
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe role of surface deformation on responsivity of the pillared layer metal–organic framework DUT-8(Ni)†
Leila
Abylgazina
 a,
Irena
Senkovska
*a,
Mariia
Maliuta
a,
Christopher
Bachetzky
b,
Marcus
Rauche
b,
Kathrin
Pöschel
c,
Johannes
Schmidt
d,
Mark
Isaacs
ef,
David
Morgan
eg,
Michal
Otyepka
hi,
Eva
Otyepkova
j,
Matthias
Mendt
l,
Yogeshwar D.
More
l,
Robin
Buschbeck
m,
Andreas
Schneemann
a,
Alla
Synytska‡
c,
Andreas
Pöppl
k,
Lukas M.
Eng
mn,
Jin-Chong
Tan
l,
Eike
Brunner
b and
Stefan
Kaskel
*a
a,
Irena
Senkovska
*a,
Mariia
Maliuta
a,
Christopher
Bachetzky
b,
Marcus
Rauche
b,
Kathrin
Pöschel
c,
Johannes
Schmidt
d,
Mark
Isaacs
ef,
David
Morgan
eg,
Michal
Otyepka
hi,
Eva
Otyepkova
j,
Matthias
Mendt
l,
Yogeshwar D.
More
l,
Robin
Buschbeck
m,
Andreas
Schneemann
a,
Alla
Synytska‡
c,
Andreas
Pöppl
k,
Lukas M.
Eng
mn,
Jin-Chong
Tan
l,
Eike
Brunner
b and
Stefan
Kaskel
*a
aChair of Inorganic Chemistry I, Technische Universität Dresden, Bergstr. 66, 01069 Dresden, Germany. E-mail: irena.senkovska@tu-dresden.de; stefan.kaskel@tu-dresden.de
bChair of Bioanalytical Chemistry, Technische Universität Dresden, Bergstr. 66, 01069 Dresden, Germany
cLeibniz Institute of Polymer Research Dresden, Hohe Str. 6, 01069 Dresden, Germany
dInstitute of Chemistry, Technische Universität Berlin, Funktionsmaterialien, Hardenbergstraße 40, 10623 Berlin, Germany
eHarwellXPS, Research Complex at Harwell, Rutherford Appleton Labs, Didcot, Oxfordshire OX110FA, UK
fDepartment of Chemistry, University College London, 20 Gower Street, Euston, London, WC1H 0AJ, UK
gSchool of Chemistry, Translational Research Hub, Cardiff University, Maindy Road, Cardiff, CF24 4HQ, UK
hRegional Centre of Advanced Technologies and Materials, Czech Advanced Technology and Research Institute (CATRIN), Palacký University Olomouc, Šlechtitelů 27, 779 00 Olomouc, Czech Republic
iIT4Innovations, VŠB – Technical University of Ostrava, 17. Listopadu 2172/15, Poruba, 708 00 Ostrava, Czech Republic
jDepartment of Physical Chemistry, Faculty of Science, Palacký University Olomouc, 17. Listopadu 12, 771 46 Olomouc, Czech Republic
kFelix Bloch Institute for Solid State Physics, Leipzig University, Linnéstr. 5, 04103 Leipzig, Germany
lDepartment of Engineering Science, University of Oxford, Parks Road, Oxford OX1 3PJ, England, UK
mInstitute of Applied Physics, Technische Universität Dresden, Nöthnitzer Straße 61, 01187 Dresden, Germany
nWürzburg-Dresden Cluster of Excellence – EXC 2147 (ct.qmat), Technische Universität Dresden, 01062 Dresden, Germany
First published on 6th March 2025
Abstract
A unique feature of flexible metal–organic frameworks (MOFs) is their ability to respond dynamically towards molecular stimuli by structural transitions, resulting in pore-opening and closing processes. One of the most intriguing modes is the “gating”, where the material transforms from the dense to the porous state. The conditions required for the solid phase structural transition are controlled by the kinetic barriers, including nucleation of the new phase commencing on the crystallite's outer surface. Thus, surface deformation may influence the nucleation, enabling deliberate tailoring of the responsivity. In the present contribution, we investigate how chemical surface treatments (surface deformation) affect the gate opening characteristics of a typical representative of gate pressure MOFs, DUT-8(Ni) ([Ni2(ndc)2(dabco)]n, ndc = 2,6-naphthalenedicarboxylate, dabco = 1,4-diazabicyclo[2.2.2]octane). A combination of various complementary advanced characterization techniques, such as NMR, nanoFTIR, terahertz, in situ XPS, in situ EPR spectroscopies, and inverse gas chromatography, are applied to unravel the changes in surface energy and mechanism of surface deformation.
Introduction
The development of new functional materials is important for many fields, including energy conversion, mobility, cooling, medicine, and gas storage.1 They need to be engineered and designed for a particular application; however, not only the bulk properties need to be considered for a specific function, but also, amongst others, their morphology and shape.2,3 Moreover, in many cases, the functionality is controlled via the surface properties, therefore, the study of these is of practical interest for many current research fields.4–6In the case of crystalline porous metal–organic frameworks (MOFs), as a highly potent class of functional materials, the high internal specific surface area naturally leads to potential applications in gas storage and separation, among others.7–12 For adsorption-driven processes, however, the outer surface is considered as a barrier, influencing the adsorption performance,13,14 and playing a fundamental role in limiting the mass transport of guest molecules in nanoporous materials.15–17 Surface permeability is sensitive to the non-ideality of a surface owing to the physical and chemical properties, host–guest interaction at the surface, and the change of environment. For example, the concentration of external acidity and host–guest interaction at the surface of SAPO zeolites have a direct influence on surface permeability.16
Various synthetic and self-assembly-based surface-decoration approaches were developed to control and design MOF properties.18,19 For example, the group of Kitagawa synthesized HKUST-1 ([Cu3(btc)2]n, btc = 1,3,5-benzenetricarboxylate) through coordination modulation, changing the crystal morphology from octahedral to cuboctahedral, which potentially allows for control over surface reactivity.20 Another widely used surface modification technique is solvent-assisted linker exchange, which, amongst others, was performed on DMOF-1 ([Zn2(bdc)2(dabco)]n, bdc – 1,4-benzenedicarboxylate, dabco – 1,4-diazabicyclo[2.2.2]octane).21 In this work, a monolayer of boron dipyrromethane molecules was assembled onto the four carboxylate-terminated surfaces, leaving the two dabco-terminated surfaces unmodified.
The modification, degradation, or deformation of the surface leads to either the enhancement or deterioration of MOF properties.18,19 For example, after exposure of HKUST-1 to water vapour, surface degradation occurs, which is associated with the formation of defects and a barrier layer, ultimately hindering the uptake and release of guest molecules.14 On the contrary, surface modification by coating with polymers is widely used to improve the stability of materials against water.22 For instance, MOF-5 post-synthetically coated with PDMS (polydimethylsiloxane) via a chemical vapour deposition route shows improved hydrolytic stability under ambient conditions.23
The effect of interfaces also becomes even more important for so-called flexible MOFs, which are capable of undergoing stimuli-induced solid-state phase transitions.24–26 In particular, the so-called gating transitions (or gate-opening transitions), involving a transformation from the non-porous to the porous one, are typically accompanied by massive crystal volume changes and rely on the nucleation of the expanded phase at the outer surface of the crystals. Kinetic studies of the phase transitions in MOFs show that the overall velocity of gas uptakes in flexible MOFs is predominantly determined by the rate of the structural transition.27,28
At sub-micron scale, the surfaces seem to determine the responsive characteristics of flexible MOFs, such as rate of phase transition, transforming, in extreme cases, flexible materials into rigid ones and vice versa, implying the transformation of the porous into unporous materials, without explicitly altering the internal structure.29–31 The flexibility is affected by the interplay of factors, including not only the crystal size, morphology and defects but also crystal surface, matrix interfaces, and grain boundaries.29,32,33 But despite of this, the role of particle surface effects remains largely underexplored, particularly in flexible MOFs and, more broadly, across the MOF material class.
These effects become more complex when the surface and the interface are chemically modified, e.g., with adsorbates. Then, the interfacial tension and energy density of interfaces do not reflect the cohesive energy between the molecules in the bulk phase anymore.34 Thus, the interface itself can be treated as a thermodynamic system on its own.
To get insights into the role of interface modification/deformation on framework flexibility, we investigated a well-studied representative of flexible pillared layer MOFs – DUT-8(Ni), ([Ni2(ndc)2(dabco)]n, ndc – 2,6-naphthalenedicarboxylate) model system, consisting of paddle wheel based [Ni2(ndc)2]n square-grid layers, which are pillared by the dabco molecules, coordinated to the terminal sites of the paddle wheels (Fig. 1).35 The framework is stabilized in the as made, open pore (op) form by the guest–host interactions. Upon removal of solvent molecules from the pores, the porous op phase transforms into the thermodynamically stable, dense closed pore (cp) form, which is stabilized by dispersive linker–linker interactions.36 The reversed phase transition can be stimulated by the adsorption of various adsorptives,35,37,38 resulting in a gating adsorption isotherm (Fig. 2). Since the cp phase is dense, the cp to op transition nucleates on the crystal external surface when an adsorptive specific pressure (gate opening pressure, pgo) is reached.28 Thus, the pgo (or the relative adsorption pressure at half-maximum uptake (APHM))30,39 is related to the activation energy the system must surpass for the structural opening transition. The desorption branch does not follow the adsorption one, resulting in a hysteresis loop. It is assumed that the desorption branch of the isotherm is closer to the thermodynamic equilibrium of the switching process, as for closing the nucleation barrier should be much lower because the fluid phase in the open framework is quasi in equilibrium with the surrounding gas phase.40,41
 | ||
| Fig. 1 (a) Building units of DUT-8(Ni); (b) crystal structures of open and closed pore phases of DUT-8(Ni); (c) possible scenarios for surface termination (C in grey, O – in red, N – in blue, Ni – in green). | ||
 | ||
| Fig. 2 Nitrogen physisorption isotherms at 77 K of samples exposed to humid air: (a) 28% humidity, (c) 55% humidity, (e) samples treated with ethanol, ethanol/water mixture, PDMS; (b), (d) and (f) corresponding APHM values. | ||
Düren and co-workers demonstrated using osmotic framework adsorbed solution theory (OFAST), that the capping groups (N,N-dimethylformamide (DMF), dabco, Hndc−, fumarate) on the surface of DUT-8(Ni) slabs with a thickness of 4–7 nm (surface to surface) modify the gate-opening/closing pressures.42
In general, smaller terminating molecules reduce overall dispersive interactions of the particle and decrease the cp → op energy difference, leading to a decrease in the gate-transition pressure.
