
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photoradiolabeling of onartuzumab with 99mTc and 188Re-tricarbonyl for radiotheranostics of gastric cancer†
Jonas
Genz
 a,
Cesare
Berton
a,
Samy
Kichou
a,
Simon
Klingler
a,
Mirja C.
Nolff
b,
Henrik
Braband
a and
Jason P.
Holland
*a
a,
Cesare
Berton
a,
Samy
Kichou
a,
Simon
Klingler
a,
Mirja C.
Nolff
b,
Henrik
Braband
a and
Jason P.
Holland
*a
aUniversity of Zurich, Department of Chemistry, Winterthurerstrasse 190, CH-8057, Zurich, Switzerland. E-mail: jason.holland@chem.uzh.ch; Tel: +41 44 63 53 990 Web: https://www.hollandlab.org
bKlinik für Kleintierchirurgie, Vetsuisse-Fakultät, University of Zurich, Winterthurerstrasse 260, CH-8057, Zurich, Switzerland
First published on 13th March 2025
Abstract
The clinically relevant nuclear isomer of technetium-99 (99mTc) and the radionuclides rhenium-186/188 (186Re and 188Re) represent an almost ideal match for the development of radiotracers for applications in diagnostic imaging and molecularly targeted radionuclide therapy. Although the chemistry of Tc and Re is similar, important differences arise in both the synthesis and properties of their complexes. Here, we report the synthesis and characterization of 99mTc- and 188Re-onartuzumab by labeling of the cancer-specific mAb onartuzumab (MetMAb) with the corresponding metal-tricarbonyl complexes derived from a novel photoactivatable ligand. The acyclic tris-amine ligand L1, featuring a photoactive aryl azide (ArN3) group, was synthesized from N1-(2-aminoethyl)ethane-1,2-diamine in 5 steps with an overall yield of 32%. Radiosynthesis of the [M(CO)3L1]+ (M = 99mTc or 188Re) photoactivatable complexes was accomplished via reduction of the [MVIIO4]− species to give the intermediate 99mTcI- and 188ReI-tricarbonyl-triaquo followed by ligand substitution with L1. The light-induced photoradiosynthesis of [M(CO)3L1-azepin]-onartuzumab (M-onartuzumab; M = 99mTc or 188Re) was achieved by irradiating the [M(CO)3L1]+ complexes in the presence of onartuzumab (formulated as MetMAb), with 395 nm light for 15 minutes at room temperature. Photoradiolabeling reactions produced M-onartuzumab radioimmunoconjugates in decay-corrected radiochemical yields of 20–30%, high radiochemical purities (RCP > 95%), and in molar activities of 1.026–4.146 MBq nmol−1. Cellular binding assays confirmed the specificity of radiotracer binding toward human hepatocyte growth-factor receptor (c-MET) expression on the surface of MNK-45 gastric adenocarcinoma cells. Subsequent planar γ-ray scintigraphy imaging and ex vivo biodistribution experiments in mouse models bearing subcutaneous MKN-45 xenografts revealed specific tumor targeting compared against competitive inhibition (blocking) controls performed at 24 hours (99mTc and 188Re) and 72 hours (188Re). Tumor uptake reached 20.20 ± 4.05 %ID g−1 for 99mTc-onartuzumab and 22.13 ± 3.11 %ID g−1 for 188Re-onartuzumab after 24 hours. Blocking experiments confirmed tumor specificity, with a reduction in tumor uptake of approximately 70% for both 99mTc-onartuzumab and 188Re-onartuzumab. Experimental data also revealed the biochemical equivalence of 99mTc-onartuzumab and 188Re-onartuzumab in terms of stability and pharmacokinetics in vivo. For 188Re-onartuzumab, activity was retained in the tumor for over 72 hours, with uptake levels at 20.21 ± 1.47 %ID g−1. Overall, the experiments demonstrated that photoradiosynthesis can be employed to develop a variety of rhenium based radioimmunoconjugates for future applications in tumor targeted radioimmunotherapy. Furthermore, these results underline the high potential of rhenium and technetium radioconjugates as theranostic platforms.
Introduction
Technetium-99m (99mTc) and rhenium-188 (188Re) are radionuclides of great interest in the field of nuclear medicine for applications in diagnostic imaging and molecularly targeted radiotherapy. Their decay properties, 99mTc (t1/2 = 6.01 h, γ-ray energy Eγ = 141 keV [intensity, Iγ = 89%]) and 188Re (t1/2 = 17.01 h, beta particle energy E(βmax) = 2.12 MeV and Eγ = 155 keV [Iγ = 15.5%]), make them ideal candidates in developing radiotracers for single-photon emission computed tomography (SPECT) and beta-radiotherapy, respectively.1–4The origins of 99mTc-tricarbonyl chemistry date back to the late 1990s when researchers sought to develop 99mTc complexes with well-defined and predictable coordination environments to improve the stability and versatility of 99mTc-based radiopharmaceuticals.5,6 The tricarbonyl core, {99mTc(CO)3}+, was first synthesized in 1995 by Alberto et al., by using a novel approach that allowed for the generation of a stable and easily functionalized [99mTc(H2O)3(CO)3]+ complex.7 This discovery was significant because the tricarbonyl moiety not only provided a stable framework for 99mTc coordination but also enabled the facile attachment of a wide range of ligands and biomolecules, thus expanding the utility of 99mTc in molecular imaging.8–10
Building on the success of 99mTc tricarbonyl chemistry, researchers turned their attention to 188Re (β-emitter), a generator radionuclide with radio-therapeutic potential.11–14 It is often stated that the chemical properties of Tc- and Re-based radiotracers are essentially identical but the differences in reactivity, and thermodynamic trends between these two elements and the corresponding complexes, often impinge on the successful adaptation of Tc-radiochemistry for applications with 186/188Re.15,16 A striking difference of the chemical properties is visible in the different kit formulations for 99mTc- and 186/188Re-tricarbonyl.17,18 The Isolink® kit by Mallinckrodt Medical B.V. contains besides Na2(H3BCO2), which is also present in kit formulation for 186/188Re, the additives sodium borate and sodium tartrate.19 For the Tc-radiochemistry the pH of the reaction is ∼12. In contrast, the radiosynthesis of {188Re(CO)3}+ is performed at pH 6.5–6.8 and contains neither sodium borate or sodium tartrate. BH3NH3 is added as an additional reducing agent and H3PO4 to adjust the pH of the kit. The reducing power of Na2(H3BCO2) is insufficient for the reduction of 186/188ReVII to 186/188ReI.14 Blower et al. observed that the formation of 186/188Re(V)-DMSA needs an increase from 23 °C to 100 °C and prolongation of the reaction time from 15 min for the 99mTc to 30 min for the 186/188Re compound while using the same kit formulation.11,20 Once isolated, the biological behavior of the 188ReV-DMSA compounds resembles 99mTcV-DMSA.21,22 Recently, Cardinale et al. performed a human trial with 99mTc/188Re-PSMA-GCK01.23 Differences in switching between 99mTc and 188Re radiochemistry were evident and include: (i) a change in radiolabeling reaction solution pH from pH8.0–8.5 (99mTc) to pH2.0–3.5 (188Re), (ii) a 4-fold increase in the ligand concentration for successful 188Re-radiolabeling, and (iii) longer reaction time (99mTc: 10 min vs.188Re: 60 min). Despite the different preparative aspects, the biological behavior of both compounds was comparable.
In the domain of radiolabeled proteins, the monoclonal antibody (mAb) rituximab has been directly radiolabeled by using the 99mTc- and 188Re-tricarbonyl cores with different approaches; the labeling experiment was attempted both on the native and on the reduced form of the mAb, yielding very different results.24 The {188Re(CO)3}+ core was less reactive on the native mAb (60% RCY at 24 h) than the corresponding 99mTc complex (>95% RCY at 3 h). However, once formed, challenge assays using cysteine and histidine as competing agents revealed that the 188Re-rituximab remained 90% intact and biochemically active after 24 h, whereas the 99mTc-rituximab adduct was unstable (<5% intact at 24 h) with most activity found unbound from the protein fraction.
These radiochemical, preclinical and clinical studies exemplify how the purported “similarities” in the chemistry of Tc and Re can, in reality, be dramatically different.
