
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An N-phosphinoamidinato borasilenide: a vinyl-analogous anion containing a base-stabilised B![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) Si double bond†
Si double bond†
Si Jia Isabel
Phang
a,
Zheng-Feng
Zhang
b,
Ming-Der
Su
 *bc and
Cheuk-Wai
So
*a
*bc and
Cheuk-Wai
So
*a
aSchool of Chemistry, Chemical Engineering and Biotechnology, Nanyang Technological University, Singapore 637371, Singapore. E-mail: CWSo@ntu.edu.sg
bDepartment of Applied Chemistry, National Chiayi University, Chiayi 60004, Taiwan
cDepartment of Medicinal and Applied Chemistry, Kaohsiung Medical University, Kaohsiung 80708, Taiwan
First published on 10th March 2025
Abstract
Borasilenes, which feature a heterodinuclear Si![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) B double bond, show interesting reactivities, due to two proximal reactive sites—boron and silicon—each with distinct electronic properties. However, borasilenes remain relatively rare due to the challenge of stabilising them. To achieve a stable borasilene, both the boron and silicon centers must be supported by sterically hindered ligands, which can, however, interfere with their potential applications. In this work, we report the synthesis of an N-phosphinoamidinato potassium borasilenide (compound 3), which is a vinyl-analogous anion containing a base-stabilised BSi− double bond. This BSi− double bond is functional and exhibits new patterns of reactivity towards CuCl(PMe3), [IrCl(cod)]2, Me3SiOTf, and MeOTf, leading to the formation of a transition metal π-complex, boron–silicon-containing metallacycle, neutral borasilene and borylsilane, respectively.
B double bond, show interesting reactivities, due to two proximal reactive sites—boron and silicon—each with distinct electronic properties. However, borasilenes remain relatively rare due to the challenge of stabilising them. To achieve a stable borasilene, both the boron and silicon centers must be supported by sterically hindered ligands, which can, however, interfere with their potential applications. In this work, we report the synthesis of an N-phosphinoamidinato potassium borasilenide (compound 3), which is a vinyl-analogous anion containing a base-stabilised BSi− double bond. This BSi− double bond is functional and exhibits new patterns of reactivity towards CuCl(PMe3), [IrCl(cod)]2, Me3SiOTf, and MeOTf, leading to the formation of a transition metal π-complex, boron–silicon-containing metallacycle, neutral borasilene and borylsilane, respectively.
Introduction
Emerging silicon and boron compounds have been at the center of main-group chemistry owing to their potential in small molecule activation,1,2 catalysis3–9 and materials science.10,11 Since the successful isolation of tetramesityldisilene12 in 1981 and NHC-diborene in 2007,13 an influx of compounds containing SiSi double bonds, kinetically stabilised by sterically hindered ligands, have been isolated.14–16 Compounds possessing BB double bonds are still in the developmental stages due to the electron deficient boron centers that require stabilisation through coordination with Lewis base ligands.17–23 These homodiatomic multiple bonds exhibit unprecedented reactivity patterns toward small molecules as well as intriguing photophysical properties.24–28 It is envisioned that borasilene, composed of a heterodinuclear BSi double bond, should exhibit a different array of reactivities compared to their homodiatomic counterparts. This can be attributed to the two proximal reactive sites—boron and silicon—each with distinct electronic properties. Such hypothesis is evidenced by silylboronic esters or silylboranes,29 where the Si–B single bonds serve as versatile reactive sites for the introduction of silyl and/or boryl functionalities into organic frameworks depending on reaction conditions. As such, borasilene should be a worthwhile synthetic target. Despite their potential for synthetic applications, borasilenes are relatively rare, with only a few reported examples. To date, only one example of a stable borasilene A (Fig. 1), supported by an electron-donating amino substituent on the boron center, has been reported by Sekiguchi et al.30 Related ate and NHC complexes of borasilenes B–D have been isolated by research groups of Sekiguchi,30 Iwamoto31 and Inoue,32 respectively, which illustrate that the stabilisation of electron deficient boron centers is important. Their reactivity remains largely unexplored, likely due to the steric hindrance of the ligands on the BSi double bond, which impedes reactivity. Recently, Iwamoto et al. reported a borasilene E containing a Br substituent on the silicon center,33 whereby its reactivity with anionic nucleophiles led to complex mixtures. This highlights that the use of a functional SiB double bond in the construction of more complex SiB double bond-containing π-conjugated systems, Si–B-heterocycles and Si–B-clusters remains a challenge.
 | ||
| Fig. 1 Reported examples of compounds composed of a SiB double bond. | ||
We chose to take an alternative approach, where a borasilene derivative is made anionic, to explore its potential for metathesis reactions with a variety of main-group elements, transition metals, and organic substrates. This strategy aims to generate a more reactive SiB double bond that can undergo functionalisation or construction of more complex boron- and silicon-containing complexes, revealing new patterns of reactivity. This approach is supported by the chemistry of disilenide anions reported by Scheschkewitz et al., where they serve as versatile building blocks for synthesising novel classes of silicon-based molecules.34,35 Herein, we report the synthesis of an N-phosphinoamidinate-stabilised potassium borasilenide (compound 3), which is a vinyl anion analogue containing a BSi− double bond. It is a versatile building block for the preparation of an (η2-borasilene)-transition metal π-complex, boron–silicon-containing metallacycle, neutral borasilene and borylsilane, respectively.