However, for larger particles (>100 nm) this energetic contribution becomes negligible. Small particles have much larger external specific surface area and for large crystallites, the ratio between the external and inner specific surface areas is much smaller than that for small nanoparticles. Hence, small particles are more sensitive to the modifications of the external surface. It is reasonable to assume that larger particles can tolerate more substantial surface deformations, which can cover not only a molecular surface layer but a thicker, partially transformed network of several unit cells in thickness.
In this context surface deformation is an important subject and should be taken into account in MOF flexibility studies. Substantial surface deformation and outer layer formation could affect not only nanoparticles but also particles in an intermediate size regime (500–3000 nm) and surface deformation could also differently affect different facets. Especially, the surface deformation mechanisms of macroscopic soft porous crystals (d > 1 μm) and their impact on switchability are largely unexplored and should be studied more seriously in the future.
From the other point of view, the nucleation of the phase transition relies on the host–guest interactions on the external surface of the cp phase. If the terminating ligands become smaller in size, it reduces the adsorption enthalpy, and pgo should increase as more guest molecules are needed to overcome the cp → op activation barrier. Since the external surface area (area available for nucleation) per particle grows with the particle size, the influence of the termination on the phase transition pressure should also be visible for the large particles.
In the present work, we studied the influence and origin of the changes in the flexible behaviour of DUT-8(Ni) cp phase exposed to various chemical species: H2O vapour, alcohols, and PDMS. The macro-sized DUT-8(Ni) crystals are not responsive towards protic solvents,37 thus, the exposure to alcohols does not lead to the structural gate-opening transition. Therefore, upon treatment with alcohol, only the outer surface experiences contact with the fluid phase. The inner surface is supposed to be not affected. The water also does not provoke structural transition but is known to hydrolyze the paddle wheel based MOFs, which is the main origin of chemical degradation.43 PDMS was chosen due to its inert nature and low surface energy,44 justifying its wide use as a protective coating.23,45 The drastic effects of the surface treatment were demonstrated by analyzing the adsorption behaviour of the treated frameworks via N2 adsorption isotherm profiles at 77 K. Various complementary analytical techniques shine a light on the surface deformation mechanism and its crucial impact on the switchability of soft porous crystals.
Results and discussion
The phase purity of as made (op), and desolvated (cp) samples was identified by powder X-ray diffraction (Fig. S1†).The crystal treatment sequences and the labelling of the samples are summarised in Scheme 1. The water vapour treatment (sample series 1) was performed at different specific relative humidity (X) for a defined time (Y). In certain cases, the samples were reactivated (R) by evacuation upon heating. The second series of samples (2) was suspended in alcohol for various periods of time (Y). The third series of samples (3) was exposed to the EtOH/H2O mixture (95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 v/v). Sample 4 was exposed to PDMS (for details, see the Experimental part).
:5 v/v). Sample 4 was exposed to PDMS (for details, see the Experimental part).
 | ||
| Scheme 1 The illustration of the samples' history, showing different treatment conditions.46 | ||
Stability test
The treatment of the sample in cp form by water, ethanol, ethanol/water mixture, and PDMS upon conditions indicated in Scheme 1, does not induce any significant structural changes, detectable in PXRD (Fig. S1†), indicating that the inner crystalline structure of the parent material is well maintained.However, a stability test performed by immersing the cp or op samples in ethanol showed that op remained intact for 4 days in ethanol, while the cp phase showed some changes in the PXRD pattern already after 6 h (Fig. S2a and c†). The op phase nanocrystals exposed to ethanol for weeks slightly change the morphology (Fig. S6†) but definitely retain the crystallinity. Exposure to liquid water leads to a deterioration of the crystalline structure of the frameworks within a few hours for both op and cp phases (Fig. S2b and d†).
Adsorption behaviour
The influence of treatment procedures on structural responsivity was investigated by nitrogen physisorption at 77 K. To monitor the changes in adsorption behaviour, the relative adsorption pressure at half-maximum uptake (APHM) as a measure of the average gate opening activation energy is used.30The second parameter to monitor the changes in adsorption behaviour is αmax (Table 1), connected to the specific pore volume of the particular sample.30 As it is related to the theoretical pore volume, αmax reflects the fraction of crystallites transformed to the op phase at given conditions.30
| Sample | APHM (p/p0)a | ΔAPHM (p/p0) | α max |
|---|---|---|---|
| a Relative adsorption pressure at half-maximum uptake.30,39 b The ratio between the maximum experimental uptake to the maximum theoretical amount, which can be adsorbed (reflects the fraction of crystallites transformed to the op phase at given conditions).30,39 | |||
| Pristine DUT-8(Ni) | 0.115 | 1.03 | |
| Treated by humid air | |||
| 1_H2O-28-0.25 | 0.136 | 0.021 | 1.10 |
| 1_H2O-28-1 | 0.143 | 0.028 | 1.16 |
| 1_H2O-28-24 | 0.164 | 0.048 | 1.10 |
| 1_H2O-28-336 | 0.325 | 0.210 | 0.86 |
| 1_H2O-55-0.25 | 0.185 | 0.070 | 1.16 |
| 1_H2O-55-1 | 0.222 | 0.107 | 1.00 |
| 1_H2O-55-24 | 0.796 | 0.222 | |
| 1_H2O-55-336 | — | 0.05 | |
| Pristine DUT-8(Ni) | 0.130 | 1.07 | |
| Treated by ethanol | |||
| 2_EtOH-6 | 0.190 | 0.060 | 0.74 |
| Treated by ethanol/water mixture | |||
| 3_EtOH/H2O | 0.461 | 0.331 | 0.77 |
| Treated by PDMS | |||
| 4_PDMS | 0.410 | 0.28 | 1.06 |
Nitrogen adsorption (77 K) shows that the parent macro-sized material, obtained using the typical synthetic protocol, transforms from cp to op phase at APHM of approximately p/p0 = 0.12 in the first adsorption cycle (Fig. 2). This represents a typical value for the material desolvated from DCM.39 However, there is a certain distribution of APHM values reported,39 therefore, exact APHM was estimated for all starting materials used for particular treatments (Fig. 2b, d and f).
In the first series of treatments, the material was exposed to air with 28% relative humidity, which led to the gradual increase of APHM values within 24 h (Fig. 2a and b, Table 1). As can be seen from Fig. 2a, the most pronounced changes in the adsorption behaviour were observed for the sample exposed to humid air (28%) during 336 h (2 weeks). The APHM value was increased from 0.115 (pristine DUT-8(Ni)) to 0.325 (1_H2O-28-336) accompanied by a significant decrease in the maximum amount of nitrogen adsorbed (Table 1).
Furthermore, the material was exposed to air with higher humidity (55%), which expectedly caused severe changes in adsorption properties (Fig. 2c and d). Considering the adsorption profiles of samples exposed to humid air (55%), the APHM values of the samples treated during 0.25 h, and 1 h are considerably increased from 0.115 (pristine DUT-8(Ni)) to 0.185 and 0.222, respectively. Moreover, the treatment of samples during 24 h and 336 h resulted in a complete loss of responsivity to nitrogen at 77 K. The results revealed that the higher the humidity and the longer the exposure time, the more severe changes in the properties of DUT-8(Ni) were observed since water is able to hydrolyze the surface of paddle wheel-based MOFs.43 However, it is remarkable to recognize the stark effects on APHM after exposure to 55% RH for up to 1 h without dramatic loss of overall adsorption capacity.
The samples were also treated with other chemical species, such as ethanol, a mixture of ethanol/water, and PDMS. The adsorption profiles of treated samples change after any treatment (Fig. 2e and f). The APHM values of samples 2_EtOH (0.19), 3_EtOH/H2O (0.46), 4_PDMS (0.41) are considerably higher than that for non-treated DUT-8(Ni) (0.13). In the case of 2_EtOH and 3_EtOH/H2O, treatment procedures also caused a decrease in nitrogen uptake (Fig. 2e).
Thus, any treatment of the materials performed in this work leads to modification of the adsorption profile (Fig. 2), resulting in a shifting of the APHM to higher values and/or decrease of nitrogen uptake, suggesting an increase in the activation barrier originating from a particular surface deformation (Table 1).
Additionally, DUT-8(Ni)_cp sample was treated with other linear alcohols, apart from EtOH. All alcohols investigated affect the gating behaviour in a similar manner as EtOH does, increasing the energy barrier of the phase transition and lowering the overall nitrogen uptake (Fig. S3†). Thus, it is reasonable to hypothesize that characteristic surface deformations occur as a result of the reaction of the alcohol with the terminal groups on the surface.
