
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnlocking the reactivity of the C–In bond: alkyl indium reagents as a source of radicals under photocatalytic conditions†
Anton A.
Gladkov
ab,
Vitalij V.
Levin
 a,
Demian Y.
Cheboksarov
ab and
Alexander D.
Dilman
*a
a,
Demian Y.
Cheboksarov
ab and
Alexander D.
Dilman
*a
aN. D. Zelinsky Institute of Organic Chemistry, Leninsky Prosp. 47, 119991 Moscow, Russian Federation. E-mail: adil25@mail.ru
bLomonosov Moscow State University, Department of Chemistry, Leninskie Gory 1-3, 119991 Moscow, Russian Federation
First published on 24th February 2025
Abstract
Generation of organic radicals from organometallic compounds is a key step in metallaphotoredox cross-coupling reactions. The ability of organoindium compounds to serve as sources of alkyl radicals under light promoted oxidative conditions is described. Organoindium reagents were used in dual photocatalytic/nickel cross-coupling with aryl bromides. These reagents can be conveniently obtained from primary, secondary and tertiary alkyl bromides and chlorides using a novel indium(I) bromide/lithium bromide system. Both steps, the formation of organoindiums and their cross-coupling are insensitive towards air and moisture and tolerate a wide variety of functional groups.
Introduction
Organometallic compounds involving alkyl lithium, magnesium and zinc reagents have found widespread applications as nucleophilic reagents.1,2 However, the increased sensitivity of these reagents to moisture and air, coupled with incompatibility of lithium and magnesium reagents with crucial functional groups such as hydroxyl, carbonyl and nitrile, limits their applicability in organic synthesis (Scheme 1A). In contrast, less polar boron, silicon and tin reagents demonstrate tolerance to a multitude of functional groups and can be handled in air, but they are poorly reactive and require activation. The modern method of activation is the conversion of organometallics to a radical, which has expanded the synthetic applications of alkyl boron3,4 and zinc5–10 reagents. The range of non-polar reagents also includes organoindium compounds, which have previously been considered to be rare reagents.11 Indium is located in close proximity to tin in the periodic table, and therefore, chemical properties of their organic derivatives are expected to be similar. However, an indisputable advantage of organoindium reagents is their low toxicity.12 Another point is an opportunity to access alkyl indium reagents directly from organic halides by insertion of indium into the carbon–halide bond. However, the latent synthetic potential of these reagents as a source of radicals under photocatalytic conditions has not yet been realized due to the low polarity of the C–In bond and the limited scope of accessible alkyl indium reagents.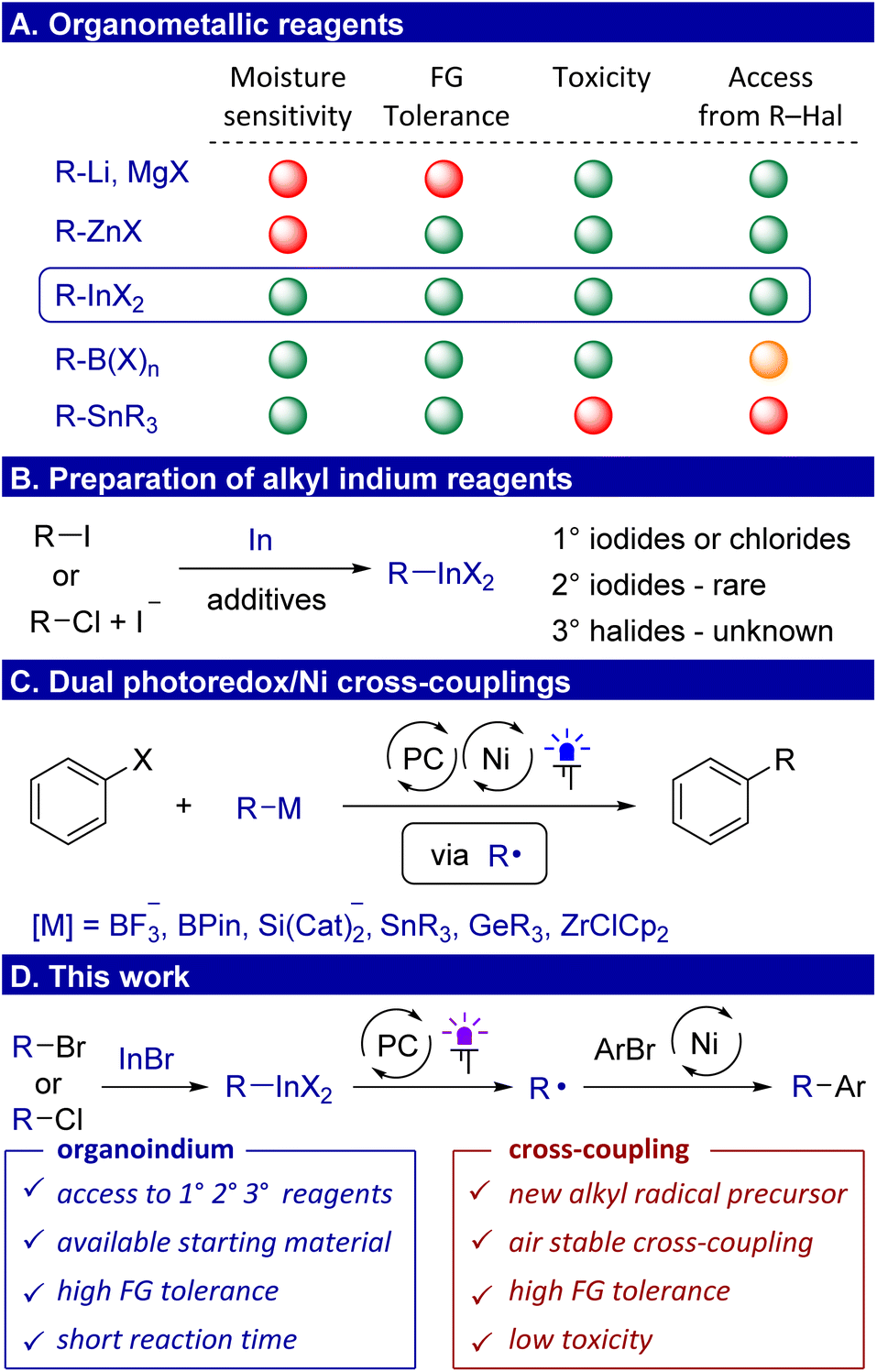 | ||
| Scheme 1 Organometallic reagents. | ||
The preparation and synthetic applications of aryl,13–17 allyl,18 allenyl,19,20 and indium enolates21 have been well studied. This is due to the ease of production and increased nucleophilicity of these compounds. Despite the progress achieved with the organoindiums mentioned above, methods for the preparation and applications of alkyl indium reagents are still limited. In earlier reports, they were prepared by transmetallation from organolithium or organomagnesium reagents.22,23 Later, the reaction of alkyl halides with elemental indium was used, but due to low activity of indium metal, additives have to be used such as copper,24,25 silver,26 cobalt,27 cesium28 or indium29 salts, and elemental iodine30 (Scheme 1B). Primary alkyl iodides were typically used, while the reaction of primary alkyl chlorides was performed in the presence of a lithium iodide additive, which likely serves to generate more reactive alkyl iodides via nucleophilic substitution.31 Of special note is that in all these methods, only a few secondary aliphatic alkyl indium reagents have been used, which are obtained exclusively from secondary alkyl iodides, while the preparation of tertiary reagents from tertiary alkyl halides is not known at all. In addition, these methods require long reaction times, which also reflects the low reducing power of indium metal. A more important aspect is that alkyl indium compounds have only been used in palladium catalyzed cross-couplings.32,33
Transition metal catalyzed cross-coupling reactions of organic halides with organometallic reagents have become a cornerstone of organic synthesis.34 The modern variant of cross-coupling involves the use of metallaphotoredox conditions.35 In particular, a dual photoredox/Ni catalytic system served as an analogue to palladium-catalyzed reactions.36 This approach allows us, on the one hand, to make the process cheaper by using the more earth abundant nickel. On the other hand, Ni/PC catalysis allows us to overcome the often rate-limiting transmetallation step involving organometallic reagents by its single-electron oxidation and subsequent interception on the nickel catalyst.37 As coupling reagents, trifluoroborates,37–41 and other boron reagents42,43 have originally been used, followed by alkyl derivatives of silicon44–47 (Scheme 1C). Over the past few years, there has been an active search for other organometallic radical precursors. In this vein, the activation of germanium48,49 and zirconium reagents50 under photoredox conditions was described. Additionally, metallaphotoredox cross-couplings with organotin compounds were also attempted and only realized in the case of very specific atrane-type alkyl tin derivatives.51 The reagents described above are difficult to obtain directly from alkyl halides, forcing them to be obtained from more nucleophilic alkyl magnesium/zinc reagents. In addition, a number of reagents suffer from a lack of atom efficiency due to the surrounding metallic center (for silicon, tin, and zirconium). The utilization of alkyl indiums could provide an effective solution to these issues. However, at present, there have been no universal methods for the synthesis of organoindium compounds from readily accessible alkyl bromides and chlorides.