Results and discussion
To begin with, the N-phosphinoamidinato chlorosilylene 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 36 underwent oxidative addition with PhBCl2 in toluene at room temperature for 16 h to afford the N-phosphinoamidinate-bridged borylsilane 2 (Scheme 1), which was isolated as a colorless crystalline solid (yield: 49%) from the concentrated reaction mixture. Compound 2 was characterised by NMR spectroscopy and X-ray crystallography. The 1H NMR spectrum displays a set of signals corresponding to the N-phosphinoamidinate ligand. The 31P{1H} and 11B{1H} NMR signals are at 46.35 ppm and −5.66 ppm, respectively. The latter indicates a four-coordinate boron center. The 29Si{1H} NMR signal of 2 (3.10 ppm) is broad due to the quadrupolar coupling with the B nucleus, and it is upfield shifted in comparison with compound 1 (7.96 ppm). The molecular structure obtained by X-ray crystallography shows that the ligand is bridging between the boron and silicon centers (Fig. 2). The Si1–B1 bond length is 2.027(2) Å, which is indicative of a single bond. Similar B–Cl bond oxidative addition has been reported by Cui et al.37
36 underwent oxidative addition with PhBCl2 in toluene at room temperature for 16 h to afford the N-phosphinoamidinate-bridged borylsilane 2 (Scheme 1), which was isolated as a colorless crystalline solid (yield: 49%) from the concentrated reaction mixture. Compound 2 was characterised by NMR spectroscopy and X-ray crystallography. The 1H NMR spectrum displays a set of signals corresponding to the N-phosphinoamidinate ligand. The 31P{1H} and 11B{1H} NMR signals are at 46.35 ppm and −5.66 ppm, respectively. The latter indicates a four-coordinate boron center. The 29Si{1H} NMR signal of 2 (3.10 ppm) is broad due to the quadrupolar coupling with the B nucleus, and it is upfield shifted in comparison with compound 1 (7.96 ppm). The molecular structure obtained by X-ray crystallography shows that the ligand is bridging between the boron and silicon centers (Fig. 2). The Si1–B1 bond length is 2.027(2) Å, which is indicative of a single bond. Similar B–Cl bond oxidative addition has been reported by Cui et al.37
 | ||
| Scheme 1 Synthesis of compounds 2 and 3. | ||
 | ||
| Fig. 2 X-ray crystal structure of 2 with thermal ellipsoids at 50% probability. All H atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Cl1–B1 1.9094(19), Cl2–Si1 2.0460(6), Cl3–Si1 2.0718(6), P1–B1 2.017(2), Si1–N1 1.7806(14), Si1–B1 2.027(2), N1–Si1–B1 112.96(7), P1–B1–Si1 99.17(9), N2–P1–B1 111.59(8). | ||
Compound 2 was reacted with excess KC8 in THF at room temperature for 2 h to afford the dimeric N-phosphinoamidinato potassium borasilenide 3 (Scheme 1), which was isolated as a reddish-brown crystalline solid (yield: 42%) from its concentrated toluene solution. It is stable at room temperature in solution under an inert atmosphere.
Compound 3 was characterised by NMR spectroscopy and X-ray crystallography. The 31P{1H} NMR signal (47.79 ppm) is broad, which is comparable with that of 2. The 29Si{1H} NMR signal (208.40 ppm) and 11B{1H} NMR signal (30.34 ppm) are downfield shifted compared to compound 6 (29Si{1H} NMR: 110.06 ppm; 11B{1H} NMR: 25.30 ppm, see below). A similar deshielding effect was found when comparing the 29Si{1H} NMR of lithium disilenide [(Tip)2SiSi(Tip)Li] (SiSi–: 100.5; SiSi–: 94.5 ppm) to disilene [(Tip)2SiSi(Tip)2] (53.4 ppm).34 Moreover, the 29Si{1H} NMR signal (208.40 ppm) is significantly downfield shifted in comparison with the ate- and silylene-complexes of borasilene (C: 58.8, E: 83 ppm),31,33 which comprise of planar boron and silicon centers. In addition, the 11B{1H} NMR signal (30.34 ppm) is downfield shifted in comparison with the phosphine-coordinated boron center in the N-phosphinoamidinato diborene (10.5 ppm)7 and diboravinyl cation (16.7 ppm),17 with a BB double bond. The UV-Vis spectrum shows two distinct absorption bands at λmax = 353 and 500 nm corresponding to  (HOMO → LUMO+4) and
(HOMO → LUMO+4) and  (HOMO−1 → LUMO), respectively, based on TD-DFT calculations (M06-2X/def2-TZVP). The molecular structure of compound 3 obtained by X-ray crystallography (Fig. 3) shows that it is a contact ion pair, where the Si⋯K distance (3.3320(12), 3.5479(13) Å) is longer than the sum of their covalent radii (Si: 1.11 Å; K: 2.03 Å). The coordination sphere on the K cation is further stabilised by π-type interaction with the C atoms of Ph and Dipp substituents. The N1–Si1–B1 bond angle (106.5(1)°) is smaller than that of compound 2 (112.96(7)°), indicating the presence of a lone pair of electrons on the negatively charged silicon center. The silicon atom adopts a flattened pyramidal geometry, with the Si1, N1, B1, and K1 centers lying on the same plane, while the K1A center is positioned 1.36 Å below the plane. A similar geometry around the Si atom was found in the aluminium center of an aluminyl anion reported by Alridge et al.38 In addition, the boron atom adopts a trigonal planar geometry (sum of the bond angles: 358.8°). The Si1–B1 bond length (1.889(4) Å) is slightly longer than the SiB double bond length in the ate- and silylene-complexes of borasilene (C: 1.859(2), E: 1.8882(16) Å),31,33 but is shorter than the Si–B single bond length in compound 2. All of this indicates that the Si–B bond in compound 3 has a double bond character.