Regarding the labeling of mAbs, Ogawa et al. also reported direct labeling of an IgG1 murine mAb with the {186/188Re(CO)3}+ core.25 After heating for 2 h at 43 °C they obtained the radiolabeled mAb in RCYs from 23% to 28%. Another direct labeling approach for 99mTc and 186/188Re utilizes the disulfide bonds of mAbs which can be reduced to the free thiol and then labeled with the radiometals.26–28 Antibodies can also be labeled with 99mTc and 186/188Re by using a multistep radiosynthesis involving rhenium complexation by the mercapto acetyl triglycine (MAG3) ligand to give the [186/188ReO-(MAG3)]− complex bearing a free carboxylic acid.29–31 After solvent exchange to an organic solvent and purification, the free carboxylic acid group of the [186/188ReO-(MAG3)]− complex can be converted to an active ester. The activated ester was repurified, the solvent exchanged for a biocompatible buffer for the protein labeling and then the bioconjugation performed. Overall RCYs for this lengthy method showed a high variability.
In this work we aimed to adapt 99mTcI-tricarbonyl chemistry for applications with 188Re and combine this tricarbonyl core with our recent development of light-induced labeling of mAbs.32,33 Previously, we explored light-activated protein bioconjugation with redox-inert (under biocompatible conditions) metals ions such as Ga3+, Zr4+, Lu3+, and In3+ ions.33–37 The use of photochemistry for radiolabeling reactions with 89Zr, 11C, and 18F is under investigation.38,39 To adapt this chemistry for use with Tc and Re, a major challenge that was overcome here was to adapt the redox-sensitive chemistry of the ArN3 moiety with reductive complexation methods that are required for complexation of TcI and ReI metal ions. We also report detailed spectroscopic studies on the chemical speciation of the {natRe(CO)3}+ in aqueous conditions, resolving a longstanding mystery in Re-based complexation chemistry.
Results and discussion
188Re-tricarbonyl radiochemistry
The current study focused on creating and evaluating a radiotheranostic pair based on the congener elements 99mTc and 188Re. First, the [MI(H2O)3(CO)3]+ (M = 99mTc or 188Re) precursors were prepared by starting from the [MVIIO4]− permetallate oxoanions (Fig. 1) following procedures adapted from Alberto and co-workers.17,18 Na2(H3BCO3) acts as the CO source for both metals. Due to the higher redox stability of 188ReVII compared with 99mTcVII, different reducing agents are required.40–42 With Tc, the CO releasing molecule Na2(H3BCO3) also acts as the reductant, while for 188Re, the stronger reductant BH3NH3 is needed. The reduction potential of BH3NH3 in alkaline media is reported as −1.216 V vs. SHE.43 For Na2(H3BCO2), no reduction potential is reported but as it decomposes during the reaction to [B(OH)4]−, HCOO− (−0.2 V vs. SHE), CO (−0.11 V vs. SHE), and H2 (0 V vs. SHE),44 the redox potentials of these products can be used as a guide. The different synthetic conditions employed between Tc and Re in the preparation of {MI(CO)3}+ (M = 99mTc or 188Re) from [MVIIO4]− led to the formation of different solution phase species (Fig. 1A and B). For instance, 99mTc has been proven to form the tris-aquo complex [99mTc(H2O)3(CO)3]+ identified in the reverse-phase HPLC with a retention time of 6.26 min (Fig. 1B, green radiotrace) and confirmed by standard co-elution methods against the authenticated sample of [natRe(H2O)3(CO)3]+ (Fig. 1B, red trace). The standard reaction temperature and time for the 99mTc synthesis of heating at 90 °C for 30 min were adjusted to 110 °C for 10 min for a shorter reaction time with identical yields.18 [99mTc(H2O)3(CO)3]+ is a stable and versatile radiosynthetic intermediate used in the Isolink® kit for the preparation of many 99mTc-based radiotracers.2,45 In the case of 188Re (Fig. 1C), a mixture of two species was identified with retention times of 4.0 min (∼77%) and 5.5 min (∼23%).17 The minor peak eluting with a retention time of 5.5 min was previously identified as [188Re(H2O)3(CO)3]+ by co-injection with the natRe complex (Fig. 1C). On the other hand, the major compound found in the 188/natRe-tricarbonyl synthesis has not previously been assigned.17 One hypothesis was that the molecule is the NH3 coordinated complex with unknown stoichiometry of formula [188Re(NH3)x(H2O)3−x(CO)3]+ but definitive characterization has remained elusive.14 | ||
| Fig. 1 Chemical reactions for the preparation and chromatographic characterization of {M(CO)3}+ (M = 99mTc, 188Re or natRe) species. (A) Radiosynthesis of [99mTc(H2O)3(CO)3]+ and [188Re(HPO4)(H2O)2(CO)3]− (please see ESI Schemes S1 and S2 and Fig. S1 and S2 for further details†); (B) radio-HPLC chromatograms of [99mTcO4]− (black radiotrace), [99mTc(H2O)3(CO)3]+ (green radiotrace), and [natRe(H2O)3(CO)3]+ (red, electronic absorption trace 254 nm). (C) Equivalent radio-HPLC chromatograms obtained during the synthesis of the {natRe(CO)3}+ core showing the retention times of the parent [188ReO4]− (black radiotrace), the 188Re-reaction mixture (blue radiotrace), and the mixture formed by using [natRe(H2O)3(CO)3]+ (magenta electronic absorption trace 254 nm) placed under the experimental conditions used in the 188Re radiochemistry. Discrete peaks for [188Re(HPO4)(H2O)2(CO)3]− and [188Re(H2O)3(CO)3]+ were observed at 4.0 min and 5.5 min, respectively. | ||
A recent study by Williams et al. used gel-electrophoresis to show that the chemical properties of the carbonyl species depend on the buffer system used.46 The use of citrate and phosphate buffer led to a negatively charged complex while Tris buffer led to a positively charged one. Here, we assigned the species at 4.0 min (Fig. 1C) as [188/natRe(HPO4)(H2O)2(CO)3]− by using a combination of (radio-)HPLC, mass spectrometry, and 31P NMR analysis (ESI Fig. S2–S6†).
Our experimental data are consistent with the formation of a pH-dependent equilibrium between two identified nat/188Re species in aqueous conditions in the presence of HPO42− anions (Fig. 2A). The ratio between [188ReO4]− (10%), [188Re(HPO4)(H2O)2(CO)3]− (70%) and [188Re(H2O)3(CO)3]+ (20%) after radiosynthesis is shown in Fig. 2B (black radiotrace, pH 6.8), indicating that under conditions shown in Fig. 1A, [188Re(HPO4)(H2O)2(CO)3]− is the major species present in solution but that the reaction does not go to completion – consistent with previous observations.17,47,48 The composition of the mixture can be changed by acidification to pH1 (Fig. 2B, grey trace) whereby the percentage of [188ReO4]− increases from ∼10% to ∼17%, and the dominant species is now [188Re(H2O)3(CO)3]+ at 73%. The natRe complex [natRe(HPO4)(H2O)2(CO)3]− was prepared by addition of 10 equivalents of NaH2PO4 to [natRe(H2O)3(CO)3]+ followed by a pH adjustment to 6.5–6.8, and HPLC analysis using electronic absorption detection at 254 nm (Fig. 2B, light grey trace). Under these conditions, [natRe(HPO4)(H2O)2(CO)3]− was the dominant peak (retention time 3.6 min). The coordinating phosphate molecule in [natRe(HPO4)(H2O)2(CO)3]− is spontaneously eliminated by adjusting the pH of the reaction mixture to 1 (Fig. 2B, light blue trace), whereby only the peak assigned to [natRe(H2O)3(CO)3]+ was observed at a retention time of 5.5 min. After raising the pH back to 6.5–6.8 of an aliquots of [natRe(H2O)3(CO)3]+, the monophosphate coordination complex formed again with the chromatogram appearing identical to the original composition (Fig. 2B, light grey trace). It should also be noted that the radiosynthesis of the 188Re-tricarbonyl complex can be performed in the absence of H3PO4 by using HCl to adjust the pH of the reaction, which yields solely in the formation of the [188Re(H2O)3(CO)3]+ complex (ESI Scheme S3 and Fig. S7†). Analysis by high-resolution electrospray ionization mass spectrometry (HR-MS) in the negative mode provided further evidence whereby the mass of the [natRe(HPO4)(CO)3]− anion was detected as the sole phosphate coordinated species (ESI Fig. S3†). It is assumed that during the ionization step of the mass spectrometry analysis, two weakly bound aquo ligands can readily dissociate but in solution phase, the octahedral complex [natRe(HPO4)(H2O)2(CO)3]− is likely to persist.