Reactivation
Surface deformations are related to surface defects and associated surface barriers, which in turn influence MOF performance.13 The surface of the materials, in some cases, can be repaired by dissolving the surface barriers in the synthesis solvent.14 In our case, this healing method is not applicable since immersing of material in DMF would induce the cp to op phase transition.37 Therefore, thermal treatment under vacuum was applied for material reactivation, considering the possible removal of deposited species. The adsorption profile of some samples exposed to humid air could be successfully recovered, which is evident from the APHM after thermal treatment in vacuum (Fig. S4†).Meanwhile, samples treated with ethanol, ethanol/water mixture and PDMS could be only partially restored (Fig. S5†), pointing towards stronger interaction of these molecules with the framework, potentially incorporation, or irreversible surface deformation.
Scanning electron microscopy (SEM)
Solvothermal synthesis produces relatively large, rod-shaped crystals with an average length of 48 μm and width of 17 μm.There are no pronounced differences between samples before and after treatment, according to SEM images (Fig. 3, S7 and S8†).
 | ||
| Fig. 3 SEM images of (a) pristine DUT-8(Ni) sample and treated samples (b) 2_EtOH-6, (c) 3_EtOH/H2O, (d) 4_PDMS. | ||
The surface of the materials is smooth. Only sample 3, which was treated with a mixture of ethanol/water has a rough surface. This sample also shows the most pronounced APHM shift (Fig. 2f).
Surface-sensitive techniques are required to investigate how the surface is affected upon treatments. Therefore, X-ray photoelectron spectroscopy (XPS) and nanoFTIR were applied.
Nanoscale Fourier-transform infrared spectroscopy (nanoFTIR)
The application of near-field infrared nanospectroscopy for the investigation of framework stabilization and guest confinement phenomena is well-documented in the literature.47,48 A standardized protocol was employed to assess the impact of humidity level and exposure duration. Well-dispersed particles, heated for 2 h at 100 °C before measurements, were selected for scattering-type scanning near-field optical microscopy (s-SNOM) studies (Fig. S9†). The pristine DUT-8(Ni) in cp phase, samples exposed to humid air 1_H2O-28-336 and 1_H2O-55-24 (Fig. 2) were chosen for investigations. Additionally, DUT-8(Ni) was exposed to higher humidity of 70% and 95% during 24 h and 4 h, respectively (1_H2O-70-24, 1_H2O-95-4).Pristine DUT-8(Ni) microsized particles are characterized by sharp edges (Fig. 4a). Subsequent nanoFTIR measurements revealed a consistent peak pattern in the mid-infrared region, with prominent absorption bands centred around 769 cm−1 and 795 cm−1, along with a less intense band at 816 cm−1 (Fig. 4b) which matches closely with ATR-FTIR bulk scale spectral feature (Fig. S10†). These vibrational bands have been depicted in the 3D contour plot derived from near-field measurements at region-specific point scans along the line on the pristine DUT-8(Ni) sample. This observation is further confirmed by various point scans performed across pristine DUT-8(Ni) samples (Fig. S11 and S12†). These findings underscore the structural integrity and chemical composition of the DUT-8(Ni) microparticles as assessed through nanoscale characterization techniques. However, incremental exposure to humidity and the time of exposure considered unveils subsequent structural disintegration or deformation as revealed by point scan along a centred line on DUT-8(Ni) nanoparticles (Fig. 4c and d), and its consequence on anisotropic structural distortion can be observed through loss of spectral specific feature for all the measurement points except the points number 1, 4, and 5 (Fig. 4c and d). These shifts are neatly spotted in 3D contours (Fig. 4f), revealing anisotropic and inconsistent structural deformation. Similar deterioration of surface and loss of spectral characteristics were observed for DUT-8(Ni) after treatment with liquid water for 5 minutes (1_H2O-100-0.08), characterized by the appearance of a new peak at 781 cm−1 (Fig. S13a and b†).
 | ||
| Fig. 4 s-SNOM imaging of individual nano/micro particles of DUT-8(Ni) and exposed to humid air. (a) Optical amplitude image for DUT-8 (Ni) pristine microsized particles viewed in AFM height topography mode, (b) near-field mid-IR spectra of DUT-8(Ni) pristine particles obtained via nanoFTIR multipoint line scans measurements for point 1–15, (c) optical amplitude image for DUT-8(Ni) sample exposed to 70% relative humidity (1_H2O-70-24) viewed in AFM height topography mode, (d) near-field mid-IR spectra of DUT-8(Ni) nanoparticles exposed to 70% relative humidity (1_H2O-70-24) obtained via nanoFTIR multipoint line scans measurements for point 1–8, (e) nanoFTIR derived 3D contour plots obtained for DUT-8(Ni) microparticles, (f) nanoFTIR derived 3D contour plots obtained for samples 1_H2O-70-24. | ||
Exposure to moderate humidity (28% RH) for longer exposure time (2 weeks) also results in similar spectral changes but in an anisotropic manner, as revealed by nanoFTIR studies using multipoint scans along different directions (Fig. S14†). The measurements reveal partial or fractional disintegration of the framework, resulting in an overall loss of spectral feature for the bulk phase (Fig. S14a–c†). However, locally, in selected nano-regions of the particle, the retention of phase was observed as revealed by point scans 1–3 (Fig. S15c and d†).
3D topography view (Fig. S15a, b† and 5a) of sample 1_H2O-28-336 and optical phase image (Fig. S14a†) also reveals loss of characteristic morphological features, which were observed for pristine DUT-8(Ni). Humidity-induced morphological changes can be further seen in AFM 3D topography images shown in Fig. 5a, emphasizing that effective degradation and morphological loss are observed for samples 1_H2O-28-336 and 1_H2O-70-24. However, morphological features for sample 1_H2O-55-24 (Fig. 5a) and nanoFTIR spectral characteristics for 1_H2O_28_336 (Fig. 5b) are mainly retained for 1_H2O-55-24, indicating only high humidity (70% < RH) and long-term exposure (i.e. 2 weeks) are effectively introducing deformation in both; morphology and spectroscopic features whereas milder treatment does not have a significant impact on the structural signature (Fig. S16†). Observations obtained from scattering-type scanning near-field optical microscopy (s-SNOM) were further validated by far-infrared (terahertz) spectroscopic measurements conducted on the micrometre thick pellets (Fig. S17†) within a wavenumbers range of 150 to 650 cm−1 using a synchrotron beam. The pellets were exposed to controlled humidity to mimic the conditions deployed to the samples investigated by s-SNOM. The measurements provide valuable insights into the region of metal–linker (M–L) bond vibrations, which are responsible for structural deformation. Spectrum extracted from these samples is depicted in Fig. 5c, showing terahertz vibrational modes at α (209 cm−1), β (226 cm−1), γ (256 cm−1), δ (280 cm−1), ε (326 cm−1), σ (354 cm−1), and ρ (401 cm−1).
 | ||
| Fig. 5 AFM 3D topography, s-SNOM imaging and far-IR (terahertz) analysis of DUT-8(Ni) nano/micro particles under diverse humidity conditions: (a) AFM 3D topography for pristine DUT-8(Ni) sample, 1_H2O-28-336, 1_H2O-55-24, 1_H2O-70-24, (b) average of near-field mid-IR spectra of DUT-8(Ni) microparticles obtained via nanoFTIR multipoint line scans measurements: DUT-8(Ni), 1_H2O-28-336 ((a) parallel line scan, and (b) perpendicular line scan), 1_H2O-55-24, 1_H2O-70-24, (c) far-IR (terahertz) analysis of DUT-8(Ni) nano/micro particles under diverse humidity conditions. | ||
The experimentally obtained terahertz modes match closely with prominent modes observed in DFT calculated terahertz spectrum (Fig. S17 and S18†), corroborating α to Ni–O symmetric vibration, β to Ni–N stretching vibrations, γ to O–Ni–N dabco ring vibration, δ to Ni–O asymmetric bend, ε to aromatic dabco ring out of plane bending, besides strong bands observed at 474 cm−1 for aromatic ndc ring wagging and bending, and 488 cm−1 for dabco ring bend. Incremental humidity exposure of pristine DUT-8(Ni) results in loss of band at 488 cm−1 for dabco ring bend in 1_H2O-55-24 and 1_H2O-28-336, and finally, it disappears for 1_H2O-95-4 sample along with low energy shift for 474 cm−1 band assigned to aromatic ndc core.
Additionally, appearance of weak band at 194 cm−1 and strong band at 354 cm−1 suggests presence or relative prominence of O–H and Ni–O stretching as well as bending (i.e. σ: 354 cm−1), respectively (Fig. S17c and S18†).49,50 This hypothesis is further corroborated by incremental appearance of band ρ (at 401 cm−1) in sequence DUT-8(Ni) < 1_H2O-28-336 < 1_H2O-55-24 < 1_H2O-95-4, indicating influence of water molecules and highlighting higher amount of Ni–O linkages. Decrement in intensity of β band, indicative for Ni–N bonds, indicates either breakdown or deformation of M–L bonds between Ni2+ and dabco (Fig. 5c).
Ex situ X-ray photoelectron spectroscopy (XPS)
XPS is a quantitative spectroscopic technique that can identify the chemical state of elements and the overall electronic structure of the material.The full-range XPS spectra of DUT-8(Ni) samples show the C 1s, O 1s, N 1s, and Ni 2p peaks (Fig. S19–S22†). Specifically, the peak at 856.08 eV corresponds to the 5-fold coordinated Ni (Fig. S19†), binding to one N from dabco as well as four carboxylate oxygens from ndc2−.
