
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpontaneous conversion of pyridine N-oxide ylide covalent organic framework (COF) into biradical COF as an efficient catalyst in catalytic dehydrogenation of nitrogen heterocycles†
Wenying
Ai‡
 *ab,
Jiawei
Zou‡
a,
Zhenfeng
Cao
a,
Kun
Cui
c,
Jianbing
Gu
a,
Lina
Du
d,
Xin
Peng
d,
Mingli
Jiao
*a,
Tao
Shen
*c and
Lingbo
Qu
*b
*ab,
Jiawei
Zou‡
a,
Zhenfeng
Cao
a,
Kun
Cui
c,
Jianbing
Gu
a,
Lina
Du
d,
Xin
Peng
d,
Mingli
Jiao
*a,
Tao
Shen
*c and
Lingbo
Qu
*b
aSchool of Materials Electronics and Energy Storage, Zhongyuan University of Technology, Zhengzhou, Henan 450007, P. R. China. E-mail: 6786@zut.edu.cn; jml@zut.edu.cn
bCollege of Chemistry and Institute of Green Catalysis, Zhengzhou University, Zhengzhou, Henan Province 450001, People's Republic of China. E-mail: qulingbo@zzu.edu.cn
cFrontiers Science Center for Transformative Molecules, School of Chemistry and Chemical Engineering, Zhangjiang Institute for Advanced Study, Shanghai Jiao Tong University, Shanghai, 200240, P. R. China. E-mail: taoshen@sjtu.edu.cn
dHenan Key Laboratory of Nanocomposites and Applications, Institute of Nanostructured Functional Materials, Huanghe Science and Technology College, Zhengzhou 450006, China
First published on 2nd June 2025
Abstract
Over the past decade, covalent organic frameworks (COFs) have garnered significant attention as supporting materials for the immobilization of radical species, showing great promise in applications such as catalysis, energy storage, and dynamic nuclear polarization. While considerable progress has been made in developing monoradical COFs, the creation of COFs with embedded biradicals remains a substantial challenge. In this study, we present a novel pyridine N-oxide ylide COF, featuring an electron-withdrawing dicarboxamide group designed to facilitate the formation of a biradical COF (pyridine N-oxide biradical COF). This biradical generation occurs through a spontaneous intramolecular single-electron transfer process under ambient conditions. By integrating both electron-withdrawing and π-conjugated units into the pyridine ring, we enhance the stability and formation of biradical species. The electron paramagnetic resonance results demonstrate that the COF structure is pivotal in stabilizing and promoting biradical species formation. Further analyses, including Fourier-transform infrared, X-ray photoelectron, and 13C cross-polarization magic angle spinning nuclear magnetic resonance spectroscopies, confirm the coexistence of ylides and biradical species within the COF material. Additionally, the COF exhibits promising catalytic activity, serving as an efficient catalyst in the dehydrogenation of nitrogen heterocycles. This work bridges the gap between ylide COFs and biradical COFs, expanding our understanding of porous materials and their potential applications in advanced chemistry.
Introduction
The use of covalent organic frameworks (COFs) as supporting materials for immobilizing radical species has generated significant interest in recent years.1–5 As a novel class of crystalline porous polymers, COFs feature precisely controlled structures,6 periodic columnar π-arrays, and tightly packed stacking in the vertical direction,7–12 making them ideal candidates for stabilizing and immobilizing radical species.13,14 For instance, in 2018, Wang et al. developed a radical-containing COF2 and demonstrated its effectiveness as a polarizing agent in dynamic nuclear polarization (DNP) nuclear magnetic resonance (NMR) experiments, owing to the fixed orientation and homogeneous distribution of unpaired electrons. These radical-modified COFs have exhibited remarkable versatility, recyclability, and efficiency in various applications. However, despite significant progress in developing monoradical COFs, the creation of biradical-containing COFs remains an unsolved challenge due to the inherent instability of biradicals.15 Their short lifespans and tendency to form redox-limited dimers by self-combination or reaction with other active species have hindered their direct application as stable chemical reagents.Pyridine N-oxides and their derivatives are readily available and versatile reagents that play vital roles in synthetic chemistry.16 Single-electron transfer chemistry involving pyridine N-oxides, via pyridine N-oxy radical species, is an emerging field with significant potential to explore new chemical spaces and address key synthetic challenges.17,18 As reactive intermediates, pyridine N-oxy radicals possess unpaired electrons, which make them highly reactive. Conversion methods for pyridine N-oxides to pyridine N-oxy radicals typically fall into two categories: (1) intermolecular single-electron oxidation using chemical oxidants, electrochemical anodes, or photocatalysts;19–22 (2) photo-initiated intramolecular single-electron transfer (Scheme 1a).23,24 The former method is triggered by an intermolecular single-electron transfer process between pyridine N-oxides and oxidants, electrochemical anodes, or photocatalysts, often resulting in the formation of monoradical species.21,25 The latter relies on direct photoexcitation to induce intramolecular single-electron transfer, leading to the generation of biradical species.23 In the irradiation-induced pyridine N-oxide ylide intramolecular single-electron transfer process, numerous studies have shown that introducing electron-withdrawing functional groups or extended π-conjugation moieties on the pyridinium unit enhances the stability of the biradical species.26–28 Furthermore, in 2020, Zhang et al. observed that the deprotonation of N-alkyl-substituted pyridinium salts exhibited radical characteristics and that the introduction of electron-withdrawing groups or extended π-conjugation moieties increased the EPR signal.29 They suggested that the pyridinium moiety could offer a π* orbital “through space” to support single-electron transfer from the carbon atom to the pyridinium unit and that electron-withdrawing groups or extended π conjugation reduced the energy of the π* orbital, thus enhancing the EPR signal.
 | ||
| Scheme 1 (a) Developed methods for the single-electron transfer of pyridine N-oxides. (b) This work. | ||
Inspired by these studies, we aimed to fully exploit the unique chemical structure of COFs and incorporate electron-withdrawing functional groups and/or π-conjugation moieties into the pyridinium unit of pyridine N-oxide ylides embedded within COFs. This strategy reduces the energy of the pyridinium π* orbital sufficiently to induce the spontaneous intramolecular single-electron transfer of singlet-state pyridine N-oxide ylides, resulting in the formation of stable triplet-state pyridine N-oxy biradicals embedded in COFs (Scheme 1b). The use of COFs as supporting materials for pyridine N-oxy biradicals offers three distinct advantages: (1) the tunable structural properties of COFs facilitate the easy introduction of electron-withdrawing functional groups, which may reduce the energy of the pyridinium π* orbital and promote the formation of triplet-state pyridine N-oxy radical species. (2) The unique periodic columnar π arrays in the vertical direction and tightly packed stacking properties of COFs provide ideal pathways for efficient charge migration. (3) The rigid framework of COFs helps fix the orientation of the embedded radicals, ensuring their uniform distribution and enhancing the stability of these radical species.