Here, we overcome this problem by reacting alkyl halides with the commercially available indium(I) bromide in the presence of lithium bromide, which is more efficient than elemental indium, allowing the use of alkyl bromides and chlorides as sources for primary, secondary, and even tertiary alkyl indium reagents (Scheme 1D). We also demonstrate that the synthetic utility of alkyl indium compounds can be greatly expanded as they can undergo single-electron oxidation and serve as excellent precursors of alkyl radicals involved in the C(sp2)–C(sp3) cross-coupling under metallaphotoredox conditions.
Results and discussion
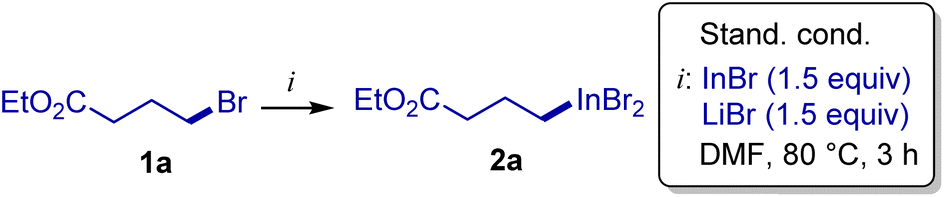
Our idea is based on the fact that indium(I) bromide in donating solvents is prone to disproportionation to In(0) and In(III).52 This combination was proposed to be effective for the following reasons. First, zero-valent metals formed as a result of reduction of their salts (such as Rieke metals) are known to be more reactive than commercial metals.53,54 Moreover, In(I) to In(0)/In(III) conversion proceeds under equilibrium conditions,55 thereby maintaining the surface of the metal fresh. Second, use of indium(I) bromide provides perfect stoichiometry upon conversion of RBr into RInBr2. This point is expected to eliminate the formation of numerous organoindium species such as dialkyl indium(III) bromide, observed when In(0) is used. Finally, the strength of the reducing indium system may be augmented with lithium halides, which are known to be instrumental in reactions of insertion of zero-valent metals into C–X bonds,56,57 and this was also applied for reactions using elemental indium.58–60 Bromide anions can also break-up alkyl indium bromide aggregates to give more reactive indium-ate species.61 In the earlier literature, examples of solvent-free interaction of indium(I) salts with simple primary alkyl halides were described,62–64 while in coordinating systems, disproportionation of indium(I) and formation of indium(II) salts were noted.62,65Ethyl 4-bromobutyrate (1a) was selected as a model substrate, and its reaction with indium(I) bromide was evaluated, and several model alkyl halides were also tested (Table 1). The reaction of 1a with 1.5 equivalent of a InBr/LiBr combination in N,N-dimethylformamide (1.0 M of 1a) was complete within 3 h at 80 °C leading to the virtually quantitative formation of reagent 2a (entry 1). The reaction performed in deuterated solvent (d7-DMF) allowed us to record 1H and 13C NMR spectra of 2a, which showed a single organoindium species, which was assigned to monoorganoindium RInBr2 based on HRMS (ESI-TOF); bis(alkyl)indium substances (such as R2InBr) were not detected (see the ESI† for details). A decrease in the reaction temperature led to some decrease in the yield, while application of the ultrasound had a positive effect (entries 3 and 4); performing the reaction at room temperature over 16 hours was also inefficient (entry 5). In other solvents, the reaction was slower than in DMF, though reasonable yields of 2a can be achieved by extending the reaction time to 16 hours (entries 7–9). Use of LiCl instead of LiBr gave a notable yield decrease (entry 10), likely due to the competitive substitution of bromide of 1a by chloride resulting in the formation of less reactive alkyl chloride (the alkyl chloride was detected by GC-MS analysis). In the absence of LiBr or the use of metallic indium led to notably decreased yields of 2a (entries 11 and 12). It was reported that benzyl (but not alkyl) halides react with elemental indium in the presence of air leading to hydroperoxides.66 It was rewarding to find that in our system, when the reaction was set up in an air atmosphere, the organoindium reagent was formed in 71% yield (entry 6). Moreover, air does not decrease the concentration of 2a in DMF-d7 over one month (stored in a vial in air and periodically checked by NMR with the internal standard; see the ESI† for details).
|
|
||
|---|---|---|
| # | Deviations from stand. cond. | Y. of 2a,a (%) |
| a Determined by 1H NMR with 1,3,5-trioxane as an internal standard. b 16 h. | ||
| 1 | None | 99 |
| 2 | InBr/LiBr (1.2 equiv.) | 97 |
| 3 | Heat at 60 °C | 80 |
| 4 | 50 °C + ultrasound | 90 |
| 5b | 20 °C | 40 |
| 6 | Air atmosphere | 71 |
| 7b | DME as solvent | 95 |
| 8b | MeCN as solvent | 77 |
| 9b | THF as solvent | 75 |
| 10 | LiCl (1.5 equiv.) instead of LiBr | 35 |
| 11 | No LiBr | 48 |
| 12 | In (1.5 equiv.) instead of InBr | 41 |
Then, we evaluated secondary and tertiary alkyl bromides (Scheme 2). After brief optimization (see the ESI† for details) we found that for secondary acyclic and cyclic bromides, a longer time (12 h) and increased loading of the InBr/LiBr system (2 equiv.) were needed to achieve high yields. Reagent 2b was also characterized by 1H and 13C NMR and HRMS (ESI-TOF). In the case of cyclohexyl bromide, lithium chloride could be used instead of lithium bromide because in this case SN2 halogen exchange (cyclohexyl bromide with chloride) is slower than the formation of the organoindium 2c-Cl. The latter protocol was applied for the preparation of the organoindium reagent from 30 mmol of cyclohexyl bromide. Preparation of tert-butyl indium reagent 2d under standard conditions was hampered by its instability at temperatures above 50 °C. Fortunately, reagent 2d could be obtained at 40 °C using ultrasonication in 63% yield. This is the first example of organoindium formation from tertiary alkyl halides through direct insertion.
