
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDrug binding disrupts chiral water structures in the DNA first hydration shell†
Ty
Santiago‡
 a,
Daniel
Konstantinovsky‡§
ab,
Matthew
Tremblay‡
ac,
Ethan A.
Perets¶
*a,
Sharon
Hammes-Schiffer
*ac and
Elsa C. Y.
Yan
*a
a,
Daniel
Konstantinovsky‡§
ab,
Matthew
Tremblay‡
ac,
Ethan A.
Perets¶
*a,
Sharon
Hammes-Schiffer
*ac and
Elsa C. Y.
Yan
*a
aDepartment of Chemistry, Yale University, New Haven, CT 06520, USA. E-mail: ethan.perets@utsouthwestern.edu; elsa.yan@yale.edu
bDepartment of Molecular Biophysics and Biochemistry, Yale University, New Haven, CT 06520, USA
cDepartment of Chemistry, Princeton University, Princeton, New Jersey 08544, USA. E-mail: shs566@princeton.edu
First published on 13th March 2025
Abstract
Knowledge of how intermolecular interactions change hydration structures surrounding DNA will heighten understanding of DNA biology and advance drug development. However, probing changes in DNA hydration structures in response to molecular interactions and drug binding in situ under ambient conditions has remained challenging. Here, we apply a combined experimental and computational approach of chiral-selective vibrational sum frequency generation spectroscopy (chiral SFG) to probe changes of DNA hydration structures when a small-molecule drug, netropsin, binds the minor groove of DNA. Our results show that chiral SFG can detect water being displaced from the minor groove of DNA due to netropsin binding. Additionally, we observe that chiral SFG distinguishes between weakly and strongly hydrogen-bonded water hydrating DNA. Chiral SFG spectra show that netropsin binding, instead of displacing weakly hydrogen-bonded water, preferentially displaces water molecules strongly hydrogen-bonded to thymine carbonyl groups in the DNA minor groove, revealing the roles of water in modulating site-specificity of netropsin binding to duplex DNA rich in adenine–thymine sequences. The results convey the promise of chiral SFG to offer mechanistic insights into roles of water in drug development targeting DNA.
Introduction
Water stabilizes DNA folding and mediates interactions of DNA with other molecules, such as proteins, small bioactive molecules, and therapeutics.1,2 Drugs targeting DNA molecules, such as chemotherapeutics3,4 and antibiotics,5 have different binding modes, including major groove binding,6 minor groove binding,7 and intercalation between base pairs.8 These binding modes differentially perturb water structures hydrating the DNA major groove, minor groove, and backbone.9 Rational drug design targeting DNA can benefit from knowledge about these distinct perturbations to DNA hydration structures. Although structural determination techniques, such as X-ray crystallography,10 NMR,11 and neutron scattering techniques,12 can provide information about hydration of DNA, they require growth of crystals, high concentration of samples, and/or low temperatures, posing challenges in studying hydration in wider experimental contexts. Optical methods, on the other hand, can overcome these barriers. For example, terahertz spectroscopy can probe collective motions of water surrounding DNA,13 Raman spectroscopy coupled with multivariate curve resolution analysis can probe hydration water,14,15 and two-dimensional infrared spectroscopy16 can use site-specific vibrational probes to observe hydration structures.17,18 Nonlinear spectroscopic techniques including sum frequency generation (SFG)19 and second harmonic generation20 have also been used to probe hydration in both inorganic21 and biological systems20,22 as well as biological function of biomolecules.23 Here, we show that chiral SFG can probe perturbations of DNA hydration structures due to DNA–molecular interactions, with selectivity to water molecules in the first hydration shell of DNA.Chiral SFG probes vibrational structures of chiral biomacromolecules,24–29 detecting chiral macroscopic arrangements of functional groups, such as amide,30–32 NH,30 and CH33 vibrational modes in protein secondary structures and CH29,34,35 and NH stretches in DNA duplexes.36 Chiral SFG requires a visible beam and an infrared beam to overlap to generate the second-order sum frequency signals (Scheme 1a).37,38 When the frequencies of the visible and SFG beams are not in resonance with an electronic transition, chiral SFG is surface specific.26,39,40 Chiral SFG can even detect achiral molecules arranged in chiral supramolecular structures, thus allowing probing of vibrational structures of water molecules in hydration shells of chiral biomacromolecules.41–43 The Petersen group showed that chiral SFG signals can detect OH stretches of water around DNA.44 However, the origin of the chiral water signals remained elusive. Our recent studies revealed that chiral SFG signals of water around proteins and DNA originate from water in the first hydration shell.25,36,41,43,45,46