In this study, we constructed a 2,6-dicarboxamide-substituted pyridine N-oxide ylide-containing COF to serve as a spontaneous transforming agent for the generation of pyridine N-oxy biradical COFs (Scheme 1b). The Fourier transform infrared (FT-IR), X-ray photoelectron spectroscopy (XPS), and electron paramagnetic resonance (EPR) spectra of this pyridine N-oxide ylide COF indicated that the pyridine N-oxy biradical COF species coexisted with the pyridine N-oxide ylide COF in a resonance form. Additionally, the EPR spectra of the pyridine N-oxide molecules and 2,6-dicarboxamide-substituted pyridine N-oxide ylide COF suggested that the covalent organic framework played a crucial role in stabilizing and facilitating the generation of biradical species.
This hybrid ylide/biradical COF also proves to be an excellent catalyst for the catalytic dehydrogenation of nitrogen heterocycles. The dehydrogenation of nitrogen heterocycles is a straightforward, simple, and atom-economic method to access the important synthons, aromatic N-heteroarenes.30,31 Traditional approaches for the dehydrogenation of nitrogen heterocycles usually involve radical reagents30,32–34 (e.g. TEMPO) or harsh oxidants33,35 (DDQ, O2 balloon, etc.) to capture H atoms from substrates. However, the difficulty of regenerating and separating these homogeneous radical reagents or oxidants limit their potential application in industry. The development of the pyridine N-oxy biradical COF in this work provides a recyclable radical reagent, and its ordered arrangement of radical active sites can improve hydrogen capture efficiency, avoiding the use of harsh oxidants in the dehydrogenation of nitrogen heterocycles. Herein, various tetrahydroquinolines and indolines were evaluated as substrates, and the desired products were obtained in moderate to high yields over a broad scope. This research is of great significance for expanding the application of COF materials in the field of green catalysis.
Results and discussion
To verify our hypothesis, we designed and synthesized a 2,6-dicarboxamide-substituted pyridine N-oxide COF material. A straightforward post-synthetic modification method was employed to generate the 2,6-dicarboxamide-substituted pyridine N-oxide COF through a one-pot oxidation of pyridine and imine units in a bis(imino)pyridine-linked COF (Bipy-COF). This approach highlights the structural advantages of COF materials, such as the ease of introducing amide electron-withdrawing substituents, extending π-electron delocalization, and ensuring robust stability. The resulting 2,6-dicarboxamide-substituted pyridine N-oxide COF (DICPO-COF) exhibits efficient electron transfer ability and can spontaneously transform into a 2,6-dicarboxamide-substituted pyridine N-oxy biradical COF through an intramolecular single-electron transfer process under natural conditions, without the need for additional oxidants, photocatalysts, electrochemical anodes, or light. FT-IR, XPS, and EPR spectra of the pyridine N-oxide ylide COF confirm that the pyridine N-oxy biradical COF species coexist with the pyridine N-oxide ylide COF in resonance form. Furthermore, this ylide/radical COF acts as an efficient catalyst for the catalytic dehydrogenation of nitrogen heterocycles, with various tetrahydroquinoline and indoline substrates yielding the desired products in moderate to high yields.In the synthesis of Bipy-COF, 1,3,5-tris(4-aminophenyl)benzene was reacted with 2,6-pyridine dialdehyde in a mixture of n-BuOH/o-DCB solvents along with a 6.0 M AcOH solution at 120 °C for 3 days (Fig. 1a). To demonstrate the oxidation of both pyridine and imine units in Bipy-COF to pyridine N-oxide and amide, respectively, we conducted a model reaction using (pyridine-2,6-diyl)bis(N-phenylmethanimine) as the starting material, diethyl ether (Et2O), and 3 equiv. m-chloroperbenzoic acid (m-CPBA) as the oxidant. The product, 2-formyl-6-(phenylcarbamoyl)pyridine 1-oxide, was obtained in 62% yield after 4 h at 0 °C. The chemical structure of this N+–O− ylide product was confirmed using high-resolution exact mass spectrometry (HRMS), FT-IR, and NMR (Fig. S1, S2, and S3†). Although one of the imine bonds in (pyridine-2,6-diyl)bis(N-phenylmethanimine) underwent hydrolysis during this oxidation due to the instability of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond in acidic conditions, we speculated that the likelihood of imine hydrolysis in the COFs may be reduced due to the stability of the COF framework. Encouraged by this model reaction, an m-CPBA-induced oxidation strategy was employed to convert Bipy-COF into the dicarbamoylpyridine-N-oxide-linked DICPO-COF, which was obtained as a dark brown powder in high yield under the same reaction conditions. The decrease in COF crystallinity after oxidation is related to partial hydrolysis of reaction sites.
N bond in acidic conditions, we speculated that the likelihood of imine hydrolysis in the COFs may be reduced due to the stability of the COF framework. Encouraged by this model reaction, an m-CPBA-induced oxidation strategy was employed to convert Bipy-COF into the dicarbamoylpyridine-N-oxide-linked DICPO-COF, which was obtained as a dark brown powder in high yield under the same reaction conditions. The decrease in COF crystallinity after oxidation is related to partial hydrolysis of reaction sites.