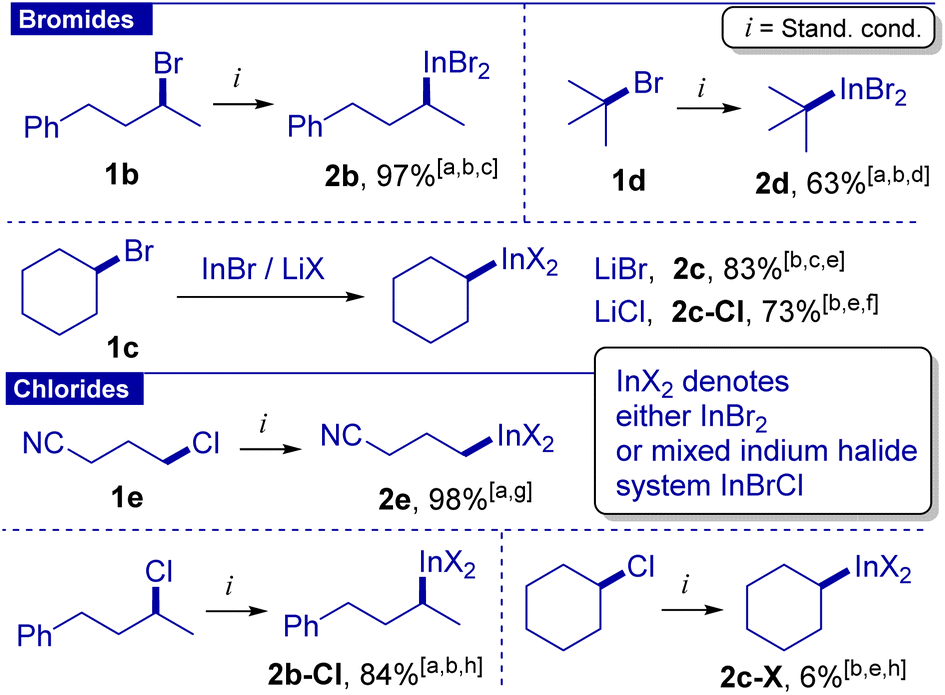 | ||
| Scheme 2 Organoindium reagents. aDetermined by 1H NMR with 1,3,5-trioxane as an internal standard. b12 h. cInBr/LiBr (2.0 equiv.). d40 °C per ultrasound. eDetermined by GC-FID with tetraline as the standard after iodination. fInBr/LiCl (2.0 equiv.). g6 h. hInBr (2.0 equiv.), LiBr (3.0 equiv.), 100 °C. | ||
Alkyl chlorides are notably less reactive compared to bromides. However, primary halides can undergo halogen exchange, as was shown for 1a (see the discussion above and entry 10). Correspondingly, we proposed that a combination of alkyl chloride with LiBr can reversibly generate alkyl bromide, which in turn can efficiently be converted into the organoindium reagent.31 Indeed, 1-chloro-3-cyanopropane gave reagent 2e in 98% yield under standard conditions but within 6 hours. The intermediate formation of 1-bromo-3-cyanobutane was confirmed by monitoring of the reaction by GS-MS analysis. Fortunately, reagent 2b-Cl was also obtained from the corresponding alkyl chloride in 84% yield by increasing the amount of lithium bromide to 3 equiv. and performing the reaction at 100 °C for 12 hours. Cyclohexyl chloride reacted slowly, apparently due to slow SN2 halogen exchange. tert-Butyl chloride did not give the indium reagent.
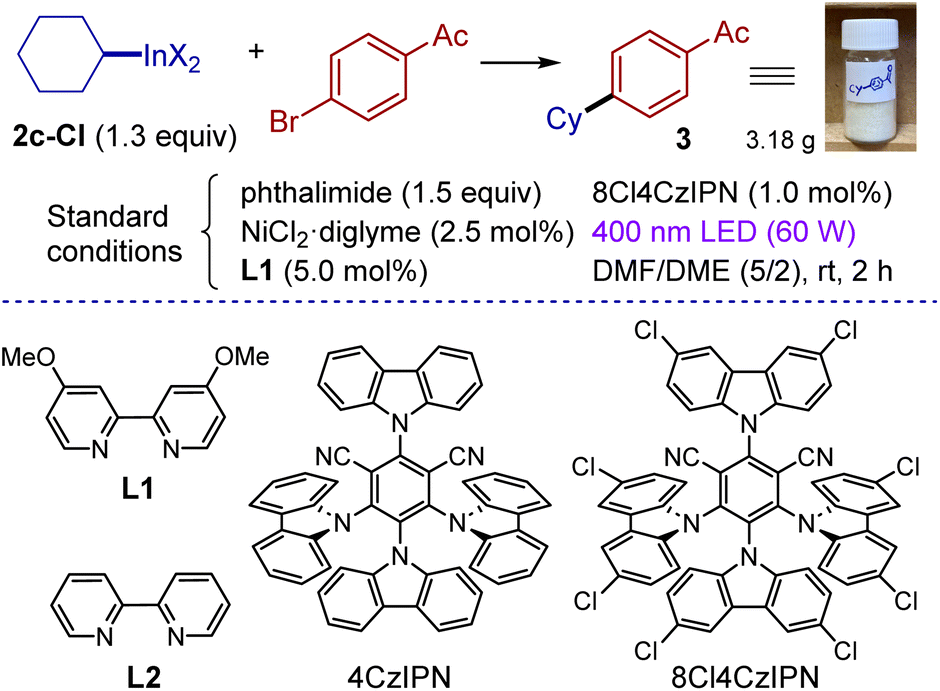
Organoindium reagents were evaluated as sources of radicals in C(sp2)–C(sp3) cross-coupling under dual photoredox/nickel catalysis. Variation of conditions for a model coupling partners (cyclohexyl indium reagent 2c-Cl and 4-bromoacetophenone) was undertaken (Table 2). The reaction is best performed using chlorinated cyanoarene photocatalyst 8Cl4CzIPN,67 a nickel dichloride-diglyme complex and 4,4′-dimethoxybipyridine (L1). Slight excess of the indium reagent (1.3 equiv.) was applied in a mixed solvent DMF/DME (5/2). The reaction was irradiated with 400 nm light and was complete within 2 hours furnishing the target product in 91% isolated yield (entry 1). Use of a stoichiometric amount of the phthalimide additive was required, which is believed to play a dual role: to suppress the side protodehalogenation of arylbromide and to hinder the formation of catalytically inactive low-valent oligomeric nickel complexes68 (entry 7). While reactions were routinely carried out under argon, running the experiment under an air atmosphere had little effect (entry 2). To evaluate sensitivity towards moisture, cross-coupling of 2c with 4-bromoacetophenone was performed in the presence of 4 equiv. of water leading to product 3 in 90% yield (see the ESI† for details). The reaction did not proceed without the nickel complex or light (entries 8 and 11); without the photocatalyst or ligand only traces of the product were detected (entries 9 and 10). Finally, the reaction can be easily scaled up, with the experiment on 17.5 mmol of aryl bromide affording gram quantities of the product (90%, 3.18 g, see the photo).