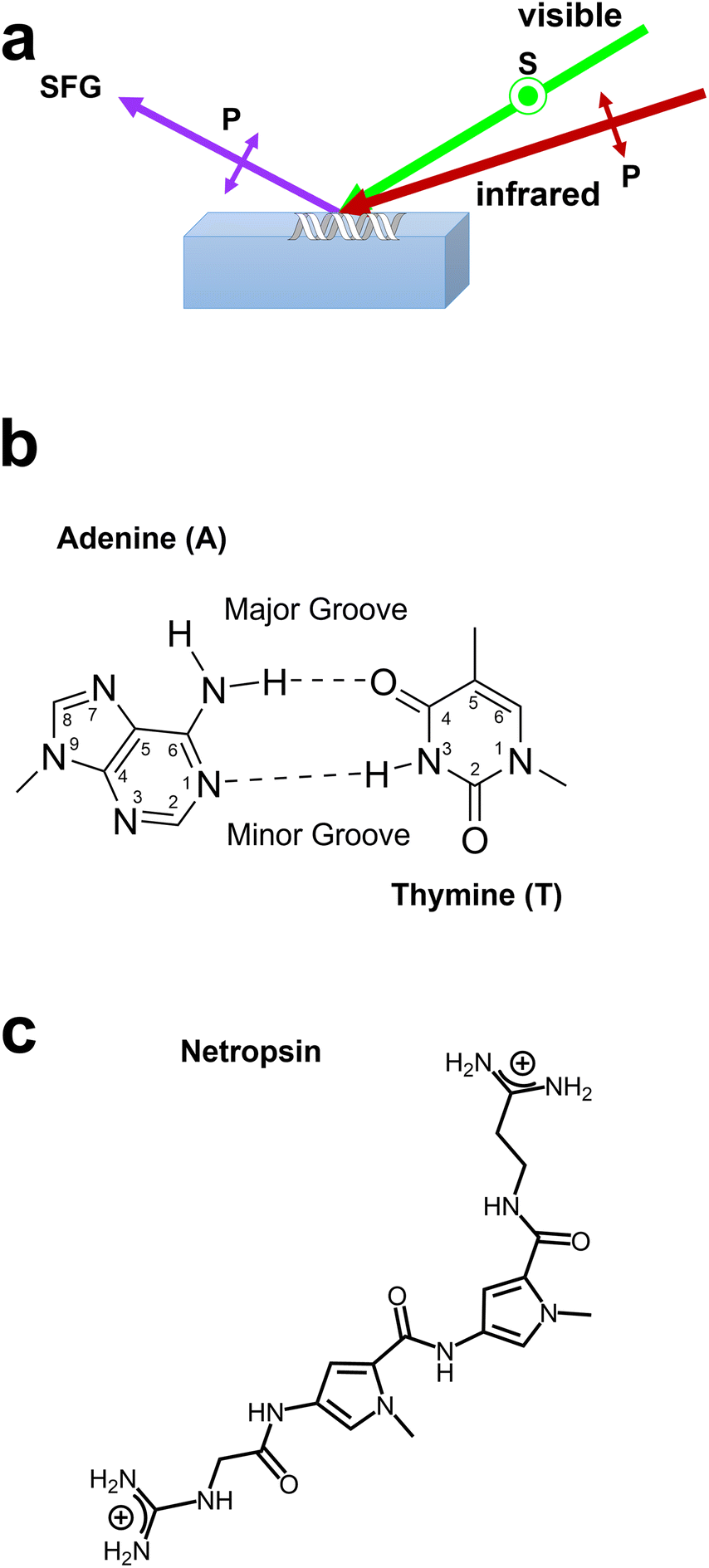 | ||
| Scheme 1 (a) Chiral SFG experimental setup for probing dsDNA using an s-polarized visible beam and a p-polarized infrared beam and detecting p-polarized SFG signals (see Methods in ESI†). (b) Adenine–thymine base pair showing minor groove and major groove chemical moieties. (c) Chemical structure of the minor groove-binding drug netropsin. | ||
In this study, we demonstrate that chiral SFG can probe biomolecular interactions by detecting changes in water structures in the first hydration shell of double-stranded DNA (dsDNA). We study (dA)12·(dT)12 dsDNA that binds an antibiotic and key drug design scaffold,47 netropsin, in the dsDNA minor groove. We obtained internal heterodyne chiral SFG spectra using dsDNA drop-casted on a quartz surface (Scheme 1a). We also performed molecular dynamics (MD) simulations on the dsDNA with its helical axis aligned with the z-axis of the simulation cell (see Methods in the ESI†) and subsequently analyzed the MD trajectories to simulate the chiral SFG response of water OH stretches using the water OH electrostatic map.48 Our experimental and simulated spectra correlate chiral SFG vibrational intensity with molecular population and demonstrate that an increasing molar ratio of netropsin to dsDNA leads to a decrease in the chiral SFG signal of water OH stretches. Our molecular modelling reveals that the decrease of the water signals originates from displacement of water molecules from the minor groove in the first hydration shell of dsDNA due to netropsin binding. Our findings demonstrate the promise of applying chiral SFG to probe binding modes of dsDNA, including minor-groove binding, major-groove binding, and intercalation. Insights from chiral SFG can potentially inform drug development and reveal molecular mechanisms of DNA structure–function correlations. Our work also illustrates that chiral SFG can monitor structural changes in the first hydration structures of folded chiral biopolymers in situ under ambient conditions, offering a unique perspective for investigating biological processes.
Results
Netropsin (Scheme 1c) binds to the (dA)12·(dT)12 dsDNA minor groove non-covalently and forms hydrogen (H) bonds with adenine N3 and thymine C2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (Scheme 1b).49–51 Netropsin requires a minimal binding site of four adenine–thymine base pairs.52 Thus, 2–3 netropsin molecules can bind to one (dA)12·(dT)12 dsDNA. We first build fully hydrated molecular models of the (dA)12·(dT)12 dsDNA bound to zero, one, and two netropsin molecules in the minor groove based on available crystal structures (see Methods in ESI†).53,54 Our previous study shows that chiral SFG is sensitive to water molecules only in the first hydration shell of the (dA)12·(dT)12 dsDNA.36Fig. 1 (right) illustrates the molecular models with all water molecules in the first hydration shell (1st row), as well as those water molecules hydrating the minor groove (2nd row), major groove (3rd row), and backbone (4th row). Fig. 1 (left) shows the corresponding simulated chiral SFG spectra of water molecules with the dsDNA binding to zero (black curve), one (red curve), and two netropsin molecules (blue curve).
O (Scheme 1b).49–51 Netropsin requires a minimal binding site of four adenine–thymine base pairs.52 Thus, 2–3 netropsin molecules can bind to one (dA)12·(dT)12 dsDNA. We first build fully hydrated molecular models of the (dA)12·(dT)12 dsDNA bound to zero, one, and two netropsin molecules in the minor groove based on available crystal structures (see Methods in ESI†).53,54 Our previous study shows that chiral SFG is sensitive to water molecules only in the first hydration shell of the (dA)12·(dT)12 dsDNA.36Fig. 1 (right) illustrates the molecular models with all water molecules in the first hydration shell (1st row), as well as those water molecules hydrating the minor groove (2nd row), major groove (3rd row), and backbone (4th row). Fig. 1 (left) shows the corresponding simulated chiral SFG spectra of water molecules with the dsDNA binding to zero (black curve), one (red curve), and two netropsin molecules (blue curve).