 | ||
| Fig. 1 (a) Preparation routes of Bipy-COF and DICPO-COF; (b) FT-IR spectra of Bipy-COF (grey line) and DICPO-COF (red line); high-resolution O 1s (c), N 1s (d), and C 1s (e) XPS spectra of DICPO-COF; (f) 13C CP/MAS NMR spectrum of Bipy-COF and DICPO-COF; (g) Pawley refinements of Bipy-COF (red line) and DICPO-COF (pink line) against their experimental PXRD patterns (black line and grey line). | ||
The structural changes in Bipy-COF were analyzed using FT-IR spectroscopy. In the FT-IR spectrum of DICPO-COF, the characteristic CN stretching band at 1664 cm−1 was significantly reduced compared to that of Bipy-COF, indicating the consumption of the starting material. Additionally, new stretching bands at 1638 cm−1 (CO) and 1560 cm−1 (N–H of amide) were observed, confirming the conversion of the imine group to an amide after oxidation.36 Moreover, a new band at 1238 cm−1, attributed to the N+–O− stretching vibration, appeared in the spectrum of DICPO-COF, providing further evidence of the oxidation (Fig. 1b).37
X-ray photoelectron spectroscopy (XPS) analysis further supported the oxidation mechanism underlying the transformation from Bipy-COF to DICPO-COF. In the unoxidized Bipy-COF, the high-resolution C 1s XPS spectrum showed two main peaks at 284.8 (ref. 38) and 286.1 eV,39 corresponding to CC and CN bonds, respectively. The N 1s XPS spectrum displayed two peaks representing pyridinic CN (398.9 eV)40 and imine CN (399.6 eV)38 bonds (Fig. 1c). After oxidation with m-CPBA, significant changes were observed in both the C 1s and N 1s XPS spectra. The C 1s XPS spectrum of DICPO-COF revealed four peaks at 284.8 eV (CC),38 288.2 eV (CO), 285.8 eV (C–N),41 and CN+–O−.37 The shift of the CN+ peak41 to a higher binding energy (288.6 eV) indicates reduced electron density around the nitrogen atoms (Fig. 1d). The N 1s XPS spectrum of DICPO-COF revealed three distinct nitrogen species: quaternary ammonium nitrogen (N+–O−) at 402.6 eV,42 amide nitrogen at 400.4 eV, and pyridinic nitrogen (CN) at 399.2 eV.41 The O 1s XPS spectrum confirmed the oxidation state of DICPO-COF, showing three types of oxygen species: CO (531.2 eV), N+–O− (531.7 eV),37 and adsorbed water or chemisorbed O2/CO2 (534.7 eV) (Fig. 1e).43
The conversion of DICPO-COF from imine to amide linkages was also validated by 13C solid-state NMR spectroscopy (13C cross-polarization (CP) magic angle spinning (MAS) NMR). In Bipy-COF, the characteristic C signal of the CN group appeared at ca. 152 ppm. After oxidation,44 the imine C signal disappeared, and a new signal appeared at ca. 160 ppm in the 13C CP/MAS NMR spectrum of DICPO-COF, corresponding to the carbon atoms in the newly formed amide bond (Fig. 1f). Additionally, the chemical shift of the pyridine-C atom shifted from ca. 153 to 138 ppm, further supporting the formation of the N+–O− ylide. Comparing the 13C CP/MAS NMR spectrum of DICPO-COF and the 13C NMR spectrum of the model compound 2-formyl-6-(phenylcarbamoyl)pyridine 1-oxide, the chemical shift of the C atom connected with the amide group in the pyridine of the model compound is 137.32 ppm, while it shifts to 138 ppm in DICPO-COF. This slight downfield shift (Δδ = +0.68 ppm) suggested the presence of more pronounced delocalization phenomenon in the pyridine ring of DICPO-COF than in the small molecule model. Such a delocalization effect leads to the reduction of the electron density on the pyridine ring, thereby causing the corresponding carbon atom shift in the 13C CP/MAS NMR spectrum.
The long-range order of DICPO-COF was confirmed by powder X-ray diffraction (PXRD) analysis. The PXRD pattern retained most of the crystallinity and atomic-scale chemical structure of DICPO-COF. The dominant peak at 5.05° and two minor peaks at 9.03° and 18.91° correspond to reflections from the (110), (300), and (011) planes, respectively, supporting the AA stacking model (Fig. 1g). The experimental PXRD data showed excellent agreement with the simulated diffraction pattern, with profile (Rp = 2.32%) and weighted profile (Rwp = 2.92%) values falling within acceptable limits.
Scanning electron microscopy (SEM) images confirmed that the oxidation of Bipy-COF did not significantly alter its morphology (Fig. 2a and b).36 The optical properties of DICPO-COF were investigated using solid-state UV-vis spectroscopy. Compared to that of Bipy-COF, the UV-vis spectrum of DICPO-COF exhibited a slight red shift, which can be attributed to the formation of amide bonds and N+–O− ylide units (Fig. 2c). DICPO-COF also displayed a characteristic absorption band around 400–600 nm, which is likely associated with the formation of biradicals.26 The optical band gap of Bipy-COF was calculated to be 2.60 eV, while that of DICPO-COF was 2.22 eV, suggesting an enhanced charge transfer capability (Fig. S19a†). The valence band position of DICPO-COF was determined to be at 1.65 eV through XPS measurements (Fig. S19b†).
 | ||
| Fig. 2 SEM images of Bipy-COF (a) and DICPO-COF (b); (c) solid-state UV-vis spectra of Bipy-COF (black) and DICPO-COF (red); (d) thermogravimetric curves of Bipy-COF (black) and DICPO-COF (red). | ||
The thermal stability of DICPO-COF was assessed by thermogravimetric analysis (TGA). The TGA curves indicated that Bipy-COF and DICPO-COF were stable up to 468 and 449 °C, respectively, under a nitrogen atmosphere (Fig. 2d), making them suitable for catalytic applications.
The permanent porosity of Bipy-COF and DICPO-COF was analyzed through nitrogen adsorption–desorption isotherms. The Brunauer–Emmett–Teller (BET) surface area of Bipy-COF was calculated to be 583 m2 g−1, whereas the surface area of DICPO-COF decreased to 351 m2 g−1 after oxidation, likely due to reduced crystallinity (Fig. 3a and b). The total pore volume of Bipy-COF was 0.536 cm3 g−1, while that of DICPO-COF was 0.485 cm3 g−1. According to the simulation results (Fig. S21†), the pore sizes of the DICPO-COF material at three distinct locations were measured as 36, 29, and 16 Å, whereas the experimentally determined pore size was 17 Å (Fig. S22†). To determine the pore size correctly, argon adsorption isotherm analysis was conducted (Fig. S23†), and revealing a predominant pore size distribution peak at 36 Å, which closely matches the largest simulated pore size distribution. This consistency between experimental and simulated results confirms that DICPO-COF possesses a highly ordered mesoporous structure with a pore size of 36 Å, thereby determining its precise pore size distribution.
 | ||
| Fig. 3 Nitrogen adsorption–desorption isotherms of Bipy-COF (a) and DICPO-COF (b). | ||
EPR spectroscopy was used to detect radical signals under four different conditions: (1) the sample was stored in a dark, anhydrous, and oxygen-free environment for 20 d and tested under the same conditions; (2) the sample was stored in an anhydrous, oxygen-free environment and exposed to natural light for 2 d and then tested; (3) the sample was stored in the dark and exposed to air for 2 d and then tested; and (4) the sample was tested under air and natural light. As shown in Fig. 4a, although the DICPO-COF material was stored in a dark, anhydrous, and oxygen-free environment for 20 d, a clear EPR signal was observed. This observation indicates that the 2,6-dicarboxamide-substituted pyridine N-oxide COF can spontaneously transform into a 2,6-dicarboxamide-substituted pyridine N-oxy biradical COF under natural conditions. Furthermore, the presence of natural light or air led to a significant enhancement in the EPR signal (Fig. 4b–d), confirming that the pyridine N-oxy biradical COF species and pyridine N-oxide ylide COF coexist in the resonance form. Based on the literature,26,45,46 the EPR signals of peak a and a′ in Fig. 4d belong to the pyridine N radical, and peak b and b′ belong to the O singlet in the biradical and monoradical, respectively. In addition, the linewidth of the O EPR signal generated in the dark without air is wider than the linewidth of the O EPR signal with air and natural light in Fig. 4d, which suggested that the structure of DICPO-COF in the dark without air mainly adopts the biradical form, and it could be oxidized to the monoradical species with air. Therefore, we could observe the linewidth changing under different conditions.