The pristine material, as well as all DUT-8(Ni) samples after different treatments, feature a divalent Ni2+ state indicated by the Ni 2p3/2 peaks, which are slightly different in binding energies.
In the O 1s region, all materials show one peak (Fig. S20†), which corresponds to the C–O bonds. This peak reveals a slight increase in the binding energies for the treated samples (2_EtOH-6, 3_EtOH/H2O, 4_PDMS). This could be caused by hydrolysis or additional oxygen bonds. In 4_PDMS, the signal is broader as the peak consists of Si–O and C–O/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds. The Si 2p spectra are shown in Fig. S23,† which confirms the presence of PDMS on the surface.
O bonds. The Si 2p spectra are shown in Fig. S23,† which confirms the presence of PDMS on the surface.
In the N 1s region (N–C in dabco) we do not observe a significant change for the treated samples (Fig. S22†). Only 3_EtOH/H2O shows a slight shift to higher binding energies. Interestingly, this coincides with the most significant change in adsorption observed for this particular sample (Fig. 2e).
In situ XPS
Since ex situ XPS data revealed changes in the chemical analysis of the MOF surface after different treatments, in situ XPS investigations of the surface of DUT-8(Ni) upon contact with a reactive atmosphere were undertaken, expecting to reveal further information on the surface deformation process.51 The pristine cp sample was exposed to 10−6 mbar EtOH for 30 min, which was dosed via a mass flow controller. Quantitative analysis of the spectra obtained upon ethanol vapour treatment reveals subtle differences in the chemical composition of the MOF surface. Following peak integration (Fig. 7), the Ni:N ratio appeared unchanged, whilst the O:Ni and C:Ni ratios increased – signifying a possible deposition of ethanol onto the sample surface.
Peak fitting of the oxygen into two species – one representing the carboxylic component of the ndc2− linker and another representing C–O bonds (consistent with ethanol adsorbed onto Cu(111)).52 Some initial trace of C–O may be seen in the fresh sample, which may be attributed to adventitious carbon,53 though the content of this species increased markedly following ethanol dosing – and when Ni:O(X) (where X = a specific oxygen environment) ratios following treatment revealed an increase in C–O content (associated with ethanol) relative to nickel content (Fig. 7e and f), and a consistent ndc2−:Ni content – suggesting that the ethanol does not remove the ndc from the sample surface, but possibly forms a terminal layer through chemisorption on the Ni centres. This may also suggest that the outer surface of pristine DUT-8(Ni) is terminated by Ni2 paddle wheels, likely with open metal sites (i.e. missing dabco) to which the ethanol coordinates. Another possibility is that it replaces one coordinative bond to the linker (Fig. 6).
 | ||
| Fig. 6 The possible coordination environment of Ni on the surface after treatment with ethanol. C – gray, O – red, Ni – green, H – white. | ||
 | ||
| Fig. 7 (a and b) Ni 2p XPS spectra, (c) content of nickel photoemission envelopes, (d) Wagner plot, (e) O 1s XPS spectra, and (f) oxygen content of fresh sample DUT-8(Ni), and sample upon EtOH dosing DUT-8(Ni)@EtOH. | ||
In order to understand changes to the surface Ni centres; modelling of the Ni 2p region using a Gupta–Sen free ion multiplets was performed (Fig. 7a and b),54 and subsequent satellite peaks representing a screened cd10L2 final-state satellite (yellow, 861 eV)55 and an unscreened cd8 satellite (blue, 865 eV) implemented.56 Upon dosing of the ethanol vapour, we see a subtle change in the satellite structure of the Ni 2p, specifically in the cd8 screened state, which noticeably increases after ethanol dosing (Fig. 7c). Previously, changes to these satellites have been suggested to indicate changes to the local structure in the form of diminished or increased charge transfer from the neighbouring O 2p sites into vacant Ni d-orbitals.57 In this case, we suggest the apparent increase in this unscreened state indicates changes to charge transfer screening processes following ethanol adsorption. An alternative analysis of the Ni 2p spectra was investigated through the use of both the differential spectral function in CasaXPS, and PCA factor generation in order to isolate potential sub-oxide formation – though the resulting spectral envelopes indicated Ni 2p emissions at >857 eV (Fig. S24†). Ni 2p emissions are rarely observed at such high energies (outside of dihalides), and as such, this interpretation was discounted.
The samples were again treated ex situ for further analysis, after which the Ni L3M4,5M4,5 auger could be properly assessed. Analysis of the kinetic energies of the three major auger emissions Ni LMMa – L3M4,5M4,5, Ni LMMb,c (indistinguishable due to the signal:noise level) – L2M2,3M4,5 and L3M2,3M4,5, and Ni LMMd,e (indistinguishable due to the signal:noise level) – L2M2,3M2,3 and L3M2,3M2,3, reveal a shift to lower kinetic energy following ethanol adsorption for Ni LMMa,b,c, i.e. those directly involving d-orbital electrons. This indicates a change in the local d-electron environment following ethanol treatment. Construction of a Wagner plot (Fig. 7d) indicates a much greater degree of covalency in the ethanol-treated materials given the low position of the treated sample on the plot – again indicating that ethanol ligands may be donating electron density into vacant Ni d-orbitals (potentially leading to distorted octahedral coordination, as also evidenced by a broadening of the Ni 2p3/2 and O KLL photoemission peaks).58 Analysis of the carbon KLL auger may be used to reveal insights into the sp2/sp3 character of the carbon species in the sample.59 Comparing the differentiated C KLL (Fig. S25†) for the fresh and dosed materials, we observe a significant increase in the d-parameter of the C KLL auger, indicating that the proportion of sp2 carbon (ndc surface terminals) decreases with respect to sp3 environments (alcohol chains).
Nuclear magnetic resonance (NMR) studies
As discussed above, treatment with ethanol, ethanol/water mixture, or PDMS leads to certain surface modifications, changing the adsorption properties. In the case of treatment with EtOH or EtOH/H2O, ethanol seems to be chemisorbed, i.e., it reacts with DUT-8 since the material cannot be reactivated in contrast to the short H2O treatment. The interpretation of the XPS spectra also points towards the donation of electrons from EtOH into vacant Ni d-orbitals. This observation leads to a possible scenario where ethanol forms a specific nickel alcoholate at the surface, which in turn changes the coordination environment of Ni. At the same time, the ndc could still be coordinated as a monodentate ligand to Ni, or even be protonated to the free acid by accepting the protons from the alcohol (Fig. 6). Since H2ndc is hardly soluble in EtOH, we can also assume the potential presence of these linkers on the surface of the particles. 13C solid-state NMR investigations were performed to visualize such possible chemical modifications. To enhance the intensity of 13C cross-polarisation (CP) MAS NMR signals of interest, the carbon atoms of the carboxylic acid groups of ndc2− were selectively 13C-labeled following a previously reported procedure.60 Note that the 13C NMR signal of carboxylate carbons in pure, unmodified linkers is observed at ca. 173 ppm.6013C-enriched carboxylic acid was then used for the synthesis of flexible DUT-8(Ni) crystals, which were desolvated from DCM, to give the cp phase (13C_DUT-8(Ni)_cp) (Fig. S27c†).It is likely that the amount of ethanol incorporated into the surface is relatively low. Therefore, the 13C_DUT-8(Ni)_cp sample was treated with 13C-labelled EtOH, giving sample 2_13C–EtOH. In bulk aqueous solution, the corresponding 13C NMR signal is found at ca. 58 ppm.
That means, changes at the linker carboxylic acid group and ethanol derivatives will be detectable with ca. 90-fold magnification compared to the other carbon atoms in the linkers and the dabco unit, i.e., the experiment will be very selective for these two functionalities. A comparison of NMR spectra for the samples before (13C_DUT-8(Ni)) and after treatment with ethanol (2_13C–EtOH) is shown in Fig. 8. The isotropic 13C NMR chemical shifts at 229 ppm and in the range between 130 and 200 ppm in both spectra are assigned to the 13C-enriched carboxylate functionalities and aromatic carbons, respectively. These signals are relatively broad, which is characteristic for the closed pore state, as reported previously.60 Note that the linker carboxylate groups exhibit pronounced paramagnetic shift effects for both the open pore and closed pore states, as studied in detail previously.60–62
 | ||
| Fig. 8 Solid-state NMR spectra of pristine 13C_DUT-8(Ni) sample (blue) and sample treated with 13C-labelled ethanol (red). The signals marked with an asterisk are rotational sidebands of the signal at 240 ppm. | ||
Strikingly, three intense new signals at chemical shifts of 134, 128, and 47 ppm are observed after ethanol treatment in the 13C CP MAS NMR spectrum. For the signals at 134 and 128 ppm, two different assignments must be considered: (i) carbon atoms originating from the linkers. If so, the signals are likely due to modified carboxylates (13C-labelled) in a coordination mode different from that of the perfect paddle wheel.
Other, non-13C-enriched linker carbons are unlikely because this would require a rather high fraction of linkers to be modified or even released from the crystal structure. (ii) Ethanol derivatives with huge paramagnetic shifts due to neighbouring Ni species. The strong downfield shift of the carboxylate signals observed in comparison to that expected for the H2ndc (173 ppm) of free linker60 could be explained by a paramagnetic (pseudocontact) shift exerted by the Ni center in perturbed paddle wheel units. Since the exact coordination environment is unknown, the values of this downshift are not predictable.
To decide between these possibilities, 13C–1H HETCOR (heteronuclear correlation) spectra were measured. These spectra corroborate interpretation (i) by linkers in perturbed environments because the signal (c) at 128 ppm correlates with 1H species at ca. 6.1 ppm. This shift is rather indicative for aromatic protons from the linkers (Fig. S27e, ESI†). These observations clearly point to ndc carboxylate carbon atoms which are in the neighbourhood of aromatic protons.