 | ||
| Fig. 1 Simulated chiral SFG response of water and molecular models of water hydrating the (dA)12·(dT)12 dsDNA bound to 0 (black), 1 (red), or 2 (blue) netropsin (NT) molecules. The simulated chiral SFG spectra (left) and MD models (right) including hydration water and the (dA)12·(dT)12 dsDNA complexed with 0, 1, or 2 netropsin molecules. (a) Water molecules in the first hydration shell and water molecules hydrating the (b) minor groove, (c) major groove, and (d) backbone. All spectra were generated by averaging over 106 frames from 100 ns of MD simulation. The simulated spectra contain chiral SFG responses of water but not the dsDNA. Spectral intensities are directly comparable and reported in arbitrary units (a.u.). See Methods in ESI† for details on selecting the first hydration shell waters and dividing them into the subsets. The spectra and structures for DNA + 0 NT were previously reported.36 | ||
The simulated chiral SFG response of water from the first hydration shell (Fig. 1a) strongly depends on the number of netropsin molecules bound to the dsDNA. Binding one netropsin reduces the signal intensities by roughly one third. Binding two netropsins almost abolishes the signals. To search for the origins of these spectral changes, we dissect these responses from the first hydration shell into the responses from water molecules hydrating the minor groove (Fig. 1b), the major groove (Fig. 1c), and the backbone (Fig. 1d). Fig. 1b shows that the signals from the minor groove reduce with the number of netropsin molecules. However, Fig. 1c and d show that the signals from the major groove and backbone do not change with netropsin binding. These results suggest that the changes in the overall chiral SFG signal of water in the first hydration shell (Fig. 1a) originate exclusively from the changes in hydration of the minor groove. These simulated results predict that netropsin binding displaces water molecules in the minor groove and thereby reduces the chiral SFG signals of water.
Guided by the computational results (Fig. 1), we obtained experimental phase-resolved chiral SFG spectra of the (dA)12·(dT)12 dsDNA at various molar ratios of netropsin to dsDNA, including 0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1:1, 1.5:1, or 2:1 (Fig. 2). Fig. 2a presents the spectra (purple) and their spectral fits (black) (see Methods for spectral fitting procedures in ESI†) with residuals of the fits (orange, top). Based on our previous studies of the (dA)12·(dT)12 dsDNA hydrated in H2O versus H218O,36 the global fitting includes three peaks at ∼3209 cm−1, ∼3347 cm−1, and ∼3400 cm−1 (Fig. 2b), which are assigned to NH stretches of the adenine NH2 (Fig. 2b, black curves). At the netropsin-to-DNA molar ratios of 1.5 and 2, the residual analysis suggests an additional peak at ∼3510 cm−1 (pink) to yield satisfactory fitting. We tentatively assign this peak to NH stretches of netropsin (Fig. 2b, pink curves), as guided by our DFT calculations (see Methods and data in the ESI†). We also identified in our previous studies of the (dA)12·(dT)12 dsDNA that chiral SFG signals of water contain two pairs of water OH stretches.36 In each pair, one peak corresponds to the symmetric stretch (solid blue curves) and the other peak corresponds to the asymmetric stretch (solid red curves) (see Methods in ESI†).36Fig. 2c shows that the first pair of water OH stretching peaks is centered at 3223 cm−1, and the second pair is centered at 3503 cm−1.
:1, 1:1, 1.5:1, or 2:1 (Fig. 2). Fig. 2a presents the spectra (purple) and their spectral fits (black) (see Methods for spectral fitting procedures in ESI†) with residuals of the fits (orange, top). Based on our previous studies of the (dA)12·(dT)12 dsDNA hydrated in H2O versus H218O,36 the global fitting includes three peaks at ∼3209 cm−1, ∼3347 cm−1, and ∼3400 cm−1 (Fig. 2b), which are assigned to NH stretches of the adenine NH2 (Fig. 2b, black curves). At the netropsin-to-DNA molar ratios of 1.5 and 2, the residual analysis suggests an additional peak at ∼3510 cm−1 (pink) to yield satisfactory fitting. We tentatively assign this peak to NH stretches of netropsin (Fig. 2b, pink curves), as guided by our DFT calculations (see Methods and data in the ESI†). We also identified in our previous studies of the (dA)12·(dT)12 dsDNA that chiral SFG signals of water contain two pairs of water OH stretches.36 In each pair, one peak corresponds to the symmetric stretch (solid blue curves) and the other peak corresponds to the asymmetric stretch (solid red curves) (see Methods in ESI†).36Fig. 2c shows that the first pair of water OH stretching peaks is centered at 3223 cm−1, and the second pair is centered at 3503 cm−1.
 | ||
| Fig. 2 Experimental data and fitting of chiral SFG spectra in the OH/NH stretching region at molar ratios of netropsin:dsDNA of 0:1, 1:1, 1.5:1, and 2:1. (a) Experimental spectra (purple) and fitting curves (black) with residuals of fitting (orange, top), (b) individual Lorentzian peaks used to fit the NH stretches from the DNA base pairs (black) and an emergent peak (pink) ascribed to NH stretches of netropsin with dotted lines marking the peak positions at 3209 cm−1, 3347 cm−1, 3400 cm−1, and 3510 cm−1, and (c) paired symmetric (blue) and asymmetric (red) peaks from water with the solid lines showing the individual peaks and dashed lines representing the total symmetric OH stretches (blue dashed line) and the total asymmetric OH stretches (red dashed line). The first pair of water peaks is centered at 3223 cm−1, and the second pair of water peaks is centered at 3503 cm−1 (a.u. = arbitrary units). | ||
Based on the fitting results, we plot the amplitude of each vibrational band as a function of the netropsin-to-DNA molar ratio (Fig. 3). The amplitude is directly correlated with the molecular population because each vibrational band shares the same peak widths and peak positions across the four spectra in our global fitting (Fig. 2). Fig. 3 shows that the amplitudes of both water OH bands (3503 cm−1 and 3223 cm−1) decrease with an increasing molar ratio (Fig. 3a). In contrast, the amplitude of the NH stretches of dsDNA does not change significantly with the molar ratio (Fig. 3b), which is consistent with the adenine NH2 moiety pointing toward the major groove and not being perturbed by netropsin binding to the minor groove. The peak at 3510 cm−1 (Fig. 3c) appears when the molar ratio reaches 1.5. This peak further increases at the molar ratio of 2 (Fig. 3c). We propose that this vibrational band is due to NH stretches of netropsin. X-ray crystal structures53,54 indicate that when netropsin binds to the DNA minor groove, it conforms to the helical structure of the minor groove. We propose that this induced chiral structure enables netropsin to generate chiral-selective SFG signals. This model is supported by the results of DFT calculations (Fig. S1a and b†) and Fourier-transformed infrared (FTIR) experiments (Fig. S1c†), as discussed in the Discussion section.