 | ||
| Fig. 4 EPR spectra of DICPO-COF in the dark and without air (a) in the dark and with air, (b) under natural light and without air (c), and under natural light with air (d). | ||
Notably, the g value of DICPO-COF was 2.0046, which is slightly lower than that of typical pyridine N-oxy biradical species,26,47 indicating that the radicals in DICPO-COF are not solely localized on the O atom but are also partially delocalized across the conjugated framework through resonance (Scheme 2a). Moreover, the EPR spectrum of DICPO-COF was significantly different from that of single free radicals, such as, DPPH radical,26 NDI,48 and MOV.49 These results show that the presence of the electron-withdrawing dicarboxamide group in DICPO-COF and the extended π conjugation on the pyridine ring could induce the generation of pyridine N-oxy biradical species with triplet ground states and enhance the stability of this biradical (Scheme 2b). EPR spectroscopy of pyridine-N-oxide, 2-formyl-6-(phenylcarbamoyl)pyridine 1-oxide, and DICPO-COF was performed to verify the role of the covalent organic framework in stabilizing and facilitating the preparation of biradical species. As shown in Fig. 5, the pyridine-N-oxide molecule showed no EPR signal, whereas 2-formyl-6-(phenylcarbamoyl)pyridine 1-oxide showed a weak EPR signal, indicating that the introduction of electron-withdrawing groups, such as amide and aldehyde groups, partly stabilized and facilitated the formation of biradical species. In contrast, DICPO-COF exhibited a strong EPR signal, suggesting that the chemical structure of DICPO-COF plays a crucial role in stabilizing and promoting the formation of biradical species. Therefore, the unique structural characteristics of COF materials endow biradical species with sufficient stability under natural conditions, which has not been achieved in small-molecule systems. Moreover, the EPR spectrum of Bipy-COF was also analysed (Fig. S26†), and it displayed a weak single peak at g = 2.0037. This peak could belong to the unpaired electrons associated with oxygen vacancies (OVs), and its g-value lies within the established range for oxygen vacancy radicals (O−·, g = 2.001–2.005).50 This result suggested that the EPR signal of DICPO-COF is not caused by the vacancies of the COF, but biradical species.
 | ||
| Scheme 2 (a) Schematic illustration of the single-electron transfer (SET) pathway of pyridine N-oxide ylide covalent organic framework (COF); (b) proposed electronic configurations of 2,6-dicarboxamide-substituted pyridine N-oxide COF (DICPO-COF) are displayed in brackets as two resonance structures. | ||
 | ||
| Fig. 5 EPR spectra of DICPO-COF, pyridine-N-oxide and small molecule model (2-formyl-6-(phenylcarbamoyl)pyridine 1-oxide). | ||
To further investigate the detailed distribution of each kind of radical species, curve-fitting and integration analysis of EPR spectra in different conditions was performed. Due to the limitations of the EPR instrument, we performed EPR curve-fitting and integration analysis using Origin software and came to the following conclusions. (1) In the dark without air (Fig. S27a†), the ca. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 N:O radical peak area ratio confirms the COF material exists in the form of a biradical with no external conditions. (2) Under natural light and in air (Fig. S27b†), significantly higher O-radical integrals than N-radical values were observed. Calculations revealed that the monoradical increment (O–N integral values) originated from photo/oxygen-synergistically generated O-radical species, indicating the coexistence of biradical/monoradical states in the system (the ratio of biradical to monoradical is about 1.2:1).
:1 N:O radical peak area ratio confirms the COF material exists in the form of a biradical with no external conditions. (2) Under natural light and in air (Fig. S27b†), significantly higher O-radical integrals than N-radical values were observed. Calculations revealed that the monoradical increment (O–N integral values) originated from photo/oxygen-synergistically generated O-radical species, indicating the coexistence of biradical/monoradical states in the system (the ratio of biradical to monoradical is about 1.2:1).
To evaluate the chemical stability of DICPO-COF, we subjected the material to 1 M HCl and 1 M NaOH treatments, followed by FT-IR, EPR, and PXRD analyses. EPR analysis suggested that under alkaline conditions, the ratio of mono-/biradical species remained unchanged, while acidic conditions predominantly retained biradical species. Both treatments caused a significant reduction in total radical intensity (Fig. S28†), likely due to radical quenching under harsh acidic/alkaline environments. FT-IR data (Fig. S29†) confirmed the preservation of the overall chemical structure. PXRD analysis (Fig. S30†) suggested that the material could maintain its long-range periodicity after acid or base treatment. In future studies, we will continue to focus on how to enhance the stability of radical-based COFs under extreme conditions.
The generation of this pyridine N-oxy biradical species significantly affects the chemical and physical properties of DICPO-COF, particularly enhancing its catalytic hydrogen atom transfer efficiency and reactivity. Therefore, DICPO-COF was examined in the dehydrogenation of nitrogen heterocycles, which is an important organic transformation in the preparation of fine chemicals, to evaluate its potential as a chemical catalyst.31,51,52 After optimization of the reaction conditions, various heterocyclic nitrogen substrates were investigated to explore the scope of the catalytic system. As shown in Fig. 6, tetrahydroquinoline substrates bearing either electron-withdrawing or electron-donating groups were transformed into the corresponding quinolines in good to excellent yields. Furthermore, the reactivity of DICPO-COF was observed in the dehydrogenation of indoline compounds, and all indoline substrates were smoothly transformed into indole products under standard conditions.