|
|
||
|---|---|---|
| # | Deviations from stand. cond. | Y. of 3,a (%) |
| a Determined by 1H NMR with CH2Br2 as an internal standard. b Isolated yield. | ||
| 1 | None | 92 (91)b |
| 2 | Open to air | 87 |
| 3 | 455 nm LED | 82 |
| 4 | 4CzIPN as PC (455 nm LED) | 80 |
| 5 | Without DME | 76 |
| 6 | L2 as the ligand | 76 |
| 7 | No phthalimide | 64 |
| 8 | No NiCl2·diglyme | 0 |
| 9 | No L1 | <5 |
| 10 | No PC | <5 |
| 11 | No light | 0 |
Under the optimized conditions, the scope of aryl halides was first evaluated using reagent 2c-Cl (Scheme 3). The reaction tolerates many functional groups such as acetyl, ester, nitrile, aldehyde, amide, sulfonamide, and the (pinacolato)boryl fragment. Comparison of bromides bearing an ester group at para, meta and ortho positions suggested that para and meta substrates have similar reactivity (products 4 and 18), while the ortho substrate afforded product 19 in a diminished yield of 51%, which is likely associated with the steric effect. Based on this observation, selective substitution of one bromine in 1,2-dibromobenzene was carried out (product 30). Aryl bromides bearing triflate, mesylate and tosylate were coupled (products 9–11) (triflate and mesylate groups can be slowly affected under our conditions in the case of electron depleting arenes; see the mechanistic discussion below). It should be pointed out that in palladium cross-couplings, aryl triflates are known to be more reactive compared to bromides. In a similar vein, the chlorine atom remained unaffected (product 15). The reaction tolerates fragments with active hydrogen such as phenol (8) and trifluoroacetanilide (13). Various bromo-substituted heterocycles were evaluated. Thus, the reaction can work with derivatives of pyridine, pyrimidine, indole, thiophene and benzothiophene. The 2-fluoropyridine fragment, which is prone to fluoride substitution, withstands the reaction conditions (product 21). A fragment of vinyl bromide was also successfully involved in cross-coupling affording a mixture of isomers in good yield (product 27). Despite impressive functional group tolerance, several types of substrates were problematic. In particular, 4-bromoaniline showed very low reactivity, with 30% conversion after 16 hours. In reactions of 4-bromonitrobenzene and 4-bromostyrene, no products were formed (see bottom of Scheme 3). A similar outcome was obtained with oxadiazole-containing substrates, which suffered from decomposition of this fragile heterocycle; in these experiments, complete conversion of starting bromides was noted along with the detection of benzonitrile by GC-MS analysis.
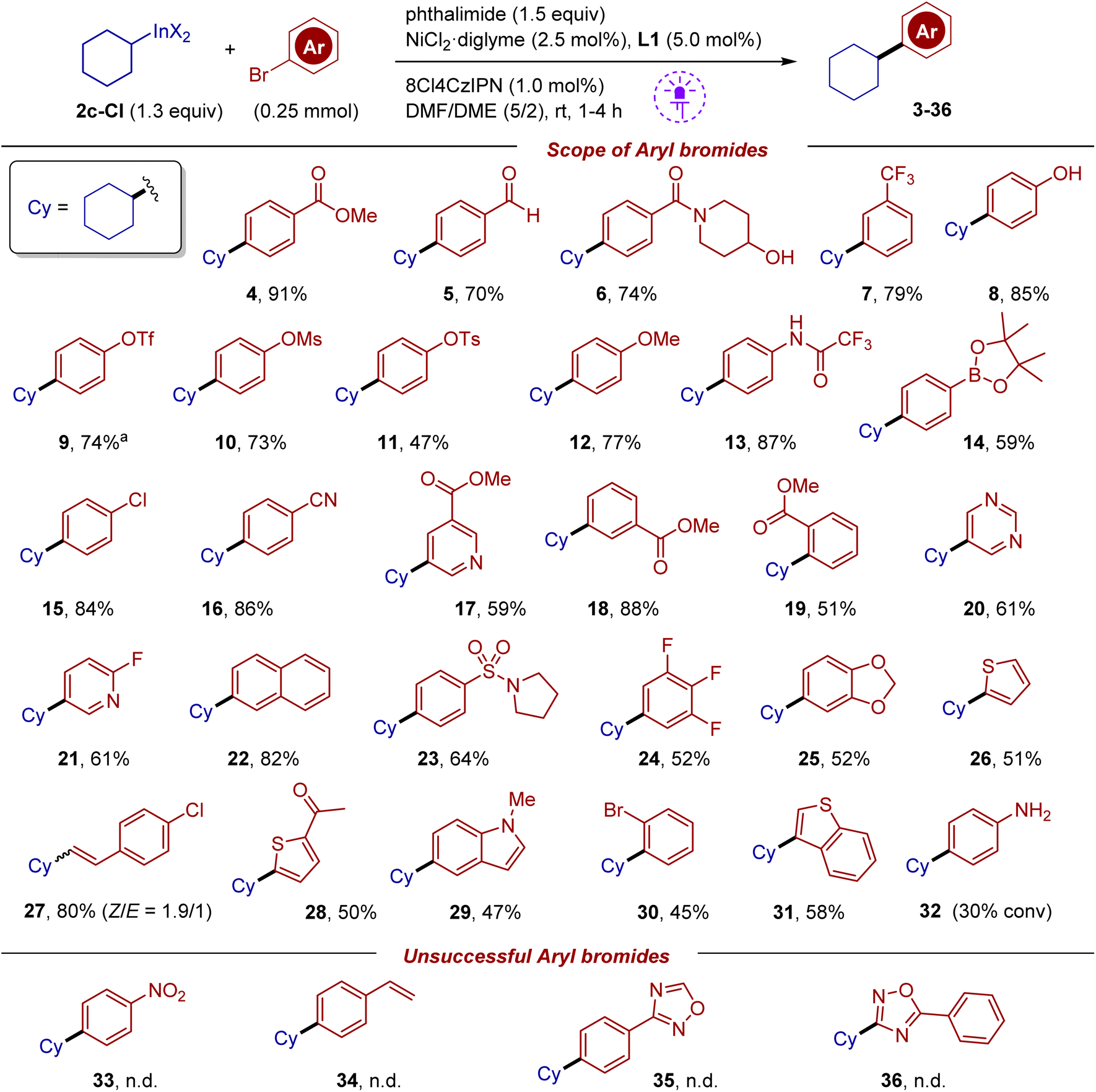 | ||
| Scheme 3 Scope of (hetero)aryl bromide coupling partners. Isolated yields are shown. | ||
Then we focused on the variation of the alkyl indium component (Scheme 4). The indium reagent was prepared from 2 equiv. of alkyl bromide or chloride followed by the addition of 1,2-dimethoxyethane co-solvent, aryl bromide (1 equiv.) and other components. The reaction proceeded smoothly with primary and secondary alkyl indium reagents containing N-Boc protected cyclic amine, ester and cyano groups, phosphonate and dioxolane fragments, as well as an unprotected hydroxy group. Various heterocycles within the indium component such as thiophene, furan, indole and triazole were tolerated. Products 42 and 44 could be obtained starting from both alkyl bromides and chlorides, though reactions from bromides gave slightly higher yields. The reaction of the tert-butyl indium reagent was unsuccessful, which is in accordance with literature reports requiring different conditions for the adoption of tertiary radicals in photoredox/Ni systems.69,70 An indium reagent prepared from 4-bromobut-1-ene gave a decreased yield (product 55), likely due to the presence of the terminal double bond. A moderate yield was also observed for the reaction of 1-bromo-3-chloropropane (product 63) because of competitive Cl/Br exchange. Benzyl bromides can also be easily converted into the indium reagents under our conditions, which required only 30 min, followed by cross-coupling (products 58–60). It is interesting to note that a reagent derived from para-bromobenzyl bromide gave the coupling product 59 suggesting that the C–Br of 4-bromoacetophenone undergoes faster activation than the C–Br of the product. Several functionalized reagents could not be prepared, which is associated either with incompatibility with the InBr/LiBr system (2g) or with rapid degradation of the indium reagent (2h–j) (see bottom of Scheme 4).