 | ||
| Fig. 3 Changes in the amplitude of vibrational bands in the chiral SFG spectra of (dA)12·(dT)12 dsDNA as a function of the molar ratio of netropsinto dsDNA: (a) the OH stretching peaks centered at 3223 cm−1 and 3503 cm−1, (b) the three peaks attributed to dsDNA NH stretches, and (c) the emergent peak at 3510 cm−1 ascribed to NH stretches of netropsin. Error bars represent the standard deviation of the spectral fit amplitude for the respective peaks (a.u. = arbitrary units). | ||
Our analyses of experimental spectra (Fig. 2 and 3) demonstrate that chiral SFG has the sensitivity for detecting changes in the hydration structures of the dsDNA due to netropsin binding. Due to the strong binding of netropsin to the AT-rich dsDNA (dissociation constant, KD ∼10−9 M),55 netropsin remains bound in the minor groove of dsDNA throughout the MD simulations and likely remains bound in the hydrated film of dsDNA on the quartz substrate in our experiments. Combined with our computational modelling (Fig. 1), we conclude that the experimental observations of changes in chiral SFG signals of water are mainly due to netropsin binding that displaces water from the minor groove of the (dA)12·(dT)12 dsDNA.
Discussion
In our simulations, binding one molecule of netropsin to dsDNA displaces on average ∼12 water molecules per frame from the dsDNA minor groove. This result agrees with previous reports combining volumetric, calorimetric, and UV-melting measurements to estimate that ∼10 water molecules are displaced from the (dA)12·(dT)12 dsDNA minor groove per netropsin bound.50 Binding a second molecule of netropsin displaces an additional ∼7 water molecules from the dsDNA minor groove. Each binding event diminishes the chiral SFG response of the (dA)12·(dT)12 “spine of hydration”36,44 by approximately one third (Fig. 1b), so that dehydration of the dsDNA minor groove by netropsin progressively reduces the chiral SFG response of minor groove water molecules (Fig. 1b). In stark contrast, the chiral SFG response of the major groove and phosphate backbone water subpopulations appear unaffected by netropsin binding (Fig. 1c and d). The predicted changes in chiral SFG signals due to displacement of water from the minor groove by netropsin agree with our experimental spectra of (dA)12·(dT)12 dsDNA at various netropsin:DNA molar ratios (Fig. 2). Analyses of these spectra reveal that chiral SFG signals of water decrease with increasing netropsin-to-DNA molar ratios (Fig. 3a).
Our residual analysis (Fig. 2, top) revealed that a peak at 3510 cm−1 appears at the netropsin-to-DNA molar ratio of 1.5 and continues to grow at the molar ratio of 2.0 (Fig. 3c). This peak is unlikely to be due to hydration water as it can be fit with a single narrow Lorentzian peak.45 The peak is also unlikely to be due to dsDNA because the DFT simulations in our previous report showed that dsDNA NH stretches of the A–T base pairs occur below 3450 cm−1.36 We propose that this peak originates from NH stretches of netropsin when bound to the (dA)12·(dT)12 dsDNA minor groove, where netropsin can adopt a chiral conformation. To examine this proposition, we obtained an experimental infrared spectrum and simulated the infrared and Raman vibrational spectra of netropsin by performing DFT calculations in both implicit water and gas phase (Fig. S1†). The simulated spectra of netropsin show vibrational resonances from 3000 cm−1 to 3600 cm−1 due to NH stretches of netropsin. The peaks at around 3550 cm−1 are due to amidine stretches in the end group of the netropsin, which also appear in the experimental infrared spectrum (Fig. S1c†). Netropsin by itself does not produce significant chiral SFG signal (Fig. S4†), so any signal that is generated when it complexes with DNA must arise from chiral induction. These results support our proposed molecular model that netropsin binds to the DNA minor groove and adopts a chiral structure that gives rise to chiral-selective SFG signals of NH stretches at 3510 cm−1 (pink, Fig. 2b).
Our experimental data reveals contributions from two pairs of OH water stretches centered at 3223 cm−1 and 3503 cm−1. We previously identified these two OH stretching bands by globally fitting the chiral SFG spectra of the (dA)12·(dT)12 dsDNA hydrated in H2O and H218O.36 However, our simulated chiral SFG spectra of water hydrating the dsDNA (Fig. 1a and S2†) only show a single pair of water OH stretching peaks centered at ∼3400 cm−1 without significant signals at ∼3200 cm−1. The experimentally observed OH stretches at the lower frequency of 3223 cm−1 indicate strong H-bonding interactions of water molecules. This strong H-bonding interaction is consistent with water molecules in the minor groove forming H bonds with the thymine C2O group and N3 of the adenine aromatic ring (Scheme 1b). In our previous studies of protein hydration,41,43 we found that the electrostatic map used in simulating the vibrational response of water under-represents the responses from strongly H-bonded water, such as those water molecules H-bonding to the carbonyl groups of protein backbones. This issue may arise because the map was originally trained using bulk water molecules that form comparatively weak H bonds.56,57 Hence, we hypothesize that our simulation of dsDNA hydration (Fig. 1) may underestimate the contributions from strongly H-bonded water in the minor groove of dsDNA.