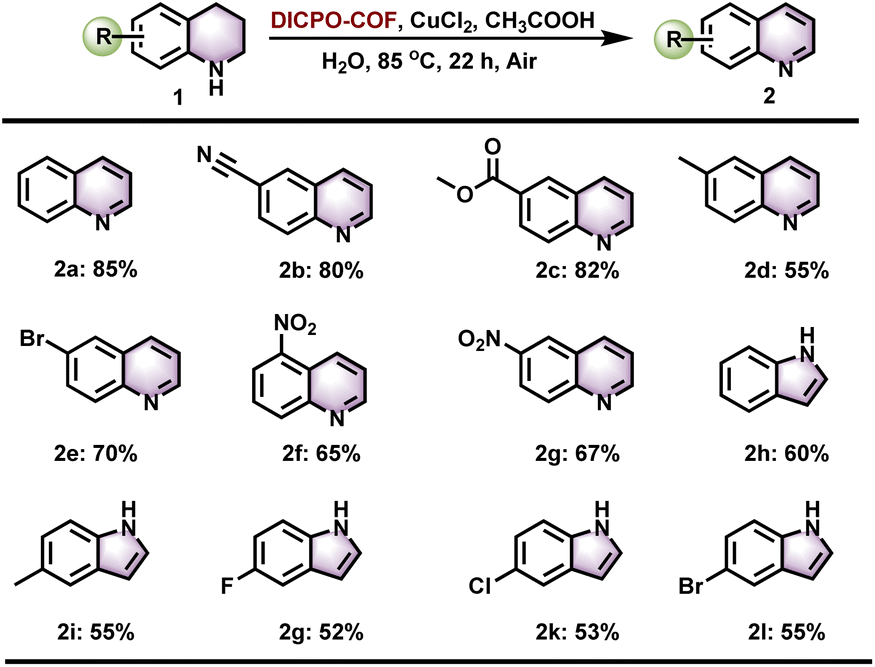 | ||
| Fig. 6 Scope of the dehydrogenation of N-heterocyclic substrate. Conditions: DICPO-COF (50 mg) N-heterocyclic substrate (50 μL, 0.40 mmol, 1 eq.), CuCl2 (10 mg, 0.07 mmol, 19 mol%), H2O (5 mL), CH3COOH (10 μL, 0.16 mmol, 0.43 eq.). | ||
An optimized reaction protocol was applied in the recycling experiments to evaluate the recyclability and practical feasibility of DICPO-COF in the dehydrogenation of 1,2,3,4-tetrahydroquinolines (THQs). In each cycle, the DICPO-COF catalyst was recovered by centrifugation, followed by washing and drying. The yield of the first cycle was 85%; however, the reaction yield decreased from the second cycle onward, and the yields stabilized at approximately 62–69% in the subsequent cycles (Fig. 7a). To explore the reasons for this phenomenon, the catalyst recovered after the fifth cycle was characterized by FT-IR, PXRD, 13C CP/MAS NMR, and EPR analyses. The FT-IR results indicated that the chemical structure of DICPO-COF was almost maintained (Fig. S34†). The PXRD characterization (Fig. S35†) showed a complete loss of crystallinity, demonstrating the disruption of long-range periodicity caused by the catalytic process. The 13C CP/MAS NMR spectra (Fig. S36†) indicated that the framework structure was maintained, though the splitting of solid-state NMR signals of DICPO-COF after the catalytic cycling process is not obvious, which may be due to incomplete removal of residual metallic copper during the cycling process. The EPR signal of DICPO-COF after the fifth cycle was significantly different from that of the sample before the catalytic reaction, exhibiting a single radical species signal (Fig. 7b). These results suggest that in the 2nd–5th cycles, the nitrogen–oxygen ylide species were oxidized by CuCl2 and converted into pyridine N-oxy monoradical species through an intermolecular single-electron transfer process (Fig. 7c), and the catalytic activity of this pyridine N-oxy monoradical species was lower than that of the biradical species. Therefore, the highest yield is obtained during the first cycle. To regenerate the biradical species, we explored reduction conditions by using Cu powder and RANEY® Ni as reducing agents to react with the generated monoradical species after catalytic reaction (Fig. S37†). However, only a very weak biradical characteristic EPR signal was detected with RANEY® Ni reduction (Fig. S38b†). Considering that the regeneration of biradical species is a very meaningful scientific problem, we will continue to work on solving this problem in future research.
 | ||
| Fig. 7 (a) Recyclability study. (b) EPR spectra of DICPO-COF before (black) and after the fifth cycle (red). (c) Generation of pyridine N-oxy monoradical species in the catalytic dehydrogenation of THQs. | ||
Several controlled experiments were conducted to explore the mechanism underlying this transformation. When the reaction was carried out in the absence of DICPO-COF or CuCl2, only low-yield products were obtained, indicating that both the COF material and CuCl2 played crucial roles in this reaction (Fig. S39b and c†). When the reaction was performed under anaerobic conditions, only a 10% yield of the product was obtained, illustrating that oxygen is also indispensable in this process. In addition, we used the known radical reagent TEMPO (2,2,6,6-tetramethylpiperidinoxyl) as a radical-type catalyst to catalyse this dehydrogenation with CuCl2 and obtained product 2a in 45% yield after 22 h of reaction (Fig. S39d†). When the commercial ylide reagent pyridine 1-oxide was used to catalyse the reaction with CuCl2, only 13% yield was obtained after 22 h (Fig. S39e†). These results suggest that the radical COF species play a more important role in this transformation than the ylide COF species. When we use pyridine N-oxide or 2-formyl-6-(phenylcarbamoyl)pyridine 1-oxide (the small molecule model of the COF's active unit) to replace DICPO-COF, the yield of product was reduced, too. These starkly contrast with the 85% yield achieved by the DICPO-COF/CuCl2 system, demonstrating that the presence of pyridine 1-oxide units (Fig. S39f and j†) or CuCl2 (Fig. S39b†) individually cannot effectively promote the reaction, and the framework structure is indispensable for high activity. We also conducted several control experiments to confirm DICPO-COF's catalytic role: twice the amount of CuCl2 (85% yield) or DICPO-COF (88% yield) separately maintained yields comparable to standard conditions (86% yield) (Fig. S40†), while using CuCl2 or DICPO-COF alone drastically reduced yields (13% and 11%, respectively) (Fig. S39b and c†). This demonstrates DICPO-COF working as a catalyst synergistically enhances catalytic efficiency with CuCl2. Notably, control experiments (Fig. S35g and h†) revealed that acetic acid, but not acetate ions, enhanced the reaction efficiency, likely by improving the water solubility of 1,2,3,4-tetrahydroquinoline. Additionally, we conducted this dehydrogenation by using Bipy-COF to replace DICPO-COF. Although Bipy-COF can catalyze the conversion of tetrahydroquinoline to quinoline, its catalytic efficiency is significantly lower than that of DICPO-COF, with a yield of only 39%. Therefore, the construction of biradical units in a COF skeleton is very important for this transformation.
Based on the experimental observations outlined above and previous studies in this field,53–57 we propose a radical species-induced mechanism for this transformation (Fig. S41†). Initially, 1,2,3,4-tetrahydroquinoline was oxidized by CuCl2 to produce radical cation intermediate 3. Further hydrogen abstraction and proton elimination of 3 by the pyridine N-oxide biradical COF resulted in intermediate 4 and DICPO-COF-2 species. Simultaneously, the –OH group in DCPO-COF-2 was oxidized by the oxygen in the air, and the biradical species DICPO-COF-1 was regenerated. Next, species 4 underwent isomerization to form intermediate 1,4-dihydroquinoline 5. Subsequently, 1,4-dihydroquinoline was further oxidized by CuCl2 and DICPO-COF-1via similar hydrogen abstraction and proton elimination steps, ultimately producing the target product, quinoline.
