TEMPO, SiO2-PTMA-5k, and SiO2-PTMA-2.5k were added to PTMA solutions of varying concentrations in 0.1 M LiTFSI in acetonitrile. The particles in the PTMA solution remained homogenously suspended throughout all the electrochemical characterizations. Fig. 5 shows cyclic voltammetry (CV) of different concentrations of PTMA (3, 6, 7.2, and 9 mM) both with and without the modified silica particles. The redox-active (i.e. TEMPO) units contributed from these modified silica particles constitute 10 mol% of the total TEMPO units in the system. For example, 10 mL of a 6 mM solution of PTMA (14.4 mg of PTMA, 6 mmol of TEMPO units) required 4.9 mg of modified silica particles (0.67 mmol of TEMPO units) for a total of 6.67 mmol TEMPO units in the solution. Due to the addition of the particles at 10 mol% of the total TEMPO units of the system, it is expected that the peak current would increase by 10%, as ip ∼ [TEMPO] for a Nernstian reaction.45
 | ||
| Fig. 5 CVs of solutions of PTMA and PTMA with added SiO2 grafted particles (SiO2-TEMPO, SiO2-PTMA-2.5k, and SiO2-PTMA-5k) at a scan rate of 10 mV s−1 in 0.1 M LiTFSI in acetonitrile. Particles contributed 10 mol% of the total TEMPO units of the system and were added to PTMA solutions of (a) 3 mM, (b) 6 mM, (c) 7.2 mM and (d) 9 mM. The working electrode was glassy carbon, the counter electrode was Pt wire, and the reference electrode was silver wire. | ||
Fig. 5a shows the CVs of 3 mM PTMA solutions with and without grafted SiO2 particles whereas Fig. 5b–d show analogous CVs for PTMA concentrations of 6 mM, 7.2 mM and 9 mM, respectively. The general observation is that there is an increase in peak current density with the addition of the modified silica particles for all PTMA solution concentrations studied. Further, a slight decrease in E1/2 from 0.22 V to 0.18 V vs. Fc0/Fc+ is observed with the addition of the modified particles, which indicates that the reaction has become more kinetically reversible. From Fig. 5b, at 6 mM PTMA (i.e., below C*), we observe the greatest increase in the peak current density (from the base 6 mM current) with the addition of SiO2-PTMA-5k particles. Specifically, the peak current increased by 17.7%, which is more than the expected 10%. This might be due to additional inter-chain charge transport pathways provided by SiO2-PTMA-5k which have sufficient chain length to bridge nearby PTMA polymer chains. In comparison, the addition of SiO2-TEMPO and SiO2-PTMA-2.5k to the PTMA solution showed the expected effect in peak current density (i.e., ∼10%). From Fig. 5c, at 7.2 mM PTMA (i.e., slightly above C*), PTMA chains begin to overlap and both SiO2-PTMA-2.5k and SiO2-PTMA-5k lead to improvements in peak current density (16.7% and 17.0%, respectively). From Fig. 5d, at 9 mM PTMA (>C*), there was no shift in E1/2 with the addition of the particles, but an increase in peak current density was observed for all cases (19–21%). The peak currents were calculated following a previous report, which, in brief, utilized the peak current relative to a tangent baseline.46 The ratio of the peak current for anodic and cathodic sweeps (Ip oxidation/Ip reduction) ranged from 1.05 to 1.1 showing the process is reversible with successive addition of modified particles and changing scan rates (see Tables S2 and S3†). This is because overlapping PTMA chains form contacts with the redox mediators, and thus the TEMPO groups on the particles can participate in redox reactions. Overall, the addition of grafted particles can lead to improvements in peak current density and PTMA activity beyond the expected ip ∼ [TEMPO] relationship.