To evaluate this hypothesis, we simulated chiral SFG spectra arising from only the subset of water molecules in the minor groove of dsDNA donating a H bond to the thymine C2O group (Fig. S3a,† gray curves). The lineshapes in these spectra indeed show contributions from two pairs of OH stretches centered at 3097 cm−1 and 3332 cm−1 (Fig. S3c†). The strongly H-bonded water molecules within this subset, corresponding to low vibrational frequencies, represent a minority within the general population of the first hydration shell. Using the current electrostatic map, the signals from these strongly H-bonded water molecules are masked by the signals from the other water molecules in the minor groove. This result aligns with our previous studies,45 where the electrostatic map used in the simulations of OH stretches appeared to underestimate the contributions from strongly H-bonded water. This underestimation is likely due to the choice of the training set used to construct the OH electrostatic map,48 which works well for simulating OH stretches of water in bulk-like environments. However, this training set has a relatively low representation of strongly H-bonded water molecules, which are more commonly found in the first hydration shell of folded biopolymers, such as DNA duplexes. Hence, expanding the training set to include water in strongly H-bonded environments in constructing the electrostatic map could potentially improve its applications to simulate vibrational responses of water in strong H-bonding environments, such as those water molecules hydrating the minor groove of dsDNA.
Intriguingly, Fig. 3a shows that the low-frequency water OH stretching band at 3223 cm−1 decreases to a larger extent than the high-frequency OH stretching band at 3503 cm−1. Because lower OH stretching frequencies correlate with stronger H-bonding interactions, this observation suggests that strongly H-bonded water molecules are more favored to be displaced by netropsin molecules. This finding aligns with our simulation results (Fig. S3†) and the crystal structures of netropsin-DNA complexes reported in the literature.53,54 Our simulations (Fig. S3a,† gray curves) show that the experimentally observed low-frequency OH stretching bands at 3223 cm−1 are likely due to water molecules that form strong H bonds with the thymine C2O group (Scheme 1b) pointing to the minor groove. Moreover, crystal structures show that netropsin binds to the minor groove of poly (dA·dT) dsDNA through forming H bonds between the netropsin's NH groups and the DNA's adenine N3 and thymine C2O carbonyls pointing toward the minor grooves (Scheme 1b).53,54 The carbonyls are relatively strong H-bond acceptors. Thus, in the absence of netropsin, these carbonyls form strong H bonds with water in the first hydration shell. When netropsin binds to the minor groove, these strongly H-bonded water molecules must be displaced. Therefore, our observed specific displacement of strongly H-bonded water is achieved by the specificity of netropsin binding to the AT-rich dsDNA. Thus, this structural information implies that the A–T sequence-selective binding of netropsin requires displacement of water molecules strongly H-bonded to the thymine C2O group, in agreement with our experimental observation of a larger decrease in the intensity of the OH stretches at lower frequency (3223 cm−1). This finding has implications for understanding the molecular mechanisms of site-specific and sequence-specific binding of small molecules to DNA duplexes. In the absence of small molecules, a DNA duplex is fully hydrated, forming strong H bonds with water in the first hydration shell. From a thermodynamic perspective, if the DNA duplex remains intact, removing strongly H-bonded water molecules will expose DNA's functional groups, creating sequence-specific sites for binding to small molecules.
Our finding has further established netropsin–dsDNA binding as a paradigm for the design of minor groove-binding drugs. Netropsin drives binding enthalpically by donating a high number of H bonds to adenine N3 and thymine C2O acceptors along the dsDNA minor groove, and entropically by displacing ordered water from the dsDNA minor groove.58,59 Crystallographic investigations of other minor groove binders have shown that many small molecules (e.g., furamidine,60 berenil,61 and DAPI62) and proteins63 can bind to the minor groove not only by displacing ordered water, but also by water-mediated H bonds to dsDNA. Our chiral SFG study could potentially be extended to investigate the structures of water-mediated H-bonding of small molecules and proteins to dsDNA.
The combination of experiments and computation offers fundamental insights into how changes in dsDNA hydration upon netropsin binding correlate with spectral perturbations. We provide the simplest interpretation based on our combined experimental and computational results, attributing the spectral perturbations to water displacement due to netropsin binding. Nonetheless, we cannot exclude the possibility that changes in electrostatic environments and DNA structures may also modulate the chiral SFG signals. Experimental chiral SFG spectra exhibit vibrational bands at various frequencies and bandwidths, which contain information about molecular structures and chemical environments. A direct comparison of simulated and experimental spectra offers quantitative assessments of the predictive power of computational approaches. Such assessments will help improve water models and develop force fields of various molecular systems for modelling important biological phenomena, such as lipid membranes interacting with biopolymers, molecular crowders impacting structures and dynamics of biopolymers,64 and denaturants (e.g., urea) destabilizing protein structures.65 Molecular orientations at interfaces can also modulate SFG spectral responses. The experimental and computational settings used herein do not probe the effect of interfacial orientation. Further development of experiments and computations that enable the analysis of the interfacial orientation effects will provide additional insights.
Our results enhance understanding of netropsin binding to adenine–thymine-rich DNA duplexes. Based on the water OH stretching frequencies, the chiral SFG spectra reveal that netropsin binding preferentially displaces strongly H-bonded water rather than weakly H-bonded water from the minor groove of adenine–thymine-rich DNA duplexes. This observation clearly demonstrates the important roles of water interactions in the first hydration shell in modulating the energetics of highly selective binding of drug molecules to DNA.
Conclusions
In this work, we demonstrate that chiral SFG can detect changes in dsDNA hydration structure resulting from minor groove binding of netropsin, a key small-molecule scaffold used in the development of drugs. We show both experimentally and computationally that increasing the concentration of netropsin depletes the chiral SFG response of water OH stretches. We also observe an emergence of a new vibrational signal that likely originates from induced chiral organization of the drug molecule. Finally, molecular dynamics simulations and experimental chiral SFG spectra show that multiple species of water exist within the minor groove of dsDNA. These results introduce chiral SFG as a chiral-selective optical method for probing specific regions and functional groups of DNA and their interactions with other molecules in situ. This capability of chiral SFG to probe the interaction of biopolymers and water in the first hydration shell provides fundamental insight into the role of water in determining structure, stability, and function of the biopolymers. Thus, chiral SFG holds promises for monitoring a wide range of biological processes, offering an alternative approach for probing biological processes in situ by detecting changes in water structures in the first hydration shell of biopolymers, including DNA, RNA, and proteins.Data availability
The data that support the findings of this study are included in the main text and the ESI.†Author contributions
T. S. data curation, formal analysis, funding acquisition, investigation, methodology, validation, visualization, writing – original draft, writing – review & editing; D. K. formal analysis, investigation, methodology, software, validation, visualization, writing – review & editing; M. T. data curation, formal analysis, investigation, methodology, validation, visualization, writing – review & editing; E. A. P. conceptualization, formal analysis, investigation, methodology, validation, visualization, writing – original draft, writing – review & editing; S. H.-S. funding acquisition, methodology, project administration, resources, supervision, writing – review & editing; E. C. Y. Y. conceptualization, funding acquisition, methodology, project administration, resources, supervision, writing – original draft, writing – review & editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the NIH (R35 GM139449 to S. H.-S.) and the NSF (CHE-1905169 to E. C. Y. Y.). D. K. was supported by these grants. M. T. was supported by NIH Grant No. R35 GM139449 (S. H.-S.). E. A. P. was supported by the NIH (5T32GM008283-31) and a John C. Tully Chemistry Research Fellowship. T. S. was supported by the NSF (CHE 1905169 to E. C. Y. Y.) and the NSF MPS-Ascend Postdoctoral Research Fellowship (CHE 2402247 to T. S.). Experimental setup was supported by the NSF (CHE 2108690 to E. C. Y. Y.).References
- J. Janin, Wet and dry interfaces: the role of solvent in protein–protein and protein–DNA recognition, Structure, 1999, 7, R277–R279 CrossRef CAS PubMed.