 | ||
| Fig. 2 Experimental data on the aqueous-phase speciation of {nat/188Re(CO)3}+ showing (A) the conversion of [nat/188Re(HPO4)(H2O)2(CO)3]− into [nat/188Re(H2O)3(CO)3]+ by acidification, and the reverse transformation of [nat/188Re(H2O)3(CO)3]+ to [nat/188Re(HPO4)(H2O)2(CO)3]− by addition of phosphate and adjustment to pH 6.8 by using aq. NaOH. (B) Radio-HPLC chromatograms of [188Re(HPO4)(H2O)2(CO)3]− (*, 4.0 min) and the minor species [188Re(H2O)3(CO)3]+ (Δ, 5.5 min) at pH 6.8 (black trace), radio-HPLC chromatograms of [188Re(H2O)3(CO)3]+ (5.5 min) and the minor species in [188Re(HPO4)(H2O)2(CO)3]− (*, 4.0 min) at pH 1 in (dark blue trace), the corresponding electronic absorption (254 nm) HPLC chromatograms of natRe species placed under the radiosynthesis conditions at pH 6.8 (grey trace) showing the presence of both [natRe(HPO4)(H2O)2(CO)3]− (*, 3.6 min) and [natRe(H2O)3(CO)3]+ (Δ, 5.1 min), and the reversible formation of [natRe(H2O)3(CO)3]+ (Δ, 5.1 min) on acidification to pH 1 (light blue trace). | ||
31P-NMR analysis of the mixture produced with the {natRe(CO)3}+ core measured in the presence of phosphate (Fig. 2B, light gray trace) revealed a multitude of species with chemical shifts in the range of 6.28 to 2.41 ppm (ESI Fig. S4A†). Similar observations were made by Williams et al.46 The addition of 200 equivalents of phosphate at pH 6.5–6.8 to an aliquot of [natRe(HPO4)(H2O)2(CO)3]− led the solution phase mixture to converge towards one main species with a resonance peak at 5.05 ppm, and two minor ones with resonance peaks at 5.78 ppm and 3.71 ppm (ESI Fig. S4B†). By acidifying an aliquot of this mixture with aq. 1 M HCl (ESI Fig. S4C†) or measuring the 31P-NMR of an aliquot at 60 °C (ESI Fig. S4D†), the previously observed species vanished and only the resonance of free phosphate remained. The temperature dependent 31P-NMR suggests an exothermic binding of the phosphate ligands. By mixing the [natRe(H2O)3(CO)3]+ complex (Fig. 2B, light blue trace) with 200 equivalents NaH2PO4 at pH 4.4, two new resonances in the 31P-NMR spectrum at 3.92 and 3.60 ppm were visible, whereas by mixing with Na2HPO4 at pH9.6 two resonances remain, a sharp one at 4.70 ppm and a broad one at 2.41 ppm, in addition to free phosphate, were visible (ESI Fig. S4E and S4F,† respectively). The 31P-NMR data indicate a complex speciation pattern exists under the aqueous-phase conditions tested. It is not clear how the observations of a singular species in the (radio)-HPLC chromatogram can be matched with the observations in the NMR experiments but it is plausible that under the chromatographic conditions either one species predominates, or that the multiple Re-phosphate species observed in NMR co-elute.
Motivated by the new findings in the 31P-NMR spectroscopy, we investigated the phosphate binding reaction by using spin saturation transfer difference NMR (SSTD NMR) experiments – similar to the methods reported by Muñoz et al.49,50 First, we determined the binding equilibrium constant at 298 K (Keq = 1.41) measured by virtue of the slow NMR exchange regime at 202 MHz for 31P, then we measured the exchange kinetics between bound and free phosphate by using a dynamic experiment. In brief, the singlet of the bound phosphate at 5.83 ppm (ESI Fig. S5†) was saturated for a given amount of time and the peak of the free phosphate was detected immediately after presaturation (Fig. 3A). The time evolution of the area under the signal is indicative of the magnetization residing on the free phosphate, which is directly connected to the exchange rate constant between free and bound phosphate. Fitting of these data to a pseudo first-order rate equation and accounting for the concentration of the reactive partners gave an observed rate constant for phosphate exchange of kobs = 0.24 ± 0.02 M−1 s−1 (Fig. 3B).
 | ||
Fig. 3 Time evolution of the 31P-NMR spectra (202 MHz, H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :D2O, phosphate buffers) measuring the exchange rate of PO43− with presaturated [natRe(HPO4)(H2O)2(CO)3]−. (A) Difference spectra highlighting the change in the intensity of the free phosphate peak found at 0.0 ppm with respect to presaturation time. (B) Integral of the signals found in (A) plotted against the presaturation time of the bound phosphate (in seconds). The solid line represents the best fit to model given by ESI eqn (5).† :D2O, phosphate buffers) measuring the exchange rate of PO43− with presaturated [natRe(HPO4)(H2O)2(CO)3]−. (A) Difference spectra highlighting the change in the intensity of the free phosphate peak found at 0.0 ppm with respect to presaturation time. (B) Integral of the signals found in (A) plotted against the presaturation time of the bound phosphate (in seconds). The solid line represents the best fit to model given by ESI eqn (5).† | ||
The kobs parameter is a convolution between the forward (k1 = 0.133 ± 0.013 M−1 s−1) and reverse (k−1 = 0.107 ± 0.012 M−1 s−1) rate constants. These parameters are calculated from the law of mass action applied on Keq together with the definition of kobs (ESI eqn (6) and (7)†) and are comparable with previous works on ligand exchange kinetics on [natRe(H2O)3(CO)3]+ carried out by Alberto and co-workers, and summarized by Helm.51,52
Synthesis of a bifunctional photoactivatable ligand for {M(CO)3}+ (M = 99mTc, 188/natRe) complexation
The bifunctional ligand DETA-ArN3 (L1) was synthesized starting from N1-(2-aminoethyl)ethane-1,2-diamine (compound 1; Scheme 1). The synthesis is shown in detail in the ESI Schemes S4–S10 and analytics Fig. S11–S32.† The triamine-binding scaffold was chosen based on the research of Rattat et al. who compared various N, O, and S donor sets.53 First, compound 1 was Boc protected at the primary amine groups to yield compound 2. Michael-addition on the central nitrogen atom of compound 2 using methyl acrylate, followed by saponification of the methyl ester, gave compound 3 in >99% yield. Amide bond formation by using 4-azidobenzylamine (compound 9) and a HATU coupling introduced the photoactivatable aryl azide into our ligand scaffold yielding compound 4 in 83% yield. Removal of the Boc protecting groups with conc. HCl revealed the free primary amines, furnishing the desired tris-amine ligand L1 in 5 steps, with an overall yield of 32% and a purity over 99% as determined by HPLC and 1H-NMR spectroscopy. | ||
| Scheme 1 Synthesis scheme of the ligand L1 and the complex natRe-L1+ starting from diethylenetriamine 1. | ||
The non-radioactive reference complex [natRe(CO)3(DETA-ArN3)]+ (natRe-L1+) was synthesized by reacting the readily available starting material [natRe(CO)5Br] with ligand L1 in a 1:5 v/v mixture of MeOH and THF at 40 °C for 1 hour (Scheme 1 and ESI Scheme S11†). The identity of the natRe complex was confirmed by NMR, HPLC, and HR-MS (ESI Fig. S33–S37†). Radiolabeling L1 with [99mTc(H2O)3(CO)3]+ was performed with a ligand concentration of 0.3 mM at pH 12 (Fig. 4A) yielding the radiolabeled complex [99mTc(CO)3(DETA-ArN3)]+ (99mTc-L1+) in high radiochemical conversion (RCC, >99%) and radiochemical purity (RCP, >99%) as analyzed by radio-HPLC (Fig. 4B, black trace). For further details, see ESI Scheme S12 and Fig. S38.† Upon purification by a C8-cartridge which is described in detail in the ESI Scheme S12,† the complex 99mTc-L1+ was obtained in an RCP > 99% and RCY of 49%. The retention time of 99mTc-L1+ measured under the same conditions as the precursor, shifted to 14.55 min, indicating an increase in lipophilicity due to the ligand. Co-injection with the authenticated natRe-L1+ complex confirmed the identity of the radioactive species (Fig. 4B, grey trace).