(HOMO−1 → LUMO), respectively, based on TD-DFT calculations (M06-2X/def2-TZVP). The molecular structure of compound 3 obtained by X-ray crystallography (Fig. 3) shows that it is a contact ion pair, where the Si⋯K distance (3.3320(12), 3.5479(13) Å) is longer than the sum of their covalent radii (Si: 1.11 Å; K: 2.03 Å). The coordination sphere on the K cation is further stabilised by π-type interaction with the C atoms of Ph and Dipp substituents. The N1–Si1–B1 bond angle (106.5(1)°) is smaller than that of compound 2 (112.96(7)°), indicating the presence of a lone pair of electrons on the negatively charged silicon center. The silicon atom adopts a flattened pyramidal geometry, with the Si1, N1, B1, and K1 centers lying on the same plane, while the K1A center is positioned 1.36 Å below the plane. A similar geometry around the Si atom was found in the aluminium center of an aluminyl anion reported by Alridge et al.38 In addition, the boron atom adopts a trigonal planar geometry (sum of the bond angles: 358.8°). The Si1–B1 bond length (1.889(4) Å) is slightly longer than the SiB double bond length in the ate- and silylene-complexes of borasilene (C: 1.859(2), E: 1.8882(16) Å),31,33 but is shorter than the Si–B single bond length in compound 2. All of this indicates that the Si–B bond in compound 3 has a double bond character.
 | ||
| Fig. 3 X-ray crystal structure of 3 with thermal ellipsoids at 50% probability. All H atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): K1–Si1 3.3320(12), K1–Si1A 3.5479(13), P1–N2 1.648(3), P1–B1 1.878(4), Si1–N1 1.866(3), Si1–B1 1.889(4), N1–Si1–B1 106.53(15), N2–P1–B1 113.96(16), P1–B1–Si1 113.05(19), K1–Si1–K1A 78.97(3). | ||
DFT calculations (M06-2X/def2-TZVP) were performed to elucidate the electronic structure of compound 3. The highest occupied molecular orbital (HOMO) corresponds to the π orbitals of the SiB bonds (Fig. 4). The Wiberg Bond Index (WBI) of 1.586 suggests that the Si–B bond has a double bond character. Accordingly, the natural bond orbital (NBO) analysis illustrates that the Si1–B1 π bond arises from the mixing of p orbitals on the boron and silicon atoms. It is slightly polarised toward the B1 atom (56.4% B1 + 43.6% Si1). The Si1–B1 σ bond is formed by the overlapping of the sp2.42 hybrids on the Si1 atom and the sp1.79 hybrids on the B1 atom. The Si1 atom has no interaction with the K atom, but it has a σ-type lone pair of electrons (sp0.56, electron occupancy: 1.87 e). These theoretical data illustrate that compound 3 is a vinyl anion analogue containing a BSi− double bond, being consistent with the conclusion deduced from NMR spectroscopic and X-ray crystallographic data.
 | ||
| Fig. 4 Highest occupied molecular orbitals (HOMO) of compounds 3–6 (M06-2X/def2-TZVP). | ||
Despite the synthesis of disilenide and digermenide anions [R2EE(R)Li] (E = Si or Ge) by Scheschkewitz et al.,34,35 the preparation of heterodinuclear vinyl anion derivatives [R2E′E(R)M] (M = group 1 metal) remains challenging. Scheschkewitz et al. reported the only potassium silagermenide (R2SiGeR′K),39 while Apeloig et al. reported the synthesis of silenide and germenide anions.40–43 Compound 3 is anticipated to be a highly effective reagent for transferring the SiB double bond to various organic and inorganic substrates to yield boron–silicon-based heterocycles, clusters and complexes.
First, the reaction of compound 3 with CuCl(PMe3) in benzene at room temperature for 4.5 h quantitatively afforded the (bis(borasilenyl)copper)–copper π-complex 4 (Scheme 2). In comparison with homodiatomic counterparts, an η2-disilene–copper complex has yet to be reported,44 while Braunschweig et al. reported some diborene–copper π-complexes.45 It is proposed that in the reaction, compound 3 reacts with CuCl(PMe3) to form a potassium bis(borasilenyl)cuprate IntI, which further undergoes metathesis reaction and π-coordination with another molecule of CuCl(PMe3) to form compound 4.
 | ||
| Scheme 2 Synthesis of compound 4 and proposed mechanism for the formation of the compound. | ||
Compound 4 was isolated as brown crystals from the concentrated reaction mixture, which were characterised by NMR spectroscopy and X-ray crystallography. It is interesting to note that the 29Si{1H} NMR signal of 4 (234.96 ppm) is in the low-field region very similar to that of 3. The 11B{1H} NMR signal (23.80 ppm) is upfield shifted in comparison to compound 3. A similar upfield shift was observed from the bis(PMe3)bis(9-anthryl)diborene (22 ppm) to its CuCl complex (17.1 ppm).45 The UV-Vis spectrum shows one distinct absorption band at λmax = 394 nm, which is majorly contributed by  (HOMO → LUMO+2), based on TD-DFT calculations, and a long absorption tail from 463 to 694 nm.