- G. Bischoff and S. Hoffmann, DNA-binding of drugs used in medicinal therapies, Curr. Med. Chem., 2002, 9, 321–348 CrossRef CAS PubMed.
- C. A. Frederick, L. D. Williams, G. Ughetto, G. A. Van der Marel, J. H. Van Boom, A. Rich and A. H. Wang, Structural comparison of anticancer drug-DNA complexes: adriamycin and daunomycin, Biochemistry, 1990, 29, 2538–2549 CrossRef CAS PubMed.
- K. Cheung-Ong, G. Giaever and C. Nislow, DNA-damaging agents in cancer chemotherapy: serendipity and chemical biology, Chem. Biol., 2013, 20, 648–659 CrossRef CAS PubMed.
- A. Finlay, F. Hochstein, B. Sobin and F. Murphy, Netropsin, a new antibiotic produced by a Streptomyces, J. Am. Chem. Soc., 1951, 73, 341–343 CrossRef CAS.
- P. L. Hamilton and D. P. Arya, Natural product DNA major groove binders, Nat. Prod. Rep., 2012, 29, 134–143 RSC.
- D. E. Wemmer and P. B. Dervan, Targeting the minor groove of DNA, Curr. Opin. Struct. Biol., 1997, 7, 355–361 CrossRef CAS PubMed.
- A. Rescifina, C. Zagni, M. G. Varrica, V. Pistarà and A. Corsaro, Recent advances in small organic molecules as DNA intercalating agents: Synthesis, activity, and modeling, Eur. J. Med. Chem., 2014, 74, 95–115 CrossRef CAS PubMed.
- T. V. Chalikian, G. E. Plum, A. P. Sarvazyan and K. J. Breslaver, Influence of Drug Binding on DNA Hydration: Acoustic and Densimetric Characterizations of Netropsin Binding to the Poly(dAdT)·Poly(dAdT) and Poly(dA)·Poly(dT) Duplexes and the Poly(dT)·Poly(dA)·Poly(dT) Triplex at 25 °C, Biochemistry, 1994, 33, 8629–8640 CrossRef CAS PubMed.
- M. Egli, V. Tereshko, M. Teplova, G. Minasov, A. Joachimiak, R. Sanishvili, C. M. Weeks, R. Miller, M. A. Maier and H. An, X-ray crystallographic analysis of the hydration of A-and B-form DNA at atomic resolution, Biopolymers, 1998, 48, 234–252 CrossRef CAS PubMed.
- D. R. Kearns and T. L. James, NMR studies of conformational states and dynamics of DNA, Crit. Rev. Biochem. Mol. Biol., 1984, 15, 237–290 CAS.
- U. Dahlborg and A. Rupprecht, Hydration of DNA: a neutron scattering study of oriented NaDNA, Biopolymers, 1971, 10, 849–863 CrossRef CAS PubMed.
- D. F. Plusquellic, K. Siegrist, E. J. Heilweil and O. Esenturk, Applications of terahertz spectroscopy in biosystems, ChemPhysChem, 2007, 8, 2412–2431 CrossRef CAS PubMed.
- J. G. Davis, B. M. Rankin, K. P. Gierszal and D. Ben-Amotz, On the cooperative formation of non-hydrogen-bonded water at molecular hydrophobic interfaces, Nat. Chem., 2013, 5, 796–802 CrossRef CAS PubMed.
- X. Lang, L. Shi, Z. Zhao and W. Min, Probing the structure of water in individual living cells, Nat. Commun., 2024, 15, 5271 CrossRef CAS PubMed.
- X.-X. Zhang, S. L. Brantley, S. A. Corcelli and A. Tokmakoff, DNA minor-groove binder Hoechst 33258 destabilizes base-pairing adjacent to its binding site, Commun. Biol., 2020, 3, 525 CrossRef CAS PubMed.
- T. Elsaesser, J. Schauss, A. Kundu and B. P. Fingerhut, Phosphate vibrations probe electric fields in hydrated biomolecules: spectroscopy, dynamics, and interactions, J. Phys. Chem. B, 2021, 125, 3899–3908 CrossRef CAS PubMed.
- M. Yang, Ł. Szyc and T. Elsaesser, Decelerated water dynamics and vibrational couplings of hydrated DNA mapped by two-dimensional infrared spectroscopy, J. Phys. Chem. B, 2011, 115, 13093–13100 CrossRef CAS PubMed.
- S. Nihonyanagi, T. Ishiyama, T.-k. Lee, S. Yamaguchi, M. Bonn, A. Morita and T. Tahara, Unified Molecular View of the Air/Water Interface Based on Experimental and Theoretical χ(2) Spectra of an Isotopically Diluted Water Surface, J. Am. Chem. Soc., 2011, 133, 16875–16880 CrossRef CAS PubMed.
- R. J. Tran, K. L. Sly and J. C. Conboy, Applications of surface second harmonic generation in biological sensing, Annu. Rev. Anal. Chem., 2017, 10, 387–414 CrossRef CAS PubMed.