 | ||
| Fig. 4 Radiosynthesis of the M-L1+ complexes (M = 99mTc, 188/natRe) showing (A) the synthetic procedures employed and (B) (radio-)HPLC chromatograms of 99mTc-L1+, 188Re-L1+, and natRe-L1+ (UV trace, 254 nm). | ||
Radiosynthesis of the corresponding 188Re-L1+ complex started from the mixture of [188Re(HPO4)(H2O)2(CO)3]− and [188Re(H2O)3(CO)3]+ produced at pH 6.8 (see ESI Scheme S13 and Fig. S39†) and used the ligand L1 at 10 mM concentration (Fig. 4A). The reaction yielded a single species, identified as the desired complex 188Re-L1+ (Fig. 4B, blue trace). Similar to the 99mTc labeled complex, the retention time of 188Re-L1+ shifted to 14.85 min. Identity of the radioactive complex was confirmed by co-injection with natRe-L1+ (Fig. 4B, grey trace). The shift in the retention times of the natRe and 188Re labelled complex is due to the serial arrangement of the UV- and radio-detector. Upon purification by a C8-cartridge eluted with a 1:4 v/v mixture of acetone/ethanol (ESI Fig. S40†), the 188Re-L1+ complex was obtained in an RCP > 99% and RCY of 92 ± 2% (n = 2). Here we note that the varying reactivities of the Tc and Re metal ions are evident leading to an increased ligand requirement, viz. 30-fold increase for the 5d metal compared with the 4d metal, to drive the 188Re-radiochemical conversion toward completion.
Radiochemical stability studies on the [99mTc/188Re-L1]+
The radiochemical stability with respect to ligand dissociation or complex degradation of purified samples of 99mTc-L1+ and 188Re-L1+, free of excess ligand (as proven by HPLC measurements, ESI Fig. S40†), were tested in the following two challenge conditions: (i) PBS at pH 7.4 at 37 °C for 24 h, and (ii) a mixture containing 1 mM cysteine and 1 mM histidine at 37 °C, pH 7.4 for 24 h (ESI Fig. S41 and S42†). Note, for 188Re the measurements were extended to 72 h. Cysteine and histidine were chosen as challengers since both offer a suitable donor set for the tricarbonyl core, are abundant in vivo, and may also be present in formulation buffers or as reactive amino acid residue on protein.54 Analysis by radio-HPLC of 99mTc-L1+ and 188Re-L1+ showed no oxidative/hydrolytic degradation in PBS as confirmed by the absence of [99mTcO4]− or [188ReO4]−. In addition, no evidence for ligand exchange was observed with histidine or cysteine indicating that both the 99mTc-L1+ and 188Re-L1+ complexes are of suitable stability for further use in the development of radiolabeled proteins.Electronic absorption (UV/vis) analysis and photodegradation studies
The electronic absorption characteristics and photodegradation kinetics of the ligand L1, the photoactive complex natRe-L1+, and the non-photoactive complex [natRe(CO)3(diethylenetriamine)]+ (used as a control; for the synthesis and characterization see ESI Scheme S14 and Fig. S43 and S44†) were analyzed by UV/vis spectroscopy (ESI Fig. S45†). The coordination of the {Re(CO)3}+ core to the ligand L1 induces a hyperchromic shift with an increase in molar absorption coefficient (ε/M−1 cm−1) at 255 nm from 7.6 × 103 to 10.8 × 103 M−1 cm−1. The molar absorption coefficients, determined by a dilution series (ESI Fig. S46 and S47†), for the ligand L1 and the natRe-L1+ complex were determined at 255, 283 and 290 nm and are 7.6 × 103, 1.8 × 103 and 1.4 × 103 M−1 cm−1 for the ligand and 10.8 × 103, 2.7 × 103 and 2.0 × 103 M−1 cm−1 for the complex (ESI Fig. S48 and S49†). The natRe-L1+ complex showed an overall hyperchromic shift between 200–350 nm compared with the electronic absorption spectrum of the non-photoactive complex [natRe(CO)3(diethylenetriamine)]+, indicating that the spectroscopic features in this wavelength range are likely associated with the ArN3 moiety.Subsequently, the rate constants of the photolysis at 395 nm of L1 and natRe-L1+ were determined by comparing the relative change of concentration of the starting reagent versus time (ESI Fig. S50†). The photolysis was done at a concentration of 0.1 mM, 25 °C and in water. Data obtained were fitted with a first-order exponential and the rate constant of the photodegradation at 395 nm was determined as k = 0.76 ± 015 min−1 for natRe-L1+ and 0.63 ± 0.15 min−1 for the ligand L1. The increase in photolysis rate constant observed for the natRe-L1+versusL1 (∼1.21 increase) is consistent with the difference in molar absorption coefficients observed on complexation of ReI ions (∼1.45 increase).
As metal-carbonyl bonds are possibly susceptible to photolysis under UV-light irradiation,55 we also investigated the photolytic stability of the nat/188Re-tricarbonyl core by exposing the model complex [nat/188Re(CO)3(diethylenetriamine)]+ (for the synthesis and characterization of the 188Re-labeled complex see: ESI Scheme S15 and Fig. S51†) to 395 nm light. No photodegradation was observed (ESI Fig. S52 and S53†).
Photoradiosynthesis of labeled proteins using 99mTc/188Re-L1+
The photochemical protein ligation using ArN3-based ligands was performed as previously reported in our group (Fig. 5).32 Thorough optimization of the photoradiolabeling conditions has been performed previously in our group.32 With this procedure, the ArN3 group is photoactivated and forms an electrophile which reacts chemoselectively with the abundant lysine residues that are available on the protein surface.56 The major byproduct of the photoreaction results from hydrolysis of the ketenimine ring to form the azepin-2-ol, which is removed efficiently from the radiolabeled antibody fraction by manual SEC.57 Extensive mechanistic data, theoretical calculations and data can be found in our previous publications.33,56 A photograph of the reaction setup is shown in ESI Photograph S1.† | ||
| Fig. 5 Chromatographic data on the photoradiosynthesis of 99mTc-onartuzumab (purple traces) and 188Re-onartuzumab (black traces) showing (A) iTLC chromatograms, (B) elution profiles recorded from manual elution of a PD-10 size-exclusion gel column, and (C) SEC-HPLC radiochromatograms for the purified samples of each radiotracer. Note ‘*’ indicates the elution of high molecular weight protein aggregates (dimers and higher order multimeric species). Equivalent chromatograms of the crude reaction mixtures are presented in ESI (Fig. S62–S64 and S65–S67†). | ||
The photoradiolabeling was first tested on the model protein human serum albumin (HSA) with 188Re-L1+ (ESI Scheme S16†). HSA (100 μL) with an initial protein concentration of 60 mg mL−1 in water was mixed with 188Re-L1+ (∼10 MBq) in 0.025 M borate buffer (pH 8–8.1, 250 μL) and irradiated with 395 nm light at 23 °C for 15 min. The crude reaction mixture was analyzed by instant thin-layer chromatography (iTLC) which showed a peak at Rf = 0 indicating protein bound activity (ESI Fig. S54†). Protein binding was further evidenced by analysis of the crude reaction mixture with manual size-exclusion PD-10 chromatography which gave a peak in the 0.0 to 1.6 mL fraction giving an RCC ∼ 65% (ESI Fig. S55†). To fully elute the byproducts which formed during the reaction an elution with 22 mL was necessary (ESI Fig. S56†). In addition, analysis by automated size-exclusion chromatography (SEC-HPLC) showed a signal that coincided with the retention time of HSA at about 15 min (ESI Fig. S57†). A dark control reaction of 188Re-L1+ with HSA which was analysed by PD-10 SEC showed no unspecific binding or non-light mediated binding (ESI Fig. S58†). As a further control, an analytical PD-10 SEC chromatogram was obtained for the complex and the photolysis products of 188Re-L1+ and for [99mTcO4]− and [188ReO4]− (ESI Fig. S59 and S60†). The tested small molecules elute from 3 mL upwards.