In contrast, signal (b) does not have such a correlation. Its 13C chemical shift of 134 ppm is, however, close to signal (c) at 128 ppm. That means signal (b) may represent a linker in perturbed paddle wheel units, which is more mobile than the one giving rise to signal (c). However, strongly adsorbed ethanol on perturbed paddle wheels with a large paramagnetic shift would also be a plausible explanation which cannot be ruled out. Finally, line (d) at 47 ppm has a chemical shift close to the bulk value of EtOH and may thus represent strongly bound EtOH.
Electron paramagnetic resonance (EPR) spectroscopy
Due to its high sensitivity, NO is successfully used as a magnetic EPR probe to identify the local adsorption sites.63To monitor the changes, sensitive to the NO adsorption, the pristine sample, as well as the sample treated with EtOH were subjected to investigations.
Both samples show a broad signal at g = 2.19 at room temperature (Fig. S28†), which might be assigned to some Ni2+–NO species or low spin Ni+ or Ni3+ defects.64 Its large line width indicates that they are either ill-defined due to structural disorder or are in high local concentration. However, the relative amount of these species is about six times larger in the sample treated with ethanol than in the non-treated sample.
The assignment of the additional broad underlying signal between 10 mT and 500 mT with negative amplitude, which occurs in both spectra, might be assigned to some antiferromagnetically coupled nickel ion species.
Both samples show at T = 14 K the typical signal of NO binding axially to open metal sites of a Ni2+ defect64 in DUT-8(Ni) (Fig. S29†). Actually, the observed signal is a superposition of an axially symmetric species gxx,yy = 2.179; gzz = 2.388 and a second species with rhombic symmetry (gxx = 2.142; gyy = 2.223; gzz = 2.355)64 and verified by spectral simulations (Fig. S29†). The intensity of this signal is much higher than that of the signals observed at room temperature. This indicates an adsorption process of NO into the pores, as already reported for NO adsorption over DUT-8(Ni).64 Therefore, the majority of these species are presumably formed within the pores and not at the outer surface of the material. The intensity of this signal is roughly the same in both samples, so there is no indication that ethanol molecules block these sites. More interesting are the EPR signals from physisorbed NO molecules, which are not directly coordinating to the Ni(II) ions and are observed for both samples at magnetic fields above 330 mT (Fig. S30†).65
For the non-treated DUT-8(Ni), only a broad signal of such physisorbed NO is observed extending up to 600 mT. Such a broad signal feature indicates the interaction of NO with a large variety of very weak adsorption sites.65 However, in the sample treated by ethanol, at least one more NO signal with distinct resolved spectral features is present, indicating the adsorption of the nitric oxide molecule at a clearly defined stronger adsorption site. Based on these observations, it can be assumed that the surface deformation in the course of ethanol treatment leads to the formation of preferred sites for NO physisorption.
Raman spectroscopy
Vibrational spectroscopy allows to investigate the phase purity, stability, structural switchability, and host–guest interactions in MOFs.66–68 Therefore, Raman spectra were collected to investigate the influence of treatment procedures on the vibrational modes of DUT-8(Ni). The spectra of the sample exposed to humid air 1-H2O-55-24 and the sample treated with ethanol 2-EtOH-6 compared to the untreated DUT-8(Ni) are shown in Fig. S31.†We observe slight differences in spectra before and after treatment. An additional peak at around 600 cm−1 was detected in the ethanol-treated sample. The characteristic band at 501 cm−1 is detected in pristine material, which is overlapped with a new signal in sample 2_EtOH-6, giving a broad peak in the range of 471–501 cm−1.
Contact angle measurement
By means of XPS and SEM, it was shown that the treatment of samples with EtOH, EtOH/H2O mixtures, and PDMS leads to surface modification and/or deformation. Contact angle measurements were performed on the pelletized samples to study the changes in the hydrophobicity/hydrophilicity of the surfaces upon treatment.69Pellets of DUT-8(Ni) were treated in the same way as the powdered material, resulting in samples 2_pellet-EtOH and 4_pellet-PDMS (Scheme 1). To confirm the coherence with the powdered samples, nitrogen physisorption at 77 K was measured on the pelletized samples, showing that the pellets after treatment behave similarly in the adsorption (Fig. S33b†).
The probe fluids for contact angle measurements have to be selected in such a way that structural opening is prevented, limiting the choice. Aprotic polar solvents induce the phase transition from cp to op, and protic solvents (alcohols) modify the surface as discussed above; therefore, both had to be excluded.37 Since the surface of the DUT-8(Ni) pellet is hydrophobic, the probe fluids with surface tension of less than 50 mN m−1 are not suitable. Therefore, diiodomethane was chosen because of the inability to open the framework and the suitable surface tension value (50.8 mN m−1).
The static contact angle (CA) measured by diiodomethane droplets constitutes 23° for the pristine DUT-8(Ni) pellet (Fig. 9a, Table S1†). After ethanol treatment, the surface of sample 2_EtOH_pellet becomes more hydrophilic, leading to the spreading of the probe fluid across the surface, maximizing the contact, and therefore, the CA decreases to 9° (Fig. 9b). It points to the growing dispersive components on the surface since CH2I2 is predominantly sensitive to dispersive interactions.
 | ||
| Fig. 9 Contact angle measurements using diiodomethane droplets (a–c) and water droplets (d–f), (a and d) pristine pellet, (b and e) 2_pellet-EtOH, (c and f) 4_pellet-PDMS. | ||
The results point rather to the incorporation of ethanol as a competitor to the ndc ligand. The coordination to the open metal site, replacing dabco, should intuitively lead to a more hydrophobic surface.
After deposition of PDMS on DUT-8(Ni), CA expectedly increases up to 57° (Fig. 9c), confirming the successful hydrophobization of the surface (sample 4_pellet-PDMS).
The surface tension of water is higher than that of diiodomethane, which makes water a more suitable probe fluid. CA measurements performed by water droplets exhibit the same trend of changes as observed for diiodomethane (Table S1,†Fig. 9d–f). However, water-based results must be considered with care due to the instability of DUT-8(Ni) against water, as discussed above.
Surface energy determination by inverse gas chromatography
In addition to the contact angle measurements, the surface energy of the materials was examined by inverse gas chromatography (IGC). IGC measures the surface properties of materials by analyzing the interactions between a solid surface and various probe gases and provides valuable insights into the surface energetics,70 including information about surface free energy, non-polar and polar contributions to the surface energy, and distribution of surface free energy, just to name a few examples of this versatile technique.71–73The surface properties of DUT-8(Ni)_cp, as well as samples treated by EtOH, an EtOH/H2O mixture and PDMS, were probed by IGC at 303 K (see section analytical methods for details). The surface thermodynamic properties of the samples, especially the dispersive and acid–base thermodynamic surface free energies, were estimated using n-hexane, heptane, and octane as non-polar solvent probes and ethyl acetate and dichloromethane as polar probes. The total free surface energies of all samples are dominated by dispersive forces, contributing 1–10% to the total surface free energy (Fig. 10).
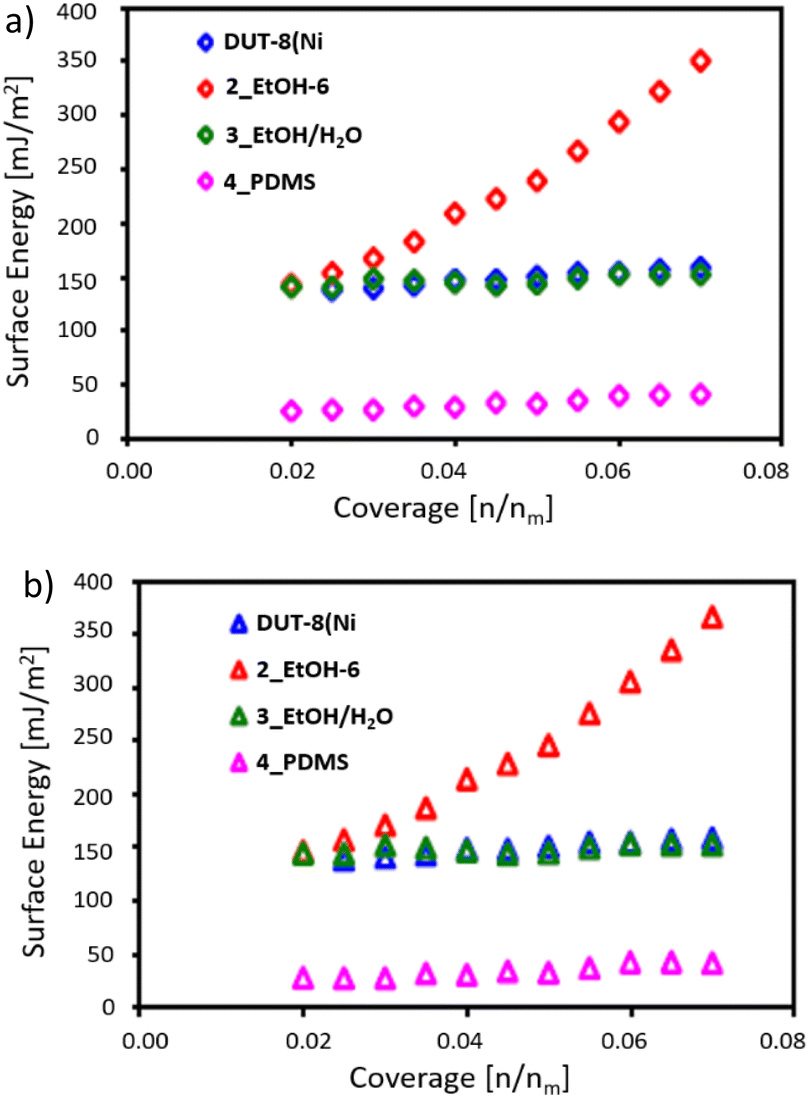 | ||
| Fig. 10 (a) Dispersive and (b) total surface free energy profiles of untreated DUT-8(Ni) (blue) and DUT-8(Ni) treated with EtOH (red), EtOH/water (green), and PDMS (pink). | ||
DUT-8(Ni) displays the lowest acid–base surface free energy, amounting to ∼1%, the samples treated by EtOH and EtOH/H2O mixture have higher acid–base surface free energies, amounting to ∼4% (at low surface coverage), and the acid–base surface free energy of the sample treated by PDMS equals to ∼2% (Fig. S34†).