- H. Wang, W. Chen, J. C. Wagner and W. Xiong, Local Ordering of Lattice Self-Assembled SDS@2β-CD Materials and Adsorbed Water Revealed by Vibrational Sum Frequency Generation Microscope, J. Phys. Chem. B, 2019, 123, 6212–6221 CrossRef CAS PubMed.
- X. Chen, W. Hua, Z. Huang and H. C. Allen, Interfacial Water Structure Associated with Phospholipid Membranes Studied by Phase-Sensitive Vibrational Sum Frequency Generation Spectroscopy, J. Am. Chem. Soc., 2010, 132, 11336–11342 CrossRef CAS PubMed.
- H. I. Okur, J. Hladílková, K. B. Rembert, Y. Cho, J. Heyda, J. Dzubiella, P. S. Cremer and P. Jungwirth, Beyond the Hofmeister Series: Ion-Specific Effects on Proteins and Their Biological Functions, J. Phys. Chem. B, 2017, 121, 1997–2014 CrossRef CAS PubMed.
- E. C. Y. Yan, Z. Wang and L. Fu, Proteins at interfaces probed by chiral vibrational sum frequency generation spectroscopy, J. Phys. Chem. B, 2015, 119, 2769–2785 CrossRef CAS PubMed.
- E. C. Y. Yan, E. A. Perets, D. Konstantinovsky and S. Hammes-Schiffer, Detecting Interplay of Chirality, Water, and Interfaces for Elucidating Biological Functions, Acc. Chem. Res., 2023, 22421–22426 Search PubMed.
- E. C. Y. Yan, L. Fu, Z. Wang and W. Liu, Biological macromolecules at interfaces probed by chiral vibrational sum frequency generation spectroscopy, Chem. Rev., 2014, 114, 8471–8498 CrossRef CAS PubMed.
- J. Wang, X. Chen, M. L. Clarke and Z. Chen, Detection of chiral sum frequency generation vibrational spectra of proteins and peptides at interfaces in situ, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 4978–4983 CrossRef CAS PubMed.
- S. Hosseinpour, S. J. Roeters, M. Bonn, W. Peukert, S. Woutersen and T. Weidner, Structure and dynamics of interfacial peptides and proteins from vibrational sum-frequency generation spectroscopy, Chem. Rev., 2020, 120, 3420–3465 CrossRef CAS PubMed.
- G. Y. Stokes, J. M. Gibbs-Davis, F. C. Boman, B. R. Stepp, A. G. Condie, S. T. Nguyen and F. M. Geiger, Making “sense” of DNA, J. Am. Chem. Soc., 2007, 129, 7492–7493 CrossRef CAS PubMed.
- L. Fu, J. Liu and E. C. Y. Yan, Chiral sum frequency generation spectroscopy for characterizing protein secondary structures at interfaces, J. Am. Chem. Soc., 2011, 133, 8094–8097 CrossRef CAS PubMed.
- L. Fu, G. Ma and E. C. Y. Yan, In situ misfolding of human islet amyloid polypeptide at interfaces probed by vibrational sum frequency generation, J. Am. Chem. Soc., 2010, 132, 5405–5412 CrossRef CAS PubMed.
- B. Zhang, J. Tan, C. Li, J. Zhang and S. Ye, Amide I SFG spectral line width probes the lipid–peptide and peptide–peptide interactions at cell membrane in situ and in real time, Langmuir, 2018, 34, 7554–7560 CrossRef CAS PubMed.
- E. A. Perets, P. E. Videla, E. C. Y. Yan and V. S. Batista, Chiral Inversion of Amino Acids in Antiparallel β-Sheets at Interfaces Probed by Vibrational Sum Frequency Generation Spectroscopy, J. Phys. Chem. B, 2019, 123, 5769–5781 CrossRef CAS PubMed.
- E. A. Perets, K. B. Olesen and E. C. Y. Yan, Chiral Sum Frequency Generation Spectroscopy Detects Double-Helix DNA at Interfaces, Langmuir, 2022, 38, 5765–5778 CrossRef CAS PubMed.
- S. R. Walter and F. M. Geiger, DNA on stage: showcasing oligonucleotides at surfaces and interfaces with second harmonic and vibrational sum frequency generation, J. Am. Chem. Soc., 2010, 1, 9–15 CAS.
- E. A. Perets, D. Konstantinovsky, T. Santiago, P. E. Videla, M. Tremblay, L. Velarde, V. S. Batista, S. Hammes-Schiffer and E. C. Y. Yan, Beyond the “spine of hydration”: Chiral SFG spectroscopy detects DNA first hydration shell and base pair structures, J. Chem. Phys., 2024, 161, 095104 CrossRef CAS PubMed.
- Y. R. Shen, Surface properties probed by second-harmonic and sum-frequency generation, Nature, 1989, 337, 519–525 CrossRef CAS.
- H.-F. Wang, Sum frequency generation vibrational spectroscopy (SFG-VS) for complex molecular surfaces and interfaces: Spectral lineshape measurement and analysis plus some controversial issues, Prog. Surf. Sci., 2016, 91, 155–182 CrossRef CAS.
- A. J. Moad and G. J. Simpson, A unified treatment of selection rules and symmetry relations for sum-frequency and second harmonic spectroscopies, J. Phys. Chem. B, 2004, 108, 3548–3562 CrossRef CAS.
- G. J. Simpson, Molecular origins of the remarkable chiral sensitivity of second-order nonlinear optics, ChemPhysChem, 2004, 5, 1301–1310 CrossRef CAS PubMed.
- E. A. Perets, D. Konstantinovsky, L. Fu, J. Chen, H.-F. Wang, S. Hammes-Schiffer and E. C. Y. Yan, Mirror-image antiparallel β-sheets organize water molecules into superstructures of opposite chirality, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 32902–32909 CrossRef CAS PubMed.
- E. A. Perets and E. C. Y. Yan, The H2O Helix: The Chiral Water Superstructure Surrounding DNA, ACS Cent. Sci., 2017, 3, 683–685 CrossRef CAS PubMed.