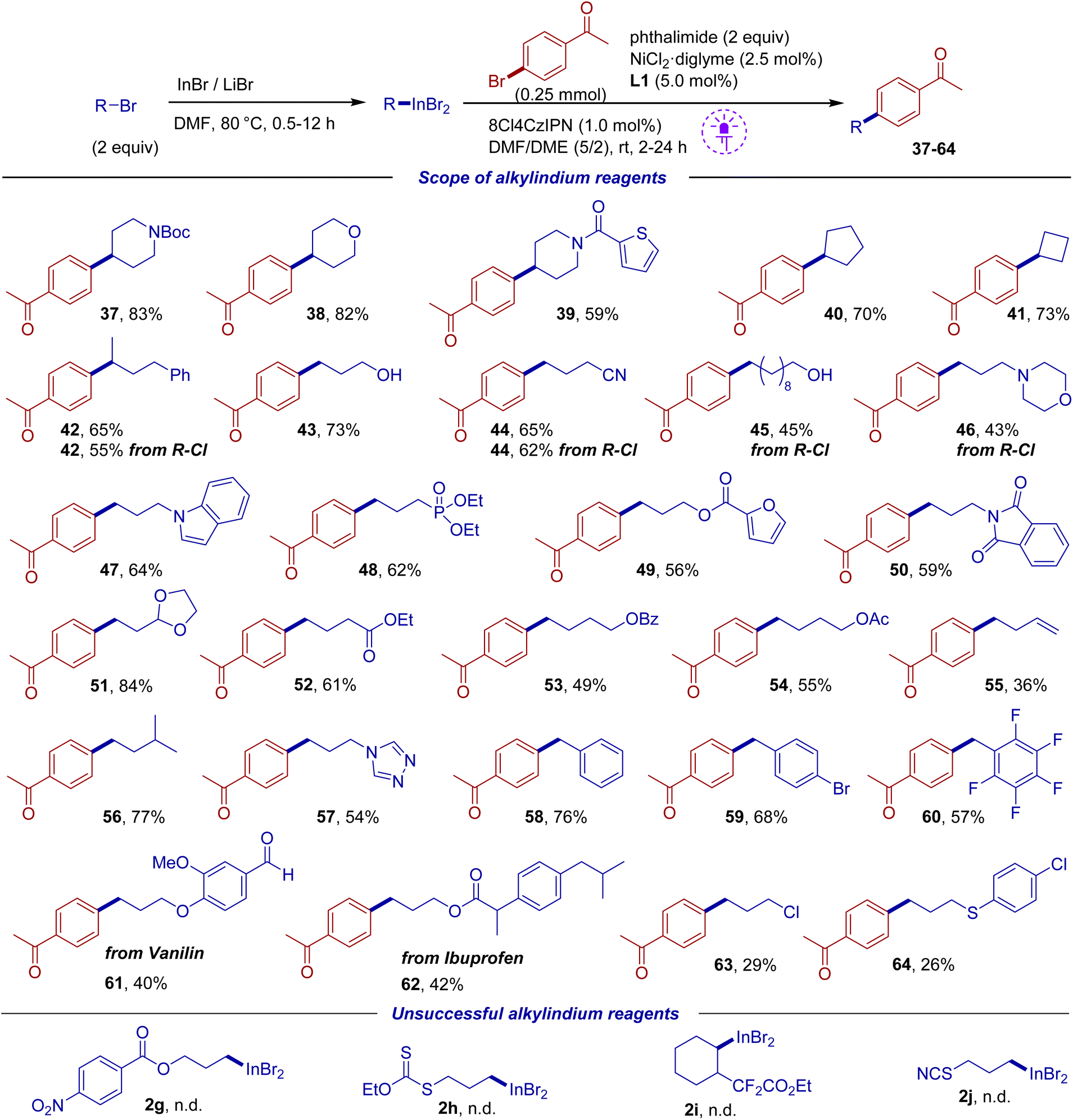 | ||
| Scheme 4 Scope of organoindium coupling partners. Isolated yields are shown, which are based on aryl bromide. | ||
α-(Trifluoromethyl)styrenes are another class of typical radical-accepting substrates working under redox conditions.71 Indeed, alkylindiums cannot be viewed as C-nucleophiles towards α-(trifluoromethyl)styrenes (confirmed by a blank experiment, see the ESI† for details). On the other hand, interaction of these styrenes with organoindiums should proceed via generation of alkyl radicals (by oxidation of organoindiums), radical addition at the styrene, SET reduction and fluoride elimination (Scheme 5). The reaction of α-(trifluoromethyl)styrenes with organoindium reagents was catalyzed by 8Cl4CzIPN and, for tertiary and secondary reagents, was complete within 30 min! Here, the tertiary indium reagent was the most effective (in line with the formation of the most stable tertiary radical). On the other hand, for a primary alkyl group, two equivalents of the indium reagent were necessary together with a long reaction time of 16 hours.