To understand the effect of particle concentration in the PTMA solution, modified silica particles were successively added to PTMA solutions at concentrations below and above PTMA's C* (i.e., at 6 mM and 7.2 mM). The particles were added in 5 mol% increments (from 0 to 15 mol%) by total TEMPO solution concentration. Fig. S14† shows a collection of CVs of the successive addition of SiO2-PTMA-5k particles. Adding the SiO2-PTMA-5k particles (up to a concentration of 15% of the TEMPO units) led to increased peak currents of 18% and 20% for 6 mM and 7.2 mM PTMA solutions, respectively, relative to the respective solutions without particles. Because the increase in peak current was consistently greater than the expected increase (0, 5, 10, 15%), we again confirm the benefit of the additional particles in forming charge transfer pathways. For SiO2-PTMA-2.5k, a similar trend was observed (Fig. S15†). In contrast, for the successive addition of SiO2-TEMPO to the PTMA solutions, the peak current density increased, particularly for the 7.2 mM PTMA solution, as shown in Fig. S16†
The redox kinetics of the 6 mM PTMA solutions with modified particles were examined using b-value analysis of CVs to further probe the redox kinetics of the composite system. Fig. 6a shows CV curves at scan rates from 2 mV s−1 to 100 mV s−1 for a 6 mM PTMA solution with SiO2-TEMPO particles, where the TEMPO units from the particles contribute 5 mol% of total TEMPO units in the solution. With the addition of the particles, peak broadening was observed and peak-to-peak separation (ΔEp) increased from 100 mV to 110 mV (at 10 mV s−1). This indicates that the reaction is slightly less reversible with the addition of the SiO2-TEMPO particles, possibly due to incomplete interchain pathways brought from the relatively insulating SiO2 cores. In comparison, when the other two types of particles are added (SiO2-PTMA-2.5k and SiO2-PTMA-5k, Fig. 6b and c), ΔEp remained at 100 mV and 95 mV, respectively, which indicates no change in the reversibility of the redox reaction. Further, the b-values of both oxidation and reduction for all three particles (SiO2-TEMPO, SiO2-PTMA-2.5k and SiO2-PTMA-5k) were ∼0.5 suggesting that the redox reactions were diffusion controlled irrespective of type of particles added.