- D. Konstantinovsky, E. A. Perets, T. Santiago, L. Velarde, S. Hammes-Schiffer and E. C. Y. Yan, Detecting the first hydration shell structure around biomolecules at interfaces, ACS Cent. Sci., 2022, 8, 1404–1414 CrossRef CAS PubMed.
- M. L. McDermott, H. Vanselous, S. A. Corcelli and P. B. Petersen, DNA's chiral spine of hydration, ACS Cent. Sci., 2017, 3, 708–714 CrossRef CAS PubMed.
- D. Konstantinovsky, T. Santiago, M. Tremblay, G. J. Simpson, S. Hammes-Schiffer and E. C. Y. Yan, Theoretical basis for interpreting heterodyne chirality-selective sum frequency generation spectra of water, J. Chem. Phys., 2024, 160, 055102 CrossRef CAS PubMed.
- D. Konstantinovsky, E. C. Y. Yan and S. Hammes-Schiffer, Characterizing interfaces by Voronoi tessellation, J. Phys. Chem. Lett., 2023, 14, 5260–5266 CrossRef CAS PubMed.
- C. J. Suckling, I. S. Hunter and F. J. Scott, Multitargeted anti-infective drugs: resilience to resistance in the antimicrobial resistance era, Future Drug Discovery, 2022, 4, Fdd73 CrossRef PubMed.
- S. Corcelli, C. Lawrence and J. Skinner, Combined electronic structure/molecular dynamics approach for ultrafast infrared spectroscopy of dilute HOD in liquid H2O and D2O, J. Chem. Phys., 2004, 120, 8107–8117 CrossRef CAS PubMed.
- G. Burckhardt, H. Votavova, J. Sponar, G. Luck and C. Zimmer, Two binding modes of netropsin are involved in the complex formation with poly(dA-dT).poly(dA-dT) and other alternating DNA duplex polymers, J. Biomol. Struct. Dyn., 1985, 2, 721–736 CrossRef CAS PubMed.
- L. A. Marky and D. W. Kupke, Probing the hydration of the minor groove of A:T synthetic DNA polymers by volume and heat changes, Biochemistry, 1989, 28, 9982–9988 CrossRef CAS PubMed.
- N. N. Degtyareva, B. D. Wallace, A. R. Bryant, K. M. Loo and J. T. Petty, Hydration changes accompanying the binding of minor groove ligands with DNA, Biophys. J., 2007, 92, 959–965 CrossRef CAS PubMed.
- C. Zimmer, G. Luck and I. Fric, Duplex structure formation between oligo(dA)'s and oligo(dT)'s generated by thymine-specific interaction with netropsin, Nucleic Acids Res., 1976, 3, 1521–1532 CrossRef CAS PubMed.
- M. L. Kopka, C. Yoon, D. Goodsell, P. Pjura and R. E. Dickerson, Binding of an antitumor drug to DNA, Netropsin and C-G-C-G-A-A-T-T-BrC-G-C-G, J. Mol. Biol., 1985, 183, 553–563 CrossRef CAS PubMed.
- X. Chen, S. N. Mitra, S. T. Rao, K. Sekar and M. Sundaralingam, A novel end-to-end binding of two netropsins to the DNA decamers d(CCCCCIIIII)2, d(CCCBr5CCIIIII)2and d(CBr5CCCCIIIII)2, Nucleic Acids Res., 1998, 26, 5464–5471 CrossRef CAS PubMed.
- L. A. Marky and K. J. Breslauer, Origins of netropsin binding affinity and specificity: correlations of thermodynamic and structural data, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 4359–4363 CrossRef CAS PubMed.
- P. A. Pieniazek, C. J. Tainter and J. L. Skinner, Interpretation of the water surface vibrational sum-frequency spectrum, J. Chem. Phys., 2011, 135, 044701 CrossRef CAS PubMed.
- B. Auer and J. Skinner, Vibrational sum-frequency spectroscopy of the water liquid/vapor interface, J. Phys. Chem. B, 2009, 113, 4125–4130 CrossRef CAS PubMed.
- B. Nguyen, S. Neidle and W. D. Wilson, A role for water molecules in DNA-ligand minor groove recognition, Acc. Chem. Res., 2009, 42, 11–21 CrossRef CAS PubMed.
- S. Neidle, L. H. Pearl and J. V. Skelly, DNA structure and perturbation by drug binding, Biochem. J., 1987, 243, 1 CrossRef CAS PubMed.
- A. Guerri, I. J. Simpson and S. Neidle, Visualisation of extensive water ribbons and networks in a DNA minor-groove drug complex, Nucleic Acids Res., 1998, 26, 2873–2878 CrossRef CAS PubMed.
- D. Brown, M. Sanderson, J. Skelly, T. Jenkins, T. Brown, E. Garman, D. Stuart and S. Neidle, Crystal structure of a berenil-dodecanucleotide complex: the role of water in sequence-specific ligand binding, EMBO J., 1990, 9, 1329–1334 CrossRef CAS PubMed.
- T. A. Larsen, D. S. Goodsell, D. Cascio, K. Grzeskowiak and R. E. Dickerson, The structure of DAPI bound to DNA, J. Biomol. Struct. Dyn., 1989, 7, 477–491 CrossRef CAS PubMed.
- Z. Morávek, S. Neidle and B. Schneider, Protein and drug interactions in the minor groove of DNA, Nucleic Acids Res., 2002, 30, 1182–1191 CrossRef PubMed.
- R. Harada, Y. Sugita and M. Feig, Protein crowding affects hydration structure and dynamics, J. Am. Chem. Soc., 2012, 134, 4842–4849 CrossRef CAS PubMed.
- P. Ganguly, J. Polák, N. F. van der Vegt, J. Heyda and J.-E. Shea, Protein stability in TMAO and mixed urea–TMAO solutions, J. Phys. Chem. B, 2020, 124, 6181–6197 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental methods, spectral fitting parameters, computational methods, additional experimental and computational spectra (PDF). See DOI: https://doi.org/10.1039/d4sc08372e |
| ‡ Equal contribution. |
| § Current address: Department of Chemistry, Columbia University, New York, New York 10027, USA. |
| ¶ Current address: Department of Molecular Biology, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA. |
| This journal is © The Royal Society of Chemistry 2025 |