 | ||
| Scheme 5 Reaction of α-(trifluoromethyl)styrenes. Isolated yields are shown. a2.0 equiv. of organoindium reagents was used; reaction time 16 h. | ||
Mechanistic considerations
To gain deeper insight into the reaction mechanism, several experiments were carried out. First, addition of TEMPO completely blocked the cross-coupling under standard conditions along with the formation of a trapping product between TEMPO and the cyclohexyl radical (Scheme 6A). To perform a radical clock experiment, 1-hexen-6-yl bromide was subjected to the reaction with the InBr/LiBr system, which gave a mixture of open/cyclic indium reagents (Scheme 6B). Their cross-coupling gave a mixture with an increased content of the cyclic product. In the literature, a reaction of 1-hexen-6-yl bromide in a nickel photoredox cross-coupling gave a 2.5/1 mixture of open/cyclic products.72 These experiments support the radical character of cross-coupling. To support the intermediacy of low valent nickel species in the catalytic cycle, the reaction was performed using zero-valent complex Ni(COD)(DQ)73 (DQ = duroquinone) as a source of nickel (instead of NiCl2-diglyme), which gave the expected product 3 in good yield (Scheme 6C). To identify species interacting with the photocatalyst, Stern–Volmer fluorescence quenching was evaluated (Scheme 6D). It was found that there was virtually no quenching of the photocatalyst excited state (PC*) with phthalimide and aryl bromide. On the other hand, lithium bromide, as well as organoindium reagent 2a served as fluorescence quenchers. As follows from the slopes of Stern–Volmer plots, for the reagent 2a, the quenching was most efficient. This indicates that reagent 2a undergoes single-electron oxidation by the PC*. Fluorescence quenching by using a solution of lithium bromide suggests possible oxidation of bromide by the photocatalyst, which is in accordance with redox potentials [E(Br−/Br) = +0.80 V vs. SCE;74 for 8Cl4CzIPN, E(PC*/PC˙−) = +1.58 V vs. SCE63]. Measurement of the redox potential of reagent 2a in DMF solution by cyclic voltammetry gave a potential of +0.89 V vs. SCE, with the curve resembling that of bromide anions (see the ESI† for details). The quantum yield of a standard reaction (2c with 4-bromoacetophenone) was determined to be 0.023. Finally, to estimate the influence of various leaving groups of the aryl component on reaction efficiency, several experiments were carried out (Scheme 6E). Thus, chloro-, bromo-, and iodo-substituted acetophenones were compared, among which the bromine derivative gave the highest yield. While the low reactivity of aryl chloride was expected, the low yield and especially low conversion of aryl iodide was surprising. It can be tentatively proposed that iodide anions formed as the reaction progresses can exert an inhibiting effect. We noted that aryl triflate and mesylate fragments remained unaffected in the reaction of 4-tryflyloxy- and 4-mesyloxy-bromobenzenes (see Scheme 3, products 9 and 10). Correspondingly, we tested highly electron deficient triflate and mesylate derived from 4-cyanophenol. These substrates bearing the electron depleting group reacted slowly, and even after 16 hours only low yields were observed. This means that the reaction is selective for aromatic bromides, thereby providing opportunities for performing orthogonal transformations.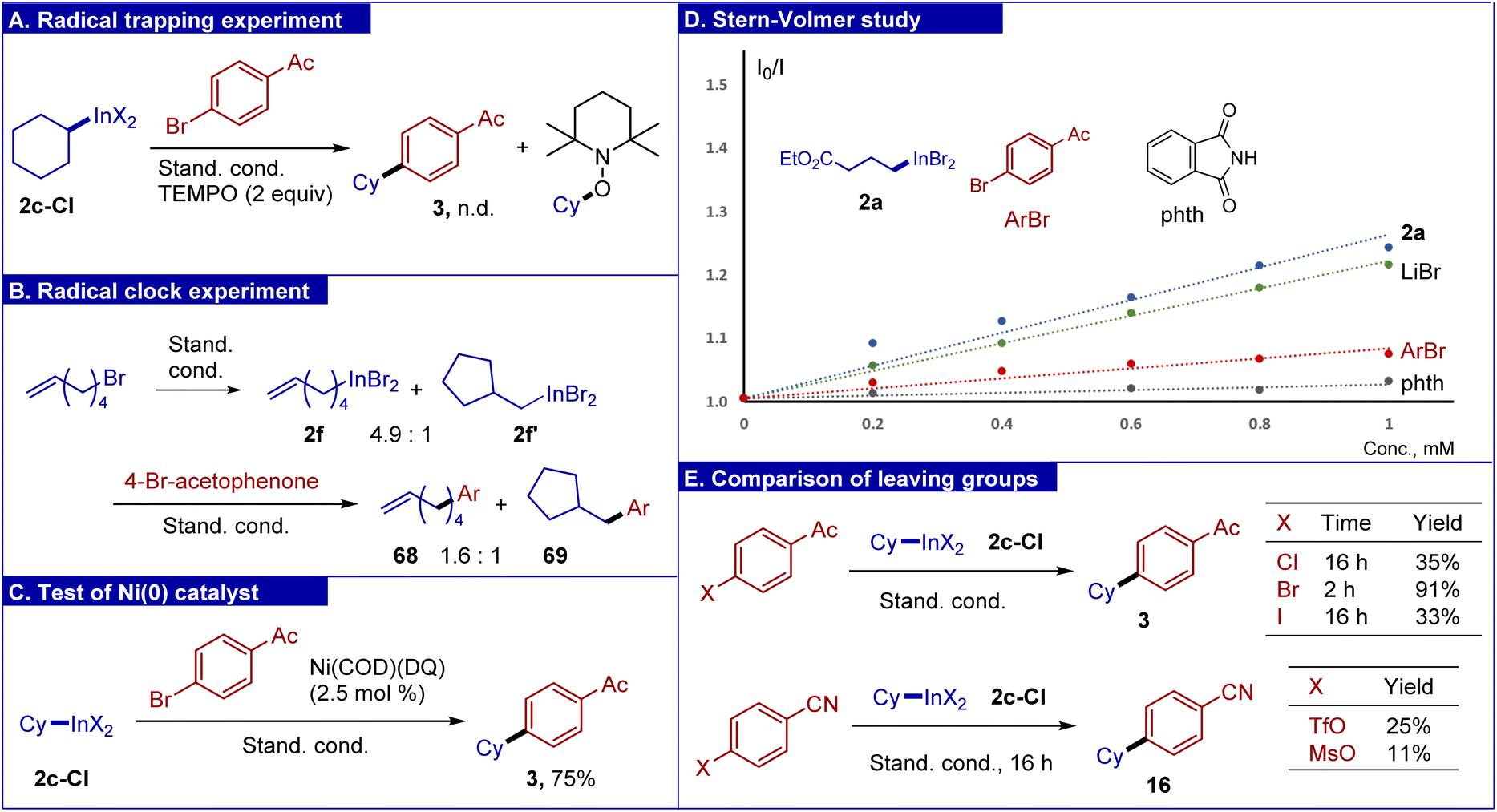 | ||
| Scheme 6 Mechanistic experiments. | ||
Based on the obtained experimental data, the following mechanism can be proposed (Scheme 7). Two types of photocatalytic cycles may be operative. Thus, the organoindium reagent is oxidized by a photoexcited catalyst to generate the alkyl radical (path A). Alternatively, bromide is oxidized to generate a bromine radical, which in turn attacks at the indium center of organoindium to liberate the alkyl radical (path B). For the nickel cycle, the reduced state of the photocatalyst generates Ni(0) with its oxidative addition into the C–Br bond to give the arylnickel(II) intermediate. Radical addition at the nickel and reductive elimination from Ni(III) completes the catalytic cycle.