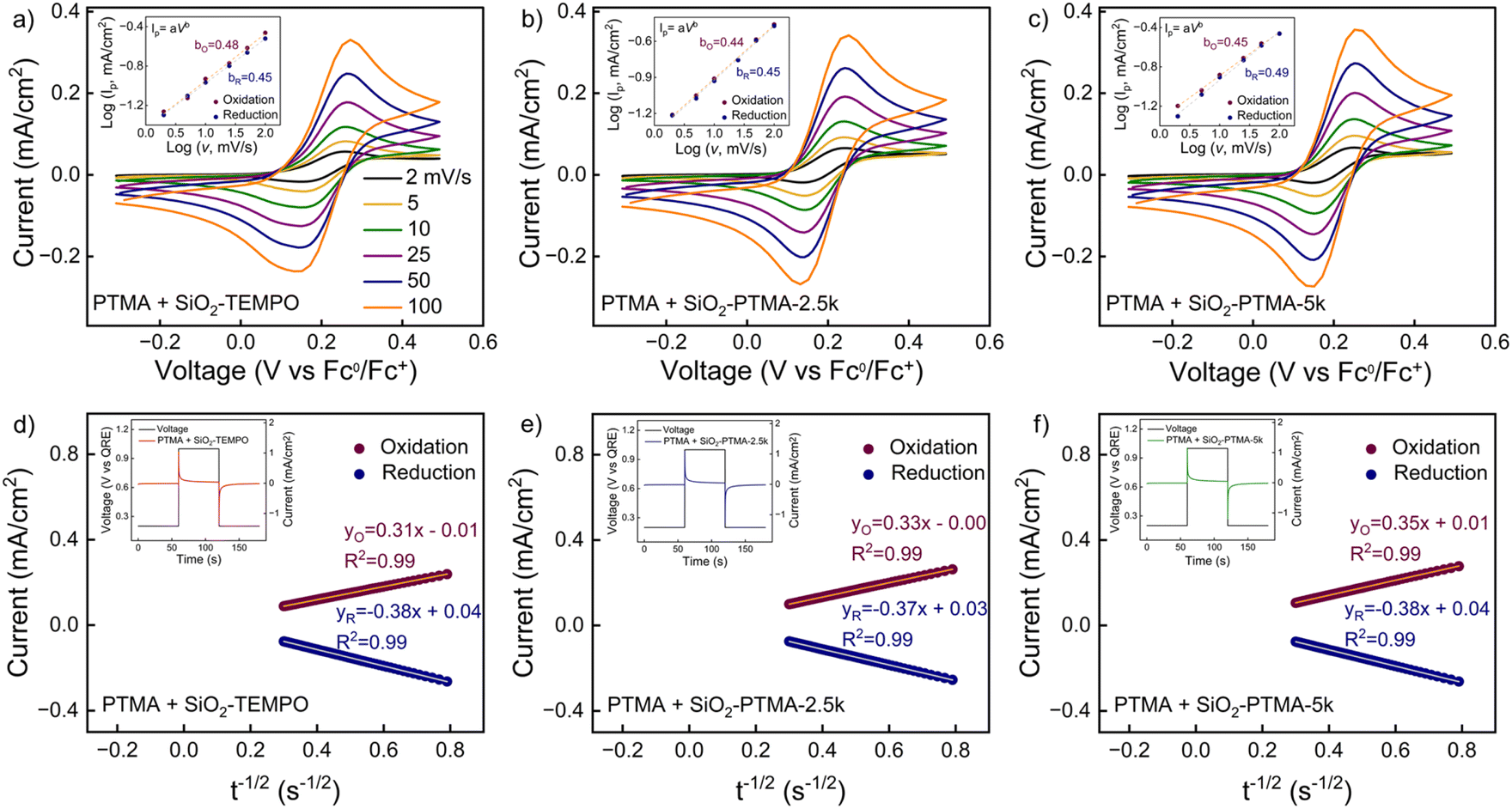 | ||
| Fig. 6 CVs at different scan rates for 6 mM PTMA solution with added: (a) SiO2-TEMPO, (b) SiO2-PTMA-2.5k and (c) SiO2-PTMA-5k particles. Insets show the peak current density vs. scan rate. Cottrell plots (d–f) from chronoamperometry of 6 mM PTMA solution added with different grafted SiO2 particles: (d) SiO2-TEMPO, (e) SiO2-PTMA-2.5k and (f) SiO2-PTMA-5k particles. The applied potential step was from 0.2 to 1 V versus QRE. Solution-state measurements were carried out in 0.1 M LiTFSI in acetonitrile with glassy carbon as the working electrode, Pt wire as the counter electrode, and Ag wire as the QRE. SiO2 grafted particles were added to the PTMA solution such that 5 mol% of the total TEMPO repeat units of the system come from the particles. | ||
Chronoamperometry was applied to obtain the apparent diffusion coefficient (Dapp) using Cottrell plots (Fig. 6d–f, S17–21, Tables S4 and S5†). For the 6 mM PTMA solution, the largest improvement in the kinetics was observed upon adding SiO2-PTMA-5k particles at 5 mol% TEMPO concentration by repeat units; specifically, Dapp improved from 9.033 ± 0.039 × 10−7 cm2 s−1 to 1.041 ± 0.009 × 10−6 cm2 s−1 (an increase of 15.2%). However, additional SiO2-PTMA-5k particles did not yield further gains in Dapp. We calculated kex,app using the Dahms–Ruff equation assuming no physical diffusion (Dphys = 0) of polymer chains and a constant distance between redox centres. With the addition of SiO2-PTMA-5k particles at 5% TEMPO concentration by repeat units, kex,app improved from 1.411 × 1011 ± 0.004 × 1011 L mol−1 s−1 to 1.546 × 1011 ± 0.012 × 1011 L mol−1 s−1 (an increase of 9.5%). We also calculated the k0 using the Nicholson method; k0 increased from 4.432 ± 0.008 × 10−4 cm s−1 to 5.526 ± 0.023 × 10−4 cm s−1 (increased by 24.6%) after addition of the SiO2-PTMA-5k particles.