Conclusions
We leveraged the unique chemical structure of COF materials and constructed a 2,6-dicarboxamide-substituted pyridine N-oxide COF that could spontaneously transform into pyridine N-oxy biradical COFs without any additional conditions. Additionally, FT-IR, XPS, EPR, and 13C CP/MAS NMR demonstrated the coexistence of ylide and biradical species in this COF material. Furthermore, the EPR spectra of the pyridine N-oxide molecules and DICPO-COF indicate that the unique structure of COF materials plays a crucial role in stabilizing and promoting the formation of biradical species. We also evaluated catalytic performance in the dehydrogenation of nitrogen heterocycles, achieving moderate-to-high yields of the target products. Given that biradical-containing COFs can be easily and diversely transformed into other functional structures, we believe that our research will boost the development of novel applications of COF materials.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
W. A. and J. Z. contributed equally to this work. W. A., T. S., L. Q. and M. J. designed and planned this study. J. Z., Z. C. and K. C. contributed to the preparation of the samples and characterization. J. G., L. D., and X. P. analyzed the data and conducted the theoretical structure simulations. W. A. and T. S. wrote and edited the manuscript. All the authors have discussed, reviewed and agreed the results and conclusions of this manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We are grateful for the financial support of the National Natural Science Foundation of China (No. 22202238, No. 22301179, and No. 22471156), Program for Innovative Research Team (in Science and Technology) of University of Henan Province (23IRTSTHN019), Young Backbone Teacher of Zhongyuan University of Technology (2024XQG01), Key Research Project Plan for Higher Education Institutions in Henan Province (23A150014), Fundamental Research Funds for the Central Universities (24X010301678, 23X010301599).Notes and references
- Q. Gu, X. Lu, C. Chen, R. Hu, X. Wang, G. Sun, F. Kang, J. Yang, X. Wang, J. Wu, Y. Y. Li, Y.-K. Peng, W. Qin, Y. Han, X. Liu and Q. Zhang, Thermally Induced Persistent Covalent-Organic Frameworks Radicals, ACS Nano, 2023, 17, 23903–23912 CrossRef.
- W. Cao, W. D. Wang, H.-S. Xu, I. V. Sergeyev, J. Struppe, X. Wang, F. Mentink-Vigier, Z. Gan, M.-X. Xiao, L.-Y. Wang, G.-P. Chen, S.-Y. Ding, S. Bai and W. Wang, Exploring Applications of Covalent Organic Frameworks: Homogeneous Reticulation of Radicals for Dynamic Nuclear Polarization, J. Am. Chem. Soc., 2018, 140, 6969–6977 CrossRef PubMed.
- X. Li, J. Zhou, W. Yin, B. Xie, Z. Liu and J.-J. Liu, Fully conjugated radical-doped COF for near-infrared photothermal conversion, C-3 thiocyanation of indoles and oxidative coupling of amines, J. Catal., 2024, 437, 115640–115649 CrossRef.
- Z. Chen, Y. Su, X. Tang, X. Zhang, C. Duan, F. Huang and Y. Li, Manipulating Grain Boundary Defects in π-Conjugated Covalent Organic Frameworks Enabling Intrinsic Radical Generation for Photothermal Conversion, Sol. RRL, 2021, 5, 2100762–2100769 CrossRef.
- F. Frezza, A. Matěj, A. Sánchez-Grande, M. Carrera, P. Mutombo, M. Kumar, D. Curiel and P. Jelínek, On-Surface Synthesis of a Radical 2D Supramolecular Organic Framework, J. Am. Chem. Soc., 2024, 146, 3531–3538 CrossRef PubMed.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Porous, Crystalline, Covalent Organic Frameworks, Science, 2005, 310, 1166–1170 CrossRef PubMed.
- R. Gao, R. Shen, C. Huang, K. Huang, G. Liang, P. Zhang and X. Li, 2D/2D Hydrogen-Bonded Organic Frameworks/Covalent Organic Frameworks S-Scheme Heterojunctions for Photocatalytic Hydrogen Evolution, Angew. Chem., Int. Ed., 2025, 64, e202414229 CrossRef PubMed.
- L. Zou, D. Si, S. Yang, Z. Chen, Y. Huang and R. Cao, Induced Charge-Compensation Effect for Boosting Photocatalytic Water Splitting in Covalent Organic Frameworks, Angew. Chem., Int. Ed., 2025, 64, e202418319 CrossRef PubMed.
- H. Wang, H. Wang, Z. Wang, L. Tang, G. Zeng, P. Xu, M. Chen, T. Xiong, C. Zhou, X. Li, D. Huang, Y. Zhu, Z. Wang and J. Tang, Covalent organic framework photocatalysts: structures and applications, Chem. Soc. Rev., 2020, 49, 4135–4165 RSC.
- M. Deng, J. Chakraborty, G. Wang, K. S. Rawat, L. Bourda, J. Sun, I. Nath, Y. Ji, P. Geiregat, V. Van Speybroeck, X. Feng and P. Van Der Voort, Transforming 2D Imine into 3D Thiazole Covalent Organic Frameworks by Conjugated Connectors: Fully Conjugated Photocatalysts, J. Am. Chem. Soc., 2025, 147, 10219–10230 CrossRef PubMed.
- C.-H. Hsueh, C. He, J. Zhang, X. Tan, H. Zhu, W.-C. M. Cheong, A.-Z. Li, X. Chen, H. Duan, Y. Zhao and C. Chen, Three-Dimensional Mesoporous Covalent Organic Framework for Photocatalytic Oxidative Dehydrogenation to Quinoline, J. Am. Chem. Soc., 2024, 146, 33857–33864 CrossRef PubMed.
- T.-X. Luan, L.-B. Xing, N. Lu, X.-L. Li, S. Kong, W. W. Yu, P.-Z. Li and Y. Zhao, Donor–Acceptor-π-Acceptor–Donor-Type Photosensitive Covalent Organic Framework for Effective Photocatalytic Aerobic Oxidation, J. Am. Chem. Soc., 2025, 147, 12704–12714 CrossRef PubMed.
- L. Luo, C. Li, Y. Wang, P. Chen, Z. Zhou, T. Chen, K. Wu, S.-Y. Ding, L. Tan, J. Wang, X. Shao and Z. Liu, Multi-Functional 2D Covalent Organic Frameworks with Diketopyrrolopyrrole as Electron Acceptor, Small, 2024, 20, 2402993–2403003 CrossRef PubMed.