(HOMO → LUMO+2), based on TD-DFT calculations, and a long absorption tail from 463 to 694 nm.
The molecular structure of compound 4 obtained by X-ray crystallography (Fig. 5) shows that the Cu1 atom is bonded to the two silicon atoms of the SiB double bonds with the Cu1–Si1/2 bond lengths (2.3192(7), 2.3364(7) Å) longer than the lithium bis(disilenyl)cuprate (2.2412(8), 2.2458(8) Å),46 and comparable with the potassium bis(silyl)cuprate (Ph3Si)2Cu[K(18-crown-6)THF2] (2.3480(7) Å).47 The Cu1–Si1/2 bond lengths are shorter than the sum of their covalent radii (Cu: 1.32, Si: 1.11 Å). The Si1–B1 and Si2–B2 bond lengths (1.921(3), 1.928(2) Å) are, on average, 0.036 Å longer than those in compound 3, but significantly shorter – by an average of 0.102 Å – than those in compound 2. In addition, the planarity of the SiB double bonds is retained (sum of the bond angles, excluding Cu2, Si1: 355.73; Si2: 354.67; B1: 357.07; B2: 353.85°). The Cu2–B1/2 (2.265(3), 2.264(3) Å) and Cu2–Si1/2 (2.4210(7), 2.3971(7) Å) bond lengths are longer than that of η2-diborene–copper complexes (Cu–B: 2.133(3)–2.149(3) Å)45 and the Cu1–Si1/2 bond lengths of compound 4, respectively. All of this indicates that the SiB π-electrons donate to the Cu2 center, while the Cu2 center with a d10-valence electron count exhibits a small degree of π-back bonding to the π*-MO of the SiB double bonds in the Dewar–Chatt–Duncanson model. In this context, the Cu2 center forms a π-complex with two SiB double bonds. The Cu1–Cu2 distance (2.7229(4) Å) is in accordance with a weak intramolecular d10–d10 interaction. DFT calculations show that the HOMO is mainly the SiB π orbitals (Fig. 4), and the NBO analysis illustrates that the Si–B bond possesses a π orbital with a WBI of 1.52, indicating that the Si–B bond is a double bond. Based on both experimental and theoretical data, compound 4 is a bis(η2-borasilene)–copper π-complex.
 | ||
| Fig. 5 X-ray crystal structure of 4 with thermal ellipsoids at 50% probability. All H atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): B1–Si1 1.921(3), B2–Si2 1.928(2), B1–Cu2 2.265(3), B2–Cu2 2.264(3), Cu1–Si1 2.3192(7), Cu1–Si2 2.3364(7), Cu2–Si2 2.3971(7), Cu2–Si1 2.4210(7), N1–Si1–B1 109.45(10), N1–Si1–Cu1 119.61(7), B1–Si1–Cu1 126.67(8), N3–Si2–B2 111.98(10), N3–Si2–Cu1 121.84(6), B2–Si2–Cu1 120.85(8), C28–B1–P1 121.61(17), C28–B1–Si1 124.45(17), P1–B1–Si1 111.01(13), C61–B2–P2 120.10(16), C61–B2–Si2 123.83(17), P2–B2–Si2 109.92(12). | ||
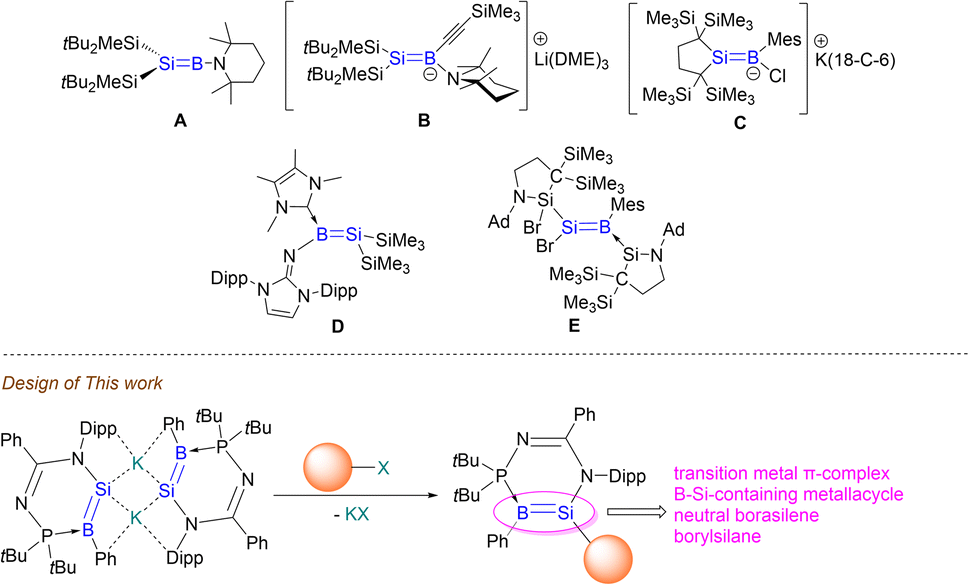
Besides copper, compound 3 could also react with heavy transition metal electrophiles, namely iridium. As a proof-of-principle, the reaction of compound 3 with [Ir(cod)Cl]2 in benzene at room temperature for 30 min quantitatively affords the B–Si–Ir metallacyclic complex 5 (Scheme 3). We propose that the monomeric derivative of compound 3 first reacts with a unit of [Ir(cod)Cl]2 to form an iridium borasilenide intermediate, Int1. Following this, it undergoes a C–H insertion reaction with the iPr of Dipp substituent viaTS1 to form a neutral borasilene and (cod)hydridoiridium(I) moieties, where the BSi double bond coordinates with the iridium center viaTS2 to afford an (η2-borasilene)iridium intermediate, Int2. It subsequently undergoes another C–H insertion reaction with the tBu substituent viaTS3 to form compound 5 and H2.