These results indicate that at 6 mM PTMA (i.e., below C*), adding a small amount of polymer-grafted particles improves kinetics by creating additional charge transport pathways among the PTMA chains in solution. We speculate that adding more particles did not further improve the kinetics because of the inert and non-conductive nature of the silica core and also because of increases in solution viscosity. For example, adding any quantity of SiO2-TEMPO particles (which are ∼81 wt% silica) diminishes the kinetic parameters for the PTMA solution; in this case, the TEMPO groups on the particle surface cannot effectively interact with the PTMA chains in solution to form pathways, and the silica core presents a physical barrier to electron diffusion. At 7.2 mM PTMA (at C*, Table S3†), adding the modified particles generally decreased the redox kinetics, likely because at this polymer concentration, the polymer chains already formed pathways for electron transport and the particles did not provide additional ones.
To investigate the impedance response of the PTMA solutions with and without the modified nanoparticles, EIS was performed at the half-wave potential (E1/2) for each system (see Fig. 7 and S22–25†). The Nyquist and Bode plots are shown in Fig. 7 for 6 mM PTMA solutions (below C*) with and without the modified particle solution, such that TEMPO units contributed of the particles constituted 10 mol% of the total TEMPO units in the system. The addition of both SiO2-TEMPO (Fig. 7a and d) and SiO2-PTMA-2.5k (Fig. 7b and e) led to increased charge transfer resistance in the high frequency region relative to the 6 mM PTMA solution (i.e., compared to without particles). However, the addition of SiO2-PTMA-5k particles yielded a decrease in charge transfer resistance compared to the polymer solution alone (Fig. 7c and f). Taken together, these trends in the charge transfer resistance support the conclusion that SiO2-TEMPO particles, with their small hydrodynamic diameter ∼60 nm, are not effective in creating charge transport pathways in the PTMA solution. The effect is less pronounced for SiO2-PTMA-2.5k particles likely because there is a balance between the short polymer chains interacting with PTMA in solution, but not as effectively as particles with the 5k grafts. In contrast, the SiO2-PTMA-5k particles can form effective interchain electron transport pathways with PTMA chains to manifest in a lower solution resistance.
 | ||
| Fig. 7 Nyquist (a–c) and Bode (d–f) plots from electrochemical impedance spectroscopy (EIS) of 6 mM PTMA solutions with and without: (a and d) SiO2-TEMPO, (b and e) SiO2-PTMA-2.5k and (c and f) SiO2-PTMA-5k. EIS was carried out at the half-wave potential with a 10 mV alternating voltage in 0.1 M LiTFSI in acetonitrile. Glassy carbon was used as the working electrode, Pt wire as the counter electrode, and Ag wire as the quasi-reference electrodes. SiO2 grafted particles were added to the PTMA solution such that TEMPO units of the particles constituted 10 mol% of total TEMPO units in the mixture. | ||
We next examined the effect of PTMA concentration and particle concentration. For comparison, EIS response was studied at different PTMA concentrations without added particles, as shown in Fig. S22,† demonstrating a decrease in the charge transfer resistance with increasing PTMA concentration. Fig. S23–S25† present changes in the EIS response for 6 mM and 7.2 mM PTMA solutions with the successive addition of the modified particles. For 6 mM PTMA solutions, the general observation is that charge transfer resistance successively increases when SiO2-TEMPO particles are added. The charge transfer resistance slightly decreases and then increases when SiO2-PTMA-2.5k particles are added to the solution. Interestingly, at 7.2 mM (above C*), the charge transfer resistance is relatively insensitive to addition of the particles. Taken together, these data demonstrate that the grafted particles have the most prominent effect on charge transfer mediation effects when the polymer solution is below C*.
The prior results considered the addition of grafted SiO2 particles to solutions of a constant PTMA concentration, such that the total concentration of TEMPO units in the mixture was changing. For a comparison, we also investigated mixtures in which the total TEMPO concentration in the solution was kept constant, but relative concentrations of PTMA and grafted particles varied. For this case, the peak current density is expected to remain constant. Fig. S26† presents a comparison of the electrochemical response of a 6.35 mM PTMA solution and a mixture of PTMA solution with the addition of 5 mol% SiO2-PTMA-5k particles (total TEMPO concentration = 6.35 mM). The CVs in Fig. S26a† illustrate that the presence of the grafted particles led to a 9.7% increase in the oxidation peak current density. EIS spectra in Fig. S26b† indicate a reduction in the charge transfer resistance for the sample with grafted particles, whereas Cottrell plots show that Dapp increased by 19% (Fig. S26c†). These data support that the enhanced electrochemical performance brought about by SiO2-PTMA-5k particles can be attributed to the formation of new interchain pathways for electron transfer and not changes in the TEMPO unit concentration.