The dispersive surface free energies of all samples are rather surface coverage insensitive, but the sample treated by EtOH displays an unexpected increase of the dispersive surface free energy with the surface coverage (Fig. 10).71,72
Therefore, we carefully analyzed the primary chromatograms (Fig. S35†). The chromatograms of hexane confirm changes in the surface properties of DUT-8(Ni) after treatment. The sample treated by EtOH displays significant tailing, indicating an additional interaction inside the sample bed, and the sample treated by EtOH/H2O mixture shows apparent fronting, which usually comes from a stronger probe–probe than probe–surface interaction.74 The hexane peaks are symmetric for the sample treated by PDMS, and the dispersive surface energy considerably decreases after PDMS deposition on the DUT-8(Ni) surface.
The obtained data are in agreement with contact angle measurements, revealing that sample 4_PDMS is hydrophobic (contact angle 57°, Fig. 9c), therefore, the surface energy is low (Fig. 10). In the case of the samples treated with ethanol 2_EtOH-6, the surface is hydrophilic with a low contact angle value of 9° (Fig. 9b). As a result, the surface energy of 2_EtOH-6 is increased in comparison with reference DUT-8(Ni) and 4_PDMS (Fig. 10).
Although a direct correlation between surface energy and adsorption energy is generally assumed,75,76 the gate opening pressure is shifted to the higher values in case of any treatment, obviously leading to the positive (EtOH) and negative (PDMS) contribution to the surface energy.
Discussion
Treatment of the flexible DUT-8(Ni) with substances able to react with or graft onto the surface (such as EtOH, water, and PDMS) leads to significant surface deformation and corresponding changes in the gating nitrogen adsorption profile. In general, the deformation enhances the activation barrier for molecules entering the pore system, leading to a shift in the gate-opening pressure to higher values. The structure and composition of this surface layer were investigated by various methods.The XPS reveals changes in the chemical composition of the surface upon treatment with EtOH, which most probably can be interpreted as the incorporation of the ethanol-like species. Since the nitrogen spectrum does not experience changes, according to the XPS, the results can be interpreted as coordination of the EtOH to the open metal site, created by dabco vacancy in the parent material, or the attachment to one coordination site, typically occupied by ndc. The NMR results, however, show new species, which are also preserved in the solution after the digestion of the MOF, pointing to chemical reaction and possible formation of nickel-alcoholate species formed upon deprotonation of alcohol.
In situ EPR data, involving NO as a probe molecule, indirectly confirms the incorporation of EtOH, creating an increasing number of defects and a new weak adsorption site for NO. The new adsorption site is also attractive for hexane, according to inverse gas chromatography. The contact angle measurement and inverse gas chromatography studies point on a more hydrophilic character of the surface after EtOH treatment and coverage increase in the surface energy.
Water treatment obviously leads to the physical adsorption of water on the MOF surface in the first stage, but prolonged exposure leads to irreversible transformation and hydrolysis, as is clearly visible from nanoFTIR investigations.
The deposition of PDMS on the surface of DUT-8(Ni) leads to the expected hydrophobization, which is deduced from the contact angle measurements, and PDMS lowers the surface energy.
However, despite the fact that more adsorption sites are created upon treatment, and the deformation can lead to positive or negative changes in the surface energy (according to contact angle measurements and inverse gas chromatography), the activation energy of the structural transition (associated with the shift in gate opening pressure to the higher values) increases in all the cases. This finding indicates that the newly formed activation barrier relies not on the adsorptive/adsorbate interactions of the outer surface but on the internal structure of the deformed layer, affecting mass transport and phase nucleation kinetics. Another aspect is that the deformed layer may hamper mass transport due to its limited porosity. Any deformation layer acts as a mechanical barrier, as phase nucleation has to tear apart this outer envelope to accomplish the volume expansion similar to a matrix effect.
Conclusion
Our findings provide significant insights into the dynamic behaviour of flexible metal–organic frameworks (MOFs), specifically in understanding and controlling their flexible behaviour. By demonstrating how surface constitution influences the kinetic barriers associated with structural transitions and gate-opening characteristics of DUT-8(Ni), our work highlights the importance of outer surface deformation originating from sample treatments (handling), which are often overlooked, particularly regarding the responsivity of flexible MOFs. Even macroscopic particles, several micrometres in diameter, are affected in their dynamics by external surface deformations. Severe alterations may occur when samples are exposed to humid air or solvents, obscuring reproducibility in data acquisition, especially if metadata are not well documented.The findings also open new avenues for tailoring MOFs' responsivity to molecular stimuli through deliberate surface engineering.
As nano- and mesoscopic particles are more sensitive to their environment, since surface deformation may alter a larger fraction of a crystallite, compared to the macroscopic counterpart, it may be valuable to reinvestigate shape memory effects observed mainly in nano/meso particles77,78 and the role of surface deformation in such systems more in detail in future. The broader implications of surface deformation effects extend to various fields, including gas storage, separation technologies, and sensing applications, where precise control over MOF response and porosity is critical.
Experimental
Analytical methods
Demodulation of the optical signal at higher harmonics of the tip resonance frequency eliminated background contributions to yield the near-field signal, comprising amplitude and phase of the scattered wave originating from the illuminated AFM tip. Employing a pseudo-heterodyne interferometric detection module, the complex optical response for designated point- and line scans on the single particle was determined at a ∼20 nm spatial resolution.79 The real part of the nanoFTIR spectrum denotes the reflectance, while the imaginary part (loss) represents the absorption spectrum.
Each integrated spectrum was acquired from an average of 14 Fourier-processed interferograms with a spectral resolution of 6.5 cm−1, 1024 points per interferogram, and an integration time of 20 ms per pixel. The sample spectrum was normalized to a reference spectrum measured on a silicon substrate region that did not contain any particles.
In situ XPS measurements were performed using a Kratos Axis ULTRA spectrometer fitted with a monochromated Al-Kα X-ray source (1486.68 eV, 700 × 300 μm analysis area). Charge compensation was achieved using a low-energy electron flood gun. The instrument was calibrated to gold metal (Au 4f, 83.95 eV) and dispersion adjusted to give a BE of 932.6 eV for the Cu 2p3/2 line of metallic copper. Ag 3d5/2 line FWHM at 10 eV pass energy was 0.544 eV. The source resolution for monochromatic Al Kα X-rays is ∼0.3 eV. The instrumental resolution was determined to be 0.35 eV at 10 eV pass energy using the Fermi edge of the valence band for metallic silver. Resolution with charge compensation system on <1.33 eV FWHM on PTFE. High-resolution spectra were obtained using a pass energy of 40 eV, step size of 0.1 eV and sweep time of 60 s, resulting in a line width of 0.696 eV for Au 4f7/2. Survey spectra were obtained using a pass energy of 160 eV. Heat and gas treatments were achieved using the Kratos high-pressure treatment chamber with vapor flows controlled using Brooks mass flow controllers, dosing a total of 1800 Langmuir before subsequent measurement.
The EPR experiments were performed with a Bruker EMX micro (X-band, 9.4 GHz) spectrometer equipped with an ER 4119 HS cylindrical cavity and an Oxford Instruments He cryostat ESR 900. Spectral simulations of the EPR spectra were performed using the MATLAB simulation package EasySpin.65
Chemicals
Ni(NO3)2·6H2O (99%), 2,6-H2ndc (99%), dabco (99%) were purchased from Sigma-Aldrich. All solvents were at least of analytical grade, purchased from commercial suppliers and used without further purification.Synthesis
DUT-8(Ni) samples were synthesized according to procedures reported earlier.30,83:Ni(NO3)2·6H2O (434 mg, 1.50 mmol), 2,6-H2ndc (294 mg, 1.36 mmol) and dabco (0.336 g, 3.00 mmol). A cloudy suspension was obtained after mixing, which was transferred to a Teflon-lined autoclave and placed into a preheated oven at 408 K for 72 h. After cooling, the product was isolated by decanting the mother liquor and washed with fresh DMF. The sample was washed by EtOH several times for TEM measurements.
The treatment under the humid air with humidity 55% was performed in the desiccator. The required humidity of the air was reached using an oversaturated water solution of Mg(NO3)2·6H2O. 232 g of Mg(NO3)2·6H2O was dissolved in water under heating until 310 K. After cooling to room temperature, the oversaturated solution was placed into the desiccator equipped with a moisture meter for humidity control. A vial containing DUT-8(Ni) powder was placed in the desiccator for 15 min, 60 min, 24 h and 2 weeks. In Scheme 1 the parameters of the treatment and corresponding names are given.
For IR measurements, humidity treatment was performed additionally by using a humidity chamber having regulated humidity measurement as a function of temperature and prepared with saturated NaCl solution in deionized water (concentration: 0.4 g mL−1) and monitored with a humidity sensor in a closed atmosphere, resulting 1_H2O-70-24 and 1_H2O-95-4.