 | ||
| Scheme 3 Synthesis of compound 5 and proposed mechanism for the formation of the compound. | ||
Compound 5 was isolated as orange crystals from the concentrated reaction mixture, which were characterised by NMR spectroscopy and X-ray crystallography. The 11B{1H} NMR signal (−58.93 ppm) is in the high field region similar to four-coordinate boron metal complexes such as the (CAAC)(dicyano)borylgold complex (−26.2 ppm).48 The 29Si NMR of compound 5 could not be obtained, due to the presence of quadrupolar coupling with the 11B and 193Ir nuclei. The UV-Vis spectrum shows a distinct absorption band at 350 nm corresponding to dz2 → pπ + dxy (HOMO−3 → LUMO), based on TD-DFT calculations.
The molecular structure of compound 5 obtained by X-ray crystallography shows that the Si1–B1 bond (1.989(7) Å) is slightly shorter than that of compound 2 (2.027 Å) by 0.038 Å (Fig. 6). In addition, the planarity of the borasilene moiety was not retained, where both boron and silicon centers adopt a trigonal pyramidal geometry (sum of the bond angles, excluding the Ir center, Si1: 337.9; B1: 343.2°). The Ir1–B1 bond (2.529(6) Å) is longer than the Ir–Bboryl bond in PBP-pincer iridium complexes (1.97–2.14 Å),49 while the Ir1–Si1 bond (2.371(2) Å) is comparable with that of the octahedral Ir-silyl complexes50 (ca. 2.39 Å), and shorter than that of the iridium silene complex [Cp*(PMe3)Ir(η2-CH2SiMe2)(H)][B(C6F5)4] (2.439(8) Å).51 This indicates that the Ir center, with a d8-valence electron count, exhibits a large degree of π-back bonding to the π*-MO of the borasilene. The shorter Ir–Si bond length reflects the greater degree of covalent bonding character, therefore elucidating that compound 5 is a metallacyclopropane, with its molecular structure revealing a three-membered B–Si–Ir ring rather than the classical π-complex. DFT calculations support the conclusion as the HOMO is composed of the Si and B pπ orbitals overlapped with the Ir dx2–y2 orbital (Fig. 4), and the NBO analysis illustrates that the Si1–B1 bond possesses solely a σ orbital (Si sp2.19 + B sp3.45) with a WBI of 1.08, indicating that the Si–B bond is a single bond. In this context, compound 5 is a B–Si–Ir metallacycle. Comparing compounds 4 and 5 with other heavier alkene analogues, the latter also demonstrate intriguing coordination modes with transition metals.44 For example, Berry et al. showed that a tungsten complex with an unhindered disilene Me2SiSiMe2 exhibits properties between those of a π-complex and metallacycle, analogous to the bonding situation observed in transition-metal complexes of olefins.52 Scheschkewitz et al. demonstrated that the SiGe double bond in a disilagermirene–nickel complex did not follow the Dewar–Chatt–Duncanson model, as the Si–Ge σ-electrons, rather than the π-electrons, donate to the nickel center.53
 | ||
| Fig. 6 X-ray crystal structure of 5 with thermal ellipsoids at 50% probability. All H atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): B1–Si1 1.989(7), B1–Ir1 2.529(6), Ir1–Si1 2.3714(16), B1–P1 1.920(7), N1–Si1 1.791(5), N2–P1 1.681(5), N1–Si1–C1 100.1(3), N1–Si1–B1 113.6(3), C1–Si1–B1 124.2(3), C27–B1–P1 123.0(4), C27–B1–Si1 118.1(4), P1–B1–Si1 102.1(3). | ||
To show that compound 3 can generate a free neutral borasilene, Me3SiOTf was reacted with compound 3 in benzene at room temperature for 15 min, resulting in the quantitative formation of compound 6 (Scheme 4). Compound 6 was isolated as orange crystals from the concentrated reaction mixture, which were characterised by NMR spectroscopy and X-ray crystallography. The 11B{1H} NMR signal (25.30 ppm) and 29Si{1H} NMR signal (110.06 ppm) are upfield shifted in comparison with that of compound 3. The UV-Vis spectrum shows three distinct absorption bands at λmax = 279, 372, and 434 nm corresponding to 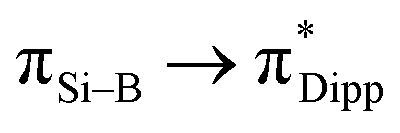 (HOMO → LUMO+3),
(HOMO → LUMO+3),  (HOMO → LUMO+1) and
(HOMO → LUMO+1) and  (HOMO → LUMO), respectively, based on TD-DFT calculations.