Conclusions
This work demonstrated the effects of charge transport in PTMA solutions with TEMPO- or PTMA-grafted silica particles with enhanced kinetics observed when particles are added to solutions of polymer below the critical concentration. SI-ATRP was used for the grafting of PTMA brushes from silica particles, producing samples with polymers of 2.5 kDa and 5.0 kDa molar mass and grafting densities of 0.378 to 2.88 chains nm−2, respectively. PTMA grafts exhibited a semi-dilute brush conformation and were less densely packed than individual TEMPO grafts. The different TEMPO-modified particles were added to solutions of PTMA below, at, and above the C*. We observed that Dapp, kex,app and k0 increased by 15.2%, 9.5% and 24.6%, respectively, with the addition of SiO2-PTMA-5k particles (consisting of 5 mol% of total TEMPO units) to a solution of polymer below the C* (6 mM PTMA). The results indicate that PTMA grafts of 5 kDa result in effective interchain charge transport pathways between PTMA chains in solution. These findings offer a promising approach to enhancing redox kinetics in polymer solutions while avoiding challenges associated with high polymer concentrations, such as chain entanglement and increased viscosity. Despite the enhanced charge transfer facilitated by PTMA grafted particles, the insulating silica particle cores do not contribute to energy storage, and are notably impactful when lower molecular weight PTMA grafts are used. Future studies should address the use of smaller particle cores (or longer polymer grafts) or the use of conductive particles to enable more efficient and scalable redox-active organic materials for sustainable energy storage technologies.Data availability
Data is available in the ESI file.†Author contributions
E. B. P., J. L. L., and M. A. conceived the study. M. A. performed the synthesis and characterizations. R. M. T. carried out the electrochemical measurements. M. A., R. M. T., E. B. P., and J. L. L. discussed the results and wrote the manuscript. E. F. helped the EPR study. E. B. P., and J. L. L., supervised the project, acquired funding for this research, and edited the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the U.S. National Science Foundation (NSF) under award numbers 2104179 and 2119672. Portions of this research were conducted with the advanced computing resources provided by Texas A&M High-Performance Research Computing. Use of the Texas A&M Department of Chemistry NMR/ESR facility is acknowledged. Use of the Texas A&M University Soft Matter Facility (RRID:SCR_022482) and contribution of Dr Wei are acknowledged. Use of the Texas A&M University Microscopy and Imaging Center FSEM/TEM facility is acknowledged.References
- Y. Tan, S.-N. Hsu, H. Tahir, L. Dou, B. M. Savoie and B. W. Boudouris, Electronic and spintronic open-shell macromolecules, Quo Vadis?, J. Am. Chem. Soc., 2022, 144(2), 626–647 CrossRef CAS PubMed.
- S. M. Mitchell, K. Niradha Sachinthani, R. Pulukkody and E. B. Pentzer, 100th Anniversary of Macromolecular Science Viewpoint: Polymerization of Cumulated Bonds: Isocyanates, Allenes, and Ketenes as Monomers, ACS Macro Lett., 2020, 9(7), 1046–1059 CrossRef CAS PubMed.
- T. Janoschka, M. D. Hager and U. S. Schubert, Powering up the future: radical polymers for battery applications, Adv. Mater., 2012, 24(48), 6397–6409 CrossRef CAS.
- N. Goujon, N. Casado, N. Patil, R. Marcilla and D. Mecerreyes, Organic batteries based on just redox polymers, Prog. Polym. Sci., 2021, 122, 101449 CrossRef CAS.