- K. T. Tan, S. Ghosh, Z. Wang, F. Wen, D. Rodríguez-San-Miguel, J. Feng, N. Huang, W. Wang, F. Zamora, X. Feng, A. Thomas and D. Jiang, Covalent organic frameworks, Nat. Rev. Methods Primers, 2023, 3, 1–19 CrossRef.
- G. E. Rudebusch, J. L. Zafra, K. Jorner, K. Fukuda, J. L. Marshall, I. Arrechea-Marcos, G. L. Espejo, R. Ponce Ortiz, C. J. Gómez-García, L. N. Zakharov, M. Nakano, H. Ottosson, J. Casado and M. M. Haley, Diindeno-fusion of an anthracene as a design strategy for stable organic biradicals, Nat. Chem., 2016, 8, 753–759 CrossRef PubMed.
- A. V. Kutasevich, V. P. Perevalov and V. S. Mityanov, Recent Progress in Non-Catalytic C–H Functionalization of Heterocyclic N-Oxides, Eur. J. Org Chem., 2021, 2021, 357–373 CrossRef.
- V. Satheesh and Y. Deng, Recent Advances in Synthetic Methods by Photocatalytic Single-Electron Transfer Chemistry of Pyridine N-Oxides, J. Org. Chem., 2024, 89, 11864–11874 CrossRef CAS.
- C.-Y. Cai, S.-J. Chen, R. R. Merchant, Y. Kanda and T. Qin, C3 Selective Hydroxylation of Pyridines via Photochemical Valence Isomerization of Pyridine N-Oxides, J. Am. Chem. Soc., 2024, 146, 24257–24264 CrossRef CAS.
- Ł. W. Ciszewski and D. Gryko, Pyridine N-oxides as HAT reagents for photochemical C–H functionalization of electron-deficient heteroarenes, Chem. Commun., 2022, 58, 10576–10579 RSC.
- M. Schlegel, S. Qian and D. A. Nicewicz, Aliphatic C–H Functionalization Using Pyridine N-Oxides as H-Atom Abstraction Agents, ACS Catal., 2022, 12, 10499–10505 CrossRef CAS.
- J.-h. Xu, W.-b. Wu and J. Wu, Photoinduced Divergent Alkylation/Acylation of Pyridine N-Oxides with Alkynes under Anaerobic and Aerobic Conditions, Org. Lett., 2019, 21, 5321–5325 CrossRef CAS PubMed.
- S. L. Rössler, B. J. Jelier, P. F. Tripet, A. Shemet, G. Jeschke, A. Togni and E. M. Carreira, Pyridyl Radical Cation for C–H Amination of Arenes, Angew. Chem., Int. Ed., 2019, 58, 526–531 CrossRef PubMed.
- B. Wang, C. Ascenzi Pettenuzzo, J. Singh, G. E. McCabe, L. Clark, R. Young, J. Pu and Y. Deng, Photoinduced Site-Selective Functionalization of Aliphatic C–H Bonds by Pyridine N-oxide Based HAT Catalysts, ACS Catal., 2022, 12, 10441–10448 CrossRef CAS.
- T. Kubota, K. Nishikida, H. Miyazaki, K. Iwatani and Y. Oishi, Electron Spin Resonance and Polarographic Studies of the Anion Radicals of Heterocyclic Amine N-Oxides, J. Am. Chem. Soc., 1968, 90, 5080–5090 CrossRef CAS.
- H. Li, F. Xie and M.-T. Zhang, Metal-Free Electrocatalyst for Water Oxidation Initiated by Hydrogen Atom Transfer, ACS Catal., 2021, 11, 68–73 CrossRef CAS.
- N.-N. Zhang, Y.-F. Han, C. Sun, M.-S. Wang and G.-C. Guo, Enhanced Photoinduced Electron Transfer and Stability of Diradicals in Neutral Extended Pyridine N-Oxides, J. Phys. Chem. C, 2019, 123, 24670–24675 CrossRef CAS.
- S. Ahn, J. H. Jang, J. Kang, M. Na, J. Seo, V. Singh, J. M. Joo and H. R. Byon, Systematic Designs of Dicationic Heteroarylpyridiniums as Negolytes for Nonaqueous Redox Flow Batteries, ACS Energy Lett., 2021, 6, 3390–3397 CrossRef CAS.
- R. S. Fernandes, A. Tiwari, S. Kanungo and N. Dey, Formation of stable naphthalenediimide radical anion: substituent-directed synergistic effects of hydrogen bonding and charge transfer interactions on chromogenic response towards hydrazine, J. Mol. Liq., 2023, 387, 122238–122250 CrossRef CAS.
- F. Liao, W. Huang, B. Chen, Z. Ding, X. Li, H. Su, T. Wang, Y. Wang, H. Miao, X. Zhang, Y. Luo, J. Yang and G. Zhang, Are pyridinium ylides radicals?, Chem. Commun., 2020, 56, 11287–11290 RSC.
- S. Bera, A. Bera and D. Banerjee, Nickel-Catalyzed Dehydrogenation of N-Heterocycles Using Molecular Oxygen, Org. Lett., 2020, 22, 6458–6463 CrossRef CAS.
- S. Chakraborty, W. W. Brennessel and W. D. Jones, A Molecular Iron Catalyst for the Acceptorless Dehydrogenation and Hydrogenation of N-Heterocycles, J. Am. Chem. Soc., 2014, 136, 8564–8567 CrossRef CAS.
- P. P. Fu and R. G. Harvey, Dehydrogenation of polycyclic hydroaromatic compounds, Chem. Rev., 1978, 78, 317–361 CrossRef CAS.
- A. E. Wendlandt and S. S. Stahl, Modular o-Quinone Catalyst System for Dehydrogenation of Tetrahydroquinolines under Ambient Conditions, J. Am. Chem. Soc., 2014, 136, 11910–11913 Search PubMed.
- Y. Wu, H. Yi and A. Lei, Electrochemical Acceptorless Dehydrogenation of N-Heterocycles Utilizing TEMPO as Organo-Electrocatalyst, ACS Catal., 2018, 8, 1192–1196 CrossRef CAS.
- R. Yang, S. Yue, W. Tan, Y. Xie and H. Cai, DMSO/t-BuONa/O2-Mediated Aerobic Dehydrogenation of Saturated N-Heterocycles, J. Org. Chem., 2020, 85, 7501–7509 CrossRef CAS.
- R. Xue, Y.-S. Liu, M.-Y. Wang, H. Guo, W. Yang, J.-X. Guo and G.-Y. Yang, Rational Conversion of Imine Linkages to Amide Linkages in Covalent Organic Frameworks for Photocatalytic Oxidation with Enhanced Photostability, ChemSusChem, 2024, 17, e202400732 CrossRef CAS PubMed.