(HOMO → LUMO), respectively, based on TD-DFT calculations.
 | ||
| Scheme 4 Synthesis of compounds 6 and 7 and proposed mechanism for the formation of compound 7. | ||
The molecular structure of compound 6 obtained by X-ray crystallography shows that the planes of the planar boron center (sum of the bond angles: 359.7°) and silicon center (359.9°) form a dihedral angle of 9.9° (Fig. 7). The B1–Si1 bond (1.865(6) Å) is comparable to that of compound 3. The electronic structure of compound 6 was elucidated by DFT calculations. The HOMO and HOMO−1 correspond to the BSi π and σ orbitals respectively (Fig. 4 and S50†). The WBI of 1.66 suggests that the SiB double bond has a stronger double bond character than that in compound 3. Accordingly, NBO analysis illustrates that the Si1–B1 π bond arises from the mixing of p orbitals on the boron and silicon atoms with even contribution (48.9% B1 + 51.1% Si1), whereas the SiB double bond in compound 3 is slightly polarised. The Si1–B1 σ bond is formed by the overlapping of the sp1.28 hybrids on the Si1 atom and the sp1.87 hybrids on the B1 atom. The introduction of the SiMe3 functionality could open the possibility of using compound 6 as a BSi double bond transfer reagent, based on the recent work of a Lewis base-stabilised phosphaborene with a trimethylsilyl functionality reported by Andrada et al.54
 | ||
| Fig. 7 X-ray crystal structure of 6 with thermal ellipsoids at 50% probability. All H atoms are omitted for clarity. Selected bond lengths (Å) and angles (°) of 6: B1–Si1 1.865(6), B1–P1 1.880(7), N1–Si1 1.814(4), N2–P1 1.647(4), Si1–Si2 2.349(2), C28–B1–Si1 118.2(5), C28–B1–P1 134.3(5), Si1–B1–P1 107.2(3), N1–Si1–B1 116.1(3), N1–Si1–Si2 120.99(15), B1–Si1–Si2 122.8(2). | ||
It is anticipated that a less sterically hindered substituent could enable the BSi double bond to undergo further addition reaction or cycloaddition. To prove our hypothesis, compound 3 was reacted with two equivalents of MeOTf in benzene at room temperature for 50 min. We propose that MeOTf reacts with compound 3 to form Intα composed of a less sterically hindered methyl substituent, where the BSi double bond undergoes further addition reaction with the second molecule of MeOTf to quantitatively form compound 7 (Scheme 4). It should be noted that compound 7 is prone to decompose over time in solution. The molecular structure obtained by X-ray crystallography shows that the N-phosphinoamidinate ligand is in a P,N-chelate fashion, bridged across the B–Si bond to form a four-membered ring. The B–Si bond length (2.036(4) Å) is comparable with that of 2, indicating that it is a single bond (Fig. 8).
 | ||
| Fig. 8 X-ray crystal structure of 7 with thermal ellipsoids at 50% probability. All H atoms are omitted for clarity. Selected bond lengths (Å) and angles (°) of 7: B1–P1 2.033(4), B1–Si1 2.036(4), N2–P1 1.714(3), N2–Si1 1.815(3), P1–Si1 2.7149(12), P1–B1–Si1 83.70(14), P1–N2–Si1 100.54(13), N2–P1–B1 89.24(14), N2–Si1–B1 86.41(13). | ||
Conclusions
In conclusion, the N-phosphinoamidinato potassium borasilenide 3 was synthesised, which is a vinyl anion derivative containing a BSi− double bond. It is a versatile SiB double bond transfer agent to a variety of organic and inorganic substrates, namely CuCl(PMe3), [IrCl(cod)]2, Me3SiOTf, and MeOTf, forming an (η2-borasilene)-transition metal π-complex, boron–silicon-containing metallacycle, neutral borasilene and borylsilane, respectively.
Data availability
The data supporting this article have been included as part of the ESI.† Deposition numbers 2394597 for 2, 2394598 for 3, 2394599 for 4, 2394600 for 5, 2394601 for 6, and 2394602 for 7 contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.Author contributions
S. J. I. Phang designed and performed the experimental study. Z.-F. Zhang designed and performed the computational study. M.-D. Su and C.-W. So prepared the manuscript.Conflicts of interest
There is no conflict of interest among the authors.Acknowledgements
This work is financially supported by the Ministry of Education Singapore, AcRF Tier 1 (RG72/21) and A*STAR MTC Individual Research Grants (M21K2c0117). M.-D. Su acknowledges the National Center for High-Performance Computing of Taiwan for generous amounts of computing time, and the Ministry of Science and Technology of Taiwan for the financial support.Notes and references
- C. Shan, S. Yao and M. Driess, Chem. Soc. Rev., 2020, 49, 6733–6754 RSC.