 | ||
| Scheme 7 Proposed mechanism. | ||
Conclusions
The utilization of photoredox catalysis has enabled the exploration of novel reaction pathways for alkyl indium compounds, which serve as sources of alkyl radicals upon light promoted single electron oxidation. Organoindium reagents were incorporated into the nickel/photoredox catalyzed cross-coupling with aryl bromides, as well as in reactions with α-(trifluoromethyl)styrenes. The synthetic utility of these reactions was enhanced with the advent of a universal methodology for the synthesis of alkyl indium reagents. Both the preparation of organoindium reagents and the metallaphotoredox cross-coupling are insensitive to air and moisture, and tolerate a wide variety of sensitive functional groups.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Investigation: A. A. Gladkov and D. Y. Cheboksarov; conceptualization: A. A. Gladkov and V. V. Levin; data curation: A. A. Gladkov and D. Y. Cheboksarov; writing – review & editing: A. A. Gladkov, V. V. Levin and A. D. Dilman.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Russian Science Foundation (Project 23-13-00130).Notes and references
- Organometallics in Synthesis: Third Manual, ed. M. Schlosser, John Wiley & Sons, Hoboken, New Jersey, 2013 Search PubMed.
- Handbook of Functionalized Organometallics: Applications in Synthesis, ed. P. Knochel, WILEY-VCH, Weinheim, 2005 Search PubMed.
- K. Duan, X. Yan, Y. Liu and Z. Li, Adv. Synth. Catal., 2018, 360, 2781–2795 CrossRef CAS.
- P. Renaud, A. Beauseigneur, A. Brecht-Forster, B. Becattini, V. Darmency, S. Kandhasamy, F. Montermini, C. Ollivier, P. Panchaud, D. Pozzi, E. M. Scanlan, A.-P. Schaffner and V. Weber, Pure Appl. Chem., 2007, 79, 223–233 CrossRef CAS.
- T. Akindele, K.-i. Yamada and K. Tomioka, Acc. Chem. Res., 2009, 42, 345–355 CrossRef CAS.
- M. Kubisiak, K. Zelga, W. Bury, I. Justyniak, K. Budny-Godlewski, Z. Ochal and J. Lewiński, Chem. Sci., 2015, 6, 3102–3108 Search PubMed.
- A. A. Gladkov, G. N. Chernov, V. V. Levin, V. A. Kokorekin and A. D. Dilman, Org. Lett., 2021, 23, 9645–9648 Search PubMed.
- A. A. Gladkov, V. V. Levin and A. D. Dilman, J. Org. Chem., 2023, 88, 1260–1269 Search PubMed.
- A. A. Gladkov, V. V. Levin and A. D. Dilman, Adv. Synth. Catal., 2023, 365, 3387–3391 CrossRef CAS.
- A. A. Gladkov, V. V. Levin and A. D. Dilman, J. Org. Chem., 2024, 89, 11826–11835 CrossRef CAS.
- Z.-L. Shen, S.-Y. Wang, Y.-K. Chok, Y.-H. Xu and T.-P. Loh, Chem. Rev., 2013, 113, 271–401 CrossRef CAS.
- J. Burgess, Chem. Soc. Rev., 1996, 25, 85–92 RSC.
- V. I. Supranovich, V. V. Levin and A. D. Dilman, Org. Lett., 2024, 26, 4537–4541 CrossRef CAS PubMed.
- Y.-H. Chen and P. Knochel, Angew. Chem., Int. Ed., 2008, 47, 7648–7651 CrossRef CAS.
- V. Papoian and T. Minehan, J. Org. Chem., 2008, 73, 7376–7379 CrossRef CAS.
- L. Adak and N. Yoshikai, J. Org. Chem., 2011, 76, 7563–7568 CrossRef PubMed.
- A. A. Rafaniello, R. Kumar, R. C. Phillips and M. J. Gaunt, Angew. Chem., Int. Ed., 2024, 63, e202408287 Search PubMed.
- V. Nair, S. Ros, C. N. Jayan and B. S. Pillai, Tetrahedron, 2004, 60, 1959–1982 CrossRef.
- P. H. Lee and K. Lee, Angew. Chem., Int. Ed., 2005, 44, 3253–3256 CrossRef PubMed.
- S. A. Babu, M. Yasuda, I. Shibata and A. Baba, J. Org. Chem., 2005, 70, 10408–10419 CrossRef.
- S. A. Babu, M. Yasuda, I. Shibata and A. Baba, J. Org. Chem., 2005, 70, 10408–10419 CrossRef PubMed.
- H. C. Clark and A. L. Pickard, J. Organomet. Chem., 1967, 8, 427–434 CrossRef.
- I. Pérez, J. P. Sestelo and L. A. Sarandeses, J. Am. Chem. Soc., 2001, 123, 4155–4160 CrossRef PubMed.
- Z.-L. Shen, K. K. K. Goh, Y.-S. Yang, Y.-C. Lai, C. H. A. Wong, H.-L. Cheong and T.-P. Loh, Angew. Chem., Int. Ed., 2011, 50, 511–514 CrossRef PubMed.
- P. Wang, B.-Z. Chen, Y.-C. Guo, W. Rao and Z.-L. Shen, Tetrahedron Lett., 2019, 60, 151288 CrossRef.
- Y. Yu, Y. Feng, W. Ma, H. Li, M. Yang, G. Zhang and Y. Yang, ChemistrySelect, 2022, 7, e202202061 CrossRef.
- P. Wang, X.-D. Song, B.-Z. Chen, W. Rao and Z.-L. Shen, Catal. Commun., 2019, 132, 105824 CrossRef CAS.
- X.-X. Feng, Z. Wu, Q.-D. Wang, B.-Z. Chen, W. Rao, J.-M. Yang and Z.-L. Shen, Appl. Organomet. Chem., 2019, 33, e5110 CrossRef.
- B.-Z. Chen, C.-X. Wang, Z.-H. Jing, X.-Q. Chu, T.-P. Loh and Z.-L. Shen, Org. Chem. Front., 2019, 6, 313–318 RSC.
- M.-L. Zhi, B.-Z. Chen, W. Deng, X.-Q. Chu, T.-P. Loh and Z.-L. Shen, J. Org. Chem., 2019, 84, 3017–3023 CrossRef CAS.
- B.-Z. Chen, M.-L. Zhi, C.-X. Wang, X.-Q. Chu, Z.-L. Shen and T.-P. Loh, Org. Lett., 2018, 20, 1902–1905 CrossRef CAS PubMed.
- K. Zhao, L. Shen, Z.-L. Shen and T.-P. Loh, Chem. Soc. Rev., 2017, 46, 586–602 RSC.
- M. M. Martínez, J. Pérez Sestelo and L. A. Sarandeses, in Adv. Organomet. Chem., ed. P. J. Pérez, Academic Press, 2023, vol. 80, pp. 177–253 Search PubMed.
- C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef.
- A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Chem. Rev., 2022, 122, 1485–1542 CrossRef PubMed.