- J. Kim, J. H. Kim and K. Ariga, Redox-active polymers for energy storage nanoarchitectonics, Joule, 2017, 1(4), 739–768 CrossRef CAS.
- T. Hagemann, M. Strumpf, E. Schröter, C. Stolze, M. Grube, I. Nischang, M. D. Hager and U. S. Schubert, (2, 2, 6, 6-tetramethylpiperidin-1-yl) oxyl-containing zwitterionic polymer as catholyte species for high-capacity aqueous polymer redox flow batteries, Chem. Mater., 2019, 31(19), 7987–7999 Search PubMed.
- T. Hagemann, J. Winsberg, M. Grube, I. Nischang, T. Janoschka, N. Martin, M. D. Hager and U. S. Schubert, An aqueous all-organic redox-flow battery employing a (2, 2, 6, 6-tetramethylpiperidin-1-yl) oxyl-containing polymer as catholyte and dimethyl viologen dichloride as anolyte, J. Power Sources, 2018, 378, 546–554 CrossRef CAS.
- E. Sánchez-Díez, E. Ventosa, M. Guarnieri, A. Trovò, C. Flox, R. Marcilla, F. Soavi, P. Mazur, E. Aranzabe and R. Ferret, Redox flow batteries: Status and perspective towards sustainable stationary energy storage, J. Power Sources, 2021, 481, 228804 CrossRef.
- M. Rubinstein and R. H. Colby, Polymer physics, Oxford university press, 2003 Search PubMed.
- L. Bello and C. E. Sing, Mechanisms of diffusive charge transport in redox-active polymer solutions, Macromolecules, 2020, 53(18), 7658–7671 CrossRef CAS.
- D. W. Walker and C. E. Sing, Effect of Hydrodynamic Interactions and Flow on Charge Transport in Redox-Active Polymer Solutions, J. Phys. Chem. B, 2024, 128(7), 1796–1811, DOI:10.1021/acs.jpcb.3c07657.
- D. N. Blauch and J. M. Saveant, Dynamics of electron hopping in assemblies of redox centers. Percolation and diffusion, J. Am. Chem. Soc., 1992, 114(9), 3323–3332, DOI:10.1021/ja00035a025.
- Y. A. Perez Sirkin and M. Tagliazucchi, Revisiting the Mechanisms of Charge Transport in Solutions of Redox-Active Molecules Using Computer Simulations: When and Why Do Analytical Theories Fail?, J. Phys. Chem. B, 2023, 127(13), 2968–2978, DOI:10.1021/acs.jpcb.2c06956.
- K. Sato, R. Ichinoi, R. Mizukami, T. Serikawa, Y. Sasaki, J. Lutkenhaus, H. Nishide and K. Oyaizu, Diffusion-Cooperative Model for Charge Transport by Redox-Active Nonconjugated Polymers, J. Am. Chem. Soc., 2018, 140(3), 1049–1056, DOI:10.1021/jacs.7b11272.
- J. S. Facci and M. Stolka, Redox migration mechanism of charge transport in molecularly doped polymers, Philos. Mag. B, 1986, 54(1), 1–18, DOI:10.1080/13642818608243174.
- A. M. Kosswattaarachchi and T. R. Cook, Concentration-dependent charge-discharge characteristics of non-aqueous redox flow battery electrolyte combinations, Electrochim. Acta, 2018, 261, 296–306, DOI:10.1016/j.electacta.2017.12.131.
- Y. Ding, C. Zhang, L. Zhang, Y. Zhou and G. Yu, Molecular engineering of organic electroactive materials for redox flow batteries, Chem. Soc. Rev., 2018, 47(1), 69–103, 10.1039/C7CS00569E.
- M. Li, S. A. Odom, A. R. Pancoast, L. A. Robertson, T. P. Vaid, G. Agarwal, H. A. Doan, Y. Wang, T. M. Suduwella and S. R. Bheemireddy, et al., Experimental Protocols for Studying Organic Non-aqueous Redox Flow Batteries, ACS Energy Lett., 2021, 6(11), 3932–3943, DOI:10.1021/acsenergylett.1c01675.