- M.-A. Légaré, C. Pranckevicius and H. Braunschweig, Chem. Rev., 2019, 119, 8231–8261 CrossRef PubMed.
- S. Stigler, S. Fujimori, A. Kostenko and S. Inoue, Chem. Sci., 2024, 15, 4275–4291 RSC.
- B.-X. Leong, J. Lee, Y. Li, M.-C. Yang, C.-K. Siu, M.-D. Su and C.-W. So, J. Am. Chem. Soc., 2019, 141, 17629–17636 CrossRef CAS.
- Y.-C. Teo, D. Loh, B.-X. Leong, Z.-F. Zhang, M.-D. Su and C.-W. So, Inorg. Chem., 2023, 62, 16867–16873 CrossRef CAS.
- B.-X. Leong, Y.-C. Teo, C. Condamines, M.-C. Yang, M.-D. Su and C.-W. So, ACS Catal., 2020, 10, 14824–14833 CrossRef CAS.
- J. Fan, J.-Q. Mah, M.-C. Yang, M.-D. Su and C.-W. So, J. Am. Chem. Soc., 2021, 143, 4993–5002 CrossRef PubMed.
- X. Chen, Y. Yang, H. Wang and Z. Mo, J. Am. Chem. Soc., 2023, 145, 7011–7020 CrossRef CAS PubMed.
- J. Lee, J. Fan, A.-P. Koh, W.-J. J. Cheang, M.-C. Yang, M.-D. Su and C.-W. So, Eur. J. Inorg. Chem., 2022, e202200129 CrossRef CAS.
- L. Shi, A. Boulègue-Mondière, D. Blanc, A. Baceiredo, V. Branchadell and T. Kato, Science, 2023, 38, 1011–1014 CrossRef PubMed.
- M. Hirai, N. Tanaka, M. Sakai and S. Yamaguchi, Chem. Rev., 2019, 119, 8291–8331 CrossRef CAS.
- R. West, M. J. Fink and J. Michl, Science, 1981, 214, 1343–1344 CrossRef CAS PubMed.
- Y. Wang, B. Quillian, P. Wei, C. S. Wannere, Y. Xie, R. B. King, H. F. Schaefer, P. v. R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2007, 129, 12412–12413 CrossRef CAS PubMed.
- C. Präsang and D. Scheschkewitz, Chem. Soc. Rev., 2016, 45, 900–921 RSC.
- F. Hanusch, L. Groll and S. Inoue, Chem. Sci., 2021, 12, 2001–2015 RSC.
- A. Rammo and D. Scheschkewitz, Chem.–Eur. J., 2018, 24, 6866–6885 CrossRef CAS.
- J. Fan, P.-T. Chia, Z.-F. Zhang, M.-C. Yang, M.-D. Su and C.-W. So, Angew. Chem., Int. Ed., 2022, 61, e202212842 CrossRef CAS.
- J. H. Muessig, M. Thaler, R. D. Dewhurst, V. Paprocki, J. Seufert, J. D. Mattock, A. Vargas and H. Braunschweig, Angew. Chem., Int. Ed., 2019, 58, 4405–4409 CrossRef CAS PubMed.
- T. E. Stennett, J. D. Mattock, L. Pentecost, A. Vargas and H. Braunschweig, Angew. Chem., Int. Ed., 2018, 57, 15276–15281 CrossRef CAS PubMed.
- W. Lu, Y. Li, R. Ganguly and R. Kinjo, Angew. Chem., Int. Ed., 2017, 56, 9829–9832 CrossRef CAS PubMed.
- S. R. Wang, M. Arrowsmith, J. Böhnke, H. Braunschweig, T. Dellermann, R. D. Dewhurst, H. Kelch, I. Krummenacher, J. D. Mattock, J. H. Müssig, T. Thiess, A. Vargas and J. Zhang, Angew. Chem., Int. Ed., 2017, 56, 8009–8013 CrossRef CAS.
- H. Braunschweig, I. Krummenacher, C. Lichtenberg, J. D. Mattock, M. Schäfer, U. Schmidt, C. Schneider, T. Steffenhagen, S. Ullrich and A. Vargas, Angew. Chem., Int. Ed., 2017, 56, 889–892 CrossRef CAS PubMed.
- H. Braunschweig, R. D. Dewhurst, K. Hammond, J. Mies, K. Radacki and A. Vargas, Science, 2012, 336, 1420–1422 CrossRef CAS PubMed.
- A. Fukazawa, Y. Li, S. Yamaguchi, H. Tsuji and K. Tamao, J. Am. Chem. Soc., 2007, 129, 14164–14165 CrossRef CAS PubMed.
- I. Bejan and D. Scheschkewitz, Angew. Chem., Int. Ed., 2007, 46, 5783–5786 CrossRef CAS PubMed.
- N. Hayakawa, S. Nishimura, N. Kazusa, N. Shintani, T. Nakahodo, H. Fujihara, M. Hoshino, D. Hashizume and T. Matsuo, Organometallics, 2017, 36, 3226–3233 CrossRef CAS.