- V. M. Chernyshev and V. P. Ananikov, ACS Catal., 2022, 12, 1180–1200 CrossRef.
- J. C. Tellis, D. N. Primer and G. A. Molander, Science, 2014, 345, 433–436 CrossRef.
- J. C. Tellis, C. B. Kelly, D. N. Primer, M. Jouffroy, N. R. Patel and G. A. Molander, Acc. Chem. Res., 2016, 49, 1429–1439 CrossRef.
- J. K. Matsui, Á. Gutiérrez-Bonet, M. Rotella, R. Alam, O. Gutierrez and G. A. Molander, Angew. Chem., Int. Ed., 2018, 57, 15847–15851 CrossRef.
- M. W. Campbell, J. S. Compton, C. B. Kelly and G. A. Molander, J. Am. Chem. Soc., 2019, 141, 20069–20078 CrossRef PubMed.
- J. P. Phelan, S. B. Lang, J. Sim, S. Berritt, A. J. Peat, K. Billings, L. Fan and G. A. Molander, J. Am. Chem. Soc., 2019, 141, 3723–3732 CrossRef PubMed.
- T. Wan, L. Capaldo, J. Djossou, A. Staffa, F. J. de Zwart, B. de Bruin and T. Noël, Nat. Commun., 2024, 15, 4028 CrossRef PubMed.
- M. Oliva, S. Pillitteri, J. Schörgenhumer, R. Saito, E. V. Van der Eycken and U. K. Sharma, Chem. Sci., 2024, 15, 17490–17497 RSC.
- V. Corcé, L.-M. Chamoreau, E. Derat, J.-P. Goddard, C. Ollivier and L. Fensterbank, Angew. Chem., Int. Ed., 2015, 54, 11414–11418 CrossRef PubMed.
- M. Jouffroy, D. N. Primer and G. A. Molander, J. Am. Chem. Soc., 2016, 138, 475–478 CrossRef.
- C. Lévêque, L. Chenneberg, V. Corcé, J.-P. Goddard, C. Ollivier and L. Fensterbank, Org. Chem. Front., 2016, 3, 462–465 RSC.
- B. A. Vara, M. Jouffroy and G. A. Molander, Chem. Sci., 2017, 8, 530–535 RSC.
- Q.-H. Xu, L.-P. Wei and B. Xiao, Angew. Chem., Int. Ed., 2022, 61, e202115592 CrossRef.
- A. Selmani, M. D. Schoetz, A. E. Queen and F. Schoenebeck, ACS Catal., 2022, 12, 4833–4839 CrossRef.
- Y. Gao, C. Yang, S. Bai, X. Liu, Q. Wu, J. Wang, C. Jiang and X. Qi, Chem, 2020, 6, 675–688 Search PubMed.
- Z. Zhou, J. Yang, B. Yang, Y. Han, L. Zhu, X.-S. Xue and F. Zhu, Angew. Chem., Int. Ed., 2023, 62, e202314832 CrossRef.
- J. A. J. Pardoe and A. J. Downs, Chem. Rev., 2007, 107, 2–45 CrossRef.
- A. Fürstner, Angew. Chem., Int. Ed., 1993, 32, 164–189 CrossRef.
- L. A. Garza-Rodríguez, B. I. Kharisov and O. V. Kharissova, Synth. React. Inorg. Met.-Org. Chem., 2009, 39, 270–290 CrossRef.
- G. Biedermann and T. Wallin, Acta Chem. Scand., 1960, 14, 594–608 CrossRef.
- A. Krasovskiy, V. Malakhov, A. Gavryushin and P. Knochel, Angew. Chem., Int. Ed., 2006, 45, 6040–6044 CrossRef PubMed.
- C. Feng, D. W. Cunningham, Q. T. Easter and S. A. Blum, J. Am. Chem. Soc., 2016, 138, 11156–11159 CrossRef.
- Y.-H. Chen and P. Knochel, Angew. Chem., Int. Ed., 2008, 47, 7648–7651 CrossRef.
- Y.-H. Chen, M. Sun and P. Knochel, Angew. Chem., Int. Ed., 2009, 48, 2236–2239 CrossRef PubMed.
- V. Papoian and T. Minehan, J. Org. Chem., 2008, 73, 7376–7379 CrossRef.
- J. J. Habeeb, F. F. Said and D. G. Tuck, J. Organomet. Chem., 1980, 190, 325–334 CrossRef.
- L. G. Waterworth and I. J. Worrall, J. Organomet. Chem., 1974, 81, 23–26 CrossRef.
- J. S. Poland and D. G. Tuck, J. Organomet. Chem., 1972, 42, 315–323 CrossRef.
- L. Waterworth and I. J. Worrall, J. Chem. Soc. D, 1971, 569 RSC.
- C. Peppe, D. G. Tuck and L. Victoriano, J. Chem. Soc., Dalton Trans., 1982, 2165–2168 RSC.
- Y. Hou, J. Hu, R. Xu, S. Pan, X. Zeng and G. Zhong, Org. Lett., 2019, 21, 4428–4432 CrossRef.
- F. Le Vaillant, M. Garreau, S. Nicolai, G. Gryn'ova, C. Corminboeuf and J. Waser, Chem. Sci., 2018, 9, 5883–5889 RSC.
- C. N. Prieto Kullmer, J. A. Kautzky, S. W. Krska, T. Nowak, S. D. Dreher and D. W. C. MacMillan, Science, 2022, 376, 532–539 CrossRef.
- G. Laudadio, P. Neigenfind, Á. Péter, C. Z. Rubel, M. A. Emmanuel, M. S. Oderinde, T. E.-H. Ewing, M. D. Palkowitz, J. L. Sloane, K. W. Gillman, D. Ridge, M. D. Mandler, P. N. Bolduc, M. C. Nicastri, B. Zhang, S. Clementson, N. N. Petersen, P. Martín-Gago, P. Mykhailiuk, K. M. Engle and P. S. Baran, Angew. Chem., Int. Ed., 2024, 63, e202314617 CrossRef PubMed.
- D. N. Primer and G. A. Molander, J. Am. Chem. Soc., 2017, 139, 9847–9850 CrossRef.
- F. Tian, G. Yan and J. Yu, Chem. Commun., 2019, 55, 13486–13505 RSC.
- A. Luridiana, D. Mazzarella, L. Capaldo, J. A. Rincón, P. García-Losada, C. Mateos, M. O. Frederick, M. Nuño, W. Jan Buma and T. Noël, ACS Catal., 2022, 12, 11216–11225 CrossRef.
- V. T. Tran, Z.-Q. Li, O. Apolinar, J. Derosa, M. V. Joannou, S. R. Wisniewski, M. D. Eastgate and K. M. Engle, Angew. Chem., Int. Ed., 2020, 59, 7409–7413 CrossRef PubMed.
- S. K. Kariofillis, S. Jiang, A. M. Żurański, S. S. Gandhi, J. I. Martinez Alvarado and A. G. Doyle, J. Am. Chem. Soc., 2022, 144, 1045–1055 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Procedures, compound characterization, and NMR spectra. See DOI: https://doi.org/10.1039/d4sc08521c |
| This journal is © The Royal Society of Chemistry 2025 |