- M. Burgess, K. Hernández-Burgos, J. K. Schuh, J. Davila, E. C. Montoto, R. H. Ewoldt and J. n. Rodríguez-López, Modulation of the electrochemical reactivity of solubilized redox active polymers via polyelectrolyte dynamics, J. Am. Chem. Soc., 2018, 140(6), 2093–2104 CrossRef CAS.
- G. Nagarjuna, J. Hui, K. J. Cheng, T. Lichtenstein, M. Shen, J. S. Moore and J. Rodríguez-López, Impact of Redox-Active Polymer Molecular Weight on the Electrochemical Properties and Transport Across Porous Separators in Nonaqueous Solvents, J. Am. Chem. Soc., 2014, 136(46), 16309–16316, DOI:10.1021/ja508482e.
- Y. Zhang, A. Park, A. Cintora, S. R. McMillan, N. J. Harmon, A. Moehle, M. E. Flatté, G. D. Fuchs and C. K. Ober, Impact of the synthesis method on the solid-state charge transport of radical polymers, J. Mater. Chem. C, 2018, 6(1), 111–118 RSC.
- H. J. Martin, B. K. Hughes, W. A. Braunecker, T. Gennett and M. D. Dadmun, The impact of radical loading and oxidation on the conformation of organic radical polymers by small angle neutron scattering, J. Mater. Chem. A, 2018, 6(32), 15659–15667 RSC.
- G. Inzelt, Role of polymeric properties in the electrochemical behaviour of redox polymer-modified electrodes, Electrochim. Acta, 1989, 34(2), 83–91 CrossRef CAS.
- M. A. Haque, A. D. Easley, J. G. Moncada, J. L. Lutkenhaus and M. D. Dadmun, Chain Conformations of TEMPO-Based Organic Radical Polymers with Varying Radical Loading and Temperature in Battery-Relevant Solvents, Macromolecules, 2024, 57(13), 6333–6343, DOI:10.1021/acs.macromol.4c01134.
- A. D. Easley, L. M. Vukin, P. Flouda, D. L. Howard, J. L. Pena and J. L. Lutkenhaus, Nitroxide Radical Polymer–Solvent Interactions and Solubility Parameter Determination, Macromolecules, 2020, 53(18), 7997–8008, DOI:10.1021/acs.macromol.0c01739.
- K. Hatakeyama-Sato, Y. Igarashi and K. Oyaizu, Charge-transport kinetics of dissolved redox-active polymers for rational design of flow batteries, RSC Adv., 2023, 13(1), 547–557 RSC.
- E. Schröter, C. Stolze, A. Saal, K. Schreyer, M. D. Hager and U. S. Schubert, All-organic redox targeting with a single redox moiety: Combining organic radical batteries and organic redox flow batteries, ACS Appl. Mater. Interfaces, 2022, 14(5), 6638–6648 CrossRef PubMed.
- E. Schröter, C. Stolze, J. Meyer, M. D. Hager and U. S. Schubert, Organic Redox Targeting Flow Battery Utilizing a Hydrophilic Polymer and Its In-Operando Characterization via State-of-Charge Monitoring of The Redox Mediator, ChemSusChem, 2023, 16(14), e202300296 CrossRef.
- R. Rohan, M.-K. Hung, Y.-F. Yang, C.-W. Hsu, C.-K. Yeh, Y.-L. Chang and J.-T. Lee, Enhancement of the high-rate performance of an organic radical thin-film battery by decreasing the grafting density of polymer brushes, ACS Appl. Polym. Mater., 2022, 4(4), 2365–2372 CrossRef CAS.
- H. Jia, C. Friebe, U. S. Schubert, X. Zhang, T. Quan, Y. Lu and J.-F. Gohy, Core-Shell Nanoparticles with a Redox Polymer Core and a Silica Porous Shell as High-Performance Cathode Material for Lithium-Ion Batteries, Energy Technol., 2020, 8(3), 1901040 CrossRef CAS.
- H.-C. Lin, C.-C. Li and J.-T. Lee, Nitroxide polymer brushes grafted onto silica nanoparticles as cathodes for organic radical batteries, J. Power Sources, 2011, 196(19), 8098–8103 CrossRef CAS.