The isolated decay-corrected radiochemical yield (RCY) of 188Re-HSA was 63 ± 3% (n = 3) and the lower limit of the molar activity of the product (estimated by assuming no protein losses during the reaction and purification steps) was ∼0.073 MBq nmol−1 of protein, with an activity concentration of 4.109 MBq mL−1. The radiochemical purity of the purified samples of 188Re-HSA was estimated to be >99% (measured independently by PD-10 SEC and SEC-HPLC). The stability of 188Re-HSA with regard to loss of protein bound activity was evaluated in PBS at 37 °C for up to 72 h and was found to 71 ± 2% (n = 3) stable (ESI Fig. S61A†). Analysis of the degradation products in ESI Fig. S61B† showed various small molecules with shorter retention times than the parent 188Re-L1+ complex. The major photolytic byproduct observed gave a retention time of t = 13.80 min compared to the un-photolyzed 188Re-L1+, complex, t = 14.85 min. This major byproduct from the photolysed reaction is tentatively assigned to the hydrolysis of the ketenimine intermediate to give the azepin-2-ol derivative, consistent with previously observed data using related radiotracers featuring aryl azides as reactive handles.33,57 The procedure was then adapted for the labeling of the monovalent (one-armed) engineered monoclonal antibody onartuzumab.
Onartuzumab (formulated in the clinical-grade mixture as MetMAb, Genentech/Roche)58 was functionalized directly with the 99mTc/188Re-L1+ complexes by irradiation with 395 nm light for 15 min at room temperature and a pH 8.1 in the presence of 0.1 M sodium borate buffer (Scheme 2). The protein concentration in the separate reaction mixtures was 15 and 14 mg mL−1 for 99mTc-onartuzumab and 188Re-onartuzumab, respectively. Analysis by iTLC showed a peak at Rf = 0 for both 99mTc-onartuzumab and 188Re-onartuzumab indicating successful protein radiolabeling (Fig. 5A). Further chromatographic analysis of the crude photolabeling reactions confirmed protein-bound activity as evidenced by a peak in the manual size-exclusion PD-10 chromatograms in the 0.0 to 1.6 mL range (Fig. 5B), and by a signal in the SEC-HPLC radiotrace that coincided with the retention time of onartuzumab at about 14.1 min (RCC: ∼30% for 99mTc, ∼20% for 188Re). The stated fast reaction under mild conditions prevent aggregation (<5% for both nuclides, Fig. 5C marked with *).
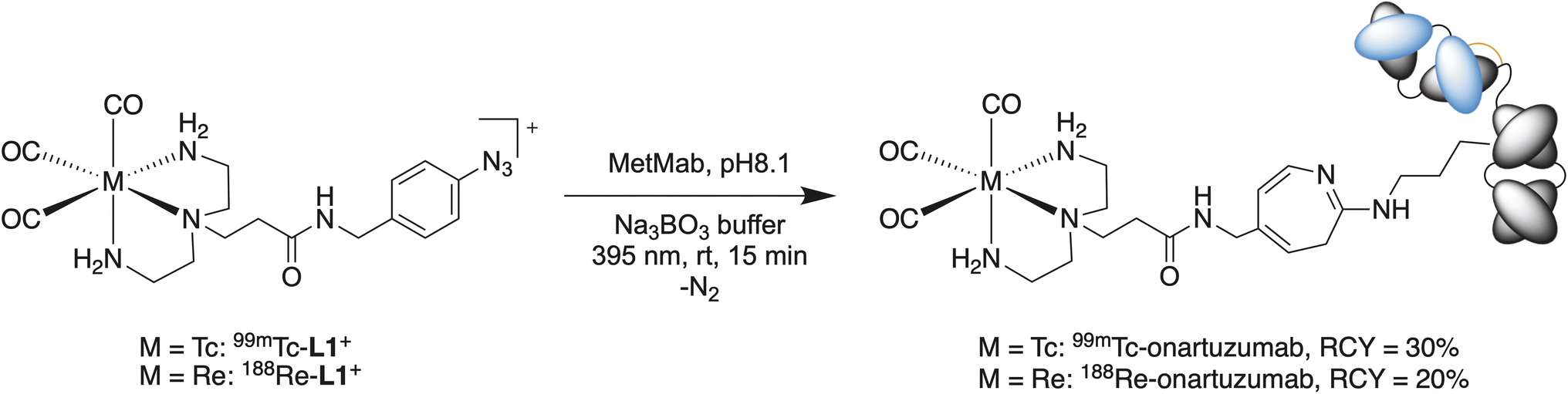 | ||
| Scheme 2 Photoradiosynthesis of 99mTc- and 188Re-onartuzumab from 99mTc- or 188Re-L1+, respectively. | ||
Purification of the radiolabeled antibody was achieved by passing the crude solution through PD-10 columns, eluting with sterile PBS and collecting the first 1.6 mL (high molecular weight). Aliquots of the crude and purified samples were retained and quality control was performed by iTLC, analytical PD-10 SEC and SEC-HPLC (Fig. 5A, B, and C, respectively). From these experiments, we observed that 99mTc-onartuzumab and 188Re-onartuzumab behave the same in these chromatographic separations. The photochemical bioconjugation gave higher yields for 99mTc compared with 188Re (30% and 20%, respectively). The lower limit of the molar activity of 99mTc- and 188Re-onartuzumab (estimated by assuming no protein losses during the reaction and purification steps) was ∼4.146 MBq nmol−1 and ∼1.026 MBq nmol−1 of protein, with an activity concentration of 77.74 MBq mL−1 and 11.54 MBq mL−1 for 99mTc-onartuzumab and 188Re-onartuzumab, respectively. Next, the biological behavior of the radioimmunoconjugates was evaluated in vitro, and in vivo by using xenograft models of human gastric adenocarcinoma with onartuzumab.
Radiochemical stability and cellular binding of 99mTc/188Re-onartuzumab
Purified samples of 99mTc-onartuzumab and 188Re-onartuzumab were incubated in PBS at pH 7.4 at 37 °C for up to 24 h and 72 h, respectively (ESI Fig. S68 and S69†). Both radiolabeled mAbs were found to be over 70% stable with respect to loss of the radionuclide from the protein fraction during the observation window. Analysis of the degradation products of 99mTc-onartuzumab by radio-HPLC, ESI Fig. S68B,† showed a small molecule with a shorter retention time of t = 13.68 min compared to the un-photolyzed parent complex 99mTc-L1+ which had a retention time of t = 14.55 min. As discussed above, we believe the most likely product formed after photolysis involved hydrolysis of the ketenimine electrophile to give the azepin-2-ol species which has been identified in previous studies on related aryl azide radiotracers.33,57The binding and specificity of 99mTc-onartuzumab and 188Re-onartuzumab to the target protein was evaluated in vitro by using intact cell binding assays with human hepatocyte growth-factor receptor (c-MET) positive and overexpressing MKN-45 gastric adenocarcinoma cells (Fig. 6). Cellular binding studies confirmed that both 99mTc-onartuzumab and 188Re-onartuzumab retained their biochemical activity and displayed specificity towards c-MET expression with 38 ± 1% (n = 4) and 35 ± 2% (n = 3) fractional binding (ESI Fig. S70 and S71†). The comparatively low values are associated with the monovalent structure of the onartuzumab and with the use of an aged ex-clinical sample of the protein, but are consistent with previous experimental studies.35,59 Blocking was possible by using a 100-fold excess of non-radioactive mAb, and the assay yielded a 2.5-fold and 5-fold decrease in binding for the 99mTc- and 188Re-onartuzumab immunoconjugates, respectively. These data confirmed the retention of bioactivity and specificity following radiolabeling of the protein and indicated that the radiotracers are suitable for further evaluation using animal models of human c-MET positive tumors.