:5 v/v) and dried in argon flow at 100 °C for 4 h with subsequent drying under dynamic vacuum, giving 3_EtOH/H2O. Analogously, the sample was soaked in 13C labelled EtOH, heated under Ar flow at 100 °C for 4 h, and evacuated for 16 h, giving 2_13C–EtOH.
Data availability
The physisorption data presented in the article have been uploaded in a digital *.aif format84 as part of the ESI.†Author contributions
I. S., S. K. – conceptualization, L. A., I. S., S. K. – formal analysis, I. S., S. K. – funding acquisition, L. A., M. M., C. B., N. B., M. R., M. I., D. M., J. S., R. B., A. S., M. M., K. P., E. O., S. K. Y. M.– investigation, I. S., S. K., – project administration, I. S., S. K., E. B., A. P., A. S., L. E., M. O., J. C. T. – validation, I. S., L. A., S. K. – writing – original draft, all – writing, review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge DFG (FOR 2433 MOF Switches, project no. 279409724, project no. 448809307) for financial support. The authors are grateful for the support from the Collaborative Research Center “Chemistry of Synthetic 2D Materials” funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) in the frame of SFB-1415 (project number 417590517). J. C. T. and Y. D. M. would like to acknowledge the ERC Consolidator Grant (PROMOFS grant agreement 771575) for funding. We also thank the Diamond Light Source for the award of beamtime SM30369, and for the technical support kindly offered by Dr Gianfelice Cinque during the far-IR measurements on Beamline B22 MIRIAM. Dr Tatiana E. Gorelik is acknowledged for the measurement of HR TEM and Nadine Bönisch for SEM.References
- J. J. de Pablo, N. E. Jackson, M. A. Webb, L.-Q. Chen, J. E. Moore, D. Morgan, R. Jacobs, T. Pollock, D. G. Schlom, E. S. Toberer, J. Analytis, I. Dabo, D. M. DeLongchamp, G. A. Fiete, G. M. Grason, G. Hautier, Y. Mo, K. Rajan, E. J. Reed, E. Rodriguez, V. Stevanovic, J. Suntivich, K. Thornton and J.-C. Zhao, npj Comput. Mater., 2019, 5, 41 CrossRef
.
-
N. Ali, M. Bilal, A. Khan, F. Ali, H. Khan, H. A. Khan and H. M. N. Iqbal, in Nanomaterials: Synthesis, Characterization, Hazards and Safety, ed. M. B. Tahir, M. Sagir and A. M. Asiri, Elsevier, 2021, pp. 55–95 Search PubMed
- U. Ulusoy, Minerals, 2023, 13, 91 CrossRef CAS
- G. A. Somorjai and Y. Li, Proc. Natl. Acad. Sci. U.S.A., 2011, 108, 917–924 CrossRef CAS PubMed
- T. Miyata, Y. K. Sato, Y. Kawagoe, K. Shirasu, H.-F. Wang, A. Kumagai, S. Kinoshita, M. Mizukami, K. Yoshida, H.-H. Huang, T. Okabe, K. Hagita, T. Mizoguchi and H. Jinnai, Nat. Commun., 2024, 15, 1898 CrossRef CAS PubMed
- J. Biener, A. Wittstock, T. F. Baumann, J. Weissmüller, M. Bäumer and A. V. Hamza, Surface Chemistry in Nanoscale Materials, Materials, 2009, 2(4), 2404–2428 CrossRef CAS
- R. Freund, O. Zaremba, G. Arnauts, R. Ameloot, G. Skorupskii, M. Dincă, A. Bavykina, J. Gascon, A. Ejsmont, J. Goscianska, M. Kalmutzki, U. Lächelt, E. Ploetz, C. S. Diercks and S. Wuttke, Angew. Chem., Int. Ed., 2021, 60, 23975–24001 CrossRef CAS PubMed
- S. Hiraide, Y. Sakanaka, H. Kajiro, S. Kawaguchi, M. T. Miyahara and H. Tanaka, Nat. Commun., 2020, 11, 3867 CrossRef CAS PubMed
- A. Ahmed, S. Seth, J. Purewal, A. G. Wong-Foy, M. Veenstra, A. J. Matzger and D. J. Siegel, Nat. Commun., 2019, 10, 1568 CrossRef PubMed
- H. Li, L. Li, R.-B. Lin, W. Zhou, Z. Zhang, S. Xiang and B. Chen, EnergyChem, 2019, 1, 100006 CrossRef PubMed
- D. Alezi, Y. Belmabkhout, M. Suyetin, P. M. Bhatt, Ł. J. Weseliński, V. Solovyeva, K. Adil, I. Spanopoulos, P. N. Trikalitis, A.-H. Emwas and M. Eddaoudi, J. Am. Chem. Soc., 2015, 137, 13308–13318 CrossRef CAS PubMed
- W. Su, Y. Xiang, Y. Dai, Y. Wang, S. Zhong and J. Li, Chem. Commun., 2024, 60, 7124–7135 RSC
- L. Heinke, Z. Gu and C. Wöll, Nat. Commun., 2014, 5, 4562 CrossRef CAS PubMed
- K. Müller, N. Vankova, L. Schöttner, T. Heine and L. Heinke, Chem. Sci., 2019, 10, 153–160 RSC
- S. Xu, K. Zheng, C.-R. Boruntea, D.-g. Cheng, F. Chen, G. Ye, X. Zhou and M.-O. Coppens, Chem. Soc. Rev., 2023, 52, 3991–4005 RSC
- M. Gao, H. Li, M. Yang, S. Gao, P. Wu, P. Tian, S. Xu, M. Ye and Z. Liu, Commun. Chem., 2019, 2, 43 CrossRef
- J. Kärger, ChemPhysChem, 2015, 16, 24–51 CrossRef PubMed
- C. V. McGuire and R. S. Forgan, Chem. Commun., 2015, 51, 5199–5217 RSC
- L. Figueroa-Quintero, D. Villalgordo-Hernández, J. J. Delgado-Marín, J. Narciso, V. K. Velisoju, P. Castaño, J. Gascón and E. V. Ramos-Fernández, Small Methods, 2023, 7, 2201413 CrossRef CAS PubMed
- A. Umemura, S. Diring, S. Furukawa, H. Uehara, T. Tsuruoka and S. Kitagawa, J. Am. Chem. Soc., 2011, 133, 15506–15513 CrossRef CAS PubMed
- M. Kondo, S. Furukawa, K. Hirai and S. Kitagawa, Angew. Chem., Int. Ed., 2010, 49, 5327–5330 CrossRef CAS PubMed
- B. V. K. J. Schmidt, Macromol. Rapid Commun., 2020, 41, 1900333 CrossRef CAS PubMed
- W. Zhang, Y. Hu, J. Ge, H.-L. Jiang and S.-H. Yu, J. Am. Chem. Soc., 2014, 136, 16978–16981 CrossRef CAS PubMed
- S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695–704 CrossRef CAS PubMed
- S. Krause, N. Hosono and S. Kitagawa, Angew. Chem., Int. Ed., 2020, 59, 15325–15341 CrossRef CAS PubMed
- I. Senkovska, V. Bon, L. Abylgazina, M. Mendt, J. Berger, G. Kieslich, P. Petkov, J. Luiz Fiorio, J.-O. Joswig, T. Heine, L. Schaper, C. Bachetzky, R. Schmid, R. A. Fischer, A. Pöppl, E. Brunner and S. Kaskel, Angew. Chem., Int. Ed., 2023, 62, e202218076 CrossRef CAS PubMed
- M. Kobalz, J. Lincke, K. Kobalz, O. Erhart, J. Bergmann, D. Lässig, M. Lange, J. Möllmer, R. Gläser, R. Staudt and H. Krautscheid, Inorg. Chem., 2016, 55, 3030–3039 CrossRef CAS PubMed
- H. Miura, V. Bon, I. Senkovska, S. Ehrling, N. Bonisch, G. Mader, S. Grunzner, A. Khadiev, D. Novikov, K. Maity, A. Richter and S. Kaskel, Adv. Mater., 2023, 35, e2207741 CrossRef PubMed
- S. Ehrling, H. Miura, I. Senkovska and S. Kaskel, Trends Chem., 2021, 3, 291–304 CrossRef CAS
- L. Abylgazina, I. Senkovska, R. Engemann, S. Ehrling, T. E. Gorelik, N. Kavoosi, U. Kaiser and S. Kaskel, Front. Chem., 2021, 9, 674566 CrossRef CAS PubMed
- L. Abylgazina, I. Senkovska, R. Engemann, N. Bönisch, T. E. Gorelik, C. Bachetzky, U. Kaiser, E. Brunner and S. Kaskel, Small, 2024, 20, 2307285 CrossRef CAS PubMed
- S. Hiraide, H. Arima, H. Tanaka and M. T. Miyahara, ACS Appl. Mater. Interfaces, 2021, 13, 30213–30223 CrossRef CAS PubMed
- J. Keupp and R. Schmid, Adv. Theory Simul., 2019, 2, 1900117 CrossRef CAS
-
A. Honciuc, in Chemistry of Functional Materials Surfaces and Interfaces, ed. A. Honciuc, Elsevier, 2021, pp. 1–3 Search PubMed