- Y. Li, Z. Jian, M. Lang, C. Zhang and X. Huang, Covalently functionalized graphene by radical polymers for graphene-based high-performance cathode materials, ACS Appl. Mater. Interfaces, 2016, 8(27), 17352–17359 CrossRef CAS.
- A. Vlad, J. Rolland, G. Hauffman, B. Ernould and J. F. Gohy, Melt-Polymerization of TEMPO Methacrylates with Nano Carbons Enables Superior Battery Materials, ChemSusChem, 2015, 8(10), 1692–1696 CrossRef CAS PubMed.
- W. Jin, T. Zhou, Z. Wang, W. Xue, C. Feng, F. Zhang, X. Huang, D. Yang, P. Théato and Y. Li, Radical polymer grafted graphene for high-performance Li+/Na+ organic cathodes, J. Power Sources, 2021, 511, 230363 CrossRef CAS.
- D. Brunel, F. Fajula, J. Nagy, B. Deroide, M. Verhoef, L. Veum, J. Peters and H. Van Bekkum, Comparison of two MCM-41 grafted TEMPO catalysts in selective alcohol oxidation, Appl. Catal., A, 2001, 213(1), 73–82 CrossRef CAS.
- Y. Sun, X. Ding, Z. Zheng, X. Cheng, X. Hu and Y. Peng, Surface initiated ATRP in the synthesis of iron oxide/polystyrene core/shell nanoparticles, Eur. Polym. J., 2007, 43(3), 762–772 CrossRef CAS.
- T. von Werne and T. E. Patten, Atom Transfer Radical Polymerization from Nanoparticles: A Tool for the Preparation of Well-Defined Hybrid Nanostructures and for Understanding the Chemistry of Controlled/“Living” Radical Polymerizations from Surfaces, J. Am. Chem. Soc., 2001, 123(31), 7497–7505, DOI:10.1021/ja010235q.
- M.-K. Hung, Y.-H. Wang, C.-H. Lin, H.-C. Lin and J.-T. Lee, Synthesis and electrochemical behaviour of nitroxide polymer brush thin-film electrodes for organic radical batteries, J. Mater. Chem., 2012, 22(4), 1570–1577 RSC.
- P. de Gennes, Conformations of polymers attached to an interface, Macromolecules, 1980, 13(5), 1069–1075 CrossRef CAS.
- S. Alexander, Adsorption of chain molecules with a polar head a scaling description, J. Phys., 1977, 38(8), 983–987 CrossRef CAS.
- J. C. Conrad and M. L. Robertson, Shaping the structure and response of surface-grafted polymer brushes via the molecular weight distribution, JACS Au, 2023, 3(2), 333–343 CrossRef CAS.
- Y. Zhang, A. M. Park, S. R. McMillan, N. J. Harmon, M. E. Flatté, G. D. Fuchs and C. K. Ober, Charge transport in conjugated polymers with pendent stable radical groups, Chem. Mater., 2018, 30(14), 4799–4807 CrossRef CAS.
- D. C. Bobela, B. K. Hughes, W. A. Braunecker, T. W. Kemper, R. E. Larsen and T. Gennett, Close packing of nitroxide radicals in stable organic radical polymeric materials, J. Phys. Chem. Lett., 2015, 6(8), 1414–1419 Search PubMed.
- K. Nakahara, S. Iwasa, M. Satoh, Y. Morioka, J. Iriyama, M. Suguro and E. Hasegawa, Rechargeable batteries with organic radical cathodes, Chem. Phys. Lett., 2002, 359(5), 351–354, DOI:10.1016/S0009-2614(02)00705-4.
- J. Allen and L. R. F. Bard, Electrochemical Methods: Fundamentals and Applications, 2000 Search PubMed.
- N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, A Practical Beginner's Guide to Cyclic Voltammetry, J. Chem. Educ., 2018, 95(2), 197–206, DOI:10.1021/acs.jchemed.7b00361.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc00051c |
| ‡ Co-first authors. |
| This journal is © The Royal Society of Chemistry 2025 |