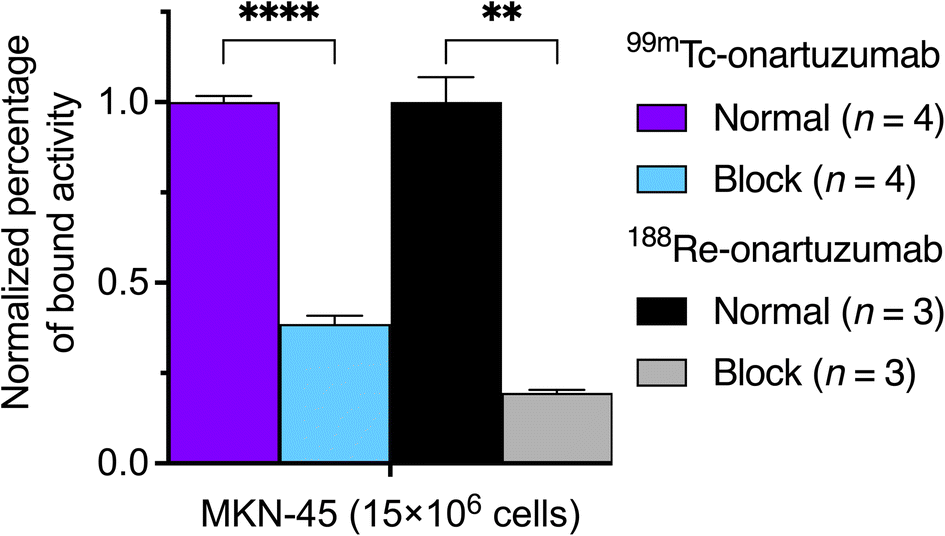 | ||
| Fig. 6 Comparison of the specific cell-bound fraction of 99mTc- and 188Re-onartuzumab on MKN-45 cells. | ||
Imaging and biodistribution studies of 99mTc- and 188Re-onartuzumab in animal models
Time-dependent γ-scintigraphy imaging was used to measure the distribution of 99mTc-onartuzumab and 188Re-onartuzumab in female athymic nude mice bearing subcutaneous MKN-45 xenografts on the right flank/shoulder. In addition, two groups of animals received a low molar activity formulation of the same batch of photoradiolabeled 99mTc-onartuzumab or 188Re-onartuzumab doped in each case with a 100-fold molar excess of non-radioactive MetMAb. These low molar activity formulations were used as control blocking groups to measure the specificity of tumor uptake in vivo.In the normal groups, the mice received an activity dose of 1.750–1.811 MBq for 99mTc for the 24 h time point, and activity doses of 0.556–0.562 MBq or 0.621–0.712 MBq for 188Re for the 24 h and 72 h time points, respectively. The normal doses contained a total protein mass of 50 μg in 150 μL of sterile PBS which was administered by intravenous (i.v.) tail-vein injection. For the blocking groups, the mice received an activity dose of 1.686–1.762 MBq for 99mTc for the 24 h time point, and activity doses of 0.571–0.629 MBq or 0.601–0.699 MBq for 188Re for the 24 h and 72 h time points, respectively. The block doses with reduced molar activity contained a total protein mass of ∼1.05 mg, also administered by i.v. tail-vein injection as a bolus injection in 150 μL of sterile PBS.
Planar γ-ray scintigraphy images were recorded 24 h post-administration (Fig. 7). Radiotracer localization in the tumor is evident for the normal groups for 99mTc- and 188Re-onartuzumab. The difference in contrast shown by these two-dimensional images is presumably due to the different geometry of the tumors (distance to the detector), low counting statistics for 188Re (Iγ = 15%) compared with 99mTc (Iγ = 89%), and does not represent a real difference as confirmed with the following biodistribution data for 99mTc- and 188Re-onartuzumab (vide infra). Nevertheless, tumor specificity was demonstrated by the reduction in signal intensity observed between the images recorded for animals assigned to the normal and blocking groups. The time progression of the distribution of the radioactive mAb between 0 and 24 h was acquired for one mouse belonging to each group (ESI Fig. S72 and S73†).
 | ||
| Fig. 7 Representative, planar γ-scintigraphy images recorded in athymic nude mice bearing MKN-45 tumors on the right flank at 24 h post-administration of the radiotracers for (left) 99mTc-onartuzumab (normal: 1.750–1.811 MBq, blocking: 1.686–1.762 MBq) and (right) 188Re-onartuzumab (normal: 0.556–0.562 MBq, blocking: 0.621–0.721 MBq). The mice received 50 μg or ∼1.05 mg of protein in the normal or blocking groups respectively. T = tumor. | ||
The first time point for biodistribution analysis was acquired at 24 h to facilitate a direct comparison between the distribution profiles of 99mTc-onartuzumab and 188Re-onartuzumab. In addition, a later time point of 72 h was used to study temporal changes in the tracer uptake/retention of the longer-lived 188Re-onartuzumab. In each case, normal and blocking groups were measured. At the specified time points, animals were euthanized after 24 h for the 99mTc and 188Re groups, and after 72 h for the 188Re group by exsanguination via cardiac puncture after isoflurane overdose and 15 tissues were harvested for activity quantification of both 99mTc and 188Re (Fig. 8 and ESI Fig. S74 and Tables S2 and S3†). A comparison between the normal groups for 99mTc/188Re at 24 h post-radiotracer administration showed no significant difference (two groups of n = 4, P = 0.7) in tumor uptake. These values were compared with the corresponding blocked groups at 24 h for 99mTc and 188Re showing a significant difference (P = 0.005 99mTc, 0.003 188Re) which confirmed the specificity of tumor targeting in vivo. The difference between tumor uptake between the 99mTc and 188Re blocked groups at 24 h was not significant (P = 0.9). Comparison between normal and blocked group for 188Re at the 72 h time point highlighted the retention of both tumor-associated activity and target specificity for 188Re-onartuzumab (P = 0.002 normal versus blocking groups).
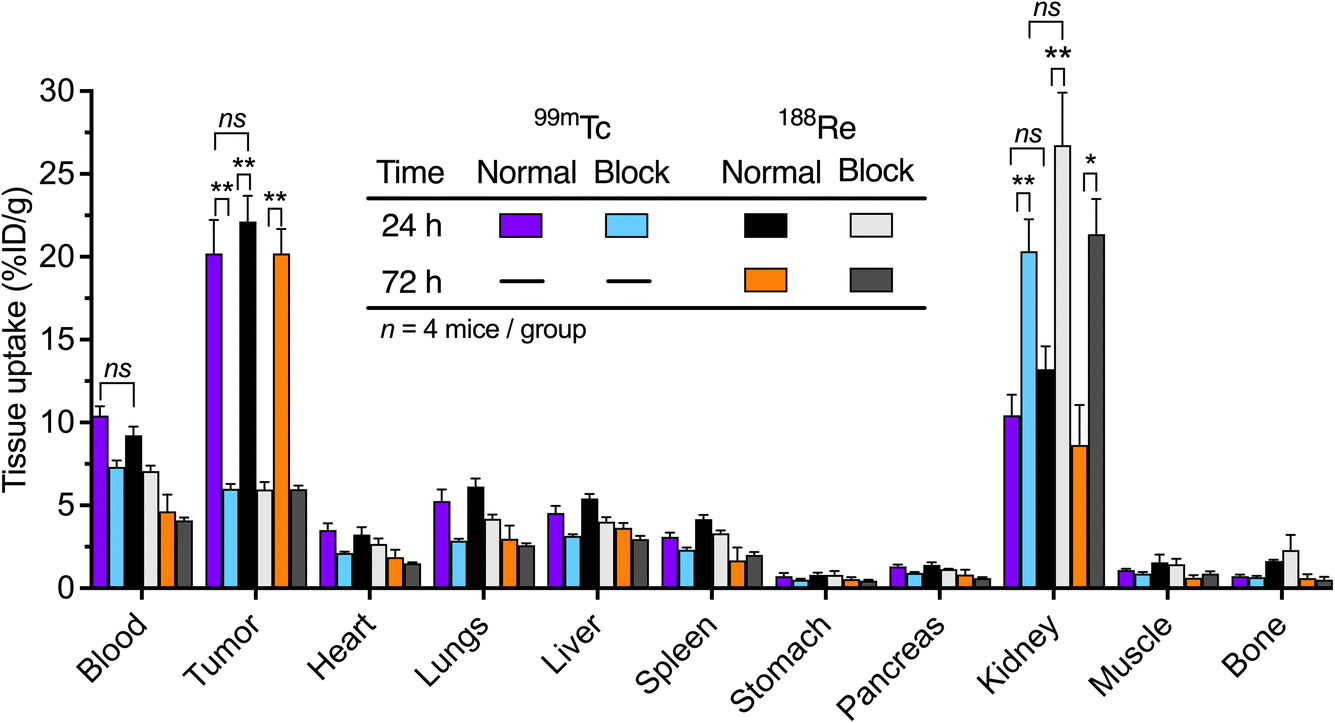 | ||
| Fig. 8 Bar chart showing ex vivo biodistribution data (% ID g−1) for the tissue uptake of 99mTc-onartuzumab (normal and blocking groups, purple and light-blue grey bars [24 h], respectively) and 188Re-onartuzumab (normal and blocking groups, black and white bars [24 h]; orange and dark grey bars [72 h], respectively) in mice bearing MKN-45 tumors at 24 h and 72 h after injection. ESI Fig. S74 and S75† provides additional information. Student's t-test: *P < 0.05, **P < 0.01, ns = not significant. | ||
No statistically significant differences were observed in the measured tumor-to-tissue contrast ratios at 24 h between 99mTc-onartuzumab and 188Re-onartuzumab for the normal groups (ESI Fig. S76†). Kidney uptake at 24 h for 99mTc/188Re, and at 72 h for 188Re, between the normal and blocking groups increased. This is a known dose-dependent phenomenon associated with the monovalent onartuzumab engineered antibody and has been observed with many different radionuclides, chelates, and conjugation strategies in the same MKN-45 xenograft models.32,35,59 Uptake in the liver and spleen was low (∼5%) for the normal and blocking groups at all time points, and for both radionuclides, indicative of the high chemical stability of the radiotracers in vivo and the low degree of protein aggregates formed by the photoradiolabeling methods used.