- T. E. Stennett, P. Bissinger, S. Griesbeck, S. Ullrich, I. Krummenacher, M. Auth, A. Sperlich, M. Stolte, K. Radacki, C.-J. Yao, F. Würthner, A. Steffen, T. B. Marder and H. Braunschweig, Angew. Chem., Int. Ed., 2019, 58, 6449–6454 CrossRef CAS PubMed.
- P. Bissinger, A. Steffen, A. Vargas, R. D. Dewhurst, A. Damme and H. Braunschweig, Angew. Chem., Int. Ed., 2015, 54, 4362–4366 CrossRef CAS PubMed.
- J.-J. Feng, W. Mao, L. Zhang and M. Oestreich, Chem. Soc. Rev., 2021, 50, 2010–2073 RSC.
- N. Nakata and A. Sekiguchi, J. Am. Chem. Soc., 2006, 128, 422–423 CrossRef CAS PubMed.
- Y. Suzuki, S. Ishida, S. Sato, H. Isobe and T. Iwamoto, Angew. Chem., Int. Ed., 2017, 56, 4593–4597 CrossRef CAS PubMed.
- D. Franz, T. Szilvási, A. Pöthig and S. Inoue, Chem.–Eur. J., 2019, 25, 11036–11041 CrossRef CAS PubMed.
- T. Koike, N. Sakurata, S. Ishida and T. Iwamoto, Angew. Chem., Int. Ed., 2024, e202411283 CAS.
- D. Scheschkewitz, Angew. Chem., Int. Ed., 2004, 43, 2965–2967 CrossRef CAS PubMed.
- A. Rammo and D. Scheschkewitz, Chem.–Eur. J., 2018, 24, 6866–6885 CrossRef CAS PubMed.
- S. J. I. Phang, Z.-F. Zhang, C.-S. Wu, Z. X. Wong, M.-D. Su and C.-W. So, Chem. Sci., 2025, 16, 4512–4518 RSC.
- L. Zhu, J. Zhang and C. Cui, Inorg. Chem., 2019, 58, 12007–12010 CrossRef CAS PubMed.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Alridge, Nature, 2018, 557, 92–95 CrossRef CAS PubMed.
- P. K. Majhi, V. Huch and D. Scheschkewitz, Angew. Chem., Int. Ed., 2021, 60, 242–246 CrossRef CAS PubMed.
- D. Bravo-Zhivotovskii, R. Dobrovetsky, D. Nemirovsky, V. Molev, M. Bendikov, G. Molev, M. Botoshansky and Y. Apeloig, Angew. Chem., Int. Ed., 2008, 47, 4343–4345 CrossRef CAS PubMed.
- L. Zborovsky, R. Dobrovetsky, M. Botoshansky, D. Bravo-Zhivotovskii and Y. Apeloig, J. Am. Chem. Soc., 2012, 134, 18229–18232 CrossRef CAS.
- D. Pinchuk, J. Mathew, A. Kaushansky, D. Bravo-Zhivotovskii and Y. Apeloig, Angew. Chem., Int. Ed., 2016, 55, 10258–10262 CrossRef CAS PubMed.
- Y. Goldshtein, Y. Glagovsky, B. Tumanskii, N. Fridman, D. Bravo-Zhivotovskii and Y. Apeloig, Angew. Chem., Int. Ed., 2022, 61, e202202452 CrossRef CAS PubMed.
- T. J. Hadlington, Chem. Soc. Rev., 2024, 53, 9718–9737 RSC.
- S. R. Wang, M. Arrowsmith, H. Braunschweig, R. D. Dewhurst, V. Paprocki and L. Winner, Chem. Commun., 2017, 53, 11945–11947 RSC.
- M. J. Cowley, K. Abersfelder, A. J. P. White, M. Majumdar and D. Scheschkewitz, Chem. Commun., 2012, 48, 6595–6597 RSC.
- J. Plotzitzka and C. Kleeberg, Inorg. Chem., 2017, 56, 6671–6680 CrossRef CAS PubMed.
- D. A. Ruiz, G. Ung, M. Melaimi and G. Bertrand, Angew. Chem., Int. Ed., 2013, 52, 7590–7592 CrossRef CAS PubMed.
- W.-C. Shih and O. V. Ozerov, Organometallics, 2017, 36, 228–233 CrossRef CAS.
- J. Y. Corey, Chem. Rev., 2016, 116, 11291–11435 CrossRef CAS PubMed.
- S. R. Klei, T. D. Tilley and R. G. Bergman, Organometallics, 2001, 20, 3220–3222 CrossRef CAS.
- D. H. Berry, J. H. Chey, H. S. Zipin and P. J. Carroll, J. Am. Chem. Soc., 1990, 112, 452–453 CrossRef CAS.
- P. K. Majhi, M. Zimmer, B. Morgenstern and D. Scheschkewitz, J. Am. Chem. Soc., 2021, 143, 8981–8986 CrossRef CAS PubMed.
- A. Koner, B. Morgenstern and D. M. Andrada, Angew. Chem., Int. Ed., 2022, 61, e202203345 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and theoretical calculations. CCDC 2394597–2394602. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc00047e |
| This journal is © The Royal Society of Chemistry 2025 |