- N. Klein, H. C. Hoffmann, A. Cadiau, J. Getzschmann, M. R. Lohe, S. Paasch, T. Heydenreich, K. Adil, I. Senkovska, E. Brunner and S. Kaskel, J. Mater. Chem., 2012, 22, 10303–10312 RSC
- P. S. Petkov, V. Bon, C. L. Hobday, A. B. Kuc, P. Melix, S. Kaskel, T. Düren and T. Heine, Phys. Chem. Chem. Phys., 2019, 21, 674–680 RSC
- N. Kavoosi, T. Savchenko, I. Senkovska, M. Maliuta, V. Bon, A. Eychmüller and S. Kaskel, Microporous Mesoporous Mater., 2018, 271, 169–174 CrossRef CAS
- V. Bon, N. Klein, I. Senkovska, A. Heerwig, J. Getzschmann, D. Wallacher, I. Zizak, M. Brzhezinskaya, U. Mueller and S. Kaskel, Phys. Chem. Chem. Phys., 2015, 17, 17471–17479 RSC
- A. De, M. Maliuta, I. Senkovska and S. Kaskel, Langmuir, 2022, 38, 14073–14083 CrossRef CAS PubMed
- R. Numaguchi, H. Tanaka, S. Watanabe and M. T. Miyahara, J. Chem. Phys., 2013, 138, 054708 CrossRef PubMed
- A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062–6096 RSC
- M. J. Thompson, C. L. Hobday, I. Senkovska, V. Bon, S. Ehrling, M. Maliuta, S. Kaskel and T. Düren, J. Mater. Chem. A, 2020, 8, 22703–22711 RSC
- K. Tan, N. Nijem, P. Canepa, Q. Gong, J. Li, T. Thonhauser and Y. J. Chabal, Chem. Mater., 2012, 24, 3153–3167 CrossRef CAS
- S. Vudayagiri, M. D. Junker and A. L. Skov, Polym. J., 2013, 45, 871–878 CrossRef CAS
- U. Eduok, O. Faye and J. Szpunar, Prog. Org. Coat., 2017, 111, 124–163 CrossRef CAS
- L. Abylgazina, I. Senkovska and S. Kaskel, Commun. Mater., 2024, 5, 132 CrossRef
- A. F. Möslein, M. Gutiérrez, B. Cohen and J.-C. Tan, Nano Lett., 2020, 20, 7446–7454 CrossRef PubMed
- Y. D. More, S. Mollick, S. Saurabh, S. Fajal, M. Tricarico, S. Dutta, M. M. Shirolkar, W. Mandal, J.-C. Tan and S. K. Ghosh, Small, 2024, 20, 2302014 CrossRef CAS PubMed
- A. Krylov, A. Vtyurin, P. Petkov, I. Senkovska, M. Maliuta, V. Bon, T. Heine, S. Kaskel and E. Slyusareva, Phys. Chem. Chem. Phys., 2017, 19, 32099–32104 RSC
- K. Nama Manjunatha and S. Paul, Appl. Surf. Sci., 2015, 352, 10–15 CrossRef CAS
- M. Kjærvik, P. M. Dietrich, A. Thissen, J. Radnik, A. Nefedov, C. Natzeck, C. Wöll and W. E. S. Unger, J. Electron Spectrosc. Relat. Phenom., 2021, 247, 147042 CrossRef
- M. Bowker and R. J. Madix, Surf. Sci., 1982, 116, 549–572 CrossRef CAS
- L. H. Grey, H.-Y. Nie and M. C. Biesinger, Appl. Surf. Sci., 2024, 653, 159319 CrossRef CAS
- R. P. Gupta and S. K. Sen, Phys. Rev. B, 1975, 12, 15–19 CrossRef CAS
- D. Alders, F. C. Voogt, T. Hibma and G. A. Sawatzky, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 7716–7719 CrossRef CAS PubMed
- M. A. van Veenendaal and G. A. Sawatzky, Phys. Rev. Lett., 1993, 70, 2459–2462 CrossRef CAS PubMed
- H. Sun, Y. Zhang, C. Wang, M. A. Isaacs, A. I. Osman, Y. Wang, D. Rooney, Y. Wang, Z. Yan, C. M. A. Parlett, F. Wang and C. Wu, Chem. Eng. J., 2022, 437, 135394 CrossRef CAS
- M. C. Biesinger, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Phys. Chem. Chem. Phys., 2012, 14, 2434–2442 RSC
- D. J. Morgan, C, 2021, 7, 51 CAS
- M. Rauche, S. Ehrling, S. Krause, I. Senkovska, S. Kaskel and E. Brunner, Chem. Commun., 2019, 55, 9140–9143 RSC
- M. Rauche, S. Ehrling, L. Abylgazina, C. Bachetzky, I. Senkovska, S. Kaskel and E. Brunner, Solid State Nucl. Magn. Reson., 2022, 120, 101809 CrossRef CAS PubMed
- J. Blahut, A. L. Lejeune, S. Ehrling, I. Senkovska, S. Kaskel, F. M. Wisser and G. Pintacuda, Angew. Chem., Int. Ed., 2021, 60, 21778–21783 CrossRef CAS PubMed
- M. Chiesa and E. Giamello, Catal. Lett., 2021, 151, 3417–3436 CrossRef CAS
- M. Mendt, F. Gutt, N. Kavoosi, V. Bon, I. Senkovska, S. Kaskel and A. Pöppl, J. Phys. Chem. C, 2016, 120, 14246–14259 CrossRef CAS
- M. Mendt, B. Barth, M. Hartmann and A. Pöppl, J. Chem. Phys., 2017, 147, 224701 CrossRef PubMed
- K. I. Hadjiivanov, D. A. Panayotov, M. Y. Mihaylov, E. Z. Ivanova, K. K. Chakarova, S. M. Andonova and N. L. Drenchev, Chem. Rev., 2021, 121, 1286–1424 CrossRef CAS PubMed
- M. Todaro, A. Alessi, L. Sciortino, S. Agnello, M. Cannas, F. M. Gelardi and G. Buscarino, J. Spectrosc., 2016, 2016, 8074297 Search PubMed
- A. Krylov, I. Yushina, E. Slyusareva, S. Krylova, A. Vtyurin, S. Kaskel and I. Senkovska, Phys. Chem. Chem. Phys., 2022, 24, 3788–3798 RSC
- L.-H. Xie, M.-M. Xu, X.-M. Liu, M.-J. Zhao and J.-R. Li, Adv. Sci., 2020, 7, 1901758 CrossRef CAS PubMed
- S. Mohammadi-Jam and K. E. Waters, Adv. Colloid Interface Sci., 2014, 212, 21–44 CrossRef CAS PubMed
- K. Jayaramulu, F. Geyer, A. Schneemann, Š. Kment, M. Otyepka, R. Zboril, D. Vollmer and R. A. Fischer, Adv.
Mater., 2019, 31, 1900820 CrossRef PubMed
- H. Saini, E. Otyepková, A. Schneemann, R. Zbořil, M. Otyepka, R. A. Fischer and K. Jayaramulu, J. Mater. Chem. A, 2022, 10, 2751–2785 RSC
- F. Gholami, M. Tomas, Z. Gholami, S. Mirzaei and M. Vakili, Electrochem, 2020, 1, 367–387 CrossRef
- R. Ho and J. Y. Y. Heng, KONA Powder Part. J., 2013, 30, 164–180 CrossRef CAS
- L. Schimka, J. Harl, A. Stroppa, A. Grüneis, M. Marsman, F. Mittendorfer and G. Kresse, Nat. Mater., 2010, 9, 741–744 CrossRef CAS PubMed
- B. Li, X. Li, W. Gao and Q. Jiang, Acta Mater., 2021, 212, 116895 CrossRef CAS
- M. Shivanna, Q.-Y. Yang, A. Bajpai, S. Sen, N. Hosono, S. Kusaka, T. Pham, K. A. Forrest, B. Space, S. Kitagawa and M. J. Zaworotko, Sci. Adv., 2018, 4, eaaq1636 CrossRef PubMed
- Y. Sakata, S. Furukawa, M. Kondo, K. Hirai, N. Horike, Y. Takashima, H. Uehara, N. Louvain, M. Meilikhov, T. Tsuruoka, S. Isoda, W. Kosaka, O. Sakata and S. Kitagawa, Science, 2013, 339, 193–196 CrossRef CAS PubMed
- D. Richards, A. Zayats, F. Keilmann and R. Hillenbrand, Philos. Trans. R. Soc., A, 2004, 362, 787–805 CrossRef PubMed
- G. M. Dorris and D. G. Gray, J. Colloid Interface Sci., 1980, 77, 353–362 CrossRef CAS
- S. C. Das, I. Larson, D. A. Morton and P. J. Stewart, Langmuir, 2011, 27, 521–523 CrossRef CAS PubMed
- C. D. Volpe and S. Siboni, J. Colloid Interface Sci., 1997, 195, 121–136 CrossRef PubMed
- S. Ehrling, E. M. Reynolds, V. Bon, I. Senkovska, T. E. Gorelik, J. D. Evans, M. Rauche, M. Mendt, M. S. Weiss, A. Pöppl, E. Brunner, U. Kaiser, A. L. Goodwin and S. Kaskel, Nat. Chem., 2021, 13, 568–574 CrossRef CAS PubMed
- J. D. Evans, V. Bon, I. Senkovska and S. Kaskel, Langmuir, 2021, 37, 4222–4226 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Additional PXRD patterns and isotherms, HR TEM, SEM, SNOM, far-IR, ATR, nanoFTIR, XPS, Raman, 13C, EPR spectra, and chromatograms. See DOI: https://doi.org/10.1039/d4sc08223k |
| ‡ Current affiliation: Functional Polymer Interfaces Research Group, University of Bayreuth, Bavarian Polymer Institute – BPI, Universitätsstr. 30, 95447 Bayreuth, Germany. |
| This journal is © The Royal Society of Chemistry 2025 |