The effective and biological half-life modeled with a one-phase decay showed high similiarty for the normal and blocking groups of 99mTc-/188Re-onartuzumab (ESI Fig. S77 and S78†). The half-life measurements for 188Re-onartuzumab were also modeled with a two phase decay, corresponding to a faster wash-out phase within the first 22 h after administration, and a second slower phase observed from 17 h up to 72 h (ESI Fig. S77A and B†).
Overall, the biochemical data collected from cellular and animal experiments showed the biochemical equivalence of the photoradiolabeled 99mTc-onartuzumab and 188Re-onartuzumab radiotracers. These observations support the further development of 188Re-based radiolabeled mAbs for applications in radioimmunotherapy, and of the concurrent use of 99mTc-mAb radiotracers as diagnostic SPECT agents for establishing dosimetry profiles.
In this work, the differences in reactivity for 99mTc and 188Re are most striking in the synthesis of the tricarbonyl precursors as they yield different species, [99mTc(H2O)3(CO)3]+ and [188Re(HPO4)(H2O)2(CO)3]− for 99mTc and 188Re, respectively. Nevertheless, upon introduction of the tris-amine ligand L1, both metal complexes transform to the analogous complexes [M(CO)3L1]+ (M = 99mTc or 188Re). The different reactivities are again evident as a 30-fold increase in ligand concentration was necessary for 188Re complexation compared with 99mTc.
In summary, we conclude that only marginal differences were observed between the performance of the 99mTc- and 188Re-onartuzumab radiotracers in vivo. Provided that the radiometal ion complexes remain stable to challenge studies, and the synthetic methods used do not reduce the biological integrity of the protein, it appears that {M(CO)3}+ chemistry is suitable for developing antibody-based 99mTc- and 188/186Re-radioimmunoconjugates a theranostics pairs.
Conclusion
We introduce new methods for the effective complexation and photoradiosynthesis of 99mTc- and 188Re-proteins using the {M(CO)3}+ core. The identity of the previously unknown aqueous-phase tricarbonyl complex of 188Re was determined which elevates the understanding of the 188Re chemistry. The novel photoactivatable complexes 99mTc-L1+ and 188Re-L1+ were synthesized in two steps from [99mTcO4]− and [188ReO4]−. 99mTc- and 188Re-onartuzumab were produced via a light-induced photochemical process using the aryl azide (ArN3) chemistry. The light-activated bond formation with the protein marks an important step for the radiolabeling of complex biomolecules like mAbs, where previously, the harsh reaction conditions required for labeling with 188Re did not allow the use of redox-sensitive groups for bioconjugation such as maleimides, isothiocyanates, or activated esters.25,26,29,60 The photochemical protein ligation reaction allows a simple and efficient conjugation of the 99mTc/188Re-tricarbonyl complexes to the mAb starting from the fully formulated antibody mixture, which is practical for clinical use. The data presented provide encouraging evidence that support the further development of 99mTc/188Re-radiolabeled proteins as radiotheranostics.Materials and methods
Full details of all experimental methods, syntheses, characterization data, and equipment used are presented in the ESI.†Ethical statement
All experiments involving mice were conducted in accordance with an animal experimentation license approved by the Zurich Canton Veterinary Office, Switzerland (Jason P. Holland).Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
The study was conceptualized by JG, CSB, and JPH. JG conducted the majority of experiments including the synthesis, characterization, radiochemistry, cellular studies and animal experiments. CSB and JG generated the 31P NMR data. JG and SaK generated the UV/vis data. SiK and JPH assisted with the animal experiments. The first draft of the manuscript was written by JG. CSB and JPH contributed to the manuscript review, editing, and data presentation. All authors approved the final submission.Conflicts of interest
No potential conflicts of interest relevant to this article exist.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme/from the European Research Council under the Grant Agreement No. 101001734, ERC-CoG-2020, PhotoPHARMA. JPH also thanks the University of Zurich (UZH) for financial support. We thank all members of the Medicinal Radiochemistry group at UZH. Special thanks go to Prof. Dr Roger Alberto for providing the Na2H3BCO2 and for the fruitful discussions regarding group 7 carbonyl chemistry.References
- N. Lepareur, F. Lacœuille, C. Bouvry, F. Hindré, E. Garcion, M. Chérel, N. Noiret, E. Garin and F. F. Russ Knapp, Front. Med., 2019, 6, 451657 CrossRef PubMed.
- A. Duatti, Nucl. Med. Biol., 2021, 92, 202–216 CrossRef CAS PubMed.
- T. T. Pham, I. N. Hungnes, C. Rivas, J. Cleaver, G. Firth, P. J. Blower, J. Sosabowski, G. J. R. Cook, L. Livieratos, J. D. Young, P. G. Pringle and M. T. Ma, J. Nucl. Med., 2024, 65, 1087–1094 Search PubMed.
- R. Alberto, Inorg. Chem., 2023, 62, 20539–20548 CrossRef CAS PubMed.
- J. R. Dilworth and S. J. Parrott, Chem. Soc. Rev., 1998, 27, 43–55 Search PubMed.
- J. Cleynhens and A. Verbruggen, Nuclear Medicine and Molecular Imaging: Volume 1-4, 2022, vol. 1, pp. 79–94 Search PubMed.
- R. Alberto, R. Schibli, A. Egli, P. August Schubiger, W. A. Herrmann, G. Artus, U. Abram and T. A. Kaden, J. Organomet. Chem., 1995, 492, 217–224 CrossRef.
- H. W. Peindy N'Dongo, P. D. Raposinho, C. Fernandes, I. Santos, D. Can, P. Schmutz, B. Spingler and R. Alberto, Nucl. Med. Biol., 2010, 37, 255–264 CrossRef PubMed.
- M. Bartholomä, J. Valliant, K. P. Maresca, J. Babich and J. Zubieta, Chem. Commun., 2009, 5, 493–512 RSC.
- G. Makris, M. Kuchuk, F. Gallazzi, S. S. Jurisson, C. J. Smith and H. M. Hennkens, Nucl. Med. Biol., 2019, 71, 39–46 CrossRef CAS PubMed.
- J. Singh, K. Reghebi, C. R. Lazarus, S. E. M. Clarke, A. P. Callahan, F. F. Knapp Jr and P. J. Blower, Nucl. Med. Commun., 1993, 14, 197–203 CrossRef CAS PubMed.
- B. A. Rhodes, C. R. Lambert, M. J. Marek, F. F. Knapp and E. B. Harvey, Appl. Radiat. Isot., 1996, 47, 7–14 CrossRef CAS PubMed.
- J. Chen, M. F. Giblin, N. Wang, S. S. Jurisson and T. P. Quinn, Nucl. Med. Biol., 1999, 26, 687–693 CrossRef CAS PubMed.
- R. Schibli, R. Alberto, J. Petrig, K. Ortner, H. Schmalle, B. Sigrist, J. Stahel, R. Schwarzbach and P. A. Schubiger, J. Labelled Compd. Radiopharm., 2001, 44, S723–S725 Search PubMed.
- J. R. Ballinger, M. S. Cooper and S. J. Mather, Eur. J. Nucl. Med. Mol. Imaging, 2004, 31, 304–305 CrossRef PubMed.
- M. D. Bartholomä, A. S. Louie, J. F. Valliant and J. Zubieta, Chem. Rev., 2010, 110, 2903–2920 CrossRef.
- R. Schibli, R. Schwarzbach, R. Alberto, K. Ortner, H. Schmalle, C. Dumas, A. Egli and P. A. Schubiger, Bioconjugate Chem., 2002, 13, 750–756 CrossRef CAS PubMed.
- R. Alberto, K. Ortner, N. Wheatley, R. Schibli and A. P. Schubiger, J. Am. Chem. Soc., 2001, 123, 3135–3136 CrossRef CAS PubMed.