- H. Pang, G. Liu, D. Huang, Y. Zhu, X. Zhao, W. Wang and Y. Xiang, Embedding Hydrogen Atom Transfer Moieties in Covalent Organic Frameworks for Efficient Photocatalytic C–H Functionalization, Angew. Chem., Int. Ed., 2023, 62, e202313520 CrossRef CAS.
- L. Guo, Y. Gao, J. Huang, J. Xue, F. Li and C. Li, Imine-linked covalent organic frameworks coordinated with nickel for ethylene oligomerization, ACS Appl. Polym., 2023, 140, e53320 CrossRef CAS.
- M. Zhang, X. Wu, Y. Xie, X. Hao, Q. Wang, Y. Zhao, J. Wu and X. Pan, Effect of COF linkage isomerism on photocatalytic hydrogen evolution performance, Mater. Chem. Front., 2023, 7, 5399–5405 RSC.
- Y. Shen, S. Liu, L. Lu, C. Zhu, Q. Fang, R. Liu, Y. Shen and S. Song, Pyridine-linked covalent triazine frameworks with bidirectional electron donor-acceptor for efficient organic pollution removal, J. Hazard., 2023, 444, 130428–130439 CrossRef CAS PubMed.
- Y. Li, X. Guo, X. Li, M. Zhang, Z. Jia, Y. Deng, Y. Tian, S. Li and L. Ma, Redox-Active Two-Dimensional Covalent Organic Frameworks (COFs) for Selective Reductive Separation of Valence-Variable, Redox-Sensitive and Long-Lived Radionuclides, Angew. Chem., Int. Ed., 2020, 59, 4168–4175 CrossRef CAS.
- Y. Song, J.-J. Zhang, Y. Dou, Z. Zhu, J. Su, L. Huang, W. Guo, X. Cao, L. Cheng, Z. Zhu, Z. Zhang, X. Zhong, D. Yang, Z. Wang, B. Z. Tang, B. I. Yakobson and R. Ye, Atomically Thin Ionic–Covalent Organic Nanosheets for Stable, High-Performance Carbon Dioxide Electroreduction, Adv. Mater., 2022, 34, 2110496–2110507 CrossRef CAS.
- S.-S. Xu, S.-W. Qiu, Z.-Y. Yuan, T.-Z. Ren and T. J. Bandosz, Nitrogen-containing activated carbon of improved electrochemical performance derived from cotton stalks using indirect chemical activation, J. Colloid Interface Sci., 2019, 540, 285–294 CrossRef CAS PubMed.
- S. Ma, Z. Li, J. Jia, Z. Zhang, H. Xia, H. Li, X. Chen, Y. Xu and X. Liu, Amide-linked covalent organic frameworks as efficient heterogeneous photocatalysts in water, Chin. J. Catal., 2021, 42, 2010–2019 CrossRef CAS.
- H. Pang, G. Liu, D. Huang, Y. Zhu, X. Zhao, W. Wang and Y. Xiang, Embedding Hydrogen Atom Transfer Moieties in Covalent Organic Frameworks for Efficient Photocatalytic C–H Functionalization, Angew. Chem., Int. Ed., 2023, 62, e202313520 CrossRef CAS PubMed.
- L. Gao, G. Wang, J. Cao, D. Yuan, C. Xu, X. Guo and S. Li, Organocatalytic decarboxylative alkylation of N-hydroxy-phthalimide esters enabled by pyridine-boryl radicals, Chem. Commun., 2018, 54, 11534–11537 RSC.
- Y.-F. Han, N.-N. Zhang, L. Li, L.-D. Xin and K.-G. Qu, Photoinduced electron transfer properties of 4-phenyl-Pyridine-N-Oxide and its coordination compound, Dyes Pigm., 2022, 197, 109917–109924 CrossRef CAS.
- L. Han, L. Qin, L. Xu, Y. Zhou, J. Sun and X. Zou, A novel photochromic calcium-based metal–organic framework derived from a naphthalene diimide chromophore, Chem. Commun., 2013, 49, 406–408 RSC.
- N. Leblanc, M. Allain, N. Mercier and L. Sanguinet, Stable Photoinduced Separated Charge State in Viologen Halometallates: Some Key Parameters, Cryst. Growth Des., 2011, 11, 2064–2069 CrossRef CAS.
- N. Zhang, X. Li, H. Ye, S. Chen, H. Ju, D. Liu, Y. Lin, W. Ye, C. Wang, Q. Xu, J. Zhu, L. Song, J. Jiang and Y. Xiong, Oxide Defect Engineering Enables to Couple Solar Energy into Oxygen Activation, J. Am. Chem. Soc., 2016, 138, 8928–8935 CrossRef CAS PubMed.
- R. Yamaguchi, C. Ikeda, Y. Takahashi and K.-i. Fujita, Homogeneous Catalytic System for Reversible Dehydrogenation–Hydrogenation Reactions of Nitrogen Heterocycles with Reversible Interconversion of Catalytic Species, J. Am. Chem. Soc., 2009, 131, 8410–8412 CrossRef CAS.
- W. Yao, Y. Zhang, X. Jia and Z. Huang, Selective Catalytic Transfer Dehydrogenation of Alkanes and Heterocycles by an Iridium Pincer Complex, Angew. Chem., Int. Ed., 2014, 53, 1390–1394 CrossRef CAS.
- Y. Shang, X. Jie, K. Jonnada, S. N. Zafar and W. Su, Dehydrogenative desaturation-relay via formation of multicenter-stabilized radical intermediates, Nat. Commun., 2017, 8, 2273–2281 CrossRef.
- D. V. Ramana and M. Chandrasekharam, Copper-Catalyzed Direct Oxidative α-Functionalization of Tetrahydroquinoline in Water under Mild Conditions, ASC, 2018, 360, 4080–4083 CAS.
- S. Elavarasan, A. Bhaumik and M. Sasidharan, An Efficient Mesoporous Cu-Organic Nanorod for Friedländer Synthesis of Quinoline and Click Reactions, ChemCatChem, 2019, 11, 4340–4350 CrossRef CAS.
- X. Li, H. Zhao, X. Chen, H. Jiang and M. Zhang, Copper-catalysed oxidative α-C(sp3)–H nitroalkylation of (hetero)arene-fused cyclic amines, Org. Chem. Front., 2020, 7, 425–429 RSC.
- S. A. Pawar, S. V. Poojari and A. Vijay Kumar, Cu2O-CD nanosuperstructures as a Biomimetic Catalyst for Oxidation of Benzylic sp C–H bonds and Secondary Amines using Molecular Oxygen: First Total Synthesis of proposed Swerilactone O, Asian J. Org. Chem., 2022, 11, e202200030 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc08236b |
| ‡ Wenying Ai and Jiawei Zou contributed equally to the manuscript. |
| This journal is © The Royal Society of Chemistry 2025 |