- R. Alberto, Eur. J. Nucl. Med. Mol. Imaging, 2003, 30, 1299–1302 CrossRef PubMed.
- N. Ramamoorthy, S. V. Shetye, P. M. Pandey, R. S. Mani, M. C. Patel, R. B. Patel, P. Ramanathan, B. A. Krishna and S. M. Sharma, Eur. J. Nucl. Med., 1987, 12, 623–628 CrossRef CAS PubMed.
- M. M. Bisunadan, P. J. Blower, S. E. M. Clarke, J. Singh and M. J. Went, Appl. Radiat. Isot., 1991, 42, 167–171 CrossRef CAS PubMed.
- K. Kothari, M. R. A. Pillai, P. R. Unni, H. H. Shimpi, O. P. D. Noronha and A. M. Samuel, Appl. Radiat. Isot., 1999, 51, 43–49 CrossRef CAS PubMed.
- J. Cardinale, F. L. Giesel, C. Wensky, H. G. Rathke, U. Haberkorn and C. Kratochwil, J. Nucl. Med., 2023, 64, 1069–1075 CrossRef CAS PubMed.
- C. R. Dias, S. Jeger, J. A. Osso, C. Müller, C. De Pasquale, A. Hohn, R. Waibel and R. Schibli, Nucl. Med. Biol., 2011, 38, 19–28 CrossRef CAS PubMed.
- K. Ogawa, H. Kawashima, S. Kinuya, K. Shiba, M. Onoguchi, H. Kimura, K. Hashimoto, A. Odani and H. Saji, Ann. Nucl. Med., 2009, 23, 843–848 CrossRef PubMed.
- G. L. Griffiths, D. M. Goldenberg, H. Diril and H. J. Hansen, Cancer, 1994, 73, 761–768 CrossRef CAS PubMed.
- E. Dadachova and S. Mirzadeh, Nucl. Med. Biol., 1997, 24, 605–608 CrossRef CAS PubMed.
- Z. Zhu, Y. Wu, Z. Zhang and Y. Liu, Radiochim. Acta, 1997, 79, 105–108 CrossRef CAS.
- J. L. Crudo, M. M. Edreira, E. R. Obenaus, M. Chinol, G. Paganelli and S. G. De Castiglia, Int. J. Pharm., 2002, 248, 173–182 CrossRef CAS PubMed.
- S. Kinuya, K. Yokoyama, K. Kobayashi, S. Motoishi, K. Onoma, N. Watanabe, N. Shuke, H. Bunko, T. Michigishi and N. Tonami, Ann. Nucl. Med., 2001, 15, 199–202 CrossRef CAS PubMed.
- G. W. M. Visser, M. Gerretsen, J. D. M. Herscheid, G. B. Snow and G. Van Dongen, J. Nucl. Med., 1993, 34, 1953–1963 CAS.
- A. Guillou, D. F. Earley, M. Patra and J. P. Holland, Nat. Protoc., 2020, 15, 3579–3594 CrossRef CAS PubMed.
- M. Patra, S. Klingler, L. S. Eichenberger and J. P. Holland, iScience, 2019, 13, 416–431 CrossRef CAS PubMed.
- R. Fay and J. P. Holland, Chem.–Eur. J., 2021, 27, 4893–4897 CrossRef CAS PubMed.
- R. Fay, M. Gut and J. P. Holland, Bioconjugate Chem., 2019, 30, 1814–1820 CrossRef CAS.
- M. Gut and J. P. Holland, Inorg. Chem., 2019, 58, 12302–12310 CrossRef CAS.
- L. S. Eichenberger, M. Patra and J. P. Holland, Chem. Commun., 2019, 55, 2257–2260 RSC.
- J. J. Zhang, L. Lou, R. Lv, J. Chen, Y. Li, G. Wu, L. Cai, S. H. Liang and Z. Chen, Chin. Chem. Lett., 2024, 35, 109342 CrossRef CAS.
- D. Lin, L. M. Lechermann, M. P. Huestis, J. Marik and J. B. I. Sap, Angew. Chem., Int. Ed., 2024, 63, e202317136 CrossRef CAS PubMed.
- C. D. Russell, Int. J. Appl. Radiat. Isot., 1982, 33, 883–889 CrossRef CAS.
- A. J. Bard and J. A. A. Ketelaar, J. Electrochem. Soc., 1974, 121, 212C CrossRef.
- M. Jovchev, H. Koch and H. Kupsch, Isotopenpraxis, 1975, 11, 369–378 Search PubMed.
- J. Yu, K. Lu, W. Lei and Q. Hao, J. Mater. Chem. A, 2022, 10, 16779–16794 RSC.
- G. Milazzo, S. Caroli and R. D. Braun, J. Electrochem. Soc., 1978, 125, 261C CrossRef.
- S. Robu, M. Schottelius, M. Eiber, T. Maurer, J. Gschwend, M. Schwaiger and H. J. Wester, J. Nucl. Med., 2017, 58, 235–242 Search PubMed.
- J. D. Williams, F. Kampmeier, A. Badar, K. Howland, M. S. Cooper, G. E. D. Mullen and P. J. Blower, Bioconjugate Chem., 2021, 32, 1242–1254 CrossRef CAS PubMed.
- J. Xia, Y. Wang, J. Yu, S. Li, L. Tang, M. Zheng, X. Liu, G. Li, D. Cheng, S. Liang and D. Yin, J. Radioanal. Nucl. Chem., 2008, 275, 325–330 CrossRef CAS.
- C. R. Dias, S. Jeger, J. A. Osso, C. Müller, C. De Pasquale, A. Hohn, R. Waibel and R. Schibli, Nucl. Med. Biol., 2011, 38, 19–28 CrossRef CAS PubMed.
- M. T. Quirós, J. Angulo and M. P. Muñoz, Chem. Commun., 2015, 51, 10222–10225 RSC.
- M. T. Quirós, C. Macdonald, J. Angulo and M. P. Muñoz, J. Visualized Exp., 2016, 117, e54499 Search PubMed.
- P. V. Grundler, B. Salignac, S. Cayemittes, R. Alberto and A. E. Merbach, Inorg. Chem., 2004, 43, 865–873 CrossRef CAS PubMed.
- L. Helm, Coord. Chem. Rev., 2008, 252, 2346–2361 Search PubMed.
- D. Rattat, K. Eraets, B. Cleynhens, H. Knight, H. Fonge and A. Verbruggen, Tetrahedron Lett., 2004, 45, 2531–2534 Search PubMed.
- D. K. Nayak, K. K. Halder, R. Baishya, T. Sen, P. Mitra and M. C. Debnath, Dalton Trans., 2013, 42, 13565–13575 Search PubMed.
- D. M. Rayner, Y. Ishikawa, C. E. Brown and P. A. Hackett, J. Chem. Phys., 1991, 94, 5471–5480 Search PubMed.
- D. F. Earley, A. Guillou, S. Klingler, R. Fay, M. Gut, F. d'Orchymont, S. Behmaneshfar, L. Reichert and J. P. Holland, JACS Au, 2022, 2, 646–664 CrossRef CAS PubMed.
- F. R. Bou-Hamdan, F. Lévesque, A. G. O'Brien and P. H. Seeberger, Beilstein J. Org. Chem., 2011, 7, 1124–1129 CrossRef CAS PubMed.
- M. Merchant, X. Ma, H. R. Maun, Z. Zheng, J. Peng, M. Romero, A. Huang, N. Y. Yang, M. Nishimura, J. Greve, L. Santell, Y. W. Zhang, Y. Su, D. W. Kaufman, K. L. Billeci, E. Mai, B. Moffat, A. Lim, E. T. Duenas, H. S. Phillips, H. Xiang, J. C. Young, G. F. Vande Woude, M. S. Dennis, D. E. Reilly, R. H. Schwall, M. A. Starovasnik, R. A. Lazarus and D. G. Yansura, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E2987 CrossRef CAS.
- A. Guillou, D. F. Earley and J. P. Holland, Chem.–Eur. J., 2020, 26, 7185–7189 CrossRef CAS PubMed.
- T. Uehara, M. Koike, H. Nakata, H. Hanaoka, Y. Iida, K. Hashimoto, H. Akizawa, K. Endo and Y. Arano, Bioconjugate Chem., 2007, 18, 190–198 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc08089k |
| This journal is © The Royal Society of Chemistry 2025 |